

Abstract
Trypanosoma brucei variant surface glycoprotein (VSG) expression is a classic example of allelic exclusion. While the genome of T. brucei contains >2,000 VSG genes and VSG pseudogenes, only one allele is expressed at the surface of each infectious trypanosome and the others are repressed. Along with recombinatorial VSG switching, allelic exclusion provides a major host evasion mechanism for trypanosomes, a phenomenon known as antigenic variation. To extend our understanding of how trypanosomes escape host immunity by differential expression of VSGs, we attempted to identify genes that contribute to VSG silencing, by performing a loss-of-silencing screen in T. brucei using a transposon-mediated random insertional mutagenesis. One identified gene, which we initially named LOS1, encodes a T. brucei MCM-Binding Protein (TbMCM-BP). Here we show that TbMCM-BP is essential for viability of infectious bloodstream-form (BF) trypanosome and is required for proper cell-cycle progression. Tandem affinity purification of TbMCM-BP followed by mass spectrometry identified four subunits (MCM4-MCM7) of the T. brucei MCM complex, a replicative helicase, and MCM8, a subunit that is uniquely co-purified with TbMCM-BP. TbMCM-BP is required not only for repression of subtelomeric VSGs but also for silencing of life-cycle specific, insect-stage genes, procyclin and procyclin-associated genes (PAGs), that are normally repressed in BF trypanosomes and are transcribed by RNA polymerase I. Our study uncovers a functional link between chromosome maintenance and RNA pol I-mediated gene silencing in T. brucei.
Introduction
Trypanosoma brucei is a protozoan parasite that causes African sleeping sickness in humans and a similar disease in livestock, in sub-Saharan Africa. T. brucei cycles between the insect vector (tsetse) and mammalian hosts. Two proliferating life-cycle forms, the insect-midgut procyclic form (PF) and the mammalian bloodstream form (BF), can be cultured and genetically manipulated. A single species of VSG is expressed at any time in each BF parasite, where VSG homogenously coats the parasite surface. All VSGs are repressed in PF, whose surface is coated with members of a small family of proteins called procyclins.
Four distinct loci contain VSGs and VSG peudogenes [1]. The majority of VSGs reside in minichromosomes and telomere-distal (sometimes called ‘chromosome-internal’) arrays, but they appear to lack promoters. VSGs in BF are transcribed by RNA polymerase I (Pol I) from about 15 subtelomeric polycistronic ‘Bloodstream-form VSG Expression Sites’ (BES) [2], [3]. These VSGs are located adjacent to telomere repeats and ∼50 kb downstream of their promoters. Only one BES is transcriptionally active and expresses one VSG in each bloodstream form cell whereas the remaining VSGs are repressed [1]. In the infectious metacyclic (tsetse) stage, VSGs are transcribed from monocistronic subtelomeric expression sites that are distinct from BESs [4], [5].
Silencing of BES promoters and VSGs appears to be regulated by factors involved in telomere dynamics and chromatin modification. Loss of TbSIR2-rp1 (a T. brucei SIR2 homologue) or TbRAP1 (a telomere-binding protein) led to derepression of silent VSGs [6], [7]. Several studies have demonstrated that chromatin remodeling and histone marks may be involved in BES promoter and/or VSG silencing; TbISWI, a member of the ISWI family of chromatin remodeling complexes [8]; TbDOT1B, a histone methyltransferase [9]; TbHAT1, a histone acetyltransferase [10]; TbDAC3, a histone deacetylase [11]; and TbSPT16, a subunit of the chromatin remodeling FACT complex [12]. Derepression of silent VSGs was also observed in BF cells depleted of chromatin assembly factors, TbASF1A and TbCAF-1b, and a linker histone, H1 [13], [14].
Thus far, studies of T. brucei antigenic variation have been largely relied on searching for homologues that have been characterized in other organisms. However, because T. brucei proteins are highly divergent from those of other model organisms, due to early separation during evolution, homologue searches have limitations. To better understand the molecular mechanisms underlying VSG silencing and to isolate components relevant to VSG gene silencing, we performed a large-scale forward genetic screen in T. brucei: a ‘loss-of-silencing’ (LOS) screen. We isolated 19 los clones that had impaired ability to repress a silent BES. One of these genes, LOS1, encodes a T. brucei MCM-Binding Protein (MCM-BP). MCM-BP is a component of a replication complex that consists of subunits of the Mini-Chromosome Maintenance (MCM) complex and of MCM-BP [15]–[17].
DNA replication initiates with binding of the Origin Recognition Complex (ORC) to replication origins and recruitment of members of the pre-replication complex (pre-RC), including CDC6, CDT1, CDC45/GINS complex and MCM complex. This activates the replication origins and proceeds to replication by DNA polymerases (reviewed in [18]). Along with these core replication proteins, several of alternative replication complexes have been identified and extensively studied in yeasts and human. Alternative replication complexes structurally resemble replication complexes and participate in the maintenance of chromosome integrity without being directly involved in DNA replication. They are either composed of specific subunits or deviate from canonical complexes by one or more subunits. For example, the 9-1-1 complex (RAD9-HUS1-RAD1), a donut-shaped heterotrimer, resembles the homotrimeric Prolierating Cell Nuclear Antigen (PCNA, a replication clamp), a replication factor that is required for the processivity of DNA polymerase [19]. PCNA loading onto DNA requires the Replication Factor C (RFC) complex, the clamp loader, which is composed of five subunits RFC1-5 [20]. RAD24 or CTF18 replaces RFC1 and forms a RAD24-RFC2-5 or CTF18-RFC2-5, and these complexes are involved in DNA damage checkpoint and chromosome segregation [21], [22]. The MCM-BP complex is the most recent addition to the list of alternative replication complexes. The MCM complex, a replication helicase, consists of six subunits, MCM2-MCM7 (reviewed in [23]). In plants, the MCM-BP, also known as the ETG1 (E2F target gene 1), interacts with all MCM subunits, MCM2-7. The ETG1 is required for DNA replication, repair, and sister-chromatid cohesion [17], [24]. In human and fission yeast, MCM-BP replaces MCM2 and forms a complex with MCM3-7 [15], [16]. An in vitro study with Xenopus egg extracts showed that MCM-BP is required for removal of the MCM complex from DNA at the end of the S-phase to ensure replication licensing for the next round [25].
DNA replication proteins have been implicated in gene silencing. Subunits of ORC, a hexameric complex, participate in heterochromatin-mediated silencing in several organisms by interacting with the heterochromatin proteins SIR2-4 and HP1 [26]–[29]. Subunits of the MCM complex are also involved in gene expression through interactions with histone H3, transcription factor STAT1, and RNA Pol II in human cells [30]–[33]. In the budding yeast temperature-sensitive mutant mcm5, subtelomeric chromatin was more loosely packed and subtelomeric genes were up-regulated at the non-permissive temperature [34]. In addition, the temperature sensitivity of mcm5 was suppressed by overexpressing TRA1, a component of the SAGA chromatin-remodeling complex [34].
In T. brucei, we are beginning to understand mechanism of DNA replication with recent identification of several subunits of ORC, components of pre-RC and MCM complex [35]–[37]. Recently, replication origins have been mapped in all 11 megabase chromosomes using chromatin immunoprecipitation (chIP) of TbORC1 [38] and BrdU labeling demonstrated that TbORC1 is required for de novo nuclear DNA synthesis both in BF and PF trypanosomes [39]. The majority of replication origins appear to overlap with start sites of polycistronic transcription units in T. brucei [38]. TbORC1 binding sites are also enriched along BESs [38] and at telomeres [39]. Similar to ORC1 in other model organisms, TbORC1 seems to have functions outside of DNA replication. Depletion of TbORC1 increased expression levels of BES-associated silent VSGs both in BF and PF trypanosomes [39], and of metacyclic VSGs in PF cells [38]. TbORC1 is also required for VSG switching, particularly a mechanism involving a transcriptional switch between the active and a silent BES [39].
In this study, we performed a forward genetic screen to isolate genes that are responsible for maintaining the repressed status of a silent BES, using a mariner-transposon random insertional mutagenesis, which led to the isolation of an allele of TbMCM-BP. TbMCM-BP is essential for cell viability in the infectious BF stage. Deficiency of TbMCM-BP is associated with derepression of silent VSGs and silent procyclic genes that are transcribed by RNA Pol I, and with a novel cell-cycle defect. Tandem affinity purification of TbMCM-BP demonstrated that TbMCM-BP is strongly associated with four T. brucei MCM-subunits, MCM4-MCM7, and MCM8, a subunit that is uniquely co-purified with MCM-BP only in T. brucei. It is notable that it is the first time that a VSG silencing factor has been selected by a phenotype-based large-scale screening approach and that T. brucei is the first organism in which MCM-BP was identified in such way.
Results
Loss-of-silencing screen and isolation of Trypanosoma brucei MCM-BP
We performed a large-scale random insertional mutagenesis screen to isolate genes that participate in the mechanism of VSG silencing. The screen was carried out in PF T. brucei, for several reasons. First, the screen requires a single-copy autonomously replicating plasmid, which is not available in BF trypanosomes. Secondly, BF trypanosomes occasionally switch their VSGs at a frequency of ∼1–2×10−5 [40], [41]. Therefore, switchers can be selected as false-positive clones. Thirdly, for a large-scale screen to work in a diploid organism (a haploid meiotic stage seems to occur only inside the insect vector [42]), it is essential to generate a sufficiently large pool of mutants with potential homozygous gene disruptions. PF cells can grow to a 10–20-fold higher density than BF cells. Finally, all VSGs, including all BES-associated VSGs, are silent in PF T. brucei. Given all these factors, PF offered a greater potential for the success of an initial screen. Once genes are isolated, they can be characterized for their roles in BF trypanosomes, where antigenic variation is physiologically relevant.
