

Abstract
Usher syndrome type I (USH1) is an autosomal recessive disorder characterized by congenital profound deafness, vestibular areflexia and prepubertal retinitis pigmentosa. The first purpose of this study was to determine the pathologic nature of eighteen USH1 putative splicing variants found in our series and their effect in the splicing process by minigene assays. These variants were selected according to bioinformatic analysis. The second aim was to analyze the USH1 transcripts, obtained from nasal epithelial cells samples of our patients, in order to corroborate the observed effect of mutations by minigenes in patient’s tissues. The last objective was to evaluate the nasal ciliary beat frequency in patients with USH1 and compare it with control subjects. In silico analysis were performed using four bioinformatic programs: NNSplice, Human Splicing Finder, NetGene2 and Spliceview. Afterward, minigenes based on the pSPL3 vector were used to investigate the implication of selected changes in the mRNA processing. To observe the effect of mutations in the patient’s tissues, RNA was extracted from nasal epithelial cells and RT-PCR analyses were performed. Four MYO7A (c.470G>A, c.1342_1343delAG, c.5856G>A and c.3652G>A), three CDH23 (c.2289+1G>A, c.6049G>A and c.8722+1delG) and one PCDH15 (c.3717+2dupTT) variants were observed to affect the splicing process by minigene assays and/or transcripts analysis obtained from nasal cells. Based on our results, minigenes are a good approach to determine the implication of identified variants in the mRNA processing, and the analysis of RNA obtained from nasal epithelial cells is an alternative method to discriminate neutral Usher variants from those with a pathogenic effect on the splicing process. In addition, we could observe that the nasal ciliated epithelium of USH1 patients shows a lower ciliary beat frequency than control subjects.
Introduction
Usher syndrome (USH) is an autosomal recessive disorder characterized by sensorineural hearing loss, retinitis pigmentosa (RP) and variable vestibular dysfunction. USH is clinically and genetically heterogeneous and is the most common form of deaf-blindness of genetic origin, representing 50% of cases [1]. This disease shows a prevalence of 3.2–6.2/100000 [2], [3], [4].
Three clinical types of USH (types I, II and III; USH1, USH2 and USH3) are recognized, mainly on the basis of the severity and progression of hearing loss, the age of onset of RP and the presence of vestibular dysfunction [5].
Usher syndrome type I (USH1) is the most severe form of the disease and it is characterized by congenital profound deafness, vestibular areflexia and prepubertal onset of retinitis pigmentosa. To date, nine loci (USH1B-USH1K) have been mapped and six genes have been identified: MYO7A (USH1B): MIM#276903; USH1C (USH1C): MIM# 605242; CDH23 (USH1D): MIM# 605516; PCDH15 (USH1F): MIM# 605514; USH1G (USH1G): MIM# 607696; and CIB2 (USH1J): MIM# 605564 [reviewed in 6], [7], [8].
Many mutations in MYO7A, USH1C, CDH23, PCDH15 and USH1G have been identified by several screenings performed in USH1 patients (http://grenada.lumc.nl/LOVD2/Usher_montpellier/). The consequences of missense, silent and intronic changes many times are unknown and additional studies are needed to know the pathogenicity of these variants.
The use of minigene assays has been shown to be a useful approach to determine the effect of these variants on the splicing process, when genes present a restricted expression profile (photoreceptors and inner hair cells, in the case of USH) and human specific tissue samples are difficult to obtain [9].
Cohn et al. [10] demonstrated the presence of eight Usher proteins in nasal ciliated epithelium using immunochemistry with fluorescent antibodies. Subsequently, Vaché et al. [11] provided evidence that splicing mutations occurring in most USH genes can be identified through ex vivo analysis of mRNA from nasal epithelial cells.
On the other hand, the cilium in photoreceptors appears ultrastructurally and molecularly very similar to the nasal ciliated epithelium. The cilia are distributed around the human body and it has been reported that an abnormality in ciliary function may be linked to the nasal cilia abnormalities, as well as to the retinal degeneration [12]. There is evidence that immotiles nasal cilial can be associated with USH1 [13].
In our cohort of patients, we identified different pathogenic variants and some putative splicing mutations in USH1 genes [14], [15], [16], [17], [18].
The first purpose of the present work was to determine the pathogenic nature of selected variants and their effect in the splicing process by minigene assays. The second aim was to analyze the USH1 transcripts, obtained from the nasal epithelium cells of our patients, in order to corroborate the observed effect of mutations by minigenes in patient’s tissues. The third goal of this study was to evaluate the nasal ciliary beat frequency in eight USH1 patients and compare it with thirty control subjects.
Materials and Methods
Ethics Statement
This study was approved by the ethic committees of the Instituto de Investigación Sanitaria IIS-La Fe (Valencia, Spain) and the General and University Hospital (Valencia, Spain). Written consent was obtained from all subjects. Clinical investigation was conducted according to the principles expressed in the Declaration of Helsinki.
Selection of Variants
The first selection of sequence variations for functional studies was carried out on the following criteria: variants found in a homozygous state or in trans with a disease-causing mutation, cosegregation with the disease, not found in 200 control chromosomes or sequence variation located in exon or introns that may affect the mRNA processing. All changes have been characterized in our cohort of patients.
Computational Analysis of Splicing Variants
To analyze the effect of variants in the splice prediction and the recognition of donor and acceptor sites, in silico analyses were performed. Four programs were used: Neural Network SPLICE (NNSplice) 0.9 from the Berkeley Drosophila Genome Project (available at http://www.fruitfly.org/seq_tools/splice.html), Human Splicing Finder (HSF)-Version 2.4 (available at http://www.umd.be/HSF/), NetGene2 (available at http://www.cbs.dtu.dk/service/NetGene2/) and Spliceview (available at http://zeus2.itb.cnr.it/~webgene/wwwspliceview_ex.html).Score-values were calculated taking into account the bioinformatic predictions, from 1 to 4, depending on how many of the four programs have predicted changes on the splice sites.
Minigene Constructions and Expression
Minigene constructions based on the pSPL3 exon trapping vector (kindly provided by Dr. I Botillo and Dr. S. Tuffery-Giraud) were used to investigate the implication of selected variants on the mRNA processing. These variants were selected based on the bioinformatic predictions.
For these putative splicing variants, the exon and intronic flanking sequences were amplified from the patient’s DNA, using the High Fidelity Phusion polymerase (Finnzymes, Espoo, Finland) with primers detailed in Table 1. Amplicons were inserted between the XhoI/NheI restriction sites, using T4 DNA ligase (Invitrogen Corporation, Carlsbad, CA). We only could obtain the wild type (WT) insert for the variant c.3717+2dupTT (PCDH15) and the mutant insert for changes c.3652G>A and c.5581C>T (MYO7A), c.1086-12G>A (USH1C), c.8722+1delG (CDH23) and c.1304_1305insC, c.2868+5G>A and c.1737C>G (PCDH15). The corresponding WT or mutant insert was obtained by site-directed mutagenesis using primers detailed in Table 2. Minigene constructions were confirmed by direct sequencing in both directions with BigDye Terminator 3.1 cycle sequencing kit from Applied Biosystems (Carlsbad, USA). Sequence reactions were analyzed on a capillary ABI 3500xl Genetic Analyzer (Applied Biosystems) according to the manufacturer’s instructions. Afterwards, minigenes were transfected into COS-7 cells as described before by Jaijo et al. [19]. RNA extraction and RT-PCR analysis was performed as previously described [20], [21]. All experiments were performed in duplicate.
