

Abstract
Autophagy is a critical cellular process required for maintaining cellular homeostasis in health and disease states, but the molecular mechanisms and impact of autophagy on cancer is not fully understood. Here, we found that Sox2, a key transcription factor in the regulation of the “stemness” of embryonic stem cells and induced-pluripotent stem cells, strongly induced autophagic phenomena, including intracellular vacuole formation and lysosomal activation in colon cancer cells. The activation occurred through Sox2-mediated ATG10 gene expression and resulted in the inhibition of cell proliferation and anchorage-independent colony growth ex vivo and tumor growth in vivo. Further, we found that Sox2-induced-autophagy enhanced cellular senescence by up-regulating tumor suppressors or senescence factors, including p16INK4a, p21 and phosphorylated p53 (Ser15). Notably, knockdown of ATG10 in Sox2-expressing colon cancer cells restored cancer cell properties. Taken together, our results demonstrated that regulation of autophagy mediated by Sox2 is a mechanism-driven novel strategy to treat human colon cancers.
Introduction
Cancer cells, but not normal cells, acquire immortalization by escaping cellular senescence [1]. Compared to normal cells, cancer cells usually require more nutrition to produce proteins and enzymes for their faster proliferation, which results in nutritional dependence of cancer cells [2]. Survival is supported by nutrients obtained through the blood supply and by self digestion of intracellular organelles in a process referred to as autophagy [3].
Autophagy is a conserved catabolic process in eukaryotes that degrades long-lived proteins, organelles, and bulk cytoplasm [4] through the lysosomal system to maintain homeostasis during starvation and normal growth control [3]. Autophagy promotes cell survival by purging the cells of damaged organelles, toxic metabolites, and intracellular pathogens and by generating the intracellular building blocks required to maintain vital functions during nutrient-limited conditions [5]. However, autophagy might also comprise an important strategy to treat cancer because autophagy can also promote cell death through excessive self-digestion and degradation of essential cellular constituents [5]. Autophagy is different from apoptosis, a process that lacks autophagosomes and autolysosomes in dying cells [6]. Notably, blockage of autophagy by knocking out beclin induces tumor development [7] and inducing autophagy by treatment with natural compounds, such as resveratrol, suppresses cancer cell proliferation and malignancy [2]. Autophagy is reportedly closely related with cellular senescence and inhibition of autophagy delays cellular senescence in mitotic human diploid cells [8]. These observations indicate that autophagy might induce senescence in cancer cells because inhibition of autophagy enhances tumorigenic properties of MCF-7 cells [9]. However, detailed mechanisms have not been clearly elucidated.
Furthermore, tamoxifen, an antagonist of the estrogen receptor in breast tissue, induces autophagy in human MCF-7 breast cancer cells [10] mediated through sphingolipid metabolites [11]. Notably, arsenic trioxide, imatinib (Gleevec) and rapamycin are compounds known to induce autophagic cell death in many human cancer cell lines [12], [13], [14]. Importantly, ovary, breast and prostate cancers are associated with monoallelic loss of beclin1 in humans. Furthermore, induction of autophagy by beclin overexpression suppresses anchorage-independent colony growth of MCF-7 cells [15] and proliferation by negative inhibition of p70S6 kinase [16], [17].
Both classical and modernized cellular reprogramming are stressful processes that activate apoptosis and cellular senescence, which are the two primary barriers to cancer development and somatic reprogramming [18]. The mTOR signaling pathway shares in the reprogramming of somatic cells, cellular senescence and autophagy formation [18]. For example, well-characterized mTOR inhibitors and autophagy activators, including PP242, rapamycin and resveratrol, notably improve the speed and efficiency of iPS cell generation [19]. Sox2, a transcription factor belonging to the HMG (High Mobility Group) superfamily, has a role in determination of cell fate, differentiation and proliferation [20]. It is also a key regulator of embryonic stem (ES) cell self-renewal and reprogramming of terminally differentiated cells to iPS cells [21]. Although the oncogenic potential of stem cell factors, such as Sox2, has been hypothesized to be based on the “stemness” similarity between stem cells and cancer cells, Sox2’s function in cancer cells is not clearly understood. Furthermore, recent studies have shown that the Sox2 protein level strongly corresponds with less differentiated basal cell-like breast carcinomas [22], and the Sox2 protein is frequently found to be down-regulated in gastric carcinomas [23]. These types of studies have caused more controversy regarding the normal function and oncogenic potential of stem cell factors. Here, we provide evidence showing that expression of Sox2 in cancer cells induces autophagy of cancer cells by up-regulating ATG10 gene expression and inducing cellular senescence, resulting in reduced malignancy of cancer cells and inhibition of tumor growth ex vivo and in vivo.
Results
Ectopic Expression of Sox2 Induces Autophagy
Recent studies indicated that ectopic expression of Sox2 by retroviral infection into MCF-7 breast cancer cells increased both the size and number of colonies formed in soft agar [24]. However, Sox2 is frequently down-regulated in gastric cancers and inhibits cell growth through cell cycle arrest and apoptosis [23]. Therefore, the role of Sox2 in cancer is controversial. To explore the role of Sox2 and other iPS factors in cancer, we ectopically expressed these factors in HCT116 human colorectal cancer cells and found that Sox2, but not Nanog, Lin28 or Oct4, induced severe vacuole formation in the cytoplasm, which is an important marker of macroautophagy [25] ( Fig. 1A ). We found that over 90% of infected cells formed different sized vacuoles in their cytoplasm and Western blotting and immunocytofluorescence assay results indicated that all the cells expressed the ectopic Sox2 protein ( Fig. 1B ). Further, we confirmed that severe vacuole formation coincided with acidic lysosomal activation in HCT116 colon cancer cells ( Fig. 1C ). Importantly, Sox2 overexpression induced LC3 (also known as ATG8b) foci formation, which is a key biomarker of autophagy ( Fig. 1D ). These results indicated that Sox2 overexpression induced autophagy.
Figure 1. Ectopic expression of Sox2 induces autophagy.

(A) Comparison of morphological changes induced by ectopic expression of iPS factors. (Upper panels) HCT116 cells were individually transduced with iPS factors, including Sox2, Nanog, Lin28 and Oct4. The cells were cultured for 5 days and changes were observed under a light microscope (X200). (Lower panels) HCT116 cells were harvested at 5 days after transduction with iPS factors and proteins extracted. The protein levels of the Sox2, Nanog, Lin28 and Oct4 were analyzed by Western blotting with specific antibodies as indicated. ß-Actin was used as an internal control to verify equal protein loading. (B) HCT116 cells forming vacuoles at 5 days after transduction were observed under light microscopy, counted and compared. Sox2 overexpression was analyzed by Western blotting and immunocytofluorescence assay (X200) using specific antibodies as indicated. ß-Actin was used as an internal control to verify equal protein loading. The cells were visualized by light microscopy (L.M.; X200). (C) Lysosomal activation analysis. HCT116 cells infected with mock or Sox2 were stained by adding lysotracker (50 nM) into the culture medium for 5 min in a 37 oC, 5% CO2 incubator. The cells were fixed with 4% formalin, washed with PBS and lysosomal activation was observed under a fluorescence microscope (X200). (D) Immunofluorescence assay of LC3b (i.e., ATG8b). HCT116 cells stably expressing mock or Sox2 were subjected to a fluorescence assay to detect LC3b. The cells were observed under a fluorescence microscope (X200). The nuclei were stained with DAPI; L.M. indicates light microscopy (X200).