To generate a reporter cell line for a loss-of-silencing (LOS) screen, BES11 (containing VSG16) was targeted with a triple-reporter cassette containing puromycin-resistance (PUR), luciferase (LUC), and emerald-GFP (emGFP) genes, adjacent to its promoter (Figure 1). Reporter cells were selected in 1 µg/ml puromycin, as a low level of transcription occurs close to a ‘silent’ BES promoter [43], allowing drug selection at a low concentration. A mariner transposase gene was integrated into the tubulin locus and insertional mutagenesis was induced by transfection of the donor-plasmid pSGL35 [44] containing a hygromycin-resistance gene (HYG) flanked by mariner inverted repeats (IRs) and a neomycin-resistance gene (NEO) for the selection of cells transformed by the plasmid. Neomycin-resistant cells (NEOR) were expanded to a population of ∼5×108 cells, prior to selection with hygromycin. Cells can only become resistant to hygromycin once the donor cassette is transposed into a transcribed chromosomal orientation.
Figure 1. Loss-of-silencing (LOS) screen.
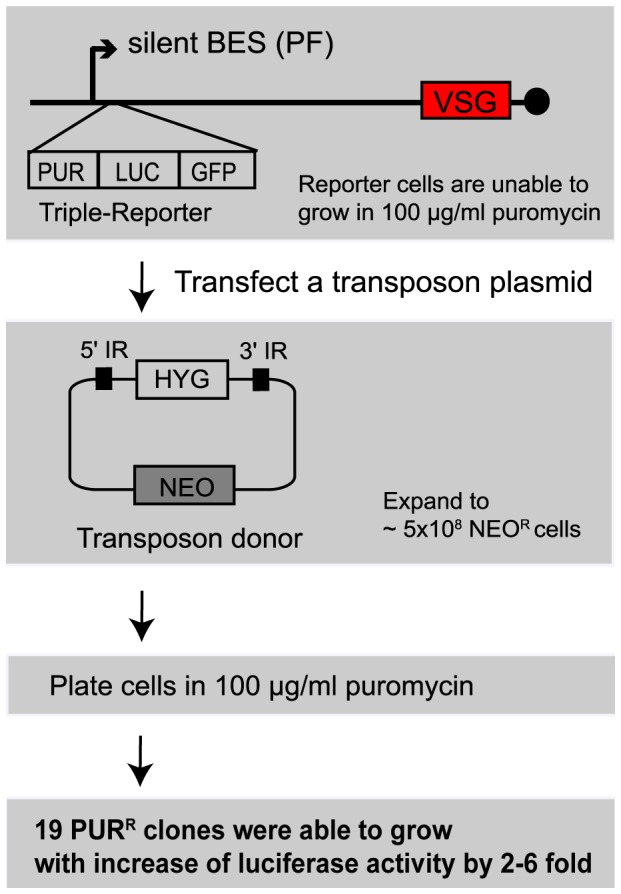
A triple reporter cassette carrying puromycin-resistance (PUR), luciferase (LUC), and emerald GFP (emGFP) genes was targeted adjacent to a silent BES promoter in procyclic form T. brucei and selected with 1 µg/ml puromycin. The reporter cells are unable to grow at a higher concentration of puromycin (100 µg/ml), due to BES promoter silencing. After stably integrating a transposase at an array of tubulin genes, cells were mutagenized using a transposon insertion. The transposon plasmid carries a mariner-donor containing HYG marker flanked by inverted repeats (IRs). Expansion of neomycin-selected cells was distributed in 96-well plates with 100 µg/ml of puromycin. Only transposon-mutant cells that had lost the ability to repress this silent BES locus can grow at this concentration of puromycin but parental cells cannot. 19 PURR clones were obtained and increased luciferase activity by 2–6-fold (Table S1). Black circle is telomere.
Transpositions that led to increased expression of PUR marker (‘los’ clones) were isolated by simultaneously selecting with 40 µg/ml of hygromycin and 100 µg/ml of puromycin. Parental cells cannot grow at this concentration of puromycin while los mutants that lost the ability to repress expression of PUR gene should. 19 PURR clones were obtained (Table S1). Luciferase expression was also increased in all clones (2–6-fold increase compared to the parental line). Transposon target sites were mapped by inverse PCR and sequencing. We were unable to identify target site(s) in three PURR clones. In some clones, the transposon-HYG donor was integrated at multiple locations, for example clones 2 and 12. In six cases, transposons were inserted at intergenic regions, which may disrupt functions of untranslated regions (UTRs) and affect expression levels of neighboring genes.
Five clones had an identical insertion in the middle of one allele of the Tb927.7.1770 (gene ID number in http://www.genedb.org and http://tritrypdb.org) open reading frame (ORF), so were not independent events, causing an about 3–6-fold increase in luciferase activity near the reporter-tagged silent BES promoter (Table S1, clone numbers are highlighted in blue). However, unlike what we expected, only one allele of Tb927.7.1770 was disrupted in all five clones. We provisionally named the disrupted gene LOS1 (loss-of-silencing gene 1).
BLAST searches with LOS1 identified sequence similarities to the MCM-Binding Protein (MCM-BP) that interacts with subunits of the MCM complex [15]–[17]. Figure 2A shows a sequence alignment of LOS1 with human, fish, worm, and plant MCM-BP, all of which contain two domains, ‘MCM Bind Superfamily’ and ‘Racemase 4 Super family’. LOS1 appears to have these domains and the transposon was inserted in the middle of the ‘Racemase 4 Superfamily’ region (Figure 2B). A functional study in fission yeast with a series of truncated MCM-BP proteins showed that the N-terminal half of MCM-BP was sufficient for the interaction with MCM4, but was unable to rescue the lethality of an mcm-BP null mutant [16]. Therefore, it is possible that the transposon-targeted los1 allele may express an N-terminal portion of TbMCM-BP, which may act as a dominant negative allele in los1 heterozygous clones, resulting in the loss of BES promoter silencing. LOS1 will be referred to as TbMCM-BP henceforth.
Figure 2. LOS1 shows sequence similarities to the MCM-Binding Protein (MCM-BP).

(A) Alignment of LOS1 with human, fish, worm and plant MCM-BP is shown. BLAST searches identify that all of MCM-BP including LOS1 contain two families of sequences, called ‘MCM Bind Superfamily’ (underlined in blue) and ‘Racemase 4 Super family’ (underlined in red). (B) A schematic diagram of T. brucei MCM-BP. A black triangle in the ‘Racemase 4 Superfamily’ domain indicates the site where a mariner transposon (Tn) was inserted.
TbMCM-BP is essential for viability of BF T. brucei
To investigate whether TbMCM-BP is required for VSG silencing in BF, where antigenic variation occurs, we attempted to make a homozygous TbMCM-BP deletion mutant in BF but we were not able to delete both alleles, suggesting that TbMCM-BP is an essential gene. To deplete the TbMCM-BP transcript by RNAi, the wild-type TbMCM-BP allele in heterozygous TbMCM-BP−/+ was first epitope-tagged in situ with myc. The TbMCM-BP−/MYC cells did not show any growth defect, verifying that the tagged version is functional. The TbMCM-BP−/MYC strain was then transfected with a tetracycline-inducible construct for TbMCM-BP dsRNA expression [45]. Two RNAi clonal cell lines showed growth defects upon tetracycline addition that correlated with the disappearance of TbMCM-BP-myc protein (Figure S1).
RNAi knockdown may have off-target effects and is sometimes not very efficient. Although TbMCM-BP depletion correlated with cell growth defect, depletion was not efficient. It was incomplete in RNAi line 2, and in cell line 1, silencing of TbMCM-BP gene took 96 hr (about 16 generations) and cells seemed to escape from RNAi silencing after 144 hr. To have a definite answer for the essentiality of TbMCM-BP in BF T. brucei, and to generate a more reliable cell line to examine cellular functions of TbMCM-BP, we constructed a conditional-knock-out (cKO) cell line (Figure 3A). One allele of TbMCM-BP was deleted using a dual positive and negative selectable PUR-TK gene fusion flanked with loxP sites. Cre-recombinase under the control of tetracycline was stably introduced to remove the PUR-TK marker. In this TbMCM-BP−/+ heterozygous strain, an endogenous TbMCM-BP allele was then replaced with a cassette that contains MCM-BP-myc followed by HYG-TK. Two loxP sites flank the cassette so that the MCM-BP-myc and HYG-TK can be removed by conditionally expressing Cre-recombinase (Figure 3A). TbMCM-BP cKO cells were harvested at 12-hr intervals for 48 hr upon Cre induction. Cell growth was arrested 12 hr after Cre induction and TbMCM-BP-myc gradually disappeared (Figure 3B and 3C), confirming that TbMCM-BP is an essential gene and demonstrating that the conditional KO system is much more reliable than RNAi-mediated gene knockdown which showed no cell growth arrest even after 144 hr of RNAi induction.
Figure 3. TbMCM-BP is essential for viability of BF T. brucei.

(A) TbMCM-BP conditional knock out (cKO) strategy using Cre-loxP. In a TbMCM-BP single KO strain, remaining wild type TbMCM-BP allele was replaced with MCM-BP-myc-HYG-TK cassette flanked by loxPs. Homozygously deleted TbMCM-BP double KO mutation can be obtained by expressing Cre-recombinase, which was stably inserted at an rDNA locus. (B) T. brucei MCM-BP is essential for cell viability of the infectious BF T. brucei. Cell growth was arrested upon TbMCM-BP removal by Cre expression. (C) Depletion of TbMCM-BP protein upon Cre expression by immunoblot. Tubulin, VSG2, and VSG3 were analyzed by immunoblot. VSG3 protein was detected in TbMCM-BP deficient cells.