Table 1. Primers used to amplify the specific insert.
| Sequence variant | Primer | Sequence 5′->3′ | Size (bp) |
| c.6_9dup (p.L4DfsX39)MYO7A | L4DfsX39_D_XhoI | AAGAAT CTCGAG AGTGGCTGAGAGAAGAATTC | 563 |
| L4DfsX39_R_NheI | AAGAAT GCTAGC GGATCCAGAGATGTGTGTAA | ||
| c.470G>A (p.S157N) MYO7A | S157N_D_XhoI | AAGAAT CTCGAG AGACTCCACCTCCCTCTTCA | 668 |
| S157N_R_NheI | AAGAAT GCTAGC GCTAATGTGAGCTTTGGTAGC | ||
| c.640G>A (p.G214R) MYO7Ac.721C>G (p.R241G) MYO7A | G214R & R241G_D_XhoI | AAGAAT CTCGAG GAAGGTGAAGGAGAGTGCG | 703 |
| G214R & R241G_R_NheI | AAGAAT GCTAGC TTCATGGTGGGATTTCCAGC | ||
| c.1097T>C (p.L366P)MYO7A | L366P_D_XhoI | AAGAAT CTCGAG TGACATGCTGGAGGGAGTTA | 608 |
| L366P_R_NheI | AAGAAT GCTAGC CTGACCAAAGCAGGCCAAAA | ||
| c.1342_1343delAG (p.S448LfsX2) MYO7A | S448LfsX2_D_XhoI | AAGAAT CTCGAG TCTCCAGGCAGAGGGAACAG | 524 |
| S448LfsX2_R_NheI | AAGAAT GCTAGC AGCCAGGCTCAGCTGACCTT | ||
| c.3508G>A (p.E1170K) MYO7A | E1170K_D_XhoI | AAGAAT CTCGAG GAGGTGCTTTATGCCCGATG | 545 |
| E1170K_R_NheI | AAGAAT GCTAGC GAGTGTGGGGAAAAGAGGTG | ||
| c.3652G>A (p.G1218R) MYO7A | G1218R_D_XhoI | AAGAAT CTCGAG ATAGATGGTGGAGCTGAGAG | 370 |
| G1218R_R_NheI | AAGAAT GCTAGC GCACTGGACACACACACACA | ||
| c.5581C>T (p.R1861X) MYO7A | R1861X_D_XhoI | AAGAAT CTCGAG ACTAGTTGCATCTGGCTGTC | 543 |
| R1861X_R_NheI | AAGAAT GCTAGC ATCCTGAGCAGCTGCAGGAT | ||
| c.1086-12G>A USH1C | USH1C_D_XhoI | AAGAAT CTCGAG ACAAGCTCGGGGACTTTGCT | 438 |
| USH1C_R_NheI | AAGAAT GCTAGC ATGGTCTCTGACCCACAGCT | ||
| c.2289+1G>A CDH23 | c.2289_D_XhoI | AAGAAT CTCGAG CAAATATTTCCTCCTCATGG | 636 |
| c.2289_R_NheI | AAGAAT GCTAGC GCAGGAGAGAAAGTATTTCA | ||
| c.6049G>A (p.G2017S) CDH23 | G2017_D_XhoI | AAGAAT CTCGAG CTTGCTCACCACTCTCATGT | 594 |
| G2017_R_NheI | AAGAAT GCTAGC GTATCGTCTCCAAGCCTCTT | ||
| c.8722+1delG CDH23 | c.8722_D_XhoI | AAGAAT CTCGAG TTCTGGGCAACACTCCGTGA | 1226 |
| c.8722_R_NheI | AAGAAT GCTAGC GGATGGACGGATAACTAATGG | ||
| c.1304_1305insC PCDH15 | c.1304_D_XhoI | AAGAAT CTCGAG CAGAAGACTGAAGCAATTAAGCC | 647 |
| c.1304_R_NheI | AAGAAT GCTAGC ATATACTGAATATCTGCAAGCTG | ||
| c.2868+5G>A PCDH15 | c.2868_D_XhoI | AAGAAT CTCGAG ATTCCAGTGGAACGGCCTTC | 533 |
| c.2868_R_NheI | AAGAAT GCTAGC TATGCTCTGTACCTGTGGATG | ||
| c.3717+2dupTT PCDH15 | c.3717_D_XhoI | AAGAAT CTCGAG CAGAATTATACCTATGTCCC | 635 |
| c.3717_R_NheI | AAGAAT GCTAGC AGTGACTAACAATCTGAGTG | ||
| c.521A>G (p.N174S) PCDH15 | N174S_D_XhoI | AAGAAT CTCGAG TTCTTCGCCCATAGCAAGA | 605 |
| N174S_R_NheI | AAGAAT GCTAGC TGAGGCATATTATACCTATG | ||
| c.1737C>G (p.Y579X) PCDH15 | Y579X_D_XhoI | AAGAAT CTCGAG AGTATTGTAACAGGACACAG | 596 |
| Y579X_R_NheI | AAGAAT GCTAGC CCTCTGATATTGTCCTCTTC |
Tails added at the beginning of the primer are indicated in bold.
Enzyme restriction sites used in this study are indicated in italics.
Table 2. Primers used for the site-directed mutagenesis.
| Sequence variants | Primer | Sequence 5′->3′ |
| c.3717+2dupTT PCDH15 WT | c.3717-D-WT | GCAAAGCCGATGTACTCGT-AAGTAGATAAAACTTCAGG |
| c.3717-R-WT | CCTGAAGTTTTATCTACTT-ACGAGTACATCGGCTTTGC | |
| c.5581C>T (p.R1861X) MYO7A MUT | R1861X-D-MUT | GCTTCCTGCAGTCCCGAAAGCACTGCCCA |
| R1861X-R-MUT | TGGGCAGTGCTTTCGGGACTGCAGGAAGC | |
| c.3652G>A (p.G1218R) MYO7A MUT | G1218R-D-MUT | AACTTCATCCACAGGGGCCCGCCCG |
| G1218R-R-MUT | CGGGCGGGCCCCTGTGGATGAAGTT | |
| c.1086-12G>A USH1C MUT | c.1086-D-MUT | CCAGTAACAGGCATGGGGATCTCATTTTAGGATTGTAG |
| c.1086-R-MUT | CTACAATCCTAAAATGAGATCCCCATGCCTGTTACTGG | |
| c.8722+1delG CDH23 MUT | c.8722-D-MUT | GGTCTTCACCATGG-TAGGGCCTGGCAGC |
| c.8722-R-MUT | GCTGCCAGGCCCTA-CCATGGTGAAGACC | |
| c.1304_1305insC PCDH15 MUT | c.1304-D-MUT | GTAGCTCTGGACAAGGACATAGAAGACTGTAAGTTAAATACATATTTTGC |
| c.1304-R-MUT | GCAAAATATGTATTTAACTTACAGTCTTCTATGTCCTTGTCCAGAGCTAC | |
| c.2868+5G>A PCDH15 MUT | c.2868-D-MUT | GAAGATGCAGACCCTCCTGTAAATAGAAGGCATTGATTAATATT |
| c.2868-R-MUT | AATATTAATCAATGCCTTCTATTTACAGGAGGGTCTGCATCTTC | |
| c.1737C>G (p.Y579X) PCDH15 MUT | Y579X-D-MUT | TGATAGTCGGGCGGACTTAGGCACTCACGG |
| Y579X-R-MUT | CCGTGAGTGCCTAAGTCCGCCCGACTATCA |
The nucleotides that have been modified are indicated in bold.
