

Abstract
Periodontal disease (PD) and atherosclerosis are both polymicrobial and multifactorial and although observational studies supported the association, the causative relationship between these two diseases is not yet established. Polymicrobial infection-induced periodontal disease is postulated to accelerate atherosclerotic plaque growth by enhancing atherosclerotic risk factors of orally infected Apolipoprotein E deficient (ApoEnull) mice. At 16 weeks of infection, samples of blood, mandible, maxilla, aorta, heart, spleen, and liver were collected, analyzed for bacterial genomic DNA, immune response, inflammation, alveolar bone loss, serum inflammatory marker, atherosclerosis risk factors, and aortic atherosclerosis. PCR analysis of polymicrobial-infected (Porphyromonas gingivalis [P. gingivalis], Treponema denticola [T. denticola], and Tannerella forsythia [T. forsythia]) mice resulted in detection of bacterial genomic DNA in oral plaque samples indicating colonization of the oral cavity by all three species. Fluorescent in situ hybridization detected P. gingivalis and T. denticola within gingival tissues of infected mice and morphometric analysis showed an increase in palatal alveolar bone loss (p<0.0001) and intrabony defects suggesting development of periodontal disease in this model. Polymicrobial-infected mice also showed an increase in aortic plaque area (p<0.05) with macrophage accumulation, enhanced serum amyloid A, and increased serum cholesterol and triglycerides. A systemic infection was indicated by the detection of bacterial genomic DNA in the aorta and liver of infected mice and elevated levels of bacterial specific IgG antibodies (p<0.0001). This study was a unique effort to understand the effects of a polymicrobial infection with P. gingivalis, T. denticola and T. forsythia on periodontal disease and associated atherosclerosis in ApoEnull mice.
Introduction
The polymicrobial nature of periodontal disease (PD) promotes chronic inflammation and orchestrates a complex disease mechanism in which inflammation results in the destruction of the periodontium (alveolar bone, cementum, periodontal ligament, and gingiva). Periodontal disease is becoming better recognized as a risk for and contributing factor to multiple systemic diseases, including atherosclerotic vascular disease (ASVD), diabetes, rheumatoid arthritis, and Alzheimer’s disease [1], [2], [3], [4], [5], [6]. Recent systematic reviews and meta-analysis of observational studies to date support an association between PD and ASVD independent of known confounders, but a casual relationship is not yet established [1], [7]. There is also a significantly increased prevalence and incidence of coronary heart disease (CHD) in patients with periodontal disease indicating that PD independently predicts CHD [8].
The existence of polymicrobial consortium in periodontal disease has been established by previous studies [9], [10]. The bacterial species Porphyromonas gingivalis (P. gingivalis), Treponema denticola (T. denticola) and Tannerella forsythia (T. forsythia) are strongly implicated in development of periodontal disease, and together are known as the “red complex” [11]. These bacteria colonize the oral biofilm late in its development, by coaggregation with the help of bridging bacteria including Fusobacterium nucleatum (F. nucleatum) [12], [13], [14], [15].
Many studies demonstrated that periodontal disease-associated bacteria enter the blood stream during mastication, brushing and flossing teeth, and during dental procedures [16]. Frequent, recurrent transient bacteremia has the potential to produce a chronic insult to the vasculature and may contribute to the injury and inflammation that initiates the development of atherosclerosis [3], [5], [6], [17]. In addition, periodontal lesions are recognized as continually renewing reservoirs for the systemic spread of bacteria and viruses, and their associated antigens, cytokines, and other proinflammatory mediators [5], [6]. Bacterial genomic 16S rDNA from numerous oral and periodontal species, including P. gingivalis, T. denticola, T. forsythia, and F. nucleatum have been detected in human clinical atherosclerotic plaque lesions [18], [19], [20], [21]. Among the microorganisms detected in atherosclerotic vessels, P. gingivalis does not appear dominant, nor does it appear consistently, and it is rarely detected without the presence of other organisms [18], [22], [23], which highlights the importance of studying how polymicrobial infection influences atherosclerosis development.
Prior in vivo studies have been limited to monobacterial infection with P. gingivalis and/or ligature models which are not truly representative of the inherent polymicrobial periodontal disease process [24], [25], [26], [27], [28], [29], [30]. Many monoinfection studies with P. gingivalis in ApoEnull mice have documented genomic bacterial DNA in inflamed aortic tissue with atheroma in the aortic root as well as detecting macrophage, T cells, and Toll-like receptors 2 and 4 (TLR-2, TLR-4), proinflammatory cytokines such as interleukin-6 (IL-6) and vascular cell adhesion molecule-1 (VCAM-1) in the aorta [24], [25], [31], [32].
Because of interspecies interactions, polymicrobial infections have the potential to result in greater deleterious effects on local oral infections and systemic infection in ASVD [1]. Furthermore, a recent report indicated that bacteria, viruses, mycoplasma, and fungi are associated with ASVD development, demonstrating that the microbiology of the atherosclerotic plaque is complex [1]. Polymicrobial infections are believed to accelerate PD, but whether a combined polymicrobial infection with P. gingivalis+T. denticola+T. forsythia will induce enhanced PD, bacteremia, systemic inflammation, and simultaneously accelerate atherosclerosis is as yet unknown.
Previous polymicrobial PD models in rats documented that the synergism of this infectious consortium results in increased alveolar bone resorption when compared to monoinfection [33]. Thus, it is apparent that periodontal disease is always polymicrobial in nature and that a reductionistic approach, using only one bacteria, will never show the true picture of events found throughout the progression of disease [34]. Therefore, the aim of this study is to develop a mouse model of periodontal disease induced via an oral infection with a polymicrobial consortium resulting in measurable effects of microbial colonization, PD progression, and pathogen dissemination through the circulation and the potential risk for invasion of the vasculature and initiation of inflammatory atherosclerosis. The primary emphasis of this study was not designed to examine the ability of an individual organism to induce periodontitis and atherosclerosis but to focus on evaluating polymicrobial infection-induced oral and systemic effects.
Materials and Methods
Microbial Strains and Inocula
P. gingivalis FDC 381, T. denticola ATCC 35404, and T. forsythia ATCC 43037 were used in this study and were routinely cultured anaerobically at 37°C as described previously [33], [35]. Bacterial concentration was determined and cells were resuspended in reduced transport fluid (RTF) at 1010 cells per mL [35]. For topical oral polymicrobial infection, P. gingivalis was mixed with an equal quantity of T. denticola for 5 min; subsequently, T. forsythia was added to the culture tubes containing P. gingivalis and T. denticola, and cells were mixed thoroughly and allowed to interact for an additional 5 min. P. gingivalis, T. denticola, and T. forsythia were then mixed with an equal volume of 4% (w/v) sterile carboxymethylcellulose (CMC; Sigma-Aldrich, St. Louis, MO) in phosphate buffered saline (PBS), and this mixture was used for oral infection (5×109 bacteria per mL) in ApoEnull mice as described previously [35].