TbMCM-BP is required for VSG silencing in BF T. brucei
To examine whether VSG silencing was lost in TbMCM-BP-depleted BF trypanosomes, we assessed the expression levels of several VSG genes in different chromosomal locations by reverse transcription quantitative PCR (RT-qPCR). We quantified derepression of three silent VSGs: two silent BES-linked subtelomeric VSGs, VSG3 (also called VSG224) and VSG13 (VSG113), and a minichromosomal VSG, VSG24 [46] (Figure 4A). We also measured expression of BSD and VSG2 from the active BES1 and of two chromosome-internal housekeeping control genes (TbTUB and TbURA3). Cre-expression was induced to knock out TbMCM-BP-myc and cells were harvested 24 and 48 hr after Cre induction. Levels of mRNA were quantified by RT-qPCR and values were normalized to the level of TbURA3 mRNA. Figure 4B shows fold changes relative to the TbMCM-BP wild type cells (0 hr). Expression of silent BES-linked VSG3 and VSG13 increased 10- and 18-fold after 24 hr and 30- and 35-fold after 48 hr upon TbMCM-BP removal. Interestingly, expression of minichromosomal VSG (VSG24) increased 2- and 12-fold after 24 and 48 hrs after TbMCM-BP removal. T. brucei contains a large number of small chromosomes called minichromosomes (MCs), which are 30–150 kb in size. According to genome-wide high-throughput sequencing analysis, it appears that there are ∼60 MC VSGs in the genome of this strain of T. brucei (GAMC, unpublished data). Mapping of 17 MCs showed a core structure consisting of a large central palindromic 177-bp repeat region and telomeric ends, some of which contain VSGs [47]. These subtelomeric VSGs do not seem to have their own Pol I promoter [47]. Depletion of TbMCM-BP increased expression levels of BES-associated subtelomeric VSGs and a minichromosomal subtelomeric VSG, but did not significantly change the expression level of the active VSG2 and BSD, or of another chromosome-internal housekeeping gene, TbTUB. Our data suggest that TbMCM-BP may have roles in silencing subtelomeric VSGs.
Figure 4. TbMCM-BP is required for VSG silencing in BF T. brucei.

(A) Diagrams of genes and their chromosomal locations: silent BES-linked VSGs (VSG3 and VSG13), a minichromosomal VSG24, the active VSG2 and BSD marker, and two housekeeping control genes (TUB and URA3). Black circles are telomere repeats. (B) TbMCM-BP deficiency derepresses silent VSG expression. After 24 and 48 hr of post-Cre-induction, mRNA levels of each genes described in (A) were measured and normalized to TbURA3 mRNA level. Fold changes relative to Cre-uninduced cells (0 hr) are plotted. Standard deviations are shown as error bars. (C) Expression of silent VSG13 protein in the VSG2 active BF cells. Cells were fixed with 1% paraformaldehyde and permeabilized with 0.2% NP40. Samples were incubated with chicken anti-VSG2 and rabbit anti-VSG13, and then incubated with secondary antibodies (green for VSG2 and red for VSG13). DNA was stained with DAPI.
We observed expression of silent VSG3 by immunoblotting after TbMCM-BP removal (Figure 3C, bottom), which demonstrated that silent VSG transcripts can be translated. We were able to detect VSG3 because it migrates slower than VSG2, eliminating the possibility that the antibody cross-reacted with the major VSG2 band. However, we were unable to detect changes in VSG13 protein expression, as the VSG13 band overlaps with that of VSG2. To ensure that elevated levels of silent VSG expression are due to loss of silencing and not due to VSG switching, we visualized the active and silent VSGs by immunofluorescence. Cells were double-stained with anti-VSG2 (active, green) and anti-VSG3 (silent, red) or VSG13 (silent, red) antibodies. We observed cells co-expressing the active VSG2 and the silent VSG3 (data not shown) and cells co-expressing the active VSG2 and the silent VSG13 (Figure 4C, yellow arrowheads) upon TbMCM-BP removal. However, silent VSGs were detectable by IF only in a subset of cells, probably because the levels of these partially derepressed VSGs were generally very low. We did not detect any switched cells that no longer expressed VSG2. Collectively, out data confirm that TbMCM-BP is required to maintain the repressed status of silent VSGs.
TbMCM-BP is required for full repression of silent RNA Pol I promoters in BF T. brucei
The loss-of-silencing phenotype of TbMCM-BP mutation was identified originally in a PF cell line with reporter genes inserted immediately downstream of a silent BES promoter. Silencing near BES promoters appears to be mechanistically distinct from silencing near telomeres (reviewed in [48]).
To examine whether TbMCM-BP also influences the silencing at a BES promoter in BF T. brucei, we inserted the same PUR-LUC-GFP triple reporter cassette at a silent BES promoter in the TbMCM-BP-conditional KO cell line and obtained two independently targeted cell lines (HSTB-683 and -684). Expression levels of PUR and LUC genes (top diagram in Figure 5A) were quantified by RT-qPCR and values were normalized to the level of TbURA3 mRNA. VSG3 derepression was used as a positive control in RT-qPCR and immunoblots (Figure 5B and 5C). TbMCM-BP depletion caused only minor effects on BES promoter silencing with maximum increases of about 4-fold in PUR and of about 5-fold in Luciferase gene expression (Figure 5B). We also observed increased expression of emGFP by flow cytometry analysis (data not shown). The derepression was not as dramatic as at the silent VSGs, probably because TbMCM-BP is required largely for subtelomeric silencing within the BES units or because, as is known [49], [50], repression is already weaker near silent promoters than closer to the telomeres. Figure 5C shows TbMCM-BP depletion and VSG3 expression after Cre induction by immunoblot analysis. Tubulin was used as a loading control.
Figure 5. TbMCM-BP is required for full repression of silent RNA Pol I promoters in BF T. brucei.

(A) Diagrams of genes and their locations: a triple reporter in a silent BES, procyclic genes under a RNA Pol I promoter, and a procyclic gene within a RNA Pol II transcription unit. (B) TbMCM-BP deficiency increased expression of silent genes transcribed from RNA Pol I promoter. After 24 and 48 hr of post-Cre-induction, mRNA levels of each genes described in (A) were measured and normalized to TbURA3 mRNA level. Fold changes relative to Cre-uninduced cells (0 hr) are plotted. Standard deviations are shown as error bars. (C) Depletion of TbMCM-BP protein upon Cre induction by immunoblot. VSG3 expression was used as a positive control for RT-qPCR and immunoblot.
Insect-midgut PF trypanosomes express several procyclic-specific genes, called procyclin (GPEET and EP variants) and procyclin-associated genes (PAGs) that are also transcribed from an RNA Pol I promoter, which is less active in BF than in PF [51]. Some PAGs are located downstream of procyclin genes under the RNA Pol I promoter (diagram in Figure 5A, middle) and some, for example PAG3 (Figure 5A, bottom), are located within RNA Pol II polycistronic units. To examine whether TbMCM-BP plays specific roles in RNA Pol I-mediated transcription, we measured expression levels of EP1 and PAGs in the absence of TbMCM-BP. As shown in Figure 5B, depletion of TbMCM-BP derepressed EP1, PAG5 and PAG4, which are transcribed by RNA Pol I, but did not significantly affect PAG3 expression directed by RNA Pol II. These results collectively indicate that TbMCM-BP is required for full repression of BES promoters and of RNA Pol I-transcribed, PF-specific genes in BF trypanosomes.
Accumulation of G2 and anucleated cells (zoids) in TbMCM-BP null mutant
MCM-BP deficiency is associated with cell-cycle arrest in G2/M and activates the G2 checkpoint in plant, human and fission yeast [16], [17], [24], [52]. In addition, MCM-BP is required for replication termination, by removing MCM at the end of S-phase in Xenopus [25]. To examine whether TbMCM-BP deficiency impairs cell-cycle progression in BF trypanosomes, we monitored cell-cycle progression after Cre-induced TbMCM-BP removal by flow cytometry, staining bulk DNA with propidum iodide. As shown in Figure 6A, control cells (grey area, 0 hr) showed normal cell cycle profiles: 2C (G1 phase, 56.5%), 4C (G2 phase, 14.9%), and a population between 2C and 4C (S phase, 8.67%). Twelve hours after Cre induction, the cell-cycle profile remained normal, but peaks representing S and G2 became higher at 24 and 36 hrs, and lower after 48 hr, indicating that TbMCM-BP-depleted cells arrest at S-G2. Interestingly, <2C content cells gradually increased over time. The accumulation and then subsequent reduction of the S-G2 population and the continuing increase of <2C cells suggest that depletion of TbMCM-BP induces a G2 arrest, and that chromosomes segregate before S-phase completes, and, therefore, that TbMCM-BP may coordinate cell cycle arrest and cytokinesis.
Figure 6. Accumulation of G2 and zoid cells in TbMCM-BP-deficient BF T. brucei.

(A) The accumulation of G2 and <2C cells upon TbMCM-BP removal by flow cytometry. (B) The accumulation of the 1N2K (G2) and zoid cells upon TbMCM-BP removal by DAPI staining.
In T. brucei, replication and segregation of kinetoplast DNA (K) precede those of nuclear DNA (N) [53], so cells at different stages can be distinguished by their NK configurations. 1N1K content indicates that cells are either in G1 or S phase. When cells are entering into late S/G2, they show 1N2K content with two closely spaced kinetoplasts. Mitotic cells have 2N2K content, with two Ks are already separated but 2N remain close to each other. Post-mitotic cells have 2N2K with separated daughter nuclei and kinetoplasts. Representatives of cells with these DNA contents are shown in Figure S2B (0 hr). To further confirm the cell cycle defects observed from the flow cytometry, we examined TbMCM-BP cKO cells stained with DAPI after Cre induction (Figure 6B). Before Cre induction (0 hr), the majority of control cells were 1N1K (78%, white bars), and G2 and M/post-M cells were 11% (light grey) and 5% (dark grey), respectively. After 12 hr of Cre induction, the G2 population increased from 11% to 31%. At 24, 36, and 48 hrs post-induction, G2 cells accumulated further.