Sox2 Induces Autophagy in Cancer Cells, but not in Normal Cells
To investigate whether Sox2 overexpression can induce vacuole formation in different colon cancer cell lines, we transduced lenti-Sox2 viral particles into CCD-18Co normal colon cells and HCT116, HT29 and WiDr human colon cancer cells. We found that all the colon cancer cell lines formed vacuoles in their cytoplasm ( Fig. 2A , arrows). However, although CCD8-18Co normal colon cells showed good expression of Sox2 after transduction with lenti-Sox2, the cells did not form vacuoles or display morphological changes ( Fig. 2A, B ). Further, additional results confirmed that vacuole formation and acidic lysosomal activation were observed in HCT116 colon cancer cells, but not in CCD-18Co normal colon cells ( Fig. 2C ). In addition, ectopic expression of Sox2 in normal mouse embryonic fibroblasts (MEFs) or human primary fibroblasts (NFDH and BJ) did not cause vacuole formation (data not shown), demonstrating that vacuole formation induced by Sox2 overexpression in HCT116 cells is indeed cancer cell-specific autophagy.
Figure 2. Cancer cell-specific lysosomal activation.

(A) CCD-18Co (CRL-1459) normal colon cells and HCT116, HT29 and WiDr colon cancer cells were cultured in the appropriate medium and transduced with Sox2 viral particles. The cells were cultured with complete growth medium for 5 days after transduction. The cells were observed under a light microscope. Cellular vacuoles are marked with an arrowhead in Sox2-expressing colon cancer cells. (B) CCD-18Co normal colon cells were cultured, transduced with mock or Sox2 viral particles and cultured with complete growth medium for 5 days. The cells were then fixed, permeabilized and hybridized with a Sox2 specific antibody and then with an Alexa 568-conjugated secondary antibody, and visualized with a fluorescence microscope (X200). Nuclei were stained with DAPI. The areas of light microscopy are the areas matched with Sox2 and DAPI fluorescence. The boxed area under low power is magnified to compare the morphology between HCT116-mock and -Sox2-expressing cells. (C) Lysosomal activation, a autophagy marker, was compared by adding lysotracker-Red (50 nM) at 5 days after transduction to CCD-18Co normal colon cells and HCT116 colorectal cancer cells infected with mock or Sox2. The cells were observed under a fluorescence microscope. L.M. indicates the same area of light microscopy corresponding to fluorescence microscopy (X200).
Sox2 Targets ATG10 to Induce Autophagy
To explore the mechanism(s) of Sox2-induced autophagy, we first used a microarray analysis of a total of 30,968 genes from cDNAs isolated from cells infected with mock or Sox2 to identify gene(s) targeted by Sox2 to induce autophagy. The results revealed 11,245 genes that were analyzed using the Significant Analysis of Microarray (SAM) program (http://www-stat.stanford.edu/~tibs/SAM). We found that 2,153 Sox2-induced genes could be classified as up-regulated and 1,575 genes were down-regulated in HCT116 cells ( Fig. 3A and Table S1). We used the Database for Annotation, Visualization and Integrated Discovery (DAVID v6.7; http://niaid.abcc.ncifcrf.gove) to further classify the genes according to their biological or molecular functions and found that gene expression levels associated with autophagy, proliferation and cell cycle regulation were substantially altered by the ectopic expression of Sox2. Altered gene expression included changes in 33 DNA repair genes, 25 DNA replication genes, 20 cell growth-related genes, 26 cell size-related genes, 191 transcription-related genes and 17 insulin signaling pathway-related genes ( Fig. 3A and Tables S1, S2 and S3). Importantly, Sox2 expression induced increased expression of ATG10, ATG3, ATG4a, ATG4D and ATG8b genes by about 2–4 fold ( Fig. 3A ). In contrast, Sox2 expression was associated with decreased expression of the ATG12, ATG16L2 and ATG9B genes ( Fig. 3A ). By searching a database containing the Sox2 consensus-binding motif in the promoter region in the genome [26], we found that the ATG10 and ATG12 promoters contain a putative Sox2 binding consensus nucleotide motif harboring 63% identity (Table S4). Comparing our microarray and the database search results, we concluded that ATG10 might be a target of Sox2 in the induction of autophagy. Our cycle-dependent RT-PCR results demonstrated that the ATG10 mRNA level from Sox2-transfected cells was increased by about 2.5 fold compared with mock ( Fig. 3B ). Notably, Western blotting results ( Fig. 3C ) and immunocytofluorescence against ATG10 ( Fig. 3D ) indicated that ATG10 and LC3 protein levels were increased by overexpression of Sox2. To determine whether the ATG10 promoter is activated by Sox2, we constructed two luciferase reporter plasmids containing −1522 to −1 (pGL3-ATG10-1522) and −711 to −1 (pGL3-ATG10-711) ( Fig. 3E ) by searching and analyzing the ATG10 promoter regions, which contain putative Sox and SRY binding motifs at −1327 and −1473 from the transcription initiation site ( Fig. 3E ). We found that that deletion of putative Sox2 binding region significantly suppressed luciferase activity ( Fig. 3F ). Notably, ATG10 promoter activity was increased by overexpression of Sox2 ( Fig. 3G ), indicating that Sox2 induces AT10 gene expression and causes autophagy.
Figure 3. Sox2 targets ATG10 to induce autophagy.
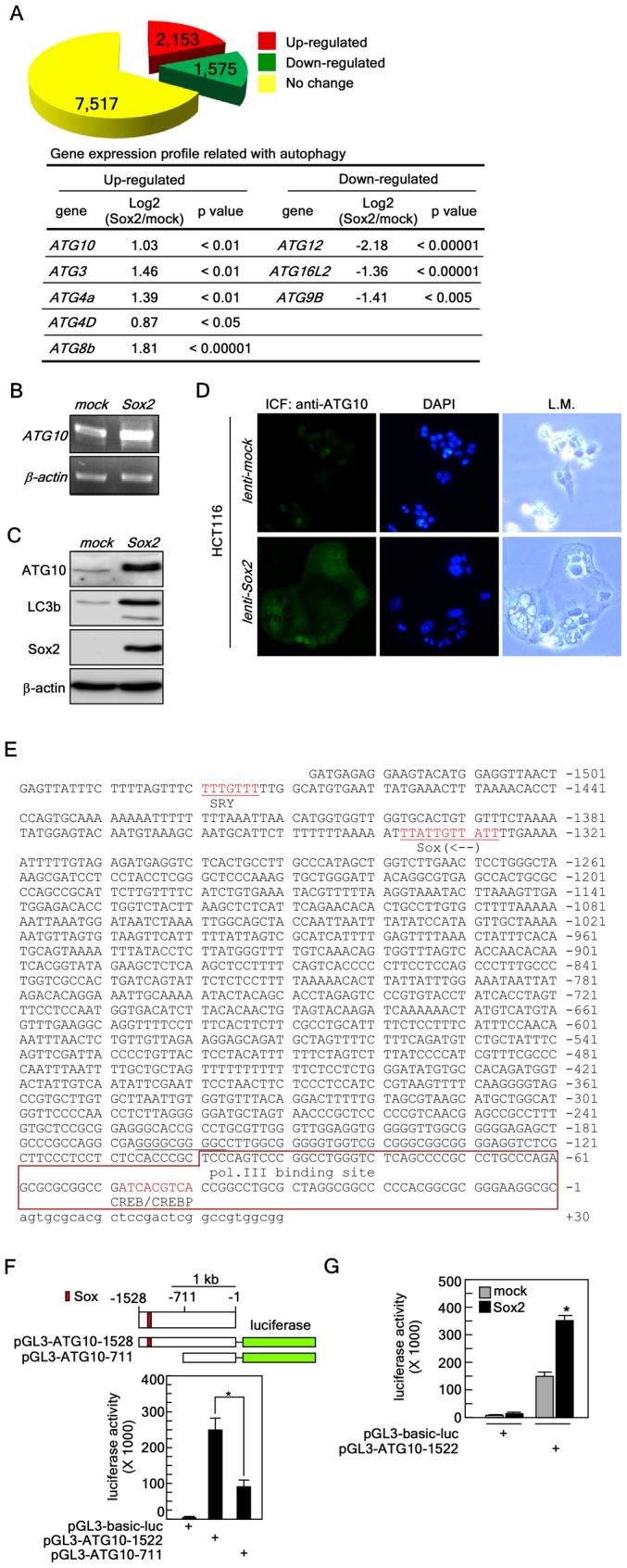