ApoEnull Mouse Infection and Oral Plaque Sampling
The polymicrobial oral infection and sampling methodology were described previously [35] (Figure 1). Briefly, proatherogenic ApoEnull mice were used as a model for atherosclerosis [35], [36] and to examine the role of oral pathogen in induction of atherosclerosis [24], [25]. Male ApoE−/− B6.129P2-Apoetm1Unc/J mice, eight-weeks-old (The Jackson Laboratories, Bar Harbor, ME) were kept in groups and housed in microisolator plastic cages. Animals were fed standard chow and water ad libitum, and were randomly distributed into two groups; one for polymicrobial infection (n = 15) and one for sham-infection (n = 10). All mouse procedures were performed in accordance with the approved protocol guidelines by the IACUC of the University of Florida (IACUC Protocol # F173). ApoEnull mice were administered sulfamethoxazole (0.87 mg per mL) and trimethoprim (0.17 mg per ml) daily for 10 days in the drinking water [35] and the mouse oral cavity was rinsed with 0.12% chlorhexidine gluconate (Peridex: 3M ESPE Dental Products, St. Paul, MN) mouth rinse [33], [37] to inhibit endogenous microorganisms and to enhance subsequent colonization of human periodontal bacteria [33]. The polymicrobial inoculum (5×109 combined bacteria per ml; 1×109 cells in 0.2 ml per mouse; 3.3×108 P. gingivalis, 3.3×108 T. denticola, and 3.3×108 T. forsythia) was administered topically to polymicrobial infection group (n = 15) for 4 consecutive days, every other week, for a total of 16 weeks to mimic chronic exposure during this period (Fig. 1). Control uninfected mice (n = 10) were inoculated with sterile 2% CMC only. Oral microbial samples were collected at 7 days postinfection [35]. In order to monitor the infection with minimal disruption of the biofilms, a total of 4 post-infection microbial samples (following weeks 2, 8, 14, and 16) were collected from all infected mice (Figure 1). The samples were collected by swabbing the oral cavity of the mice using a sterile veterinary cotton swab with a head width of 2.6 mm. The teeth and surrounding gingival tissue are swabbed and the cotton tip is immersed in 10∶1 TE buffer. The resuspended bacterial cells are used for DNA extraction and PCR.
Figure 1. Schematic diagram.

Schematic diagram of the experimental design illustrating the length of the study and highlighting important time points. Eight polymicrobial infections (Infection I through Infection VIII) are indicated at 12, 14, 16, 18, 20, 22, 24 and 26 weeks of age. Oral plaque samplings are indicated by PCR at week 2, 8, 14 and 16 following the initial infection. Blood and tissue specimen collection was performed at euthanasia following 16 weeks of infection.
All mice were monitored daily until euthanasia and mice appeared healthy throughout the experimental period. Following 16 weeks of polymicrobial infection, mice were euthanized and the blood, maxilla and mandibles, aorta, heart, spleen, liver, and kidneys were collected. Blood was collected, serum separated, and stored at −20°C for immunoglobulin G (IgG) antibody analysis [37] and serum cholesterol evaluation [25]. The mouse left maxillae and mandibular regions were resected from each mouse, autoclaved, and mechanically defleshed for evaluation of maxillary and mandibular alveolar bone loss by morphometric analysis [37]. The mouse right mandibular region was also resected from each mouse and immediately fixed in 10% neutral buffered formalin and decalcified with Immunocal (Decal Chemical Corporation, Tallman, NY) for 28 days at 4°C for histology and morphometric analysis [37], [38], [39].
Detection of P. gingivalis, T. denticola, and T. forsythia Genomic DNA in Oral Plaque
DNA was isolated from mouse oral plaque samples using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI) following manufacturer’s protocol. PCR was performed with a Bio-Rad thermal cycler using 16S rRNA gene species-specific oligonucleotide primers (P. gingivalis): 5′-TGTAGATGACTGATGGTGAAAACC-3′ (forward), 5′-ACGTCATCCCCACCTTCCTC-3′ (reverse); (T. denticola) TAATACCGAATGTGCTCATTTACAT-3′(forward), 5′-CTGCCATATCTCTATGTCATTGCTCTT-3′ (reverse); and (T. forsythia) 5′-AAAACAGGGGTTCCGCATGG-3′ (forward), 5′-TTCACCGCGGACTTAACAGC-3′ (reverse) [33], [37]. Genomic DNA extracted from these three strains served as positive controls and PCR performed with no template DNA served as negative control. PCR was performed in a 50 µl reaction mixture containing Phusion® High-Fidelity PCR Master Mix (New England Biolabs, Ipswich, MA), template DNA and 0.2 µM of oligonucleotide primers, using the following parameters: 1 cycle of initial denaturation was performed at 98°C for 30 seconds, 35 cycles of a denaturing step at 98°C for 10 seconds, a primer-annealing step at 52°C for 30 seconds and an extension step at 72°C for 30 seconds, a final extension cycle was performed at 72°C for 5 min. PCR products were separated by 1.5% agarose gel electrophoresis and the bands were visualized using a BioRad Gel Doc XR/Chemidoc Gel Documentation System (BioRad, CA, USA). Each PCR assay could detect at least 0.05 pg of DNA standard.
Serum Antibody Analysis
Sera from infected mice (n = 15) at 16 weeks were used to determine immunoglobulin G (IgG) and IgM antibody concentrations against whole cells (formalin-killed) of P. gingivalis, T. denticola, and T. forsythia measured by an enzyme-linked immunosorbent assay (ELISA) [33], [37], [38], [39]. Briefly, whole P. gingivalis, T. denticola, and T. forsythia cells were treated overnight with 0.5% formalin in buffered saline (FK cells), washed, diluted to OD600 0.3, and coated in wells of microtiter plates [33]. Diluted mice sera (1∶100 for IgG and 1∶20 for IgM) were reacted with the bacterial antigen for 2 h at room temperature. After washing, the secondary antibody goat anti-mouse IgG and IgM conjugated to alkaline phosphatase (1∶5000) (Bethyl Laboratories, Montgomery, TX) were added to the plates and the assay developed with p-nitrophenolphosphate (Sigma-Aldrich). The assay reactions were terminated by the addition of 3M NaOH and analyzed at OD405 using a Bio-Rad Microplate Reader. Mice serum antibody concentrations were assessed using a gravimetric standard curve that consisted of 8 mouse IgG and IgM concentrations (Sigma-Aldrich), which were coated onto wells, detected, and developed as described previously [38], [39].
Morphometric Analysis of Periodontal Alveolar Bone Loss
The horizontal alveolar bone resorption (ABR) area and the presence of periodontal intrabony defects were measured by histomorphometry as described previously [37], . Briefly, the maxilla and mandible (n = 10−15) were immersed in 3% (vol/vol) hydrogen peroxide overnight after autoclaving and defleshing the maxillae and mandibles and stained in an aqueous solution of 0.1% methylene blue to delineate the cemento-enamel junction (CEJ) [37], [38], [39]. Digital images of both buccal and lingual root surfaces of all molar teeth were captured under a 10×stereo dissecting microscope (SteReo Discovery V8; Carl Zeiss Microimaging, Inc, Thornwood, NY), after superimposition of buccal and lingual cusps to ensure reproducibility and consistency. The line tool was used to measure the horizontal alveolar bone resorption from the CEJ to the alveolar bone crest (ABC). The surface perimeters of CEJ and ABC were traced using the calibrated line tool (AxioVision LE 29A software version 4.6.3.). Two blinded examiners performed all measurements twice at separate times. The means of the measurements were obtained for each of the two quadrants. Periodontal intrabony defects were detected under a 10×stereo dissecting microscope (SteReo Discovery V8) by an experienced periodontist (JL). The maxillae and mandibles were tilted and stabilized with dental wax to verify the presence of the intrabony defects in buccal and lingual surfaces. Only the presence or absence of intrabony defects was detected because the crevasses in the mouse jaw are too small to measure depth and width.