The positions of deleted nucleotides are represented with (−).
Nasal Epithelial Cells Samples from Controls and Patients
A total of thirty-eight fresh nasal samples were obtained from eight USH1 patients, five patients with mutations in MYO7A gene and three patients with mutations in the CDH23 gene, and thirty control subjects. The nasal samples were obtained from the middle nasal concha using curettage without local anesthesia, in a period during which acute infection was absent [12], in the ENT service of the Hospital General de Valencia, Spain.
RNA and Sequence Analysis
RNA extraction from nasal epithelial cells and RT-PCR analysis was performed as previously described [11]. cDNA was used as template in nested PCR reactions with specific primers in order to amplify the regions containing the mutation (Table 3). PCR products were tested on 1.5% agarose gel and purified by ExoSAP. PCR products were sequenced and analyzed. All experiments were performed in duplicate.
Table 3. Primers used to amplify fragments of MYO7A and CDH23 cDNAs from nasal cells.
| Gene | Exons | Primers | Sequence 5′->3′ | Size(bp) |
| MYO7A | 1–5 | MYO_EXP_1-5-EXT-D | AGAGACAAGAGACACACACA | 628 |
| MYO_EXP_1-5-EXT-R | CTTCTTGTTGGTATACTGGC | |||
| MYO_EXP_1-5-INT-D | TAGAACGAGACTTGGAGCCA | 490 | ||
| MYO_EXP_1-5-INT-R | TTCACAGCCACCAGGATGGA | |||
| 3–9 | MYO_EXP_3-9-EXT-D | ACCATGTGTGGATGGACCTG | 937 | |
| MYO_EXP_3-9-EXT-R | CTTCGAGATCTCCCAGTTCT | |||
| MYO_EXP_3-9-INT-D | ACTCTGGGCAGGTCCAGGT | 806 | ||
| MYO_EXP_3-9-INT-R | TGTTGGCGTACTCCTGGCT | |||
| 8–14 | MYO_EXP_8-14-EXT-D | TGTTCTACTGCATGCTGGAG | 906 | |
| MYO_EXP_8-14-EXT-R | CCTGCAAAATGGTTGATGCC | |||
| MYO_EXP_8-14-INT-D | AAGAAGAAGCTGGGTTGGG | 811 | ||
| MYO_EXP_8-14-INT-R | TTGGCGTTGAGCTTGTGCT | |||
| 25–33 | MYO_EXP_25-33-EXT-D | ATGAGACCCTGGGCAAGAAG | 1136 | |
| *MYO_EXP_25-33-EXT-R | CTTCTGGGCATCAGTTCTCC | |||
| MYO_EXP_25-33-INT-D | AGGGCCAGAAGAAGAGCAGT | 924 | ||
| MYO_EXP_25-33-INT-R | CGCTCCAGGATCATCTCAGA | |||
| 38–45 | MYO_EXP_38-45-EXT-D | TCCTATGACTACTTCAGGCC | 1010 | |
| MYO_EXP_38-45-EXT-R | GGGGAAGTAGGACTTGTCCT | |||
| MYO_EXP_38-45-INT-D | TCAAGCAGGCGCTGCTCAAGA | 835 | ||
| MYO_EXP_38-45-INT-R | CGTGCACTTGTGGTAGCCTC | |||
| CDH23 | 18–24 | CDH_EXP_18-24-EXT-D | ATGAACAGATATCCAATGGGC | 790 |
| CDH_EXP_18-24-EXT-R | GGTTCTGAAAGGTGGGGTCA | |||
| CDH_EXP_18-24-INT-D | AATGACAACCCTCCCACCTT | 627 | ||
| CDH_EXP_18-24-INT-R | CACTGCTGGTGGCTTTCAGA | |||
| 43–49 | CDH_EXP_43-49-EXT-D | CATCAACGACAACGACCCTGT | 1084 | |
| CDH_EXP_43-49-EXT-R | GGTTGAGGTCGTGGTCAATG | |||
| CDH_EXP_43-49-INT-D | TCTGTGAAGGACAACCCGGA | 708 | ||
| CDH_EXP_43-49-INT-R | GAATGGTGACAATGGCTGTG | |||
| 57–63 | CDH_EXP_57-63-EXT-D | CTCATCTTGGTGGCCAGCGAC | 1015 | |
| CDH_EXP_57-63-EXT-R | CCGGCAGCCGGACAGAGAT | |||
| CDH_EXP_57-63-INT-D | TTCATCGTCAAGGCCTCCAG | 713 | ||
| CDH_EXP_57-63-INT-R | TAGCTGCTCCTTGTTCTCA |
The primer MYO_EXP_25-33-EXT-R was designed by Vaché et al. [11].
Nomenclature of Variants
The sequences obtained were compared with the consensus sequence NM 000260.3 for MYO7A, NM 153676.2 for USH1C, NM 022124.3 for CDH23 and NM 033056.3 for PCDH15.
For the protein nomenclature, we used the Mutalyzer 2.0 beta-21 program (available at https://mutalyzer.nl/).
Nasal Ciliary Beat Frequency
Ciliary beat frequency was measured as described by Armengot et al. [12] in eight USH1 patients and thirty healthy subjects. The beat pattern was observed using high-resolution digital high-speed video imaging. Values were expressed in Hertz (Hz) as the mean ± standard error of mean.
Statistical Analysis
Ciliary beat frequencies were analyzed by Kruskal-Wallis test followed by Dunns post-hoc test. When only two groups were compared, Mann-Whitney U test was used. Significance levels were set at p<0.05. Data were analyzed with statistical analysis software GraphPad Prism (Version 5.01, San Diego, USA).
Results
Computational Analysis
One hundred and eight variants identified in the USH1 genes were studied with bioinformatic tools (SpliceView, NNSplice, NetGene2 and HSF) in order to analyze the effect of these variants in the splice prediction and the recognition of donor and acceptor sites. Nine MYO7A, three CDH23, five PCDH15 and one USH1C variants were predicted to alter the splicing mechanism creating or eliminating donor/acceptor splice sites, see table 4. Considering the four programs (Splice View, NNSplice, NetGene2 and HSF) the score-values were calculated. Eight out of the eighteen variants showed the highest score (4), five of them showed a score of 3, two of the variants were observed to show a score of 2 and the three remaining changes showed a score of 1.