(A) Microarray. Top panel, HCT116 colorectal cancer cells stably expressing mock or Sox2 were subjected to microarray analysis as described in “Materials and Methods”. The yellow color indicates the number of genes that did not show a significant difference between mock and Sox2 expression. The red and green colors indicate the number of genes up- or down-regulated, respectively, in HCT116 cells stably expressing Sox2 compared with cells expressing the mock control. Bottom table, summary of autophagy-related genes obtained from the microarray analysis. Up- and down-regulation are denoted in Log2 values. (B) Expression of ATG10 induced by Sox2. Total RNA (1 µg) from HCT116 cells infected with mock or Sox2 was reverse transcribed and ATG10 was amplified by cycle-dependent PCR and the ATG10 expression level was visualized by agarose gel electrophoresis at the 21st cycle. ß-Actin was used as an internal control to verify equal utilization of cDNA for PCR. (C) Up-regulation of ATG10 and LC3b protein levels induced by Sox2 expression. Proteins were extracted from HCT116 cells stably expressing mock or Sox2 and ATG10 and LC3b proteins were visualized by Western blotting using specific antibodies. ß-Actin was used as an internal control to verify equal protein loading. (D) Confirmation of Sox2-induced ATG10 protein level. HCT116 colorectal cancer cells were infected with mock or Sox2 and cultured 5 days. The cells were fixed, hybridized with an ATG10 specific primary antibody and Alexa 488-conjugated secondary antibody. The ATG10 protein levels were observed by fluorescence microscopy. L.M. indicates the same area of light microscopy corresponding to fluorescence microscopy (X200). (E) Construction of the ATG10 promoter luciferase reporter plasmid. The nucleotide sequences of the human ATG10 promoter region were downloaded from Ensemble (http://uswest.ensemble.org). The putative promoter analysis was conducted using TFSEARCH (v1.3) (http://cbrc.jp/htbin/nph-tfsearch). The putative SRY and Sox binding consensus sequences are denoted in red and the polymerase III binding site is boxed. (F) The BAC clone (RP11-111B20) was purchased from Empire Genomics (Buffalo, NY) and 1528 and 711 bp of the ATG10 promoter region were amplified by PCR. The PCR fragment was recombined with the pGL3 basic vector to construct pGL3-AT10-1582 and pGL3-ATG10-711 luciferase reporter plasmids. The pGL3-ATG10-luc reporter plasmids were confirmed by DNA sequencing. The ATG10 promoter luciferase reporter plasmids, pGL3-AT10-1582 and pGL3-ATG10-71-luc, were transiently transfected into HCT116 cells and the firefly luciferase activity was analyzed after 24 h. The phRL-SV40 renilla luciferase reporter plasmid was co-transfected as an internal control to verify equal transfection and normalization of firefly luciferase activity (*p<0.001). (G) The pGL3-ATG10-1528 luciferase reporter plasmid was transiently co-transfected with a mock or Sox2 viral expression vector into HCT116 cells and the firefly luciferase activity was analyzed after 24 h. The phRL-SV40 renilla luciferase reporter plasmid was co-transfected as an internal control to verify equal transfection and normalization of firefly luciferase activity (*p<0.001).
Sox2-induced Autophagy is Mediated Through the Down-regulation of the Akt Signaling Pathway, but not Through Class III PI3-K Signaling
Fasting or depletion of growth factors such as insulin induces autophagy [27]. Our microarray results demonstrated that gene expression associated with the insulin signaling pathway was suppressed (Table S3). Therefore, we examined changes in protein levels associated with autophagy and insulin signaling by Western blotting. We found that Class III PI3-K (Vps34p) and beclin were not changed, indicating that the Class III PI3-K/beclin signaling pathway is not involved in Sox2-induced autophagy ( Fig. 4A ). By investigating the PI3-K-Akt signaling pathway, we found that Akt phosphorylation (Ser308) was substantially inhibited and the total protein levels of Akt, mTOR and p70S6K were decreased in Sox2-infected cells compared with mock-infected cells ( Fig. 4B ). To confirm signaling related to the inhibition of the PI3-K-Akt signaling pathway, we analyzed GSK3α/ß and PTEN. The results indicated that phosphorylation of GSK3α/ß at Ser9/Ser21 was suppressed and total GSK3α/ß protein levels were slightly increased in Sox2-infected cells compared with mock-infected cells ( Fig. 4C ). Notably, we found that PTEN phosphorylation was also increased ( Fig. 4C ), resulting in suppression of PI3-K signaling by de-phosphorylation [28]. These results indicated that Sox2 alters the PI3-K-Akt signaling pathway through GSK3α/ß and PTEN signaling, although the upstream specific kinase responsible for phosphorylation of PTEN is not known in this instance. We treated HCT116 cells expressing Sox2 or mock with insulin or LY294002, a PI3-K inhibitor. Results indicated that Sox2-induced vacuole formation was markedly inhibited by insulin treatment ( Fig. 4D left panel s, E). However, the mock-infected cells did not show vacuole formation whether or not treated with insulin in normal cell culture condition (10% FBS) or suppressed vacuole formation in starvation condition (0% FBS) ( Fig. 4D left and middle panels, E, F). We also found that mock-infected cells exhibited increased vacuole formation after treatment with LY294002, whereas Sox2 overexpression caused more vacuole formation induced by LY294002 treatment compared with mock ( Fig. 4D right panel s, E, F). Similar phenomena were observed under starvation conditions ( Fig. 4E, F ). Taken together, these results indicated that the down-regulation of the PI3-K-Akt pathway by PTEN might cooperate to induce autophagy associated with Sox2 expression.
Figure 4. Sox2-induced autophagy is mediated through the down-regulation of the Akt signaling pathway, but not through Class III PI3-K signaling.

(A–C) Protein levels of Class I signaling molecules including Akt, mTOR and p70S6K and class III PI3-K signaling molecules including Vps34p and beclin were analyzed in HCT116 cells stably expressing mock or Sox2. The individual proteins were visualized by Western blotting using specific antibodies. β-Actin was used as an internal control to verify equal protein loading. (D) Involvement of Class I PI3-K signaling in Sox2-induced autophagy. (D, left panels) HCT116 cells stably expressing mock or Sox2 were treated with insulin or LY294002 under 10% FBS-containing conditions at 2 days after infection. Vacuole formation was observed by light microscopy (X200). The boxed areas are individually magnified 2 more times (lower panels for mock and Sox2) to better visualize vacuoles. (D, right panels) The cells were transduced with Sox2 viral particles and cultured 2 days. The cell culture medium was replaced with McCoy’s 5a without FBS supplementation but including 5 µg/ml insulin or 5 µg LY294002 and then cells were cultured for 5 days. The cells were observed under light microscopy (X200). (E) Quantitative comparison of autophagy formation induced by class I PI3-K signaling. The vacuole forming cells from D and F were counted and compared in a numerate graphic (*p<0.001).