Detection of Bacterial Genomic DNA in Internal Organs
Genomic DNA was isolated from heart, liver, spleen, abdominal aorta and thoracic aorta tissue samples using the Phenol:Chloroform method of extraction [36]. Tissue cells were lysed using a 1∶50 solution mixture of DNA Extraction Buffer (10 mM Tris-base, 0.1 M EDTA and 0.5% SDS) and Proteinase K Solution (Invitrogen, Carlsbad, CA) shaking at 600 rpm, 55°C, overnight. The supernatant was collected, Phenol:Chloroform:Isoamyl Alcohol (Invitrogen, Carlsbad, CA) was added and this mixture was allowed to shake at 300 rpm, room temperature, for 2 h. The supernatant was once again collected and this step was repeated for 2 additional hours. After spinning down, the top layer was collected, 100% cold ethanol and 10 M Ammonium Acetate was added and the mixture was stored at −20°C overnight. The next day the pellet was washed with 75% cold ethanol and dissolved in molecular grade water. PCR was performed as previously described for oral plaque samples.
Morphometric Analysis of Aortic Atherosclerosis
The heart, aortic arch, thoracic aorta, and abdominal aorta were harvested from the mice after euthanasia. The aorta was cut into two equal parts, arch/thoracic (termed thoracic) and abdominal aortic lengths. The aortic root arising from the heart was also isolated as this is a site of accelerated plaque in ApoEnull mice [40]. The largest plaques are typically detected in the ascending aorta as it emerges from the heart at the level of the aortic valve. Each specimen was then cut into two sections, one for isolation of bacterial genomic DNA and the other for histology. Each section (half of the heart, aortic root, thoracic aorta, and abdominal aorta) were then fixed in 10% neutral buffered formalin, processed and paraffin embedded as previously described [36], [41], [42], [43]. Paraffin embedded samples were cut transversely into cross sections of 5 µm thick cryosections on the Leica EG 1160 Cryostat (Leica Microsystems Inc, Bannockburn, IL), two to three cross sections taken along the length of each segment. Sections were stained with Hematoxylin and Eosin (H&E) for morphometric analysis using an Olympus DP7 color video camera attached to an Olympus BX51 microscope (Olympus America, Center Valley, PA). The mean plaque area, lumen, and internal elastic lamina (IEL) area, intimal and medial thickness ratios, as well as immune cell counts were measured using the Image Pro system MC 6.0 software program (Olympus America, Center Valley, PA), with measurements adjusted to the microscopic objective. The mean total cross-sectional intimal plaque area and the mean intimal thickness normalized to the medial thickness (I/M) was calculated for each arterial section, the mean for measurements from each aorta was calculated for each mouse, and used for statistical analyses. Finally, the images were digitized and analyzed as previously described [36], [44], [45].
Fluorescent In Situ Hybridization (FISH)
FISH allows the detection of bacterial species-specific ribosomal nucleic acid sequences in cells by binding to oligonucleotide probes to their complementary target sequences for in situ detection of bacteria [46]. In situ hybridization was performed on formalin-fixed paraffin-embedded infected right mandible tissue sections using Alexafluor-568 (Invitrogen, Carlsbad, CA) 3′-labeled oligonucleotide probes specific for P. gingivalis 5′-CAATACTCGTATCGCCCGTTATTC-3′ [47], T. dentcola 5′-CATGACTACCGTCATCAAAGAAGC-3′ [48], [49], [50] or T. forsythia 5′-CGTATCTCATTTTATTCCCCTGTA-3′ [47], [51] 16S rRNA. The emission maximum for the Alexafluor fluorescent dye is 578/603 nm, red-orange fluorescence. The protocol was modified accordingly [46]. Tissue sections on slides were deparaffinized in decreasing concentrations of xylene and ethanol: 100% xylene 6 min, 50–50 xylene-ethanol, 100% ethanol, 95% ethanol, 75% ethanol, 50% ethanol 3 min each, running water 5 min. Samples were blocked with Denhardt’s reagent (Fisher, Waltham, MA) and covered with hybridization solution (900 mM NaCl, 20 mM Tris-HCl pH 7.5, 0.01% SDS, 20% formamide) containing 5 µg/ml probe, and incubated for 3 h at 46°C. The probe was rinsed off and incubated in wash buffer (20 mM Tris-HCl, pH7.5, 5 mM EDTA, 0.01% SDS, 0.225 M NaCl), at 48°C for 25 min. Blocking buffer, hybridization buffer and wash buffer all contained protectRNA (Sigma-Aldrich) to protect bacterial RNA from degradation. Tissue sections were counter-stained with DAPI and mounted with mowiol. Stained slides dried overnight before being viewed under a fluorescence microscope. Images were acquired using a spinning disk confocal system connected to a Leica DMIRB microscope with a 63X oil immersion objective, equipped with a Photometrics cascade-cooled EMCCD camera, under the control of the open-source software package µManager (http://www.micro-manager.org/).
Inflammatory Biomarker Serum Amyloid A (SAA)
Sera from polymicrobial-infected and sham-infected mice (n = 6) at 16 weeks were used to detect acute phase reactant SAA concentrations using the Mouse Serum Amyloid A ELISA kit (Kamiya Biomedical Company, Seattle, WA) [52]. The kit contained pre-coated plates containing bound anti-SAA antibody that was used for both calibrations and samples. All procedures were performed at room temperature. Diluted (1∶100) serum samples were added to pre-coated plate and incubated for 1 h. After washing the wells using washing solution from the kit, Enzyme-Antibody Conjugate was added and the plate was incubated for 30 min. After another wash TMB substrate solution from the kit was added and incubated for 10 min. The reaction was stopped using the stop solution contained in the kit. Absorbance was determined at OD450 using a Bio-Rad Microplate Reader. A second order polynomial calibration curve was made using the standards contained in the kit. The kit contained a mouse SAA calibrator of known concentration (10.5 µg/ml) and this was reconstituted and diluted to the suggested dilutions. The diluted samples of known concentrations were then included in the ELISA run along with our samples. The data was analyzed for statistical significance by Mann-Whitney t-test.
Serum Cholesterol Analysis
Serum levels of total cholesterol, triglycerides, glucose, insulin, and serum creatinine were analyzed (Shands Medical Laboratories, Rocky Point Core Lab, Gainesville, FL) after euthanasia in mice infected with periodontal pathogens and sham-infected control [25]. Serum cholesterol levels were determined using the Roche Cholesterol liquid reagent assay on a Roche/Hitachi 917 analyzer (Roche Diagnostics, IN). Serum triglyceride levels were assessed using the Roche triglyceride assay with liquid triglycerides reagents on the Roche/Hitachi 917 and Roche/Hitachi 717 analyzers (Roche Diagnostics; IN). Similarly, glucose serum level was assessed using the Roche Glucose HK liquid assay on Roche/Hitachi 917 and 717 analyzers (Roche Diagnostics; IN). Serum insulin levels were assessed using the Roche Elecsys Insulin assay with the Enzymun-Test Insulin method (Roche Diagnostics; IN). The serum creatinine determination was performed using the Roche Creatinine plus assay with a creatinine HPLC method (Roche Diagnostics; IN).