Table 4. Results from four different bioinformatic programs used to predict the effect on the splicing process.
| Sequence variants | Type ofsplicesite | NetGene2 | HSF | NNSplice | Splice View | Score |
| c.6_9dup (p.L4DfsX39) MYO7A [14] | Acceptor | Score for acceptor siteincreases from 77 to 82 | The WT consensus sequenceis not recognized | One donor site is not recognized | New acceptor sites are created and other acceptor sites are not recognized | 3 |
| c.470G>A (p.S157N) MYO7A [14] | Donor | Score for the main donorsite decreases from 93 to 60 | Score for donor site decreasesand a new acceptor site iscreated | The main donor site isnot recognized | The main donor site isnot recognized | 4 |
| c.640G>A (p.G214R) MYO7A [22] | Acceptor | Neutral | The WT consensus sequenceis not recognized | A new acceptor site is created | Neutral | 1 |
| c.721C>G (p.R241G) MYO7A [15] | Donor | Three new donor siteare created | A new acceptor siteis created | Score for the main acceptor site decreases from 81 to 59 | A new donor site is created | 4 |
| c.1097T>C (p.L366P) MYO7A [15] | Acceptor | Score for the main acceptorsite decreases from 83 to 77 | Score for the acceptorsite decreases | A new acceptor site is created | Neutral | 3 |
| c.1342_1343delAG (p.S448LfsX2) MYO7A [14] | Donor | The main donor site is notrecognized | The main donor site and theacceptor site arenot recognized | The main donor site isnot recognized | The main donor site isnot recognized | 4 |
| c.3508G>A (p.E1170K) MYO7A [23] | Acceptor | Score for the main acceptorsite decreases from 85 to 77 | Neutral | Neutral | Neutral | 1 |
| c.3652G>A (p.G1218R) MYO7A [15] | Acceptor | A new acceptor siteis created | A new acceptor siteis created | A new acceptor site is created | A new acceptor site is created | 4 |
| c.5581C>T (p.R1861X) MYO7A [22] | Acceptor | Score for the main acceptorsite decreases from 77 to 72 | Neutral | Neutral | Neutral | 1 |
| c.1086-12G>A USH1C [17] | Acceptor | A new acceptor siteis created | Score for acceptor sitedecreases | Score for the main acceptor site decreases from 48 to 45 | The main acceptor site isnot recognized | 4 |
| c.2289+1G>A CDH23 [24] | Donor | The main donor site isnot recognized | The main donor site is notrecognized | The main donor site isnot recognized | The main donor site isnot recognized | 4 |
| c.6049G>A (p.G2017S) CDH23 [25] | Donor | Neutral | The main donor site is notrecognized | The main donor site isnot recognized | Score for the maindonor site decreasesfrom 89 to 84 | 3 |
| c.8722+1delG CDH23 [16] | Donor | Neutral | The main donor site is notrecognized | The main donor site isnot recognized | The main donor site isnot recognized | 3 |
| c.521A>G (p.N174S) PCDH15 [18] | Acceptor | Score for the main acceptorsite decreases from 92 to 87 | The main acceptor site isnot recognized | Neutral | Neutral | 2 |
| c.1304_1305insC (p.T436YfsX12) PCDH15 [18] | Donor | Score for the main donorsite decreases from 63 to 51 | The WT consensus sequenceis not recognized | Score for the maindonor site decreasesfrom 99 to 94 | Score for the main donor site decreases from 81 to 79 | 3 |
| c.1737C>G (p.Y579X) PCDH15 [18] | Donor | Score for the main donorsite decreases from 52 to 37 | The main donor siteis not recognized | Neutral | Neutral | 2 |
| c.2868+5G>A PCDH15 [18] | Donor | The main donor site is notrecognized and a newdonor site is created | The main donor siteis not recognized. | The main donor site isnot recognized. | The main donor site isnot recognized. | 4 |
| c.3717+2dupTT PCDH15 Present study | Donor | The main donor site isnot recognized | The main donor siteis not recognized | The main donor site isnot recognized | The main donor site isnot recognized | 4 |
Minigene Constructions
In order to confirm the bioinformatic predictions, minigenes were constructed and the different splicing products were analyzed. The minigene assays showed that only seven of them were affecting the mRNA processing: three MYO7A variants, c.470G>A, c.1342_1343delAG and c.3652G>A, three CDH23 changes, c.2289+1G>A, c.6049G>A and c.8722+1delG, and one PCDH15 variant, c.3717+2dupTT, see Table 5.
Table 5. Effects of USH1 variants on splicing.
| Putative splicing variants | Scoreaccording tobioinformatictools | Effect on RNA level according to minigene results | Effect on protein level according to minigene results | Effect on RNA level according to nasal cells results | Effect on protein level according to nasal cells results |
| c.6_9dup (p.L4DfsX39) MYO7A | 3 | No effect on splicing | p.L4DfsX39 | No effect on splicing | p.L4DfsX39 |
| c.470G>A (p.S157N) MYO7A | 4 | Exon skipping | p.T96WfsX29 | – | – |
| c.640G>A (p.G214R) MYO7A | 1 | No effect on splicing | p.G214R | No effect on splicing | p.G214R |
| c.721C>G (p.R241G) MYO7A | 4 | No effect on splicing | p.R241G | – | – |
| c.1097T>C (p.L366P) MYO7A | 3 | No effect on splicing | p.L366P | – | – |
| c.1342_1343delAG (p.S448LfsX2) MYO7A | 4 | The mutation removes the main donorsite and creates a new donor splice-siteleading to a deletion of 24bp | p.N443_E450del | – | – |
| c.3508G>A (p.E1170K) MYO7A | 1 | No effect on splicing | p.E1170K | No effect on splicing | p.E1170K |
| c.3652G>A (p.G1218R) MYO7A | 4 | The mutation does not recognize the acceptorsite and a new acceptor splice-site is createdleading to a deletion of 103-bp in the 5′end of exon 29 | p.Y1211AfsX18 | – | – |
| c.5581C>T (p.R1861X) MYO7A | 1 | No effect on splicing | p.R1861X | No effect on splicing | p.R1861X |
| * c.5856G>A (p.K1952K) MYO7A | 4 | Exon skipping | p.A1915_K1952del | Exon skipping | p.A1915_K1952del |
| c.1086-12G>A USH1C | 4 | No effect on splicing | Neutral | – | – |
| c.2289+1G>A CDH23 | 4 | This variant does not recognize the main donorsite and a new donor splice-site is createdinserting the first 149 nucleotides of intron21+ Exon skipping | p.N765SfsX35+ p.E727KfsX9 | Transcript with an insertionof the first 149 nucleotides of intron 21 plus the last 54 nucleotides of the same intron | p.N765SfsX35 |
| c.6049G>A (p.G2017S) CDH23 | 3 | Exon skipping | p.T1976_G2017del | Probable NMD | - |
| c.8722+1delG CDH23 | 3 | Deletion of the last nucleotide of exon 60 | p.S2909AfsX43 | Deletion of the lastnucleotide of exon 60 | p.S2909AfsX43 |
| c.521A>G (p.N174S) PCDH15 | 2 | No effect on splicing | p.N174S | – | – |
| c.1304_1305insC (p.T436YfsX12) PCDH15 | 3 | No effect on splicing | p.T436YfsX12 | – | – |
| c.1737C>G (p.Y579X) PCDH15 | 2 | No effect on splicing | p.Y579X | – | – |
| c.2868+5G>A PCDH15 | 4 | No effect on splicing | Neutral | – | – |
| c.3717+2dupTT PCDH15 | 4 | c.3717+2dupTT does not recognize the maindonor site and creates a new donor splice-sitethat includes the first 52 nucleotides ofintron 27+ Exon skipping | p.V1242RfsX2+ p.A1168_L1239del | – | – |
Not performed in this study are indicated with (−).