Sox2-induced Autophagy Suppresses Proliferation and Anchorage-independent Colony Growth by Inducing Cellular Senescence in HCT116 Cells
Suppression of PI3-K activity by PTEN has an important role in cell proliferation and cancer development in the colon [29], [30], [31]. PI3-K inhibition by PTEN or wortmannin has an inverse effect compared with tumor necrosis factor- α on the balance between the p50 and p65 subunits of nuclear factor kB [31]. GM3, a ganglioside, treatment dramatically increases cyclin-dependent kinase (CDK) inhibitor (CKI) p21WAF1 expression through the accumulation of the p53 protein by the PTEN-mediated inhibition of PI3-K-Akt-MDM2 survival signaling in HCT116 colon cancer cells [29]. In human colorectal cancer, a negative correlation between PTEN protein levels and Akt phosphorylation has been observed [30]. Our results indicated that down-regulation of PI3-K-Akt signaling through PTEN might be involved in Sox2-induced autophagy ( Fig. 4 ). Our cell proliferation and cell cycle analyses indicated that Sox2 infection into HCT116 cells suppressed proliferation by inducing accumulation of cells in G0/G1 and sub-G1 and reduction of S cell cycle phases compared with mock-infected cells ( Fig. 5A ). The attenuation of cell proliferation was associated with about 90% inhibition of anchorage-independent cancer growth in soft agar, a hallmark of cancer cell properties [32] ( Fig. 5B ). To explore the signaling responsible for inducing the loss of cancer cell properties such as suppression of proliferation and soft agar growth, we examined various tumor suppressors with a known role in regulating cell cycle. We observed an increase in p53 phosphorylation (Ser15) and increases in the total protein levels of p16INK4a and p21 ( Fig. 5C ), which are well known to be strongly involved in cellular senescence [33]. We then examined senescence using an SA-ß-galactosidase assay and found that ectopic expression of Sox2 enhanced ß-galactosidase activity and protein level ( Fig. 5D , left and middle panels) and suppressed Ki-67 protein level, a cell proliferation marker ( Fig. 5D, right panel ). Notably, over 90% of cells containing vacuoles stained ß-gal positive ( Fig. 5D , graph), Taken together, these results indicated that Sox2-induced autophagy causes a loss of tumor properties in HCT116 colorectal cancer cells.
Figure 5. Sox2-induced autophagy causes senescence in HCT116 colorectal cancer cells resulting in suppressed proliferation and decreased anchorage-independent colony growth.

(A) Left panel, effect of Sox2 expression on cell proliferation. HCT116 cells (2×103) stably expressing mock or Sox2 were seeded in 96-well plates and proliferation was analyzed by MTS assay at 24 h intervals up to 96 h (*, p<0.001). Middle and right panels, effect of Sox2 expression on cell cycle distribution. HCT116 cells (4×105) stably expressing mock or Sox2 were seeded, cultured and then cell cycle distribution (middle panel) and sub-G1 accumulation (right panel) were analyzed by FACS (*, p<0.001). (B) Effect of Sox2 expression on anchorage-independent colony growth. HCT116 cells (8×103) stably expressing mock or Sox2 were subjected to a soft agar colony growth assay for 5–7 days. The cell colonies were scored using a microscope and the Image-Pro PLUS (v.6) computer software program (*p<0.001). (C) Protein profiling related to cell cycle regulation. Proteins were extracted from HCT116 cells stably expressing mock or Sox2 and subjected to Western blotting to detect the level of various proteins known to be involved in cell cycle regulation. ß-Actin was used as an internal control to verify equal protein loading. (D) Cellular senescence assay. Left panel, HCT116 cells stably expressing mock or Sox2 were analyzed for senescence using a senescence ß-galactosidase staining kit. The cells were observed under a light microscope (X200). Middle and right panels, the protein level of ß-galactosidase or Ki-67 was detected in HCT116 cells stably expressing mock or Sox2 using specific primary antibodies as indicated and an HRP-conjugated secondary antibody. Immunocytochemical detection was performed using the Sigma FASTTM 3,3′-Diaminobenzidine Terahyfrochloride with Metal Enhancer Tablet Sets (DAB peroxidase substrate). The protein levels were visualized using a light microscope (X200). Graph, indicates a comparison of ß-galactosidase positive cell population in HCT116 cells stably expressing mock or Sox2 (*p<0.001).
Knockdown of ATG10 Restores Sox2-induced Autophagy, Cellular Senescence and Cell Proliferation
Our previous results demonstrated that Sox2-mediated autophagy is mediated through ATG10 expression ( Fig. 3 ). To examine whether knockdown of ATG10 can suppress autophagy induced by Sox2 overexpression, we selected a psh-ATG10-1755 knockdown vector ( Fig. 6A ). The sh-ATG10 viral particles were infected into Sox2-preinfected cells and expression of Sox2 ( Fig. 6B , upper panels) and knockdown of ATG10 ( Fig. 6B , bottom panels) were confirmed by an immunofluorescence assay. Very importantly, the cellular morphologies of autophagy induced by Sox2, including cell flattening, nuclear size and vacuole formation, were totally converted to those observed in mock-infected HCT116 cells ( Fig. 6B, L.M.). Furthermore, we confirmed that the morphological recovery corresponded with restoration of Ki-67, ß-galactosidase, p16INK4a and p21 protein levels in the mock-infected HCT116 cells ( Fig. 6C ). Notably, we confirmed that knockdown of ATG10 in HCT116 stably expressing Sox2 recovered cell proliferation, which was inhibited by Sox2 overexpression, and the growth was even faster than that of HCT116 cells stably expressing mock control ( Fig. 6D ). These results indicated that ATG10 plays an important role in autophagy formation induced by Sox2 expression in HCT116 colorectal cancer cells.
Figure 6. Knockdown of ATG10 restores Sox2-induced autophagy, cellular senescence and proliferation.

(A) Confirmation of the knockdown efficiency of ATG10. Knockdown of ATG10 using pLKO-shATG10. The pLKO-shATG10 knockdown lenti-viral particles were infected into HCT116 cells stably expressing Sox2 and cells cultured for 36 h. The proteins were extracted and knockdown efficiency was determined by Western blot using a specific ATG10 antibody. ß-Actin was used as an internal control to verify equal protein loading. (B) Morphological changes induced by ATG10 knockdown in HCT116 cells stably expressing Sox2. HCT116-mock, -Sox2 and -Sox2/sh-ATG10 cells were analyzed for morphological changes and protein levels of Sox2 (red) and ATG10 (green) were determined by an immunofluorescence assay using specific antibodies. The cells were observed under a fluorescence or light microscope (X200). L.M. indicates light microscopy. (C) Restoration of cell cycle, cell proliferation and senescence markers by knocking down ATG10 in HCT116 cells stably expressing Sox2. HCT116-mock, -Sox2 and -Sox2/sh-ATG10 cells were seeded into a 4-chamber slide, fixed and permeabilized. Markers for cell proliferation, cellular senescence and cell cycle regulation included Ki-67, ß-galactosidase, p16INK4a and p21. These markers were analyzed by immunocytochemistry with specific antibodies as indicated using the Sigma FASTTM 3,3′-Diaminobenzidine Terahyfrochloride with Metal Enhancer Tablet Sets (DAB peroxidase substrate). The cells were observed under a fluorescence or light microscope (X200). (D) Restoration of proliferation by knocking down ATG10 in HCT116 cells stably expressing Sox2. HCT116-mock, -Sox2 and -Sox2/sh-ATG10 cells (2×103) were seeded into 96-well plates and proliferation was analyzed by MTS assay at 24 h intervals up to 96 h (*p<0.05, **p<0.001).