Histology of Gingival Inflammation
Histomorphologic analysis of the right mandible was examined for gingival inflammation according to previously established protocol [37]. Briefly, right mandibles of polymicrobial-infected (n = 15) and sham-infected (n = 10) mice were removed and fixed in 10% neutral buffered formalin for 24 h. The tissue was decalcified, embedded, sectioned and stained as described previously [38], [39]. The interproximal areas between the molars in each specimen were examined and images were taken at 20X magnification. Degree of inflammation, type of inflammatory cells, apical migration of junctional epithelium, and epithelial hyperplasia were recorded [37].
Statistical Analysis
Antibody analysis and alveolar bone resorption data are presented in figures as means ± standard deviations (SD). Unpaired, two-tailed Student’s t-test was used to compare two independent groups. Simple regression analysis was performed to evaluate the association between bacterial DNA presence and aortic plaque formation. The serum amyloid A data was analyzed for statistical significance by Mann-Whitney t-test. For all statistical analysis, Prism for Windows, version 5.0 (GraphPad Software, San Diego, CA) or Statsview statistic package were used (p<0.05 considered significant).
Results
Monitoring Oral Polymicrobial Infection
Subsequent to treatment with sulfamethoxazole and trimethoprim, ApoEnull mice were monitored for the presence of human periodontal pathogens (P. gingivalis, T. denticola and T. forsythia) by PCR using bacterium-specific primers. Oral plaque samples were obtained from both pathogen infected and sham-infected mice, in the weeks following oral polymicrobial infections (weeks 2, 8, 14, and 16) and screened by PCR. This was done to confirm colonization/infection of the bacterial inocula. PCR analysis of samples collected post-infection at week 2 resulted in (n = 10, 0, 1) detection of infected mice positive for P. gingivalis, T. denticola, and T. forsythia, respectively (Table 1). PCR analysis following week 8 of infections also detected (n = 15, 15, 15) positive mice, with detected bacterial DNA at week 14 (n = 15, 13, 15) and at week 16 (n = 13, 13, 14) in infected mice positive for these three periodontal pathogens, respectively (Table 1). No sham-infected mice were positive for P. gingivalis, T. denticola, or T. forsythia, at any of the time points examined (Table 1).
Table 1. Distribution of ApoEnull mice oral microbial samples positive for bacterial genomic DNA by PCR.
| No. of oral microbial samples positive for bacterial genomic DNA | ||||||||||||
| P. gingivalis | T. denticola | T. forsythia | ||||||||||
| 2 wka | 8 wk | 14 wk | 16 wk | 2 wk | 8 wk | 14 wk | 16 wk | 2 wk | 8 wk | 14 wk | 16 wk | |
| Infectedb n = 15 | 10 | 15 | 15 | 13 | 0 | 15 | 13 | 13 | 1 | 15 | 15 | 14 |
| Controlc n = 10 | – | – | – | – | – | – | – | – | – | – | – | – |
Indicate time points at which oral microbial samples were collected (2, 8, 14, and 16 weeks) following polymicrobial infection for determination of microbial colonization by species specific PCR analysis.
Indicate mice were infected for 8 alternate weeks with P. gingivalis, T. denticola, and T. forsythia. Samples were analyzed using appropriate specific PCR primers with positive and negative controls.
Oral microbial samples were collected from sham-infected control mice periodically and examined for P. gingivalis, T. denticola, and T. forsythia using bacteria specific primers and all mice were negative. - Indicate oral microbial samples negative by PCR analysis. Pg-indicate P. gingivalis; Td-indicate T. denticola; and Tf-indicate T. forsythia.
Antibody Response to P. gingivalis/T. denticola/T. forsythia
To provide additional confirmation of oral polymicrobial infection and to document a bacterial specific humoral response to polymicrobial infection, we evaluated pathogen-specific serum IgG and IgM levels against formalin-killed whole cells for P. gingivalis, T. denticola, and T. forsythia in mice sera from both polymicrobial-infected and sham-infected mice (Figure 2A). All mice in the polymicrobial-infected group for 16 weeks demonstrated significantly (P<0.0001) elevated IgG antibody to P. gingivalis, T. forsythia, and T. denticola compared to the levels in sham-infected control mice. However, anti-P. gingivalis IgG antibody levels were higher than anti-T. forsythia and anti-T. denticola IgG antibody levels. Among the three pathogens, P. gingivalis and T. forsythia induced levels of serum IgG antibody in polymicrobial infected mice approximately 100,000-fold compared to the levels found in the sham-infected mice. Similarly, T. denticola induced levels of serum IgG antibody in polymicrobial infected mice approximately 10,000-fold compared to the levels of sham-infected mice (Figure 2A). None of the polymicrobial-infected mice induced IgM antibody to P. gingivalis/T. forsythia/T. denticola during 16 weeks of infection (data not shown).
Figure 2. Periodontal disease parameters.

(A) Serum IgG antibody levels from ApoEnull mice collected after 16 weeks of oral polymicrobial infection with P. gingivalis, T. denticola, and T. forsythia (n = 10−15). The graph shows the results for IgG antibody reactive with each of the three bacterial species. The bars indicate mean antibody levels in serum from mice infected with consortia or from controls. Significantly higher levels of IgG antibody were seen in polymicrobial-infected mice when compared to sham-infected controls. (B) Area of horizontal alveolar bone resorption in mandibular and maxillary palatal surfaces in ApoEnull mice. Each bar indicates the mean alveolar bone resorption for three molars in each quadrant of infected (n = 15) and sham-infected mice (n = 10). The vertical line denotes standard deviation from mean. *** Asterisks indicate significantly different (P<0.0001) than the sham-infected controls. The IgG antibody levels are expressed as log10. Pg indicates P. gingivalis; Td indicates T. denticola; Tf indicates T. forsythia; Cont indicates sham-infected control mice. Pg/Td/Tf indicates polymicrobial infection with P. gingivalis, T. denticola and T. forsythia. (C, D) Fluorescent in situ hybridization of formalin-fixed paraffin-embedded right mandible section from a polymicrobial-infected mouse using Alexafluor-568 labeled 16S RNA species specific (P. gingivalis, T. dentcola and T. forsythia) oligonucleotide probes. Red fluorescence indicates the presence of bacterial 16S RNA. The white arrow in the zoomed in square points to T. dentcola spirochete morphotype. The white arrow heads point to P. gingivalis coccobacilli morphotype. (E, F) Representative images of maxilla and mandible palatal surfaces showing extensive interproximal intrabony defects in mice infected with Pg/Td/Tf. M1 indicates first molar, M2 indicates second molar and M3 indicates third molar. Black arrows indicates intrabony defects. (G) Sham-infected mouse mandible-palatal showing no intrabony defects.