NMD: Nonsense mediated decay.
c.5856G>A (p.K1952K, MYO7A) was previously analyzed by Jaijo et al. [26].
c.470G>A (p.S157N, MYO7A)
The minigene assay showed that the processing of the WT minigene generated two main fragments: the correct processing (band A) and the exon skipping (band B). The mutant minigenes only showed one fragment corresponding to the skipping of exon 5 (band B) (Fig. 1A). If this mutant transcript was translated, it would produce a truncated protein of only 123 amino acids in length, p.T96WfsX29.
Figure 1. In vitro splicing assays for the seven splicing mutations identified in the USH1 genes.

Gel electrophoresis shows the different splicing processes for WT minigene and mutants constructions. COS-7 cell transfection experiments were performed in duplicate. A. c.470G>A (p.S157N, MYO7A ). Band A is the correct transcript of exon 5 (MYO7A). Band B is the skipping of involved exon. B. c.1342_1343delAG (p.S448LfsX2, MYO7A ). Band A is the correct transcript corresponding to the exon 12 (MYO7A). Band B is the skipping of exon 12. C. c.3652G>A (p.G1218R MYO7A ). Band A is the correct transcript of exon 29 (MYO7A). The band B is the exon skipping. Band C is the heteroduplex formation from band A and band B. Band D is the aberrant splicing process that include the deletion of 103-bp of 5′end of exon 29. D. c.2289+1G>A ( CDH23 ). Band A is the normal transcript of exon 21 (CDH23). Band B is the skipping of exon 21. Band C is the aberrant splicing process that includes the first 149 nucleotides of intron 21. E. c.6049G>A (p.G2017S, CDH23 ). Band A is the correct transcript of exon 46 (CDH23). Band B is the skipping of exon 46. Band C is the heteroduplex formation from the band A and B. F. c.8722+1delG ( CDH23 ). Band A is the correct splicing process of exon 60 (CDH23). Band B is the abnormal splicing process of exon 60 that shows a deletion of the last nucleotide (G) of the involved exon. G. c.3717+2dupTT ( PCDH15 ). Band A is the correct transcript of exon 27 (PCDH15). Band B is the skipping of the involved exon. Band C is the transcript corresponding to the new donor splice site from exon 27 plus the first 52 nucleotides of the intron 27. Band D is the heteroduplex formation from the band B and the band D.
c.1342_1343delAG (p.S448LfsX2, MYO7A)
In vitro analyses confirmed that the WT transcript contained the correct exon (band A) but the c.1342_1343delAG construction generated several bands. We were only able to sequence band B that corresponded to the exon 12 with a partial deletion. The mutation removed the main donor site and created a new donor splice-site leading to a deletion of 24-bp (Fig. 1B). Thus, the new myosin VIIA protein generated, if this aberrant transcript was translated, it would be of 2207 amino acids in length, p.N443_E450del.
c.3652G>A (p.G1218R, MYO7A)
In vitro experiments showed that WT minigene generated two different transcripts: one of them corresponded to the correct transcript (band A) and the smallest and strongest transcript was the skipping of exon 29 (band B). A third band (band C) was also observed corresponding to the heteroduplex formation from the two obtained transcripts. Mutant minigene only showed one transcript corresponding to the aberrant splicing process (band D). This mutation avoided the recognition of the main acceptor site and created a new acceptor splice-site leading to a deletion of 103-bp in the 5′ end of exon 29 (Fig. 1C). If the c.3652G>A transcript was translated, it would generate a new truncated protein of 1227 amino acids in length, p.Y1211AfsX18.
c.2289+1G>A (CDH23)
The WT minigene generated a strong band corresponding to the correct transcript (band A) and a thin band corresponding to the exon skipping (band B). The mutant minigene showed two strong bands; one of them was the transcript with the exon 21 plus part of the intron 21 (band C) and the other band was the exon skipping (band B). This variant generated two different transcripts: one of them did not recognize the main donor site and created a new donor site including the first 149 nucleotides of intron 21 and the other transcript produced the skipping of exon 21. Thus, c.2289+1G>A would create two different proteins; p.N765SfsX35, of 798 amino acids in length, would correspond to the exon 21 plus 149 nucleotides of intron 21, and the other new truncated protein would be p.E727KfsX9, of 734 amino acids in length (Fig. 1D).
c.6049G>A (p.G2017S, CDH23)
Minigene assays revealed that the WT construction created two different transcripts: one of them corresponded to the correct transcript (band A) and the smallest transcript was the skipping of exon 46 (band B). A third band was also observed corresponding to the heteroduplex formation from the two smaller transcripts (band C). However, the c.6049G>A construction only showed one band corresponding to the skipping of exon 46 (band B) (Fig. 1E). If the transcript containing the c.6049G>A variant was translated, it would generate a protein of 3312 amino acids in length, p.T1976_G2017del.
c.8722+1delG (CDH23)
In vitro experiments showed that the WT construction generated one band (band A) corresponding to the correct transcript and the mutant construction showed a band with apparently the same size of the correct transcript (band B). However, when these fragments were sequenced, we could observe that the two fragments were different. The mutation produced the displacement of the donor splice site one nucleotide upstream, being the last nucleotide of the exon 60 processed as intron (Fig. 1F). If this mutant transcript was translated, it would be generating a new truncated protein of 2950 amino acids in length, p.S2909AfsX43.
c.3717+2dupTT (PCDH15)
In our series of patients, we identified a novel variant in intron 27 of the PCDH15 gene in one Spanish USH1 family in homozygous state. We confirmed by minigene assays that the WT construction generated one correct fragment (band A). However, the c.3717+2dupTT minigene showed two different transcripts: the smallest and strongest transcript corresponded to the skipping of exon 27 (band B) and transcript containing exon 27 plus part of intron 27 (band C). A bigger band was observed corresponding to the heteroduplex from the two transcripts (band D). This novel mutation generated two different transcripts: the smallest and strongest transcript that corresponded to the skipping of involved exon 27 and a second transcript corresponding to the no recognition of the main donor splice site and the creation of a new donor site that includes the first 52 nucleotides of intron 27 (Fig. 1G). Therefore, the mutation would be creating two different proteins; one of them, corresponding to the skipping of exon 27, would create an in-frame deletion of 72 amino acids in length, p.A1168_L1239del, and a new truncated protein, corresponding to the exon 27 plus the first 52 nucleotides of intron 27, of 1241 amino acids in length, p.V1242RfsX2.
Nasal Epithelial Cells
To carry out the second goal of the present study, we designed different primers to amplify particular fragments of the MYO7A and CDH23 cDNAs from the nasal epithelium cells of patients and controls, in order to corroborate the observed effect of mutations by minigenes in the patient’s tissues. We only could obtain samples from subjects carriers of eight of the nineteen variants analyzed in vitro by minigenes. These samples were obtained from five USH1 patients and two family healthy carriers of the mutations c.6_9dup (RP-1481) and c.640G>A (RP-1546) in the MYO7A gene (Table 6).