Interestingly, the number of abnormal cells greatly increased over time, reaching up to 58% 36 hr post-induction (Figure 6B, blue bars). The most prominent abnormality caused by TbMCM-BP deficiency is accumulation of anucleated cells (0N0K, 0N1K, 0N2K), also known as zoids [54], [55]. These are probably the majority of >2C cells observed in the flow cytometry analysis. As zoids started accumulating at 24 hr post Cre induction, following 1N2K (G2 or late S) accumulation at 12 hr (Figure 6B), some of G2-arrested mother cells may have segregated into a zoid and 4C progeny. Other abnormal cells were observed less frequently (Figure S2), for example, cells that appeared to have less than 1N and a fragmented nucleus, probably due to defective DNA replication. Other cells showed defective chromosome segregation, exhibiting an elongated nucleus, DNA bridging, absence of kinetoplast, and uncoupled segregation of N and K (for example cells with 2N content but 2K remained close to each other and cells with 1N content but 2 separated Ks). The cell cycle profiles of the conditional-KO mutant suggest that TbMCM-BP is required for proper cell-cycle progression, especially at S/G2 phase, and timely cell division.
PF and BF trypanosomes have different mitotic checkpoints. Deficiency of the mitotic-cyclin CYC6 increased the appearance of zoids in PF, but caused the accumulation of multinucleated endoreduplicated cells in BF [56]. Furthermore, mutations in T. brucei cell cycle and mitotic checkpoint genes encoding Polo-like kinase 1, and cdc2-related kinases [56]–[60] as well as TbTRF (a telomere-binding protein) [61] and TbSPT16 [12] all increased multinucleated cells in BF. In addition, depletion of replication factors, TbORC1 or subunits of MCM complex was not associated with accumulation of zoid cells in BF [36], [37], [39]. The accumulation of zoids observed in TbMCM-BP deficient cells is therefore a novel phenotype that has not been observed in any mutation of BF trypanosomes examined.
T. brucei MCM-BP co-purifies with MCM4-MCM8
In human and yeast, MCM-BP replaces MCM2 and forms a complex with MCM3-7 [15], [62] while plant MCM-BP interacts with all MCM2-7 [17]. To characterize the T. brucei MCM-BP complex, we first examined interaction of TbMCM-BP with two MCM subunits, MCM2 (present in plant but not in human and yeast MCM-BP purification) and MCM5 (present in all MCM-BP complex purified so far). We carried out reciprocal co-immunoprecipitations (co-IPs) using epitope-tagged proteins expressed from their endogenous loci, TbMCM-BP-myc, MCM2-flag, or MCM5-flag in BF cells. The epitopes were inserted in heterozygous backgrounds (one allele deleted) to ensure functionalities of tagged versions. Lysates were immunoprecipitated and analyzed by immunoblotting using anti-myc or anti-flag antibodies. TbMCM-BP-myc pulled down only MCM5 but not MCM2 and TbMCM-BP-myc was consistently present only in MCM5-flag IP but not in MCM2-flag IP (Figure 7A). These results suggest that TbMCM-BP replaces MCM2 to form the T. brucei MCM-BP complex, similar to human and yeast MCM-BP.
Figure 7. TbMCM-BP co-purifies with MCM4-MCM8.

(A) TbMCM-BP interacts with MCM5 but not with MCM2 by co-immunoprecipitation. (B) Allele modification in cells exclusively expressing TbMCM-BP tagged with PTP. (C) Immunoblot monitoring of TbMCM-BP-PTP tandem affinity purification in extract (Input), IgG affinity chromatography flowthrough (FT-IgG), TEV protease eluate (Elu TEV), flowthrough of the anti-ProtC affinity chromatography (FT-ProtC), and final eluate (Elu). Note that TEV protease-mediated removal of the ProtA domains led to a size reduction of tagged TbMCM-BP (‘BP-PTP’ to ‘BP-P’). Relative amounts of aliquots are specified by x values. (D) Tandem affinity purification of TbMCM-BP. Final eluate and smaller aliquots of the TEV protease eluate and extract were separated on an SDS–gradient polyacrylamide gel and stained with Coomassie blue or Sypro Ruby. (E) TbMCM-BP is localized in the nucleus throughout the cell cycle in PF.
To further characterize the TbMCM-BP complex, we isolated TbMCM-BP by tandem affinity purification (TAP) employing an approach that was developed in trypanosomes [63] and is based on the composite PTP tag consisting of a protein C epitope, a tobacco etch virus (TEV) protease site, and tandem protein A (ProtA) domains. As it is difficult to grow BF cells in a large quantity due to their sensitivity to cell density, TbMCM-BP was purified from PF trypanosomes, which grow to a 10–20-fold higher density.
We fused the PTP sequence to the 3′ end of the remaining TbMCM-BP allele in TbMCM-BP−/+ PF cells by targeted insertion of plasmid pTbMCM-BP-PTP-NEO, thereby generating a cell line that expressed only C-terminally tagged TbMCM-BP (Figure 7B). Tandem affinity purification of tagged TbMCM-BP from crude extract prepared from this cell line was highly efficient as revealed by immunoblot monitoring of individual purification steps (Figure 7C). Staining of purified proteins after separation on an SDS–gradient polyacrylamide gel revealed a strong signal for TbMCM-BP as well as an array of bands that contained sub-stoichiometric amounts of co-purified proteins (Figure 7D). Most of these proteins appear to specifically interact with TbMCM-BP because the vast majority of these bands were not detectable in comparable purifications of other nuclear protein complexes in trypanosomes (for an example see [63]). Eight MCM proteins, MCM2-MCM9, were annotated (based on sequence homology) in the T. brucei genome. Liquid chromatography-tandem mass spectrometry analysis of the entire lane showed that TbMCM-BP co-purified with MCM4-MCM8, but not with MCM2, MCM3, and MCM9 (Table 1). MCM8 was not found in any MCM-BP complex that was isolated to date, which include complexes from human, plant, frog egg and fission yeast [15]–[17], [25]. The canonical T. brucei MCM complex has been recently characterized and consists of MCM2-7 [37]. Our protein interaction data suggest that the TbMCM-BP complex has an unusual composition. Furthermore, TbMCM-BP is located in the nucleus throughout the cell cycle in PF trypanosomes (Figure 7E).
Table 1. Mass spectrometric identification of T. brucei MCM subunits that co-purified with TbMCM-BP.
| Annotation1 | Mol. mass2 | # unique peptides3 | Sequence coverage4 | Peptide count5 | Best ion score6 | Protein score7 | |
| MCM-BP | Tb927.7.1770 | 68.2 | 27 . | 49% . | 404 . | 133 . | 8663 . |
| MCM4 | Tb927.11.12250 | 93.3 | 31 . | 47% . | 70 . | 102 . | 888 . |
| MCM5 | Tb927.11.5570 | 85.5 | 45 . | 57% . | 191 . | 125 . | 2093 . |
| MCM6 | Tb927.11.11640 | 97.3 | 3 . | 5% . | 3 . | 40 . | 60 . |
| MCM7 | Tb927.11.16140 | 82.1 | 42 . | 44% . | 124 . | 87 . | 713 . |
| MCM8 | Tb927.10.10410 | 79.1 | 27 . | 30% . | 63 . | 84 . | 414 . |
Accession number of the GeneDB/TriTrypDB databases.
Molecular mass in kDa.
Number of distinct peptide sequences identified for each protein.
Maximal sequence coverage of unique peptides identified from a single gel slice.
Total number of peptides identified for each co-purified protein.
Highest ion score among identified peptides for each protein (ion scores were considered significant at ≥22 with expectation values ≤0.05).
The protein score is the sum of highest ions score for each distinct sequence.
Discussion
Loss-of-silencing (LOS) screen using random mutagenesis in T. brucei
We performed our loss-of-silencing screen in PF, due to technical problems in BF and also due to some of advantages that the PF system offers, although monoallelic VSG expression is relevant in BF T. brucei. T. brucei is a diploid organism and transposon mutagenesis relies on subsequent second allele disruption by gene conversion (GC) from the first targeted allele to generate a pool of homozygous mutants, which is particularly critical for isolation of ‘loss-of-function’ alleles. It is therefore important that a screen should start with a sufficiently large pool of workable number of cells for the success of large-scale searches. Because VSGs are also repressed in PFs, which can grow to a 10–20 fold higher density than BF cells, PFs offered better chances for success of our initial screen.
Transposon insertional mutagenesis was induced by transfecting a transposon-donor plasmid [44] containing a HYG gene flanked by mariner inverted repeats (IRs) and NEO for the selection of cells transformed by the plasmid. NEO-resistant cells were expanded to a population of ∼5×108 cells, prior to selection with hygromycin. Cells can only become resistant to hygromycin once the donor cassette is transposed into a transcribed chromosomal orientation. HYG transposition appears to be about 5% [44], suggesting that 2.5×107 of 5×108 NEO-resistant cells should be HYG-resistant, having transposon insertion somewhere in the genome. Previously, two independent homozygous glycosylation mutants were isolated from a large population of trypanosomes mutagenized with a mariner-transposon and the second allele seemed to have been disrupted by gene conversion by a mariner-targeted allele with an apparently significantly elevated GC rate, ∼2.5×10−3 [44]. This is about 250-fold higher than the GC rate measured at the TbURA3 locus (∼1×10−5) [40]. If the elevated GC rate is a common feature of transposon-targeted loci, 6.25×104 of 2.5×107 HYG-resistant cells should be homozygous mutants, which is more than enough to cover the entire genome of T. brucei (∼8×103 genes).