Alveolar Bone Resorption and Intrabony Defects
The progression of PD resulting from polymicrobial infection with P. gingivalis/T. denticola/T. forsythia was examined by measuring the effects of polymicrobial infection on ABR. The mandible and maxilla of polymicrobial-infected and sham-infected ApoEnull mice were collected at necropsy and morphometric analysis was performed in order to confirm ABR (Figure 2B). The findings demonstrated significantly higher (P<0.0001) palatal horizontal ABR in both the mandible and maxilla of polymicrobial infected mice when compared to sham-infected mice. The measured mean alveolar bone loss area found in the mandible of polymicrobial-infected mice was 0.90 mm2 compared to only 0.66 mm2 in sham-infected mice. The mean bone loss area measured in the maxilla of polymicrobial-infected mice resulted in 0.82 mm2 compared to only 0.55 mm2 in sham-infected mice (Figure 2B, P <0.0001). The presence or absence of intrabony defects was also examined (Figure 2E, F and G). We found that 53.33% of the total surface of both the mandible and maxilla of polymicrobial infected mice contained intrabony defects compared to only 22.5% of the teeth surfaces of sham-infected mice (Table 2).
Table 2. Polymicrobial infection-induced periodontal intrabony defects in ApoEnull mouse.
| Jaw/Surfaces | Poly-microbial Infection | Sham-infected Control |
| Maxilla | 58/90* a (64.44%)** b | 19/60 (31.67%) |
| Mandible | 38/90 (42.22%) | 8/60 (13.33%) |
| Buccal | 38/90 (42.22%) | 11/60 (18.33%) |
| Palatal | 58/90 (64.44%) | 16/60 (26.67%) |
| Total | 96/180 (53.33%) | 27/120 (22.5%) |
aThe frequency was calculated by tooth surfaces containing periodontal intrabony defects out of total tooth surfaces. Calculation of total tooth surface: polymicrobial-infected group has 90 tooth surfaces [15 mice×3 molars×2 sides (buccal, palatal)] and control has 60 (10 mice×3 molars×2 sides).
bNumbers in the parenthesis indicate the percentage of tooth surface with periodontal intrabony defects.
Microbial Systemic Invasion and Detection of Genomic DNA from Aorta, Heart, Liver, and Spleen
The heart, aorta, liver, and spleen were harvested post euthanasia and examined for the presence of P. gingivalis, T. denticola, and T. forsythia genomic DNA via PCR with bacterium-specific primers. Phenol:chloroform DNA extraction was performed on one half of each mouse heart for both polymicrobial-infected and sham-infected mice. PCR analysis revealed 1 out of 15 polymicrobial-infected mice were positive for the existence of P. gingivalis genomic DNA in the heart (Table 3). PCR analysis of thoracic and abdominal aortic samples revealed the presence of a polymicrobial infection for both areas of the aorta in several of the polymicrobial-infected mice. In the thoracic and abdominal aorta 9 out of 15 and 10 out of 15 mice were found to contain P. gingivalis genomic DNA, respectively. T. denticola genomic DNA was found in the thoracic aorta of 12 out of 15 polymicrobial-infected mice. Similarly, analysis of the thoracic and abdominal aorta resulted in a positive finding of T. forsythia DNA in 12 out of 15 and 9 out of 15 mice, respectively. No bacterial genomic DNA was detected in any of the organs sampled from the sham-infected mice (Table 3).
Table 3. Detection of P. gingivalis, T. denticola, T. forsythia genomic DNA in ApoEnull mouse tissue.
| Thoracic Aorta | Abdominal Aorta | Heart | Liver | Spleen | |
| Pg/Td/Tf | Pg/Td/Tf | Pg/Td/Tf | Pg/Td/Tf | Pg/Td/Tf | |
| Infected n = 15 | 9/12/12 | 10/0/9 | 1/0/0 | 7/1/5 | 0/1/0 |
| Control n = 10 | 0/0/0 | 0/0/0 | 0/0/0 | 0/0/0 | 0/0/0 |
Post-euthanasia the thoracic aorta, abdominal aorta, heart, liver and spleen were harvested in liquid nitrogen. In order to assess systemic infection with P. gingivalis, T. denticola and/or T. forsythia, total DNA from each perspective organ was isolated and examined for bacterial genomic DNA by PCR using species specific primers. The numbers indicated with forward slash correspond to the number of mice positive for Pg/Td/Tf respectively.
Fluorescent In situ Hybridization
All three organisms were detected in infected mouse gingival tissues by fluorescent in situ hybridization with oligonucleotide probes to P. gingivalis, T. denticola, and T. forsythia. Clusters of both P. gingivalis and T. denticola are seen (Figure 2C and D). Individual cells of P. gingivalis can be seen (white arrow heads, Figure 2C) as well as individual spiral shaped T. denticola (white arrow Figure 2C). T. forsythia could not be identified in gingival tissues. The presence of these pathogens clearly demonstrates that they invaded gingival epithelium and possibly entered circulation and thus complements the genomic DNA data results (Table 3).
Histomorphometric Analysis of Atherosclerotic Plaque
Atherosclerotic plaque was measured by two experienced researchers (AL, LL) blinded to the experimental groups. Histologic cross sections of the ascending aorta demonstrated increased inflammatory cell invasion in the intimal and adventitial layers of polymicrobial-infected mice and associated increased plaque growth when compared to sham-infected mice. The presence of increased plaque area was proportional to an increase in the number of bacterial species found. The presence of polymicrobial infection produced a trend in increased plaque area (Figure 3A). Intimal/medial thickness ratios indicated a similar trend but were not significant with increased intimal thickness ratios in polymicrobial infections (Figure 3B). The number of invading mononuclear cells in the adventitial (Figure 3C) and intimal (Figure 3D) layers was similarly increased in mice with polymicrobial infections. With polymicrobial infection, mean ascending aortic plaque area was increased with invading foam cell macrophage (p<0.05) (Figure 3E) when compared to sham-infected controls (Figure 3F).
Figure 3. Atherosclerotic vascular disease parameters.

(A) The total aortic plaque area was measured and an increase in plaque area (mm2) was found for polymicrobial-infected ApoEnull mice when compared to sham-infected control mice (P<0.05). (B) Intimal/medial thickness ratios were determined and an increase was observed in the polymicrobial-infected mice compared to sham-infected mice. Polymicrobial infection-induced macrophage infiltration in ApoEnull mice. 4 µm thick paraffin embedded sections of the aortic arch were cut and stained with H&E; (C) Number of macrophage per total adventitial area. (D) Number of macrophage per total intimal area. Control indicates sham-infected ApoEnull mice and Pg+Td+Tf indicate polymicrobial-infected mice. An increase in macrophages was seen in both the adventitial and intimal layers in the polymicrobial-infected mice when compared to the sham-infected mice. Histologic evaluation of 4 µm thick paraffin embedded aortic arch sections stained using H? plaque area is flanked with black arrows, macrophage infiltration is indicated using white arrow with open head and cholesterol crystals are indicated using white arrows with filled heads. (E) Sham-infected control mice tissue (Magnification 40×); (F) Pg, Td, and Tf infection mice tissue (Magnification 40×) containing larger plaque area, increased macrophage infiltration and increased presence of cholesterol crystals, suggesting polymicrobial infection-induced aortic plaque and aortic wall thickness in ApoEnull mice.