Table 6. Genotypes of the five USH1 patients and the two family healthy carriers of USH1 mutations presented in this study.
| Patient | Gene | Allele 1/Allele 2 |
| RP-1481* | MYO7A | c.6_9dup (p.L4DfsX39) (exon2)/+ |
| RP-1546* | MYO7A | c.640G>A (p.G214R) (exon7)/+ |
| RP-115 | MYO7A | c.3508G>A (p.E1170K) (exon 28)/c.3238A>T (p.K1080X) (exon 25) |
| RP-1479 | MYO7A | c.5581C>T (p.R1861X) (exon 40)/c.5581C>T (p.R1861X) (exon 40) |
| RP-280 | MYO7A | c.5856G>A (p.K1952K) (exon 42)/c.1190C>A (p.A397D) (exon 11) |
| RP-1534 | CDH23 | c.2289+1G>A (intron 21)/c.6049G>A (p.G2017S) (exon46) |
| RP-928 | CDH23 | c.8722+1delG (intron 60)/c.6511delC (exon48) |
Family healthy carriers are indicated with an asterisk (*).
Only in four of the eight studied variants (c.5856G>A in the MYO7A gene, c.2289+1G>A, c.6049G>A and c.8722+1delG in the CDH23 gene), we could observe an abnormal splicing process ex vivo (Table 5). In the remaining four samples both WT and mutant alleles were amplified showing that the presence of the mutations did not affect the splicing process.
c.5856G>A (p.K1952K, MYO7A)
Jaijo et al. [26] analyzed the c.5856G>A variant by minigene constructions. It showed the skipping of exon 42. The analysis of this change in the patient RP-280 replicate 1 revealed the presence of two transcripts: a fragment corresponding to the expected size, 835-bp also in the control sample (band A), and a fragment of 721-bp (band B). However, in the patient’s replicate 2, we only could amplify the smallest fragment. Sequencing of the products showed that the fragment of 835-bp corresponded to the normal allele. However, the product of 721-bp was the transcript without exon 42; if the transcript containing the p.K1952K variant was translated, it would create a new protein of 2177 amino acids in length, p.A1915_K1952del (Fig. 2A).
Figure 2. Transcript analysis of USH1 variants in nasal epithelial cells.
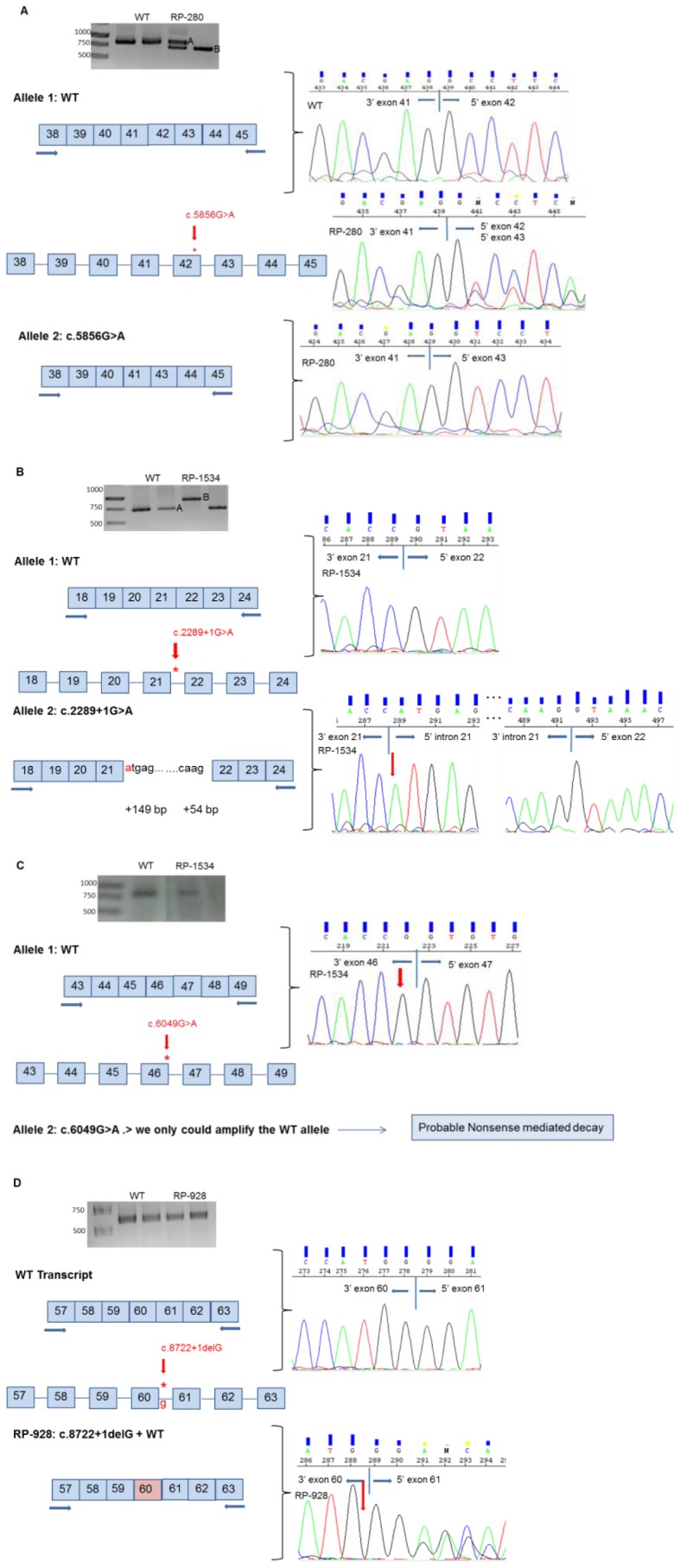
Gel electrophoresis shows the RT-PCR products obtained for USH1 patients and control samples. Electropherograms of the transcripts obtained show the molecular characterization of the effect of the studied variants. All experiments were performed in duplicate. A. c.5856G>A (p.K1952K, MYO7A ). Band A is the correct splicing process of the exons 38–45 of the MYO7A gene. Band B is the skipping of exon 42. RP-280 replicate 1 shows two transcripts corresponding to the WT allele (band A) and mutant allele (band B). RP-280 replicate 2 shows only the band B corresponding to the aberrant transcript. B. c.2289+1G>A ( CDH23 ). Band A is the transcript corresponding to the normal RNA processing of the exons 18–24 of the CDH23 gene. Band B is the aberrant transcript that includes the first 149 nucleotides and the last 54 nucleotides of intron 21. C. c.6049G>A (p.G2017S, CDH23 ). RP-1534 shows only the WT allele. D. c.8722+1delG ( CDH23 ). RP-928 shows the heterozygous transcript from WT allele and the mutant allele. It presents a deletion of the last nucleotide (G) of exon 60 of CDH23 gene.
c.2289+1G>A CDH23
xamination of the RT-PCR product of the patient RP-1534 showed the presence of two transcripts, band A (observed in the replicate 2) and band B (observed in the replicate 1). However, in the control sample only one transcript was observed (band A). Sequencing of the band A showed the correct transcript of 627-bp. The patient’s replicate 1 amplified an abnormal transcript that included the first 149 nucleotides of intron 21 and the last 54 nucleotides of the same intron (band B) (Fig. 2B). Therefore, the mutation would be creating a new truncated protein of 798 amino acids in length, p.N765SfsX35.
c.6049G>A (p.G2017S, CDH23)
The patient RP-1534 also carried the p.G2017S. In this case, we only could amplify and sequence the normal allele (Fig. 2C).
c.8722+1delG (CDH23)
Analysis of the RT-PCR product of patient RP-928 showed only one band. However, sequencing clearly revealed the presence of two distinct transcripts: a normal spliced transcript and an abnormal transcript in which the splice site is modified causing the removal of one nucleotide (G) in the exons 60 and 61 boundary (Fig. 2D). Thus, if this mutant transcript was translated, the new protein would be of 2950 amino acids in length, p.S2909AfsX43.