Sox2-induced Autophagy Suppresses Cancer Growth in a Xenograft Mouse Model
Our previous results demonstrated that Sox2-induced autophagy suppressed cell proliferation and anchorage-independent colony growth ( Fig. 5A, B ). To determine whether Sox2 overexpression suppresses tumor growth in vivo, we examined xenograft tumor growth in athymic nude mice. HCT116 cells (3×106) infected with mock or Sox2 were injected subcutaneously into the right flank of athymic nude mice. Tumor growth was measured twice a week. At the 30-day endpoint, HCT116 cells infected with mock vector showed significantly larger tumor growth compared to HCT116 cells infected with Sox2 ( Fig. 7A ). The first measurable tumor was observed at day 21 after injection of HCT116 cells infected with Sox2 compared with day 7 in HCT116 cells infected with mock vector ( Fig. 7B ). Furthermore, tumor growth was markedly attenuated in athymic nude mice injected with HCT116 cells infected with Sox2 compared with HCT116 cells infected with mock vector ( Fig. 7C ). To examine Sox2, ATG10 and ATG8b protein levels in tumor tissues, we conducted an immunofluorescence assay by confocal microscope using Sox2, ATG10 and ATG8b antibodies. We found that tumor tissues from HCT116 cells infected with Sox2 displayed a higher Sox2 protein level compared with tumors from HCT116 cells infected with mock vector ( Fig. 7D ). Furthermore, the tumor tissues formed from HCT116 cells infected with Sox2 exhibited higher ATG10 and ATG8b protein levels compared with tumors from the HCT116 cells infected with mock vector ( Fig. 7D ). Taken together, these results demonstrated that Sox2-induced autophagy suppresses cancer growth in an in vivo xenograft mouse model.
Figure 7. Sox2-induced autophagy suppresses cancer growth in a xenograft mouse model.

(A) Xenograft in vivo tumor growth. HCT116 cells (3×106) stably expressing mock (n = 9) or Sox2 (n = 10) were injected subcutaneously into the dorsal right flank of athymic nude mice. At the endpoint (30 days) after injection, mice were euthanized and necropsied. (B) Sox2 expression delayed tumor growth in athymic nude mice. Tumor incidence of HCT116 cells stably expressing mock or Sox2 was analyzed at first measurable tumor growth after injection and compared (*p<0.05, *p<0.01). (C) Sox2 expression suppresses in vivo tumor growth. Average tumor growth of HCT116 cells stably expressing mock or Sox2 was measured and compared. Tumor volume was calculated from measurements of 2 diameters of the individual tumor according to the formula: tumor volume (mm3) = [longer diameter × shorter diameter2]/2 (*p<0.05, *p<0.01). (D) Immunohistofluorescence analysis of tumor tissues. The largest tissues formed from HCT116 cells stably expressing mock or Sox2 were necropsied, fixed in Zamboni’s fixative and tissue slides were prepared. The tissues were hybridized with specific antibodies as indicated and visualized with Cy3 (red color) to detect Sox2 and Cy2 (green color) for detection of ATG10 and ATG8b by laser scanning confocal microscopy (X200; NIKON C1si Confocal Spectral Imaging System, NIKON Instruments Co.).
Discussion
Monitoring autophagy is outlined recently in “Guidelines for the use and interpretation of assays for monitoring autophagy [34]. Although each autophagy assay is not absolutely limited to monitoring autophagy, the guidelines suggest several diverse methods, including transmission electron microscopy, Atg8-LC3 detection and quantification, SQSTM1/p62 and LC3 binding protein turnover, and assessing mTOR, AMPK and Atg1/ULK1 expression. Our results demonstrated that Sox2 overexpression induced vacuole formation and lysosomal activation ( Fig. 1 ). Importantly, Sox2 induced LC3 accumulation visualized by Western blotting ( Fig. 3C ) and foci formation visualized by immunocytofluorescence ( Fig. 1D ). Moreover, our results demonstrated that the Sox2 induced ATG10 gene expression ( Fig. 3B–F ). Thus, we hypothesized that Sox2-induced morphological and cellular functional changes are caused by autophagy.
Sox2 expression induced autophagy in all colon cancer cell lines examined. When we produced and infected lenti-Sox2 into cells, nearly 100% of the cells were infected with Sox2 and over 90% formed vacuoles in the cytoplasm ( Fig. 1B ). The cells exhibiting intracellular vacuoles have undergone flattening of the cytoplasm and nuclear enlargement. The Sox2 protein levels in flattened cells were lower compared with cells not flattened. Moreover, cells having small nuclei and high level of Sox2 expression also exhibited small vacuoles and continued growing ( Fig. 2C ). The population of cells undergoing cellular senescence induced by Sox2 overexpression had extensive vacuole formation (i.e., over 90%) and proliferation was actually stopped. Because of this, we conducted all experiments with new transduction each time. Importantly, ectopic expression of Sox2 induced multiple nuclei (i.e., aneuploidy), which is an important biological phenomenon that we are currently studying. ( Fig. 1D , 3D , 6B ).
Although recent studies have increased our understanding of the molecular mechanisms and biological functions of autophagy, whether autophagy acts fundamentally in a cell survival or cell death pathway or both is not clear. In this study, we found that autophagy inhibited cell proliferation ( Fig. 5A ) and tumor growth ex vivo ( Fig. 5B ) and in vivo ( Fig. 7A–C ). Because the role of autophagy is likened to a double-edged sword [3], a complete understanding of the role of autophagy in cancer cell growth might help in the search for clues to find strategies to treat cancer. For example, chemically-induced liver cancer cells exhibited lower autophagic activity compared with their non-neoplastic liver counterparts and transformed cell lines showed lower protein degradation rates during nutritional stress compared with nontransformed control cells [35]. Very interestingly, several chemopreventive flavonoids and other phytochemical compounds, including saponins, curcumin, genistein, quercetin, resveratrol, sulforaphane, tocotrienols and vitamins, stimulate autophagic vacuolation in many cancer cell types [2]. Furthermore, many anticancer drugs or compounds including tamoxifen, arsenic trioxide, imatinib and rapamycin induce autophagic cell death in many human cancer cell lines [10], [12], [13], [14]. Importantly, monoallelic loss of the beclin1 gene is genetically associated with human cancers such as ovary, breast and prostate cancer [7]. Moreover, beclin-induced autophagy formation suppresses MCF-7 breast cancer cell colony growth under anchorage-independent conditions [15] and also inhibits cell proliferation by inhibition of p70 S6kinase activity [16], [17].
Many human cancers exhibit an enhanced activation of mTOR, resulting in reduced autophagy. Elegant in vivo experiments demonstrated that constitutively active Akt inhibits the induction of autophagy in vitro and in vivo, but accelerates the growth of tumors in vivo, in particular in cells that lack expression of Bax and Bak [36]. Conversely, tumor suppressor gene products upstream of mTOR signaling, such as PTEN (phosphatase and tensin homolog deleted on chromosome ten) and TCSs (tuberous sclerosis complex TSC1 and TSC2), antagonize PI3-K activation and promote autophagy by down-regulation of mTOR [37]. These studies are in agreement with our ex vivo results showing that ectopic expression of Sox2 reduced PTEN phosphorylation ( Fig. 4C ), resulting in subsequent reduction of phosphorylated and total Akt1 and downstream signaling molecules, such as mTOR and p70S6 kinase ( Fig. 4B ). Importantly, insulin stimulation partially rescued Sox2-induced autophagy under normal cell culture conditions ( Fig. 4D left panel s, E) and also starvation conditions ( Fig. 4D right panel s, E), indicating that Sox2-induced autophagy might correspond with negative regulation of Akt-mTOR signaling mediated through PTEN. Moreover, we suggest that constitutively active Akt (Myr-Akt) might rescue the autophagy formation induced by Sox2 overexpression. Although the molecular mechanism explaining how Sox2 modulates PTEN phosphorylation is not clearly understood, our results suggested that Sox2-induced autophagy might be applicable in suppressing colon cancer.