Polymicrobial Infection and the Effect of Atherosclerosis Risk Factors
We determined whether established atherosclerosis risk factors were present in order to document whether periodontal pathogens are able to modulate risk factors during 16 weeks of oral infection (Table 4). The serum total cholesterol level for infected mice (n = 13) was significantly higher than sham-infected mice (P<0.05). Similarly, serum total triglyceride levels were also significantly (P<0.001) higher in polymicrobial-infected (n = 15) when compared to sham-infected control mice. The serum glucose level for polymicrobial-infected mice (n = 5) was lower than sham-infected mice but did not reach significance. However, there are no observed differences in insulin or serum creatinine levels between polymicrobial-infected and sham-infected mice (n = 5) (Table 4).
Table 4. Determination of polymicrobial oral Infection-induced risk factors for atherosclerosis.
| Risk Factors | Poly-microbial Infection | Sham-infected Control |
| Total cholesterol, mg per dLa | 508.8±19.27* | 442.4±13.94 |
| Total triglyceride, mg per dLb | 208.7±9.765** | 155.0±8.747 |
| Glucose, mg per dLc | 181.6±16.93 | 207.2±24.52 |
| Insulin, ng per mLc | <0.5 | <0.5 |
| Creatinine, mg per dLc | 0.1–<0.1 | <0.1 |
Values are mean±SD;
indicates P<0.05;
indicates P<0.001;
n = 13 mice per group;
n = 15 mice per group;
n = 5 mice per group.
Polymicrobial Infection-induced Inflammatory Biomarker SAA
Since Serum Amyloid A (SAA) is one of the major acute phase proteins mainly associated with high density lipoproteins, a quantitative determination of SAA was performed in the polymicrobial-infected mice. The results showed a significant (P<0.05) increase (110-fold) in SAA production in polymicrobial-infected mice when compared to the sham-infected mice (Figure 4A).
Figure 4. Serum Amyloid A and Linear regression plot.
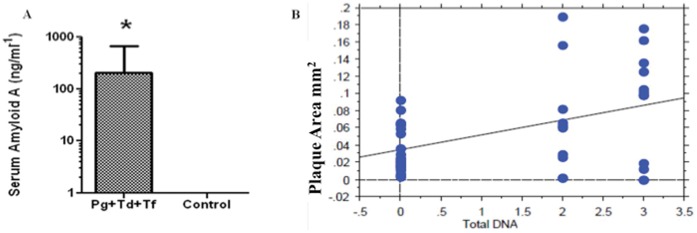
(A) ELISA analysis of antibody levels to Serum Amyloid A (SAA). Pg/Td/Tf infected mice showed approximately 110-fold increase in SAA levels compared to sham-infected mice (P<0.05). (B) A simple regression analysis was performed using the STATview software of the aortic plaque area in mm2 versus the number of bacterial species found by PCR, using species specific primers, in the aorta of polymicrobial infected mice. The simple regression plot demonstrates a positive correlation between the number of detected bacterial species and plaque size in the aorta (R2 = 0.192, P<0.005).
Correlation between Number of Bacterial Species Detected and Atherosclerotic Plaque Area
Regression analysis was performed to assess the association between the presence of polymicrobial species and the progression of atherosclerotic plaque. The total number of species detected by PCR, using species specific primers, (0-controls with no bacterial species found, 1-single bacterial species found, 2-two species found, or 3-three species found) was plotted versus the total aortic plaque area as measured in the aorta for each mouse (Figure 4B). A linear correlation between the number of total bacterial species detected in the aortic tissues and an increase in atherosclerotic plaque area was found (R2 = 0.192, P<0.005) (Figure 4B).
Histology of Gingival Inflammation
Minimal differences were observed in the degree of inflammation, type of inflammatory cells, apical migration of junctional epithelium, and epithelial hyperplasia in right mandible of polymicrobial-infected and sham-infected mice (Data not shown).
Discussion
Observational studies to date support an association between PD and ASVD independent of known confounders [1], [2], [3], [4], [5], [6]. Recent mechanistic in vivo studies clearly demonstrated the plausibility of a direct link between P. gingivalis monoinfections and atherogenesis in an ApoEnull mouse model and have documented biological pathways in induction of inflammatory atherosclerosis [24], [53]. However, human PD is a chronic infection that is exclusively initiated by complex subgingival biofilm including P. gingivalis, T. denticola, T. forsythia, F. nucleatum, and P. intermedia. Moreover, genomic DNA from nine periodontal bacteria and three viruses has been detected in inflammatory ASVD lesions, suggesting that the true nature of atherosclerotic lesions is of a polymicrobial nature. Prior monoinfection studies will not have direct relevance to the polymicrobial nature of periodontitis or serve as a precise model to examine the link between periodontitis and atherosclerosis induction. Thus, several periodontal pathogens as co-infection model are more appropriate indices to examine the role of their ability to induce vascular inflammation and ASVD. Although PD subgingival biofilms are more complex and polymicrobial, we infected with three well-characterized pathogens to examine their synergistic ability to induce periodontitis, dissemination, vascular inflammation, and initial stages of atherosclerosis. Furthermore, the primary emphasis of this study was to focus on evaluating polymicrobial infection-induced oral and systemic effects.
Successful oral colonization of mice with P. gingivalis/T. denticola/T. forsythia is a critical event in pathogenicity. Polymicrobial colonization in the ApoEnull mice oral cavity is consistent with our previous findings in the rat model of polymicrobial PD [33]. These three pathogens as a polymicrobial infection also induced greater ABR and more intrabony defects. Moreover, the alveolar bone crests showed rough and irregular margins which indicates that periodontitis was in progression. These results also provide direct evidence that these three pathogens colonized in the mice oral cavities and induced ABR similar to our previous observations in rats [33].
The polymicrobial oral infection elicited highest profiles for serum IgG antibodies in ApoEnull mice. Among the three pathogens, P. gingivalis infection elicited the highest levels of IgG antibody followed by T. forsythia while the levels of T. denticola antibodies were slightly lower than T. forsythia. These clearly indicate robust colonization of these pathogens and an induction of humoral immune response in infected ApoEnull mice. Furthermore, ApoEnull mice IgG antibody levels induced by these pathogens are significantly higher than rats [33] indicating higher colonization capacity of the individual pathogen in the polymicrobial consortium. Since previous in vivo studies were published with P. gingivalis in a monoinfection models using different infection protocols [24], [25], [26], we could not compare our polymicrobial infection immune response profiles, ABR, and development of atherosclerosis to previous monoinfection data.
Presence of genomic DNA of P. gingivalis/T. denticola/T. forsythia in internal organs such as thoracic and abdominal aorta and liver clearly indicate P. gingivalis/T. denticola/T. forsythia gained access to systemic circulation and to the aorta (bacteremic episodes) from gingival tissues and may have induced vascular wall lesions and promoted the formation of early atheromatous plaque in the aortic arch. Simple regression analysis found a positive correlation (p<0.05) between the number of species detected by genomic DNA of bacteria detected in the aorta and plaque size in thoracic aorta. The linear correlation derived from the presence of multiple species of bacteria and increased aortic plaque size pointed towards the cooperativity existing in the polymicrobial consortium towards initiation of pathogenesis, but defining the degree of cooperation was not the objective of our current study. Enhanced serum acute phase reactant SAA, a biomarker of inflammation [54], [55] demonstrated the role of periodontal pathogens in CVD. In addition, alteration of total cholesterol and triglycerides demonstrate the pathogen’s ability to modulate atherosclerosis risk factors [25]. Furthermore, detection of highly specific molecular signals with pathogen specific oligonucleotide probes provide strong evidence that these pathogens primarily invaded gingival tissues and entered systemic circulation and thereby initiated events inducing atherosclerosis.