Ciliary Beat Frequency
The ciliary beat frequencies obtained from each patient and control were summarized in Table 7. Ciliary beat pattern was normal in all the patients. We analyzed whether USH1 patients showed differences in ciliary beat frequency compared to controls. As shown in Fig. 3A, the ciliary beat frequency was significantly reduced in these patients (9.68±0.49 Hz, Mann Whitney U test, p = 0.031) compared to controls (10.88±0.25 Hz).
Table 7. Nasal ciliary beat frequency of USH1 patients and controls.
| MYO7A patients | Nasal Ciliary Beat Frequency (Hz) |
| 1 | 8.5 |
| 2 | 12 |
| 3 | 11.5 |
| 4 | 9.5 |
| 5 | 9.5 |
| CDH23 patients | Nasal Ciliary Beat Frequency (Hz) |
| 1 | 8 |
| 2 | 9 |
| 3 | 9.5 |
| Control subjects | Nasal Ciliary Beat Frequency (Hz) |
| 1 | 11.58 |
| 2 | 10 |
| 3 | 10 |
| 4 | 12 |
| 5 | 11.35 |
| 6 | 10.2 |
| 7 | 13 |
| 8 | 9 |
| 9 | 10.6 |
| 10 | 12.3 |
| 11 | 11 |
| 12 | 12 |
| 13 | 12.5 |
| 14 | 10.8 |
| 15 | 9.1 |
| 16 | 14 |
| 17 | 10.7 |
| 18 | 9.5 |
| 19 | 9.5 |
| 20 | 11.8 |
| 21 | 11 |
| 22 | 9.5 |
| 23 | 9 |
| 24 | 10.8 |
| 25 | 14 |
| 26 | 9.75 |
| 27 | 9.75 |
| 28 | 11.75 |
| 29 | 10.5 |
| 30 | 10.5 |
| Disease Group | Nasal ciliary beat frequency (Hz), mean ± SD |
| MYO7A (n = 5) | 10.20±1.44 |
| CDH23 (n = 3) | 8.33±0.76 |
| USH1 (n = 8) | 9.68±1.38 |
| Control (n = 30) | 10.88±1.36 |
Figure 3. Nasal ciliary beat frequency in five MYO7A and three CDH23 patients, and thirty controls.

A. Comparison between USH1 group and Control group. The nasal ciliary beat frequency was significantly different between these groups (Mann Whitney Test, p = 0.031). B. Comparison between the MYO7A group and CDH23 group, MYO7A and Control group and CDH23 group and Control group. The nasal ciliary beat frequency was significantly different between CDH23 group and Control group (P<0.05).
We also evaluated whether ciliary beat frequency were different between MYO7A and CDH23 patients. No statistical differences were found between MYO7A patients (10.20±0.66 Hz) and CDH23 patients (8.83±0.44 Hz, Kruskal-Wallis test followed by Dunńs test). However, nasal ciliary beat frequency was significantly lower in CDH23 patients than in controls (p<0.05, Kruskal-Wallis Test followed by Dunńs test) (Fig. 3B).
Discussion
A high number of mutations responsible for USH have been identified by screenings performed in USH patients. The effect of variants that lead to premature stop codons is not questioned; however, it is complicated to predict the consequences of missense, silent and intronic changes, in order to discriminate neutral variants from those with pathogenic effect. Variants putative to affect the splicing process are usually considered pathogenic on the basis of their conservation in the canonical splice site, their absence in control samples, cosegregation with the disease in families and results from bioinformatic predictions [26].
Four bioinformatic programs (NNSplice, SpliceView, HSF and NetGene2) were used to predict the damaging effect of the variants identified in the USH1 genes of our cohort and eighteen changes were selected as putative to affect the splicing process (Table 4). These results are only computational predictions so, additional studies are necessary to confirm the effect on the mRNA processing. Minigenes and RNA assays were performed to achieve this goal.
Minigene assays revealed that only seven of the eighteen studied variants were affecting the splicing process (Table 5). The bioinformatic analysis of these seven variants had shown high scores. A maximum score of 4, for five of them: c.470G>A, c.1342_1343delAG and c.3652G>A in the MYO7A gene, c.2289+1G>A in CDH23 gene and c.3717+2dupTT in PCDH15 gene. The other two variants (c.6049G>A and c.8722+1delG in the CDH23 gene) showed a score of 3. However, other studied variants that also reached a high score (≥3) did not show any effect in the splicing process when they were analyzed by minigenes. According to these results, all types of putative pathogenic variants should be analyzed by bioinformatic tools, and, when high scores are observed, minigene analysis should be performed to avoid false positive. See Table 5.
However, the splicing processes observed by minigenes would not necessarily reflect the real processing in affected tissues. In our study, we could analyze RNA from nasal epithelial cells from patients or carriers of eight variants previously analyzed by minigene constructions, to determine their effect on splicing process in patient’s tissues. Analysis of the RT-PCR amplification of nasal cells allowed us to confirm that four of the eight studied variants, one MYO7A (c.5856G>A) and three CDH23 (c.2289+1G>A, c.6049G>A and c.8722+1delG), were affecting the splicing process. The same results were obtained by minigene constructions for the eight studied variants, except for two CDH23 changes, c.2289+1G>A and c.6049G>A.
For the c.2289+1G>A variant the different results obtained by both methods are due to the minigene construction did not contain the entire intron 21 of the CDH23 gene.
In the study of the c.6049G>A variant by analysis of RT-PCR amplification from nasal cells we were not able to amplify the mutant transcript. This fragment could be degraded by nonsense mediated decay mechanism (NMD), decreasing the mRNAs levels and therefore, the protein levels. However, by minigene construction we had observed in this case the skipping of exon 46.
In three cases (c.5856G>A (MYO7A), c.2289+1G>A (CDH23) and c.6049G>A (CDH23)), we could only amplify one of the two alleles due to the low USH1 genes expression level in this tissue. In fact, these USH transcripts are only detectable by nested PCR. Accordingly, it is advisable to detect the presence of heterozygous changes by sequencing, the studied mutation or other SNPs, to confirm the amplification of the two alleles and avoid incorrect conclusions when apparently only one fragment is obtained after PCR.
Therefore, minigenes are a good approach to ascertain the pathogenic nature of splice variants when is difficult to obtain RNA from patients’ tissues, as in the case of USH genes, that present a restricted expression profile associated with photoreceptors and inner hair cells. These constructions are expressed in living cells where the splicing machinery remains intact.
In addition, we can confirm that the analysis of nasal epithelial cells is an alternative method to discriminate neutral Usher variants from those with a pathogenic effect on the splicing process.
Finally, we have also demonstrated that the nasal ciliated epithelium of USH1 patients has a lower ciliary beat frequency than control subjects. Armengot et al. [12] suggested that mutations in the USH1 and USH2 genes could be responsible for the lower ciliary beat frequency. They observed a low beat frequency in USH2 patients. However, the ciliary activity was sufficient to operate normally and no clinical consequences were observed in these patients.