However, we were unable to identify any known genes that affect BES promoter silencing in PF, such as TbISWI, TbNLP, or TbDAC3 [8], [11], [64], [65]. In addition, only one allele was disrupted when we examined 8 transposon insertion sites in los clones (highlighted in yellow in Table S1), including the five TbMCM-BP-targeted los1 clones. Therefore, the screen was not saturated and the previously observed high GC rate at the transposon-inserted site [44] was probably due to its position, rather than due to a universal effect of mariner transposition. If transposon-targeted regions have typical GC rates (∼1×10−5) [40], 2.5×107 NEOR HYGR cells should represent about 250 homozygous los mutants, covering only 3.1% of the genome.
One of the differences between the previous proof-of-principle transposon screen that isolated two homozygous glycosylation mutants and our LOS screen is that, in the former, a pool of transposon-mutagenized trypanosomes was strongly selected using eight sequential treatments with increasing amount of concanavalin A (conA, a drug that counter-selects glycosylation mutants), which may have allowed further enrichment of homozygous mutants. On the other hand, a trypanosome pool was treated one time with 100 µg/ml puromycin in our LOS screen, which may have limited the chance to accumulate homozygous mutants in the pool.
Despite the shortfall, it is noteworthy that our screen isolated a true positive clone that affects VSG silencing. An N-terminal half of fission yeast MCM-BP is sufficient for MCM4 interaction [16] and the transposon was targeted downstream of putative MCM binding domain in our los1 clones. Therefore, it is possible that a C-terminally truncated Tbmcm-BP protein is expressed and acts as a dominant negative allele by competing with wild-type MCM-BP for the interaction with MCM subunits. In addition, in six clones, transposons were inserted at intergenic regions, which could disrupt functions of untranslated regions (UTRs) and affect expression levels of neighboring genes. In fact, in one los clone (clone 10), Tb927.4.4520 expression was increased 5 fold proving that a transposon insertion can interfere with gene expression near its target site. Spontaneous loss of heterozygosity in Leishmania major has been reported to be 10−4∼10−6 and increased 20–1,000-fold after mutagen treatments [66]. Therefore, it may be possible to significantly increase the size of homozygous mutants in the pool to cover the entire genome of T. brucei, by sequentially treating transposon-targeted trypanosomes with increasing amount of puromycin together with mutagens. We conclude that our transposon-mediated random mutagenesis can be a useful tool for a large-scale genetic screen once properly tuned and it could isolate not only ‘loss-of-function’ mutants, but also ‘gain-of-function’ and overexpression mutants.
MCM-BP in gene silencing in bloodstream form T. brucei
A number of recent studies demonstrated that chromosome maintenance factors are required for silencing of BES promoters and VSGs, and transcriptional VSG switching (in situ switching) in T. brucei. Depletion of TbORC1, a protein involved in replication initiation, derepressed BES-associated silent VSGs and metacyclic VSGs in BF trypanosomes and increased the in situ switching rate [35], [36], [38], [39]. Depletion of TbSCC1, a subunit of cohesin involved in chromosome segregation, also increased in situ VSG switching [67]. Derepression of silent VSGs was also observed in BF cells depleted of chromatin assembly factors, TbASF1A and TbCAF-1b, and the linker histone H1 [13], [14]. BF trypanosomes treated with aphidicolin, an inhibitor of DNA replication, increased expression of genes adjacent to a silent BES promoter in a dosage dependent manner but did not significantly change the expression level of silent VSGs [68]. At a high dose, aphidicolin induces global chromosome aberrations [69]. Interestingly, several of chromatin remodeling proteins showed similar phenotypes as an aphidicolin treatment. For example, TbISWI, TbSPT16, TbDAC3 and TbNLP showed strong effects near silent BES promoters without significantly affecting silencing near telomeres [8], [11], [12], [65]. Because we have limited mechanistic understanding of trypanosome DNA replication, chromatin assembly, and antigenic variation, it is not clear how these factors involved in BES and/or VSG silencing are linked. However, because TbMCM-BP-depleted cells showed stronger derepression near telomeres than silent BES promoters, TbMCM-BP should have roles in VSG silencing independently of chromatin remodeling proteins.
Recently, replication origins have been mapped along all 11 megabase chromosomes by TbORC1 chromatin IP [38]. Most replication origins appear to be located at the start of RNA pol II polycistronic transcription units. TbORC1 is also enriched along BES, including subtelomeric VSG regions [38], and TbORC1 associates with telomeres [39]. It is not known whether any of TbORC1 binding sites in BESs are utilized as replication origins but, given the high density of binding sites, it is likely that some of them might be required for cellular functions other than replication initiation. Human MCM-BP interacts with DBF4, a regulatory subunit of an early S-phase specific kinase complex DDK, which also contains CDC7 protein kinase [70]. Phosphorylation of MCM helicase by DDK is required for replication initiation, and MCM-BP partially inhibits phosphorylation of MCM by DDK [70] suggesting that MCM-BP has roles in replication initiation. In addition, overexpression of MCM-BP in fission yeast inhibited replication in S-phase and accumulated cells with less than 2C content and fragmented nuclei which could be due to a replication defect and/or to the uncoupling of completion of DNA replication and cell division [16]. On the other hand, MCM-BP is required specifically for replication termination by removing MCM helicase complex at the end of S-phase in Xenopus [25]. It is not clear whether TbMCM-BP is also required for replication initiation and termination. However, while silencing of TbORC1, MCM subunit genes, and TbMCM-BP affected G2 progression in BF cells, only TbMCM-BP depletion caused significant accumulation of anucleated cells (zoids). Therefore, the role of TbMCM-BP seems to be specific at least during cell cycle progression. It appears that TbMCM-BP is mostly chromatin-unbound because we were not able to immunoprecipitate much DNA with TbMCM-BP (data not shown) suggesting that TbMCM-BP plays its role in chromosome maintenance via transient interaction with chromatin or via indirect interaction with chromatin.
Topoisomerase 2α (Top2 in yeasts) is required for centromere maintenance and for replication termination [71]–[73]. Telomere entanglement in taz1Δ (a fission yeast homologue of TRF, a telomere-binding protein) can be relieved by a top2 mutant [74]. In Xenopus, MCM-BP is required at the late S phase to remove MCM, probably to block re-replication [25]. Similarly, ICRF193, an inhibitor of Top2, inhibited MCM dissociation at the end of replication [25]. Interestingly, TbTOPO2α co-purified with TbMCM-BP (Table S2 and Table S3). As only four unique peptides of TbTOPO2α were identified by mass spectrometry, the interaction between TbTOPO2α and TbMCM-BP might occur transiently at a specific phase of the cell cycle. One possibility is that TbMCM-BP may be required for the coordination of replication termination and chromosome segregation by interacting with TOPO2α at the end of S-phase. Catenated sister chromosomes and/or entangled telomeres may induce modification of chromatin structure and influence transcriptional status of silent loci.
Derepression was much stronger at subtelomeric VSGs than at silent BES promoters. Therefore, TbMCM-BP may not have a direct effect on the activity of silent BES promoters. Transcription initiates at silent BESs but the transcripts are attenuated [43], thereby creating a gradient of levels of transcripts along silent BESs with less transcripts produced near telomeres. Disruption of telomere-binding protein TbRAP1 has a stronger effect on genes near telomeres in silent BESs [7]. RNAs from derepressed silent VSGs in TbMCM-BP-depleted cells seem to be polyadenylated, because they were readily identified in oligo-dT-reverse transcribed cDNA, and VSGs expressed from silent BESs were detected by immunoblot and IFs. We do not know whether the role of TbMCM-BP in VSG silencing is dependent on TbRAP1, but it is possible that TbMCM-BP functions with some chromatin binding proteins with greater affinities to telomeres or subtelomeric regions. Chromatin changes along silent BESs may facilitate transcription elongation, leading to a disproportional increase of transcripts from the most distally positioned VSG gene.
Depletion of TbMCM-BP also increased the levels of procyclin and PAG RNAs, which are transcribed from an RNA pol I promoter that is repressed in BF. This transcription unit is located between two convergent RNA Pol II polycistrons. Chromatin remodellers, TbISWI and TbNLP, are also required for repression of procyclin and PAGs [64], [65]. Therefore, TbMCM-BP seems to work in a separate pathway from TbISWI and/or TbNLP regarding the silencing of VSG loci, but might work together with these chromatin factors in silencing of a chromosome-internally located RNA Pol I-transcription unit. Depletion of TbCAF-1b led to derepression of silent VSGs specifically in late-S phase and G2 [13] and depletion of TbSPT16 caused G2/M-specific derepression of silent BES promoters [12]. However, derepression of silent VSGs was observed during all cell cycle in TbASF1A-depleted cells [13]. TbMCM-BP is required for S/G2 cell cycle progression, but silent VSG derepression does not seem to depend on the phase of the cell cycle, or on abnormalities in cell cycle. However, we were able to detect silent VSG expression only in a subset of TbMCM-BP-depleted cells by IF, and a more quantitative method would be necessary to firmly determine if there is a correlation between TbMCM-BP-mediated VSG silencing and cell cycle. Collectively, data from various genes suggest that networks of multiple complex pathways are involved in silencing VSG and procyclin transcription loci in BF trypanosomes. It has been difficult to perform complicated genetic analysis in T. brucei, partly because generation of mutations has relied solely on sequential transfections using limited number of selection markers, due to inability to reproduce meiotic stages in culture. With our loxP-Cre and conditional KO systems, we can now analyze, relatively easily, genetic interactions between multiple key factors by creating double or triple mutants and this will be helpful to get more detailed molecular understandings on complex mechanisms, such as antigenic variation.
This is the first time that a VSG silencing factor has been selected by a phenotype-based large-scale screening approach, and T. brucei is the first organism in which MCM-BP was identified in such way. TbMCM-BP is strongly associated with four T. brucei MCM-subunits, MCM4-MCM7, and MCM8, a subunit that is uniquely co-purified only with MCM-BP in T. brucei. Whether or not this TbMCM-BP's unique composition has special roles in antigenic variation in T. brucei remains to be determined.