In summary, our findings clearly showed (i) polymicrobial colonization/infection in the oral cavity of mice, (ii) specific serum IgG antibody response, (iii) increased ABR and intrabony defects, (iv) in vivo invasion in gingival tissues by FISH, (v) P. gingivalis/T. denticola/T. forsythia genomic DNA in aorta, (vi) induction of serum inflammatory marker SAA, (vii) alteration of serum cholesterol and triglycerides, (viii) induction of atherosclerotic plaque in aortic root, and (ix) the creation of a polymicrobial periodontal disease model in ApoEnull mice. This polymicrobial mouse model system serves as a better platform to further study the effects of putative periodontal pathogens in systemic diseases including atherosclerosis, rheumatoid arthritis, Alzheimer’s disease, and diabetes. Therefore, this study attempts to provide a newer experimental model to evaluate the cascade of events in polymicrobial consortium as well as it adds strong evidence for the involvement of major periodontal pathogens in CVD.
Funding Statement
This study was funded by NIH NIDCR RO1 DE020820 to Kesavalu Lakshmyya. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Lockhart PB, Bolger AF, Papapanou PN, Osinbowale O, Trevisan M, et al.. (2012) Periodontal Disease and Atherosclerotic Vascular Disease: Does the Evidence Support an Independent Association?: A Scientific Statement From the American Heart Association. Circulation. [DOI] [PubMed]
- 2. Friedewald VE, Kornman KS, Beck JD, Genco R, Goldfine A, et al. (2009) The American Journal of Cardiology and Journal of Periodontology Editors’ Consensus: periodontitis and atherosclerotic cardiovascular disease. Am J Cardiol 104: 59–68. [DOI] [PubMed] [Google Scholar]
- 3. Paquette DW, Brodala N, Nichols TC (2007) Cardiovascular disease, inflammation, and periodontal infection. Periodontol 2000 44: 113–126. [DOI] [PubMed] [Google Scholar]
- 4. Mustapha IZ, Debrey S, Oladubu M, Ugarte R (2007) Markers of systemic bacterial exposure in periodontal disease and cardiovascular disease risk: a systematic review and meta-analysis. J Periodontol 78: 2289–2302. [DOI] [PubMed] [Google Scholar]
- 5. Zelkha SA, Freilich RW, Amar S (2010) Periodontal innate immune mechanisms relevant to atherosclerosis and obesity. Periodontol 2000 54: 207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kebschull M, Demmer RT, Papapanou PN (2009) “Gum bug, leave my heart alone!”–epidemiologic and mechanistic evidence linking periodontal infections and atherosclerosis. J Dent Res 89: 879–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Humphrey LL, Fu R, Buckley DI, Freeman M, Helfand M (2008) Periodontal disease and coronary heart disease incidence: a systematic review and meta-analysis. J Gen Intern Med 23: 2079–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bahekar AA, Singh S, Saha S, Molnar J, Arora R (2007) The prevalence and incidence of coronary heart disease is significantly increased in periodontitis: a meta-analysis. Am Heart J 154: 830–837. [DOI] [PubMed] [Google Scholar]
- 9. Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL Jr (1998) Microbial complexes in subgingival plaque. J Clin Periodontol 25: 134–144. [DOI] [PubMed] [Google Scholar]
- 10. Dashper SG, Seers CA, Tan KH, Reynolds EC (2011) Virulence factors of the oral spirochete Treponema denticola. J Dent Res 90: 691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Socransky SS, Haffajee AD (2005) Periodontal microbial ecology. Periodontol 2000 38: 135–187. [DOI] [PubMed] [Google Scholar]
- 12. Kolenbrander PE (2009) Multispecies communities: interspecies interactions influence growth on saliva as sole nutritional source. Int J Oral Sci 3: 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kolenbrander PE, Andersen RN (1989) Inhibition of coaggregation between Fusobacterium nucleatum and Porphyromonas (Bacteroides) gingivalis by lactose and related sugars. Infect Immun 57: 3204–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kolenbrander PE, Andersen RN, Moore LV (1989) Coaggregation of Fusobacterium nucleatum, Selenomonas flueggei, Selenomonas infelix, Selenomonas noxia, and Selenomonas sputigena with strains from 11 genera of oral bacteria. Infect Immun 57: 3194–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kolenbrander PE, Parrish KD, Andersen RN, Greenberg EP (1995) Intergeneric coaggregation of oral Treponema spp. with Fusobacterium spp. and intrageneric coaggregation among Fusobacterium spp. Infect Immun 63: 4584–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Forner L, Larsen T, Kilian M, Holmstrup P (2006) Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J Clin Periodontol 33: 401–407. [DOI] [PubMed] [Google Scholar]
- 17. Iwai T (2009) Periodontal bacteremia and various vascular diseases. J Periodontal Res 44: 689–694. [DOI] [PubMed] [Google Scholar]
- 18. Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ (2000) Identification of periodontal pathogens in atheromatous plaques. J Periodontol 71: 1554–1560. [DOI] [PubMed] [Google Scholar]
- 19. Kozarov E, Sweier D, Shelburne C, Progulske-Fox A, Lopatin D (2006) Detection of bacterial DNA in atheromatous plaques by quantitative PCR. Microbes Infect 8: 687–693. [DOI] [PubMed] [Google Scholar]
- 20. Nonnenmacher C, Stelzel M, Susin C, Sattler AM, Schaefer JR, et al. (2007) Periodontal microbiota in patients with coronary artery disease measured by real-time polymerase chain reaction: a case-control study. J Periodontol 78: 1724–1730. [DOI] [PubMed] [Google Scholar]
- 21. Ott K, Wuhr E (1982) [Clinical study of functional disorders in marginal periodontitis]. Dtsch Zahnarztl Z 37: 634–639. [PubMed] [Google Scholar]
- 22. Fiehn NE, Larsen T, Christiansen N, Holmstrup P, Schroeder TV (2005) Identification of periodontal pathogens in atherosclerotic vessels. J Periodontol 76: 731–736. [DOI] [PubMed] [Google Scholar]
- 23. Figuero E, Sanchez-Beltran M, Cuesta-Frechoso S, Tejerina JM, del Castro JA, et al. (2011) Detection of periodontal bacteria in atheromatous plaque by nested polymerase chain reaction. J Periodontol 82: 1469–1477. [DOI] [PubMed] [Google Scholar]
- 24. Gibson FC, 3rd, Hong C, Chou HH, Yumoto H, Chen J, et al (2004) Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation 109: 2801–2806. [DOI] [PubMed] [Google Scholar]
- 25. Lalla E, Lamster IB, Hofmann MA, Bucciarelli L, Jerud AP, et al. (2003) Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol 23: 1405–1411. [DOI] [PubMed] [Google Scholar]
- 26. Miyamoto M, Ishihara K, Okuda K (2006) The Treponema denticola surface protease dentilisin degrades interleukin-1 beta (IL-1 beta), IL-6, and tumor necrosis factor alpha. Infect Immun 74: 2462–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brodala N, Merricks EP, Bellinger DA, Damrongsri D, Offenbacher S, et al. (2005) Porphyromonas gingivalis bacteremia induces coronary and aortic atherosclerosis in normocholesterolemic and hypercholesterolemic pigs. Arterioscler Thromb Vasc Biol 25: 1446–1451. [DOI] [PubMed] [Google Scholar]