Growth arrest of senescent cells is initiated with the activation of p53 by inhibition of its degradation through the involvement of p14ARF, a tumor suppressor that sequesters the MDM2 protein [38]. Growth arrest can also occur through p53 acetylation mediated by the promyelocyte leukemia (PML) tumor suppressor in Ras-induced accelerated senescence [39], [40]. Cells that undergo senescence cannot divide even if stimulated by mitogens, but they remain metabolically and synthetically active and show characteristic changes in morphology, such as enlarged and flattened cell shape and increased granularity [1]. Although global and progressive epigenetic alterations are proposed to play a crucial role in oncogene-induced senescence [26], a recent study demonstrates that overexpression of ULK3, a human ortholog of ATG1, induces autophagy and senescence and inhibition of autophagy delays the senescence phenotype [8]. Our findings indicated that Sox2 overexpression induced autophagy through positive regulation of ATG10 expression, and knockdown of ATG10 in Sox2 overexpressing cells restored Sox2-induced autophagy and senescence phenotypes, which included vacuole formation, flattened cell morphology, and cellular senescence and proliferation markers, including ß-galactosidase, Ki-67, p16INK4a and p21, to normal cell morphology ( Fig. 6A, B ). Importantly, Sox2-mediated suppression of cell proliferation was more substantially enhanced compared with parental HCT116-mock cells ( Fig. 6C ). These results demonstrated that the Sox2-ATG10 signaling pathway induces autophagy and then promotes cellular senescence. Furthermore, in our in vivo experiment, we found that HCT116 parental cells grew well in the xenograft mouse model ( Fig. 7A–C ). However, Sox2 overexpressing HCT116 cells grew very poorly in these mice ( Fig. 7A–C ), indicating that Sox2-induced senescence mediated through autophagy inhibits tumor growth.
Because mitotic cells cannot redistribute and eliminate damaged organelles, proteins and aggregates through cell division, basal levels of autophagy might be particularly essential for post-mitotic cells [41]. Unlike the ubiquitin-proteasome system, which is another major protein degradation system, autophagic breakdown of substrates is not only limited by steric considerations, but also uniquely capable of degrading whole organelles such as mitochondria, peroxisomes, and endoplasmic reticulum [41]. Thus, basal- and induced-autophagy are important for the physiological control of the number and quality of organelles in diverse phyla and function to eliminate superfluous, aged and damaged organelles and proteins [41]. For example, reactive oxygen species (ROS), critical inducers of autophagy, forces to initiate cardiovascular differentiation programs [42], [43], indicate that autophagy might play an important role in the ES cell self-renewal program by elimination of oxidized or damaged proteins or subcellular organelles for quality control of ES cells. Sox2 plays a key role in ES cell self-renewal and iPS production [21], [44]. The Sox2 protein level is dramatically decreased during the ES cell differentiation process [45]. Our results indicated that Sox2 overexpression induced autophagy in HCT116 colorectal cancer cells ( Fig. 1 and 2 ). However, whether ES cells exhibit higher autophagic activity compared with differentiated cells is not known. In this study, we provided evidence showing that Sox2 induced autophagy through the up-regulation of ATG10 gene expression and cellular senescence, resulting in inhibition of tumor growth ex vivo and in vivo. We also suggest that autophagy mediated by Sox2 results in loss of malignancy in colon cancer cells ex vivo and in vivo. Overall, our study could be critical to our understanding of the underlying roles of Sox2 in cancer.
Materials and Methods
Reagents
Chemical reagents, including Tris, NaCl, and SDS for molecular biology and buffer preparation were purchased from Sigma-Aldrich (St. Louis, MO). Restriction enzymes and some modifying enzymes were purchased from New England BioLabs, Inc. (Beverly, MA). The Taq DNA polymerase was obtained from Qiagen, Inc. (Valencia, CA). The DNA ligation kit (v. 2.0) was purchased from TAKATA Bio, Inc. (Otsu, Shiga, Japan). The lentiviral expression vectors including pSin-EF2-Sox2, -Nanog, -Oct4 and -Lin28 and packaging vectors including pMD2.0G and psPAX were purchased from Addgene Inc. (Cambridge, MA). Antibodies against beclin and ATG8b (LC3-b) were from ABGENT (San Diego, CA) and p16INK4a and p21 were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Nanog, Oct4, Lin28 and ATG10 antibodies were purchased from Abcam (Cambridge, MA). Lysotracker was purchased from Invitrogen (Carlsbad, CA). Antibodies against Sox2, Class III PI3-K, phosphorylated p53 (Ser15), total and phosphorylated Akt, mTOR, p70 S6 kinase and ß-galactosidase were purchased from Cell Signaling Technology, Inc. (Danvers, MA). The SA-ß-galactosidase assay kit for assessing cellular senescence was purchased from Cell Signaling Technology Inc. and CellTiter 96® Aqueous One Solution for cell proliferation analysis was from Promega (Madison, WI). Dulbecco’s modified Eagle’s medium (DMEM) and McCoy’s 5a medium were purchased from Cellgro (Manassas, VA).
Cell Culture and Transfections
HEK293T, HCT116, CCD-18Co, HT29, and WiDr cells were purchased from ATCC and maintained according to ATCC suggested conditions. Briefly, HEK293T cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics at 37°C in a 5% CO2 incubator. HCT116 colorectal cancer cells were cultured in McCoy’s 5a medium supplemented with 10% FBS and antibiotics at 37°C in a 5% CO2 incubator. To prepare Sox2, Oct4, Nanog, Lin28 viral particles, each of the viral vectors and packaging vectors (pMD2.0G and psPAX) was transfected into HEK293T cells following Addgene’s suggested protocols and using JetPEI (Q-Biogene, West Chester, PA) also following the manufacturer’s suggested protocol. The transfection medium was exchanged at 4 h after transfection and then cells were cultured an additional 36 h. The viral particles were harvested by filtration using a 0.45 µm sodium acetate syringe filter and used for infection into 60% confluent HCT116 cells using 8 µg/ml of polybrane (Millipore, Billerica, MA) overnight. The cell culture medium was replaced with fresh complete growth medium and cells incubated for 24 h and then selected with 1.5 µg/ml of puromycine for 36 h.
MTS Assay
To estimate proliferation, HCT116 cells stably expressing mock or Sox2 were trypsinized, counted and then seeded into 96-well plates (2×103 cells/well). After culturing for 2 h, 20 µl of the CellTiter 96® Aqueous One Solution (Promega) were added to each well and cells were then incubated for 1 h at 37°C and 5% CO2. To stop the reaction, 25 µl of a 10% SDS solution were added and absorbance was measured at 492 and 690 nm. Cell proliferation was measured at 24-h intervals.
Cell Cycle Analysis
HCT116 cells (4×105) stably expressing mock or Sox2 were seeded into 60-mm dishes and cultured for 16 h at 37°C in a 5% CO2 incubator. The medium was changed and cells cultured for 24 h and then cells were trypsinized, fixed with methanol, stained with propidium iodide (PI) and then analyzed for cell cycle phase using the FACS Calibur (Becton Dickinson, Franklin Lakes, NJ).