Materials and Methods
Trypanosome strains
Trypanosoma brucei bloodstream forms (strain Lister 427 antigenic type MITat1.2 clone 221a) were cultured in HMI-9 at 37°C [75]. The cell lines constructed for this study are listed in Table S4, and bloodstream form (BF) trypanosomes were either wild type or of ‘single marker’ (SM) background expressing T7 RNA polymerase and Tet repressor (TetR) [45]. Procyclic trypanosomes (PF427) were from the same Lister 427 strain and cultured in SDM-79 at 27°C [76]. Stable clones of BF and PF trypanosomes were in HMI-9 and SDM-79 media, respectively. BF clones were maintained in HMI-9 media containing necessary antibiotics at the following concentrations, unless otherwise stated: 2.5 µg/ml G418 (Sigma); 5 µg/ml blasticidin (Invivogen); 5 µg/ml hygromycin (Sigma); 0.1 µg/ml puromycin (Sigma); 1 µg/ml phleomycin (Invivogen). Stable clones of procyclic trypanosomes were obtained using electroporation and maintained in SDM-79 media containing necessary antibiotics at the following concentrations: 15 µg/ml G418 (Sigma); 40 µg/ml hygromycin (Sigma); 1 µg/ml puromycin (Sigma).
Triple-reporter cell line (HSTB-10): PF427 was transfected with a linearized plasmid (pHJ1) containing a puromycin-resistance (PUR), luciferase (LUC), and emerald-GFP (emGFP) genes, and targeting sequences immediate downstream of a BES promoter. The targeted BES was identified by PCR analyses using a primer set specific to the insert and ESAG7 located downstream of a BES promoter and using a polymorphism of ESAG6 [77]. The clone that had the reporter insertion at the BES11 (HSTB-10) was obtained.
Reporter cell line expressing the mariner transposase (HSTB-39): The HSTB-10 was transfected with a pHJ2 to stably express mariner transposase.
TbMCM-BP knock out (KO) cell lines: one allele of TbMCM-BP was deleted (HSTB-317) using PUR conjugated with Herpes simplex virus thymidine kinase gene (HSVTK or TK) flanked by loxP sites so that the markers can be removed by expressing Cre-recombinase and be reused [78]. To delete the second allele, a fragment of pSY45 digested with Sal I and Not I containing 650 bp upstream homology sequences, HYG-TK, and 1042 bp downstream homology sequences were transfected either into wild type or HSTB-317.
TbMCM-BP RNAi cell lines (HSTB-464 and -465): the wild-type MCM-BP allele in the sKO (HSTB-317) was epitope-tagged in situ with 3xmyc using one-step PCR epitope tagging plasmid pMOTag53M [79] to make HSTB-459. The HSTB-459 was transfected with pHJ35 containing 437 bp RNAi sequences specific to TbMCM-BP to make HSTB-464 and -465.
TbMCM-BP conditional knock out (cKO) cell line (HSTB-660): HSTB-317 (TbMCM-BP sKO, mcm-BPΔPUR-TK/MCM-BP-wt) was transfected with pLEW100-Cre-EP1 [78] to make inducible Cre expression cell line (HSTB-645). The PUR-TK marker was removed by expressing Cre recombinase to obtain HSTB-647. The wild type allele of TbMCM-BP in the HSTB-647 was then replaced with a TbMCM-BP cKO cassette containing MCM-BP-myc followed by HYG-TK and flanked by loxP sites (pDS24), to make HSTB-660.
TbMCM-BP cKO cell line with a silent BES marked with a triple reporter (HSTB-683 and -684): HSTB-660 was transfected with pHJ1 to randomly target one of silent BESs with markers, PUR-LUC-emGFP. Targeting was confirmed by PCR analyses and puromycin sensitivity at 10 µg/ml.
To make a PF cell line expressing TbMCM-BP-PTP, HSTB-586 (mcm-BPΔHYG-TK/MCM-BP) was transfected with pDS15 (pMCM-BP-PTP-NEO). The tag sequence was fused to the C-terminus of an endogenous TbMCM-BP allele by targeted insertion of the plasmid pDS15. Cells expressing MCM-BP-3xmyc, MCM2-flag, and MCM5-flag were constructed using one-step PCR epitope tagging method [79]. The sequences of primers used here are available upon request.
Construction of plasmids
Plasmids used for this study are listed in Table S5. Detailed construction information and maps are available upon request.
Loss-of-silencing (LOS) screen
To make a loss-of-silencing reporter cell line (HSTB-10), PF trypanosomes were transfected with a pHJ1, a triple-reporter plasmid containing a puromycin-resistance (PUR), luciferase (LUC), and emerald-GFP (emGFP) genes, and targeting sequences immediate downstream of a BES promoter. The HSTB-10 was transfected with a pHJ2 to stably express mariner transposase to make HSTB-39. We titrated puromycin concentration for the screen and confirmed that the 100 µg/ml effectively kills the reporter cells with the rate of spontaneous mutation ∼3×10−7. Random insertional mutagenesis was induced by transfecting cells with the donor-plasmid pSGL35 [44] containing a hygromycin-resistance gene (HYG) flanked by mariner inverted repeats (IRs) and a neomycin-resistance gene (NEO) to select cells transformed by the plasmid. The plasmid-transfected cells were expanded under G418 selection until the population reached 5×108 and were distributed in the 96-well plates containing 100 µg/ml puromycin and 40 µg/ml hygromycin. 19 PURR HYGR clones were isolated and examined for luciferase activity (Promega). The transposon target sites were mapped by inverse-PCR followed by sequencing [44].
mRNA preparation and reverse-transcription-quantitative real-time PCR
Total mRNA was prepared using RNA STAT60 (TEL-Test) as described in manufacturer's protocol and cDNAs were generated using oligo dT20 and reverse-transcriptase (Stratagene). RNA was amplified using primers specific to individual genes by quantitative PCR using the LightCycler 480 (Roche). Amplified double-stranded DNA product during 40 cycles was detected by SYBR Green I. All measurements were in triplicate and compared with a 1000-fold range of serially diluted standard genomic DNA prepared from the wild-type strain. The sequences of primers are available upon request.
Co-immunoprecipitation and immunoblot
About 108 cells were lysed in lysis buffer (25 mM Tris–HCl (pH 8.0), 1 mM EDTA, 0.5% NP-40, 10% glycerol, 1 mM phenylmethylsulfonylfloride (PMSF), 1 mM dithiothreitol (DTT), protease inhibitor cocktails (Sigma)). Cell lysates were immunoprecipitated and analyzed by immunoblot using with anti-myc (Sigma and in-house monoclonal antibody core facility) or anti-flag antibodies (Sigma). TAP immunoblot monitoring of TbMCM-BP-PTP was carried out with the monoclonal anti-protein C antibody HPC4 (Roche).
Tandem affinity purification of TbMCM-BP and identification of co-purified proteins by mass spectrometry
Tandem affinity purification of TbMCM-BP-PTP was performed exactly as described in the standard protocol [63]. Purified proteins were separated on an SDS-10 to 20% polyacrylamide gradient gel and first stained with Sypro Ruby (Invitrogen) and then with Coomassie blue (Gelcode Coomassie stain; Pierce) according to the manufacturers' protocols. For the identification of co-purified proteins, the gel lane of the final eluate was cut into 15 pieces, and peptides derived from trypsin-digested proteins were separated by liquid chromatography and analyzed by tandem mass spectrometry using an Ultimate 3000 HPLC system (Dionex) and a nanospray LTQ Orbitrap XL (Thermo Scientific) mass spectrometer. Proteins were identified using Mascot and NCBI non-redundant protein sequence database of eukaryotes, with carbamidomethyl (C) as static and oxidation (M) as variable modifications. Peptide Mass tolerance was set to ±25 ppm and fragment mass tolerance to ±0.8 Da. Peptides were considered as identified when their score was ≥22 and expectation values were ≤0.05.
Immunofluorescence and DNA staining
Immunofluorescence was carried out as described previously [80] with minor modifications. Briefly, cells were fixed with 1% paraformaldehyde for 5 minutes and permeabilized with 0.2% NP40 in PBS. Cells were incubated with chicken anti-VSG2 and either rabbit anti-VSG3 or VSG13, and then incubated with secondary antibodies (Alexa 488 for chicken and Texas Red for rabbit antibodies). TbMCM-BP-PTP was detected with a polyclonal anti-protein A antibody (Sigma) followed by an Alexa 594-conjugated anti-rabbit secondary antibody (Invitrogen). DNA was stained with 0.5 µg/ml DAPI. Images were captured using a Zeiss Axioplan 2 or AxioVert 200 fluorescence microscope and edited with Adobe Photoshop.
Flow cytometry
Cells were fixed with 70% ice-cold ethanol and stained with 0.5 µg/ml propidium iodide to examine cell cycle progression by flow cytometry.
Supporting Information
Depletion of TbMCM-BP by RNAi caused cell growth defect. The TbMCM-BP−/MYC strain was transfected with a construct containing an RNAi cassette specific to TbMCM-BP, which can be induced by adding tetracycline. Depletion of TbMCM-BP by RNAi showed a growth defect, which correlated with TbMCM-BP-myc disappearance. Two independent cell lines were examined and tubulin was used as a loading control. * indicates breakdown product of tubulin.
(TIF)
TbMCM-BP deficiency accumulated anucleated (zoid) cells and abnormalities in cell cycle and division. (A) Profiling of abnormal cells by DAPI staining. Anucleated cells greatly accumulated upon TbMCM-BP removal. Other abnormalities observed less frequently are categorized. (B) Examples of DAPI stained cells with the DNA content of 1N1K, 1N2K, and 2N2K, and of abnormal NK contents.
(TIF)
Mapping of transposon-targeted regions in LOS clones.