- 28. Mkonyi LE, Bletsa A, Bolstad AI, Bakken V, Wiig H, et al. (2012) Gingival Lymphatic Drainage Protects Against Porphyromonas gingivalis-Induced Bone Loss in Mice. Am J Pathol 181: 907–916. [DOI] [PubMed] [Google Scholar]
- 29.Adamowicz K, Wang H, Jotwani R, Zeller I, Potempa J, et al.. (2012) Inhibition of GSK3 abolishes bacterial-induced periodontal bone loss in mice. Mol Med. [DOI] [PMC free article] [PubMed]
- 30.Saadi-Thiers K, Huck O, Simonis P, Tilly P, Fabre JE, et al.. (2012) Periodontal and Systemic Responses in Various Mice Models of Experimental Periodontitis: Respective Roles of Inflammation Duration and Porphyromonas Gingivalis Infection. J Periodontol. [DOI] [PubMed]
- 31. Li L, Messas E, Batista EL Jr, Levine RA, Amar S (2002) Porphyromonas gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E-deficient murine model. Circulation 105: 861–867. [DOI] [PubMed] [Google Scholar]
- 32. Hayashi C, Gudino CV, Gibson FC, 3rd, Genco CA (2010) Review: Pathogen-induced inflammation at sites distant from oral infection: bacterial persistence and induction of cell-specific innate immune inflammatory pathways. Mol Oral Microbiol 25: 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kesavalu L, Sathishkumar S, Bakthavatchalu V, Matthews C, Dawson D, et al. (2007) Rat model of polymicrobial infection, immunity, and alveolar bone resorption in periodontal disease. Infect Immun 75: 1704–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kuramitsu HK, He X, Lux R, Anderson MH, Shi W (2007) Interspecies interactions within oral microbial communities. Microbiol Mol Biol Rev 71: 653–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nahid MA, Rivera M, Lucas A, Chan EK, Kesavalu L (2011) Polymicrobial infection with periodontal pathogens specifically enhances microRNA miR-146a in ApoE−/− mice during experimental periodontal disease. Infect Immun 79: 1597–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kesavalu L, Lucas AR, Verma RK, Liu L, Dai E, et al. (2012) Increased atherogenesis during Streptococcus mutans infection in ApoE-null mice. J Dent Res 91: 255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bainbridge B, Verma RK, Eastman C, Yehia B, Rivera M, et al. (2010) Role of Porphyromonas gingivalis phosphoserine phosphatase enzyme SerB in inflammation, immune response, and induction of alveolar bone resorption in rats. Infect Immun 78: 4560–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Verma RK, Bhattacharyya I, Sevilla A, Lieberman I, Pola S, et al. (2010) Virulence of major periodontal pathogens and lack of humoral immune protection in a rat model of periodontal disease. Oral Dis 16: 686–695. [DOI] [PubMed] [Google Scholar]
- 39. Verma RK, Rajapakse S, Meka A, Hamrick C, Pola S, et al. (2010) Porphyromonas gingivalis and Treponema denticola Mixed Microbial Infection in a Rat Model of Periodontal Disease. Interdiscip Perspect Infect Dis 2010: 605125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R (1994) ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb 14: 133–140. [DOI] [PubMed] [Google Scholar]
- 41. Bartee MY, Dai E, Liu L, Munuswamy-Ramanujam G, Macaulay C, et al. (2009) 10 M-T7: measuring chemokine-modulating activity. Methods Enzymol 460: 209–228. [DOI] [PubMed] [Google Scholar]
- 42. Dai E, Liu LY, Wang H, McIvor D, Sun YM, et al. (2010) Inhibition of chemokine-glycosaminoglycan interactions in donor tissue reduces mouse allograft vasculopathy and transplant rejection. PLoS One 5: e10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lucas A, Liu L, Macen J, Nash P, Dai E, et al. (1996) Virus-encoded serine proteinase inhibitor SERP-1 inhibits atherosclerotic plaque development after balloon angioplasty. Circulation 94: 2890–2900. [DOI] [PubMed] [Google Scholar]
- 44. Liu L, Dai E, Miller L, Seet B, Lalani A, et al. (2004) Viral chemokine-binding proteins inhibit inflammatory responses and aortic allograft transplant vasculopathy in rat models. Transplantation 77: 1652–1660. [DOI] [PubMed] [Google Scholar]
- 45. Liu L, Lalani A, Dai E, Seet B, Macauley C, et al. (2000) The viral anti-inflammatory chemokine-binding protein M-T7 reduces intimal hyperplasia after vascular injury. J Clin Invest 105: 1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Manz W (1999) In situ analysis of microbial biofilms by rRNA-targeted oligonucleotide probing. Methods Enzymol 310: 79–91. [DOI] [PubMed] [Google Scholar]
- 47. Sunde PT, Olsen I, Gobel UB, Theegarten D, Winter S, et al. (2003) Fluorescence in situ hybridization (FISH) for direct visualization of bacteria in periapical lesions of asymptomatic root-filled teeth. Microbiology 149: 1095–1102. [DOI] [PubMed] [Google Scholar]
- 48. Moter A, Gobel UB (2000) Fluorescence in situ hybridization (FISH) for direct visualization of microorganisms. J Microbiol Methods 41: 85–112. [DOI] [PubMed] [Google Scholar]
- 49. Moter A, Leist G, Rudolph R, Schrank K, Choi BK, et al. (1998) Fluorescence in situ hybridization shows spatial distribution of as yet uncultured treponemes in biopsies from digital dermatitis lesions. Microbiology 144 (Pt 9): 2459–2467. [DOI] [PubMed] [Google Scholar]
- 50. Okuda K, Ishihara K, Nakagawa T, Hirayama A, Inayama Y, et al. (2001) Detection of Treponema denticola in atherosclerotic lesions. J Clin Microbiol 39: 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rudney JD, Chen R, Zhang G (2005) Streptococci dominate the diverse flora within buccal cells. J Dent Res 84: 1165–1171. [DOI] [PubMed] [Google Scholar]
- 52. Yamazaki M, Honjo H, Nakagawa H, Ishiguro YS, Okuno Y, et al. (2007) Mechanisms of destabilization and early termination of spiral wave reentry in the ventricle by a class III antiarrhythmic agent, nifekalant. Am J Physiol Heart Circ Physiol 292: H539–548. [DOI] [PubMed] [Google Scholar]
- 53. Miyamoto T, Yumoto H, Takahashi Y, Davey M, Gibson FC, 3rd, et al (2006) Pathogen-accelerated atherosclerosis occurs early after exposure and can be prevented via immunization. Infect Immun 74: 1376–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kumon Y, Suehiro T, Hashimoto K, Nakatani K, Sipe JD (1999) Local expression of acute phase serum amyloid A mRNA in rheumatoid arthritis synovial tissue and cells. J Rheumatol 26: 785–790. [PubMed] [Google Scholar]
- 55. Meek RL, Urieli-Shoval S, Benditt EP (1994) Expression of apolipoprotein serum amyloid A mRNA in human atherosclerotic lesions and cultured vascular cells: implications for serum amyloid A function. Proc Natl Acad Sci U S A 91: 3186–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]