Western Blotting
Proteins were extracted with RIPA cell lysis buffer by freezing and thawing and protein concentration was measured. The same amount of protein was resolved by SDS-polyacrylamide gel electrophoresis, transferred onto PVDF membranes and blocked in 5% skim milk for 1 h at room temperature. Antibody hybridization was performed overnight at 4°C and HRP-conjugated secondary antibody binding was then conducted for 1 h at room temperature. The membranes were washed and proteins visualized using the ECL detection kit (Amersham Biosciences, Piscataway, NJ).
cDNA Microarray
The total RNAs from HCT116 cells stably expressing mock or Sox2 were extracted using an RNA extraction kit (Qiagen, Valencia, CA). The RNA purity for this experiment was over 2.0 in the ratio of OD260/OD280. The microarray was conducted by the Phalanx Biotech Group (Palo Alto, CA) and duplicate array results were obtained using the Significance Analysis of Microarrays (SAM) program from the Stanford Microarray database (http://smd.stanford.edu). SAM identified the genes with statistically significant changes in expression by performing a set of gene-specific t-tests (p<0.05). The genes were color-coded as red for up-regulation and green for down-regulation. The identified genes were annotated using the DAVID database (http://niaid.abcc.ncifcrf.gov) [46], [47].
Immunofluorescence Assay
HCT116 cells stably expressing mock, Sox2 or shATG10 were seeded in 2- or 4-chamber slides depending on the experiment and cultured 24 h. The cells were fixed with 4% formalin, permeabilized with 0.5% Triton X-100/1X PBS for 10 min, hybridized with anti-Sox2, ATG10 or ATG8b as a primary antibody and anti-mouse IgG goat-conjugated with Alexa 568 or Alexa 488 (Invitrogen) as a secondary antibody. The cells were mounted with Fluoro-GEL II containing DAPI (Electron Microscopy Sciences, Hatfield, PA) and observed under a fluorescence microscope (X200). Tumors were excised and dropped-fixed into Zamboni’s fixative [0.03% picric acid (w/v) and 2% paraformaldehyde (w/v)] for 48 h at 4°C and then transferred to a 20% sucrose solution with 0.05% sodium azide in phosphate buffered saline (PBS) for storage. Processing and staining of tumors were carried out according to a published procedure [48]. Optical sections were captured by laser scanning confocal microscopy (NIKON C1si Confocal Spectral Imaging System, NIKON Instruments Co., Melville, NY) [48].
Anchorage-independent Colony Assay
Anchorage-independent colony growth was investigated using HCT116 cells stably expressing mock or Sox2. In brief, cells (8×103/ml) were mixed in 1 ml of 0.3% McCoy’s 5a agar containing 10% FBS. The cultures were maintained in a 37°C, 5% CO2 incubator for 5–7 days and the cell colonies were scored using a microscope and the Image-Pro PLUS (v.6) computer software program (Media Cybernetics, Silver Spring, MD) as described by Colburn et al. [49].
In vivo Tumor Growth Assay
HCT116 cells stably expressing mock or Sox2 were harvested at 90% confluence and suspended (3×106) in 200 µl of McCoy’s 5a medium without FBS or antibiotics. Mice were divided into 2 groups of 9 or 10 mice each. Each individual cell line (3×106) was injected into mice (9, mock vector control; 10, Sox2). The cells were injected subcutaneously into the right flank of each mouse. The mice were weighed twice each week and monitored every day for tumor growth. When tumors appeared, they were measured twice a week by computerized caliper. Tumor volume was calculated from measurements of 2 diameters of the individual tumor according to the formula: tumor volume (mm3) = [longer diameter × shorter diameter2]/2. Mice were monitored until tumors reached 1,000 mm3 total volume at which time mice were euthanized and tumors extracted for histochemical analysis. All experiments were performed according to guidelines approved by the University of Minnesota Institutional Animal Care and Use Committee.
Ex vivo and Xenograft Animal Studies
The human cells, including HEK293T, HCT116, CCD-18Co, HT-29, and WiDr cells, used in ex vivo studies were purchased from ATCC and studies were approved by the University of Minnesota Institutional Biosafety Committee (IBC). The xenograft animal study utilizing athymic nude mice was conducted under the approval of the University of Minnesota Institutional Animal Care and Use Committee (IACUC).
Statistical Analysis
Each experiment was conducted at least two times independently in triplicate. Data are shown as means ± S.D. (standard deviation) or S.E. (standard error) of values obtained from triplicate experiments as indicated in each figure legend. Significant differences were evaluated using the Student’s t–test.
Supporting Information
Microarray of HCT116 colorectal cancer cells stably expressing Sox2 . These microarray results were compared with HCT116 cells stably expressing a mock vector control. The red color indicates up-regulated genes, green color denotes down-regulated genes, and the yellow color indicates no difference in gene expression.
(ZIP)
The microarray results are grouped as indicated using the DAVID Database ( http://niaid.abcc.ncifcrf.gov ) (1, 2) and the numbers of genes up- (red) or down-regulated (green) are denoted. The p value was obtained by t-tests comparing gene expression in HCT116 cells expressing Sox2 with HCT116 cells expressing mock control.
(ZIP)
Summary of gene expression in cellular responsiveness to Sox2 expression. Table S3 summarizes gene expression profiles (from Table S2) in HCT116 colorectal cancer cells induced by Sox2 expression compared with mock expression.
(TIF)
Identity of putative consensus sequences in autophagy-related genes. The data were obtained by a re-analysis of results from a previous publication (3) and summarized. The Sox2- associated score indicates the identity of Sox-binding consensus sequences, 5′-CATTGAT-3′ (4).
(TIF)
(DOCX)
Acknowledgments
We thank Tonya M. Poorman at The Hormel Institute, University of Minnesota for help submitting our manuscript.
Funding Statement
This work was supported in part by The Hormel Foundation and National Institutes of Health grants CA111536, CA120388, CA077646, R37CA081064 and ES016548, and by the Research Fund, M-2011-B0002-00025 of The Catholic University of Korea. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Campisi J (2001) Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol 11: S27–31. [DOI] [PubMed] [Google Scholar]
- 2. Singletary K, Milner J (2008) Diet, autophagy, and cancer: a review. Cancer Epidemiol Biomarkers Prev 17: 1596–1610. [DOI] [PubMed] [Google Scholar]
- 3. Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306: 990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8: 741–752. [DOI] [PubMed] [Google Scholar]
- 5. Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6: 463–477. [DOI] [PubMed] [Google Scholar]
- 6. Schweichel JU, Merker HJ (1973) The morphology of various types of cell death in prenatal tissues. Teratology 7: 253–266. [DOI] [PubMed] [Google Scholar]
- 7. Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 100: 15077–15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, et al. (2009) Autophagy mediates the mitotic senescence transition. Genes Dev 23: 798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oh S, Xiaofei E, Ni D, Pirooz SD, Lee JY, et al. (2011) Downregulation of autophagy by Bcl-2 promotes MCF7 breast cancer cell growth independent of its inhibition of apoptosis. Cell Death Differ 18: 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kondo Y, Kanzawa T, Sawaya R, Kondo S (2005) The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 5: 726–734. [DOI] [PubMed] [Google Scholar]
- 11. Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, et al. (2004) Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem 279: 18384–18391. [DOI] [PubMed] [Google Scholar]
- 12. Ertmer A, Huber V, Gilch S, Yoshimori T, Erfle V, et al. (2007) The anticancer drug imatinib induces cellular autophagy. Leukemia 21: 936–942. [DOI] [PubMed] [Google Scholar]
- 13. Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I (2003) Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res 63: 2103–2108. [PubMed] [Google Scholar]
- 14. Tsang CK, Qi H, Liu LF, Zheng XF (2007) Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today 12: 112–124. [DOI] [PubMed] [Google Scholar]