(DOC)
Mass spectrometric identification of non-MCM proteins that co-purified with TbMCM-BP.
(DOC)
Unique peptide sequences identified in each protein. Note: Some cases, same unique peptide sequence contained redundant peptide which show(s) different methionine oxidation site (underlined).
(DOC)
Trypanosoma brucei strains used in this study.
(DOC)
Plasmids used in this study.
(DOC)
Acknowledgments
We thank members of the Cross and Papavasiliou labs for helpful discussions, especially Danae Schulz and Monica Mugnier for help with flow cytometry analyses. We also thank Joseph Fernando and Milica Tesic Mark for mass spectrometry at the Proteomics resources center and Mike Rout lab for sharing a microscope and Zhanna Hakhverdyan for help.
Funding Statement
The work was supported by the Rockefeller University Women in Science Fellowship to HK and by grants R01 AI021729 (GAMC) and R01 AI059377 (AG) from the U.S. National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases (NIAID) or the NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Horn D, Barry JD (2005) The central roles of telomeres and subtelomeres in antigenic variation in African trypanosomes. Chrom Res 13: 525–533. [DOI] [PubMed] [Google Scholar]
- 2. Günzl A, Bruderer T, Laufer G, Schimanski B, Tu LC, et al. (2003) RNA polymerase I transcribes procyclin genes and variant surface glycoprotein gene expression sites in Trypanosoma brucei . Eukaryot Cell 2: 542–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hertz-Fowler C, Figueiredo LM, Quail MA, Becker M, Jackson A, et al. (2008) Telomeric expression sites are highly conserved in Trypanosoma brucei . PLoS ONE 3: e3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alarcon CM, Son HJ, Hall T, Donelson JE (1994) A monocistronic transcript for a trypanosome variant surface glycoprotein. Mol Cell Biol 14: 5579–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nagoshi YL, Alarcon CM, Donelson JE (1995) The putative promoter for a metacyclic VSG gene in African trypanosomes. Mol Biochem Parasitol 72: 33–45. [DOI] [PubMed] [Google Scholar]
- 6. Alsford S, Kawahara T, Isamah C, Horn D (2007) A sirtuin in the African trypanosome is involved in both DNA repair and telomeric gene silencing but is not required for antigenic variation. Mol Microbiol 63: 724–736. [DOI] [PubMed] [Google Scholar]
- 7. Yang X, Figueiredo LM, Espinal A, Okubo E, Li B (2009) RAP1 is essential for silencing telomeric variant surface glycoprotein genes in Trypanosoma brucei . Cell 137: 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hughes K, Wand M, Foulston L, Young R, Harley K, et al. (2007) A novel ISWI is involved in VSG expression site downregulation in African trypanosomes. EMBO J 26: 2400–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Figueiredo LM, Janzen CJ, Cross GAM (2008) A histone methyltransferase modulates antigenic variation in African trypanosomes. PLoS Biol 6: e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kawahara T, Siegel TN, Ingram AK, Alsford S, Cross GAM, et al. (2008) Two essential MYST-family proteins display distinct roles in histone H4K10 acetylation and telomeric silencing in trypanosomes. Mol Microbiol 69: 1054–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang QP, Kawahara T, Horn D (2010) Histone deacetylases play distinct roles in telomeric VSG expression site silencing in African trypanosomes. Mol Microbiol 77: 1237–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Denninger V, Fullbrook A, Bessat M, Ersfeld K, Rudenko G (2010) The FACT subunit TbSpt16 is involved in cell cycle specific control of VSG expression sites in Trypanosoma brucei . Mol Microbiol 78: 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alsford S, Horn D (2012) Cell-cycle-regulated control of VSG expression site silencing by histones and histone chaperones ASF1A and CAF-1b in Trypanosoma brucei . Nucl Acids Res 40: 10150–10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Povelones ML, Gluenz E, Dembek M, Gull K, Rudenko G (2012) Histone H1 Plays a Role in Heterochromatin Formation and VSG Expression Site Silencing in Trypanosoma brucei . PLoS Pathog 8: e1003010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sakwe AM, Nguyen T, Athanasopoulos V, Shire K, Frappier L (2007) Identification and characterization of a novel component of the human minichromosome maintenance complex. Mol Cell Biol 27: 3044–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ding L, Forsburg SL (2011) Schizosaccharomyces pombe Minichromosome Maintenance-binding Protein (MCM-BP) Antagonizes MCM Helicase. J Biol Chem 286: 32918–32930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takahashi N, Quimbaya M, Schubert V, Lammens T, Vandepoele K, et al. (2010) The MCM-binding protein ETG1 aids sister chromatid cohesion required for postreplicative homologous recombination repair. PLoS Genet 6: e1000817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pospiech H, Grosse F, Pisani FM (2010) The initiation step of eukaryotic DNA replication. Subcell Biochem 50: 79–104. [DOI] [PubMed] [Google Scholar]
- 19. Parrilla-Castellar ER, Arlander SJ, Karnitz L (2004) Dial 9-1-1 for DNA damage: the Rad9-Hus1-Rad1 (9-1-1) clamp complex. DNA repair 3: 1009–1014. [DOI] [PubMed] [Google Scholar]
- 20. Johnson A, O'Donnell M (2005) Cellular DNA replicases: components and dynamics at the replication fork. Annu Rev Biochem 74: 283–315. [DOI] [PubMed] [Google Scholar]
- 21. Majka J, Burgers PM (2004) The PCNA-RFC families of DNA clamps and clamp loaders. Prog Nucl Acid Res Mol Biol 78: 227–260. [DOI] [PubMed] [Google Scholar]
- 22. Mayer ML, Gygi SP, Aebersold R, Hieter P (2001) Identification of RFC(Ctf18p, Ctf8p, Dcc1p): an alternative RFC complex required for sister chromatid cohesion in S. cerevisiae . Mol Cell 7: 959–970. [DOI] [PubMed] [Google Scholar]
- 23. Forsburg SL (2004) Eukaryotic MCM proteins: beyond replication initiation. Microbiol Mol Biol Rev 68: 109–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takahashi N, Lammens T, Boudolf V, Maes S, Yoshizumi T, et al. (2008) The DNA replication checkpoint aids survival of plants deficient in the novel replisome factor ETG1. EMBO J 27: 1840–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nishiyama A, Frappier L, Mechali M (2011) MCM-BP regulates unloading of the MCM2-7 helicase in late S phase. Genes Dev 25: 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prasanth SG, Shen Z, Prasanth KV, Stillman B (2010) Human origin recognition complex is essential for HP1 binding to chromatin and heterochromatin organization. Proc Natl Acad Sci U S A 107: 15093–15098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mancio-Silva L, Rojas-Meza AP, Vargas M, Scherf A, Hernandez-Rivas R (2008) Differential association of Orc1 and Sir2 proteins to telomeric domains in Plasmodium falciparum . J Cell Sci 121: 2046–2053. [DOI] [PubMed] [Google Scholar]
- 29. Fox CA, Ehrenhofermurray AE, Loo S, Rine J (1997) The origin recognition complex, SIR1, and the S phase requirement for silencing. Science 276: 1547–1551. [DOI] [PubMed] [Google Scholar]
- 30. Ishimi Y, Ichinose S, Omori A, Sato K, Kimura H (1996) Binding of human minichromosome maintenance proteins with histone H3. J Biol Chem 271: 24115–24122. [DOI] [PubMed] [Google Scholar]
- 31. Snyder M, He W, Zhang JJ (2005) The DNA replication factor MCM5 is essential for Stat1-mediated transcriptional activation. Proc Natl Acad Sci U S A 102: 14539–14544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DaFonseca CJ, Shu F, Zhang JJ (2001) Identification of two residues in MCM5 critical for the assembly of MCM complexes and Stat1-mediated transcription activation in response to IFN-gamma. Proc Natl Acad Sci U S A 98: 3034–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holland L, Gauthier L, Bell-Rogers P, Yankulov K (2002) Distinct parts of minichromosome maintenance protein 2 associate with histone H3/H4 and RNA polymerase II holoenzyme. Eur J Biochem 269: 5192–5202. [DOI] [PubMed] [Google Scholar]
- 34. Dziak R, Leishman D, Radovic M, Tye BK, Yankulov K (2003) Evidence for a role of MCM (mini-chromosome maintenance)5 in transcriptional repression of sub-telomeric and Ty-proximal genes in Saccharomyces cerevisiae . J Biol Chem 278: 27372–27381. [DOI] [PubMed] [Google Scholar]
- 35. Godoy PD, Nogueira-Junior LA, Paes LS, Cornejo A, Martins RM, et al. (2009) Trypanosome prereplication machinery contains a single functional orc1/cdc6 protein, which is typical of archaea. Eukaryot Cell 8: 1592–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tiengwe C, Marcello L, Farr H, Gadelha C, Burchmore R, et al. (2012) Identification of ORC1/CDC6-Interacting Factors in Trypanosoma brucei Reveals Critical Features of Origin Recognition Complex Architecture. PLoS ONE 7: e32674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dang HQ, Li Z (2011) The Cdc45.Mcm2-7.GINS protein complex in trypanosomes regulates DNA replication and interacts with two Orc1-like proteins in the origin recognition complex. J Biol Chem 286: 32424–32435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tiengwe C, Marcello L, Farr H, Dickens N, Kelly S, et al. (2012) Genome-wide analysis reveals extensive functional interaction between DNA replication initiation and transcription in the genome of Trypanosoma brucei . Cell reports 2: 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Benmerzouga I, Concepcion-Acevedo J, Kim HS, Vandoros AV, Cross GAM, et al. (2013) Trypanosoma brucei Orc1 is essential for nuclear DNA replication and affects both VSG silencing and VSG switching. Mol Microbiol 87: 196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim HS, Cross GAM (2010) TOPO3alpha influences antigenic variation by monitoring expression-site-associated VSG switching in Trypanosoma brucei . PLoS Pathog 6: e1000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim HS, Cross GAM (2011) Identification of Trypanosoma brucei RMI1/BLAP75 homologue and its roles in antigenic variation. PLoS ONE 6: e25313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peacock L, Ferris V, Sharma R, Sunter J, Bailey M, et al. (2011) Identification of the meiotic life cycle stage of Trypanosoma brucei in the tsetse fly. Proc Natl Acad Sci U S A 108: 3671–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vanhamme L, Poelvoorde P, Pays A, Tebabi P, Xong HV, et al. (2000) Differential RNA elongation controls the variant surface glycoprotein gene expression sites of Trypanosoma brucei . Mol Microbiol 36: 328–340. [DOI] [PubMed] [Google Scholar]