Acknowledgments
Authors are grateful to the patients participating in the study and to their family members, and also to the FARPE, FIAPAS, ONCE (Valencia) and La Caixa-Camp de Morvedre for their help and co-operation. The exon trapping expression vector pSPL3 was kindly provided by S. Tuffery-Giraud and I. Bottillo. We are very grateful to Pilar Bañuls for her generous collaboration in the present study.
Funding Statement
This work was supported by grants SAF2011-26443, FIS CP11/00293, PI11/02618, ADE10/00020 and PI10/01825 from Spanish Ministry of Science and Innovation and grants Prometeo/2008/045 and GV/2012/028 from Regional Government (Generalitat Valenciana). CIBERER (CB/06/07/1030) and CIBERES (CB06/06/0027) are an initiative of the Institute of Health Carlos III from the Spanish Ministry of Science and Innovation. The bioanalyzer ABI3500xl was purchased with the grant PROMIS (II12/00023) from the Instituto de Salud Carlos III. MJA and GGG are recipient of a fellowship from the Ministerio de Educación (REF: AP2099-3344 and AP2008-02760, respectively). RR has a Contrato-Investigador SNS Miguel Servet (CP09/118) from Instituto de Salud Carlos III, Ministerio Ciencia e Innovación. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Vernon M (1969) Sociological and psychological factors associated with hearing loss. J. Speech Hear Res. 12: 541–563. [DOI] [PubMed] [Google Scholar]
- 2. Hope CI, Bundey S, Proops D, Fielder AR (1997) Usher syndrome in the city of Birmingham-prevalence and clinical classification. Br. J. Ophthalmol 81: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kimberling WJ, Weston MD, Möller C, van Aarem A, Cremers CW, et al. (1995) Gene mapping of Usher syndrome type IIa: localization of the gene to a 2.1–cM segment on chromosome 1q41. Am J Human Genet. 56: 216–223. [PMC free article] [PubMed] [Google Scholar]
- 4. Espinos C, Millan JM, Beneyto M, Najera C (1998) Epidemiology of Usher syndrome in Valencia and Spain. Community Genet. 1: 223–8. [DOI] [PubMed] [Google Scholar]
- 5. Davenport SLH, Omenn GS (1977) The heterogeneity of Usher Syndrome. Amsterdam Excerpta Media Foundation. International Congress ser abstr. 215: 87–88. [Google Scholar]
- 6. Millán JM, Aller E, Jaijo T, Blanco-Kelly F, Gimenez-Pardo A, et al. (2011) An update on the genetics of usher syndrome. J Ophthalmol. 2011: 417217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaworek TJ, Bhatti R, Latief N, Khan SN, Riazuddin S,et al. (2012) USH1K, a novel locus for type I Usher syndrome, maps to chromosome 10p11.21-q21.1. J Hum Genet. 10.1038/jhg.2012.79. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 8.Riazuddin S, Belyantseva IA, Giese AP, Lee K, Indzhykulian AA, et al. (2012) Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet. doi:10.1038/ng.2426. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 9. Roepman R, Wolfrum U (2007) Protein networks and complexes in photoreceptor cilia. Subcell Biochem. 43: 209–35. [DOI] [PubMed] [Google Scholar]
- 10.Cohn E, Orten DJ, Weston M, Kimberling W, Cosgrove D (2006) Nasal cytology will assist the diagnosis od specific Usher syndromes. First International Symposium on Usher syndrome and related disorders, Omaha, October 3 to 6, 2006. p. 22.
- 11. Vaché C, Besnard T, Blanchet C, Baux D, Larrieu L, et al. (2010) Nasal epithelial cells are a reliable source to study splicing variants in Usher syndrome. Hum Mutat. 31(6): 734–41. [DOI] [PubMed] [Google Scholar]
- 12. Armengot M, Salom D, Diaz-Llopis M, Millan JM, Milara J, et al. (2012) Nasal ciliary beat frequency and beat pattern in retinal ciliopathies. Invest Ophthalmol Vis Sci. 53(4): 2076–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bonneau D, Raymond F, Kremer C, Klossek JM, Kaplan J, et al. (1993) Usher syndrome type I associated with bronchiectasis and immotile nasal cilia in two brothers. J Med Genet. 30(3): 253–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jaijo T, Aller E, Oltra S, Beneyto M, Nájera C, et al. (2006) Mutation profile of the MYO7A gene in Spanish patients with Usher syndrome type I. Hum Mutat. 27(3): 290–1. [DOI] [PubMed] [Google Scholar]
- 15. Jaijo T, Aller E, Beneyto M, Najera C, Graziano C, et al. (2007) MYO7A mutation screening in Usher syndrome type I patients from diverse origins. J Med Genet. 44(3): e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oshima A, Jaijo T, Aller E, Millan JM, Carney C, et al. (2008) Mutation profile of the CDH23 gene in 56 probands with Usher syndrome type I. Hum Mutat. 29(6): E37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aparisi MJ, García-García G, Jaijo T, Rodrigo R, Graziano C, et al. (2010) Novel mutations in the USH1C gene in Usher syndrome patients. Mol Vis. 16: 2948–54. [PMC free article] [PubMed] [Google Scholar]
- 18. Jaijo T, Oshima A, Aller E, Carney C, Usami S, et al. (2012) Mutation screening of the PCDH15 gene in Spanish patients with Usher syndrome type I. Mol Vis. 18: 1719–26. [PMC free article] [PubMed] [Google Scholar]
- 19. Jaijo T, Aller E, García-García G, Aparisi MJ, Bernal S, et al. (2010) Microarray-based mutation analysis of 183 Spanish families with Usher syndrome. Invest Ophthalmol Vis Sci. 51: 1311–7. [DOI] [PubMed] [Google Scholar]
- 20. Bottillo I, De Luca A, Schirinzi A, Guida V, Torrente I, et al. (2007) Functional analysis of splicing mutations in exon 7 of NF1 gene. BMC Med Genet. 12 8: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Le Guédard-Méreuze S, Vaché C, Baux D, Faugère V, Larrieu L, et al. (2010) Ex vivo splicing assays of mutations at noncanonical positions of splice sites in USHER genes. Hum Mutat. 31(3): 347–55. [DOI] [PubMed] [Google Scholar]
- 22. Adato A, Weil D, Kalinski H, Pel-Or Y, Ayadi H, et al. (1997) Mutation profile of all 49 exons of the human myosin VIIA gene, and haplotype analysis, in Usher 1B families from diverse origins. Am J Hum Genet. 61(4): 813–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cuevas JM, Espin s C, Millan JM, Sanchez F, Trujillo MJ, et al. (1999) Identification of three novel mutations in the MYO7A gene. Hum Mutat. 14(2): 181. [DOI] [PubMed] [Google Scholar]
- 24. Astuto LM, Bork JM, Weston MD, Askew JW, Fields RR, et al. (2002) CDH23 mutation and phenotype heterogeneity: a profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am J Hum Genet. 71(2): 262–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roux AF, Faugère V, Le Guédard S, Pallares-Ruiz N, Vielle A, et al. (2006) Survey of the frequency of USH1 gene mutations in a cohort of Usher patients shows the importance of cadherin 23 and protocadherin 15 genes and establishes a detection rate of above 90%. J Med Genet. 43(9): 763–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jaijo T, Aller E, Aparisi MJ, García-García G, Hernan I, et al. (2011) Functional analysis of splicing mutations in MYO7A and USH2A genes. Clin Genet. 79(3): 282–8. [DOI] [PubMed] [Google Scholar]