- 15. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, et al. (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402: 672–676. [DOI] [PubMed] [Google Scholar]
- 16. Lee SB, Kim S, Lee J, Park J, Lee G, et al. (2007) ATG1, an autophagy regulator, inhibits cell growth by negatively regulating S6 kinase. EMBO Rep 8: 360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang RC, Levine B (2010) Autophagy in cellular growth control. FEBS Lett 584: 1417–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Menendez JA, Vellon L, Oliveras-Ferraros C, Cufi S, Vazquez-Martin A (2011) mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: A roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle 10: 3658–3677. [DOI] [PubMed] [Google Scholar]
- 19. Kuo TC, Chen CT, Baron D, Onder TT, Loewer S, et al. (2011) Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat Cell Biol 13: 1214–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kamachi Y, Uchikawa M, Kondoh H (2000) Pairing SOX off: with partners in the regulation of embryonic development. Trends Genet 16: 182–187. [DOI] [PubMed] [Google Scholar]
- 21. Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663–676. [DOI] [PubMed] [Google Scholar]
- 22. Rodriguez-Pinilla SM, Sarrio D, Moreno-Bueno G, Rodriguez-Gil Y, Martinez MA, et al. (2007) Sox2: a possible driver of the basal-like phenotype in sporadic breast cancer. Mod Pathol 20: 474–481. [DOI] [PubMed] [Google Scholar]
- 23. Otsubo T, Akiyama Y, Yanagihara K, Yuasa Y (2008) SOX2 is frequently downregulated in gastric cancers and inhibits cell growth through cell-cycle arrest and apoptosis. Br J Cancer 98: 824–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Y, Shi L, Zhang L, Li R, Liang J, et al. (2008) The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J Biol Chem 283: 17969–17978. [DOI] [PubMed] [Google Scholar]
- 25. Dunn WA Jr (1990) Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J Cell Biol 110: 1935–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Narita M, Krizhanovsky V, Nunez S, Chicas A, Hearn SA, et al. (2006) A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 126: 503–514. [DOI] [PubMed] [Google Scholar]
- 27. Altman BJ, Wofford JA, Zhao Y, Coloff JL, Ferguson EC, et al. (2009) Autophagy provides nutrients but can lead to Chop-dependent induction of Bim to sensitize growth factor-deprived cells to apoptosis. Mol Biol Cell 20: 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vazquez F, Ramaswamy S, Nakamura N, Sellers WR (2000) Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol 20: 5010–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi HJ, Chung TW, Kang SK, Lee YC, Ko JH, et al. (2006) Ganglioside GM3 modulates tumor suppressor PTEN-mediated cell cycle progression–transcriptional induction of p21(WAF1) and p27(kip1) by inhibition of PI-3K/AKT pathway. Glycobiology 16: 573–583. [DOI] [PubMed] [Google Scholar]
- 30. Colakoglu T, Yildirim S, Kayaselcuk F, Nursal TZ, Ezer A, et al. (2008) Clinicopathological significance of PTEN loss and the phosphoinositide 3-kinase/Akt pathway in sporadic colorectal neoplasms: is PTEN loss predictor of local recurrence? Am J Surg 195: 719–725. [DOI] [PubMed] [Google Scholar]
- 31. Kim S, Domon-Dell C, Wang Q, Chung DH, Di Cristofano A, et al. (2002) PTEN and TNF-alpha regulation of the intestinal-specific Cdx-2 homeobox gene through a PI3K, PKB/Akt, and NF-kappaB-dependent pathway. Gastroenterology 123: 1163–1178. [DOI] [PubMed] [Google Scholar]
- 32. Rheinwald JG, Beckett MA (1981) Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultures from human squamous cell carcinomas. Cancer Res 41: 1657–1663. [PubMed] [Google Scholar]
- 33. Zhang X, Li J, Sejas DP, Pang Q (2005) The ATM/p53/p21 pathway influences cell fate decision between apoptosis and senescence in reoxygenated hematopoietic progenitor cells. J Biol Chem 280: 19635–19640. [DOI] [PubMed] [Google Scholar]
- 34. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, et al. (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8: 445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gozuacik D, Kimchi A (2004) Autophagy as a cell death and tumor suppressor mechanism. Oncogene 23: 2891–2906. [DOI] [PubMed] [Google Scholar]
- 36. Mathew R, Karantza-Wadsworth V, White E (2007) Role of autophagy in cancer. Nat Rev Cancer 7: 961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Turcotte S, Giaccia AJ (2010) Targeting cancer cells through autophagy for anticancer therapy. Curr Opin Cell Biol 22: 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Y, Xiong Y, Yarbrough WG (1998) ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 92: 725–734. [DOI] [PubMed] [Google Scholar]
- 39. Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, et al. (2000) PML is induced by oncogenic ras and promotes premature senescence. Genes Dev 14: 2015–2027. [PMC free article] [PubMed] [Google Scholar]
- 40. Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, et al. (2000) PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406: 207–210. [DOI] [PubMed] [Google Scholar]
- 41. Kourtis N, Tavernarakis N (2009) Autophagy and cell death in model organisms. Cell Death Differ 16: 21–30. [DOI] [PubMed] [Google Scholar]
- 42. Sauer H, Neukirchen W, Rahimi G, Grunheck F, Hescheler J, et al. (2004) Involvement of reactive oxygen species in cardiotrophin-1-induced proliferation of cardiomyocytes differentiated from murine embryonic stem cells. Exp Cell Res 294: 313–324. [DOI] [PubMed] [Google Scholar]
- 43. Schmelter M, Ateghang B, Helmig S, Wartenberg M, Sauer H (2006) Embryonic stem cells utilize reactive oxygen species as transducers of mechanical strain-induced cardiovascular differentiation. FASEB J 20: 1182–1184. [DOI] [PubMed] [Google Scholar]
- 44. Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I (2008) MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 455: 1124–1128. [DOI] [PubMed] [Google Scholar]
- 45. Gu P, LeMenuet D, Chung AC, Mancini M, Wheeler DA, et al. (2005) Orphan nuclear receptor GCNF is required for the repression of pluripotency genes during retinoic acid-induced embryonic stem cell differentiation. Mol Cell Biol 25: 8507–8519. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 46. Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, et al. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4: P3. [PubMed] [Google Scholar]
- 47. Huang S, Sinicrope FA (2010) Celecoxib-induced apoptosis is enhanced by ABT-737 and by inhibition of autophagy in human colorectal cancer cells. Autophagy 6: 256–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wacnik PW, Baker CM, Herron MJ, Kren BT, Blazar BR, et al. (2005) Tumor-induced mechanical hyperalgesia involves CGRP receptors and altered innervation and vascularization of DsRed2 fluorescent hindpaw tumors. Pain 115: 95–106. [DOI] [PubMed] [Google Scholar]
- 49. Colburn NH, Wendel EJ, Abruzzo G (1981) Dissociation of mitogenesis and late-stage promotion of tumor cell phenotype by phorbol esters: mitogen-resistant variants are sensitive to promotion. Proc Natl Acad Sci U S A 78: 6912–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Microarray of HCT116 colorectal cancer cells stably expressing Sox2 . These microarray results were compared with HCT116 cells stably expressing a mock vector control. The red color indicates up-regulated genes, green color denotes down-regulated genes, and the yellow color indicates no difference in gene expression.
(ZIP)
The microarray results are grouped as indicated using the DAVID Database ( http://niaid.abcc.ncifcrf.gov ) (1, 2) and the numbers of genes up- (red) or down-regulated (green) are denoted. The p value was obtained by t-tests comparing gene expression in HCT116 cells expressing Sox2 with HCT116 cells expressing mock control.
(ZIP)
Summary of gene expression in cellular responsiveness to Sox2 expression. Table S3 summarizes gene expression profiles (from Table S2) in HCT116 colorectal cancer cells induced by Sox2 expression compared with mock expression.
(TIF)
Identity of putative consensus sequences in autophagy-related genes. The data were obtained by a re-analysis of results from a previous publication (3) and summarized. The Sox2- associated score indicates the identity of Sox-binding consensus sequences, 5′-CATTGAT-3′ (4).
(TIF)
(DOCX)