- 44. Leal S, Acosta-Serrano A, Morris JC, Cross GAM (2004) Transposon mutagenesis of Trypanosoma brucei identifies glycosylation mutants resistant to Concanavalin A. J Biol Chem 279: 28979–28988. [DOI] [PubMed] [Google Scholar]
- 45. Wirtz E, Leal S, Ochatt C, Cross GAM (1999) A tightly regulated inducible expression system for dominant negative approaches in Trypanosoma brucei . Mol Biochem Parasitol 99: 89–101. [DOI] [PubMed] [Google Scholar]
- 46. Boothroyd CE, Dreesen O, Leonova T, Ly KI, Figueiredo LM, et al. (2009) A yeast-endonuclease-generated DNA break induces antigenic switching in Trypanosoma brucei . Nature 459: 278–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wickstead B, Ersfeld K, Gull K (2004) The small chromosomes of Trypanosoma brucei involved in antigenic variation are constructed around repetitive palindromes. Genome Res 14: 1014–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Horn D, McCulloch R (2010) Molecular mechanisms underlying the control of antigenic variation in African trypanosomes. Curr Opin Microbiol 13: 700–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Horn D, Cross GAM (1997) Position-dependent and promoter-specific regulation of gene expression in Trypanosoma brucei . EMBO J 16: 7422–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Horn D, Cross GAM (1995) A developmentally regulated position effect at a telomeric locus in Trypanosoma brucei . Cell 83: 555–561. [DOI] [PubMed] [Google Scholar]
- 51. Biebinger S, Rettenmaier S, Flaspohler J, Hartmann C, Penadiaz J, et al. (1996) The PARP promoter of Trypanosoma brucei is developmentally regulated in a chromosomal context. Nucl Acids Res 24: 1202–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jagannathan M, Sakwe AM, Nguyen T, Frappier L (2012) The MCM-associated protein MCM-BP is important for human nuclear morphology. J Cell Sci 125: 133–143. [DOI] [PubMed] [Google Scholar]
- 53. Ploubidou A, Robinson DR, Docherty RC, Ogbadoyi EO, Gull K (1999) Evidence for novel cell cycle checkpoints in trypanosomes: kinetoplast segregation and cytokinesis in the absence of mitosis. Journal of Cell Science 112: 4641–4650. [DOI] [PubMed] [Google Scholar]
- 54. Cross GAM, Manning JC (1973) Cultivation of Trypanosoma brucei sspp. in semi-defined and defined media. Parasitology 67: 315–331. [DOI] [PubMed] [Google Scholar]
- 55. Robinson DR, Sherwin T, Ploubidou A, Byard EH, Gull K (1995) Microtubule polarity and dynamics in the control of organelle positioning, segregation, and cytokinesis in the trypanosome cell cycle. J Cell Biol 128: 1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hammarton TC, Clark J, Douglas F, Boshart M, Mottram JC (2003) Stage-specific differences in cell cycle control in Trypanosoma brucei revealed by RNA interference of a mitotic cyclin. J Biol Chem 278: 22877–22886. [DOI] [PubMed] [Google Scholar]
- 57. Li Z, Wang C (2006) Changing roles of aurora-B kinase in two life cycle stages of Trypanosoma brucei . Eukaryot Cell 5: 1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tu X, Wang C (2005) Pairwise knockdowns of cdc2-related kinases (CRKs) in Trypanosoma brucei identified the CRKs for G1/S and G2/M transitions and demonstrated distinctive cytokinetic regulations between two developmental stages of the organism. Eukaryot Cell 4: 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Umeyama T, Wang CC (2008) Polo-like kinase is expressed in S/G2/M phase and associated with the flagellum attachment zone in both procyclic and bloodstream forms of Trypanosoma brucei . Eukaryot Cell 7: 1582–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hammarton TC, Kramer S, Tetley L, Boshart M, Mottram JC (2007) Trypanosoma brucei Polo-like kinase is essential for basal body duplication, kDNA segregation and cytokinesis. Mol Microbiol 65: 1229–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li B, Espinal A, Cross GAM (2005) Trypanosome telomeres are protected by a homologue of mammalian TRF2. Mol Cell Biol 25: 5011–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li JJ, Schnick J, Hayles J, MacNeill SA (2011) Purification and functional inactivation of the fission yeast MCM(MCM-BP) complex. FEBS Lett 585: 3850–3855. [DOI] [PubMed] [Google Scholar]
- 63. Schimanski B, Nguyen TN, Günzl A (2005) Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryot Cell 4: 1942–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stanne TM, Kushwaha M, Wand M, Taylor JE, Rudenko G (2011) TbISWI regulates multiple polymerase I (Pol I)-transcribed loci and is present at Pol II transcription boundaries in Trypanosoma brucei . Eukaryot Cell 10: 964–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Narayanan MS, Kushwaha M, Ersfeld K, Fullbrook A, Stanne TM, et al. (2011) NLP is a novel transcription regulator involved in VSG expression site control in Trypanosoma brucei . Nucl Acids Res 39: 2018–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gueiros-Filho FJ, Beverley SM (1996) Selection against the dihydrofolate reductase-thymidylate synthase (DHFR-TS) locus as a probe of genetic alterations in Leishmania major . Mol Cell Biol 16: 5655–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Landeira D, Bart JM, Van Tyne D, Navarro M (2009) Cohesin regulates VSG monoallelic expression in trypanosomes. J Cell Biol 186: 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sheader K, Vruchte DT, Rudenko G (2004) Bloodstream form-specific up-regulation of silent VSG expression sites and procyclin in Trypanosoma brucei after inhibition of DNA synthesis or DNA damage. J Biol Chem 279: 13363–13374. [DOI] [PubMed] [Google Scholar]
- 69. Glover TW, Berger C, Coyle J, Echo B (1984) DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet 67: 136–142. [DOI] [PubMed] [Google Scholar]
- 70. Nguyen T, Jagannathan M, Shire K, Frappier L (2012) Interactions of the human MCM-BP protein with MCM complex components and Dbf4. PLoS ONE 7: e35931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fachinetti D, Bermejo R, Cocito A, Minardi S, Katou Y, et al. (2010) Replication termination at eukaryotic chromosomes is mediated by Top2 and occurs at genomic loci containing pausing elements. Mol Cell 39: 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Luo K, Yuan J, Chen J, Lou Z (2009) Topoisomerase IIalpha controls the decatenation checkpoint. Nat Cell Biol 11: 204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nitiss JL (2009) DNA topoisomerase II and its growing repertoire of biological functions. Nature reviews 9: 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Germe T, Miller K, Cooper JP (2009) A non-canonical function of topoisomerase II in disentangling dysfunctional telomeres. EMBO J 28: 2803–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hirumi H, Hirumi K (1989) Continuous cultivation of Trypanosoma brucei bloodstream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol 75: 985–989. [PubMed] [Google Scholar]
- 76. Brun R, Schonenberger M (1979) Cultivation and in vitro cloning of procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Acta Tropica 36: 289–292. [PubMed] [Google Scholar]
- 77. Becker M, Aitcheson N, Byles E, Wickstead B, Louis E, et al. (2004) Isolation of the repertoire of VSG expression site containing telomeres of Trypanosoma brucei 427 using transformation-associated recombination in yeast. Genome Res 14: 2319–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Scahill MD, Pastar I, Cross GAM (2008) CRE recombinase-based positive-negative selection systems for genetic manipulation in Trypanosoma brucei . Mol Biochem Parasitol 157: 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Oberholzer M, Morand S, Kunz S, Seebeck T (2006) A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol Biochem Parasitol 145: 117–120. [DOI] [PubMed] [Google Scholar]
- 80. Lowell JE, Cross GAM (2004) A variant histone H3 is enriched at telomeres in Trypanosoma brucei . J Cell Sci 117: 5937–5947. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Depletion of TbMCM-BP by RNAi caused cell growth defect. The TbMCM-BP−/MYC strain was transfected with a construct containing an RNAi cassette specific to TbMCM-BP, which can be induced by adding tetracycline. Depletion of TbMCM-BP by RNAi showed a growth defect, which correlated with TbMCM-BP-myc disappearance. Two independent cell lines were examined and tubulin was used as a loading control. * indicates breakdown product of tubulin.
(TIF)
TbMCM-BP deficiency accumulated anucleated (zoid) cells and abnormalities in cell cycle and division. (A) Profiling of abnormal cells by DAPI staining. Anucleated cells greatly accumulated upon TbMCM-BP removal. Other abnormalities observed less frequently are categorized. (B) Examples of DAPI stained cells with the DNA content of 1N1K, 1N2K, and 2N2K, and of abnormal NK contents.
(TIF)
Mapping of transposon-targeted regions in LOS clones.
(DOC)
Mass spectrometric identification of non-MCM proteins that co-purified with TbMCM-BP.
(DOC)
Unique peptide sequences identified in each protein. Note: Some cases, same unique peptide sequence contained redundant peptide which show(s) different methionine oxidation site (underlined).
(DOC)
Trypanosoma brucei strains used in this study.
(DOC)
Plasmids used in this study.
(DOC)