

Abstract
The aim of this paper is to investigate the physicochemical properties of binary amorphous dispersions of poorly soluble sulfonamide/polymeric excipient prepared by ball milling. The sulfonamides selected were sulfathiazole (STZ), sulfadimidine (SDM), sulfamerazine (SMZ) and sulfadiazine (SDZ). The excipients were polyvinylpyrrolidone (PVP) and polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol graft co-polymer, commercially known as Soluplus®. Co-milled systems were characterised by powder X-ray diffraction and differential scanning calorimetry. PVP was shown to form amorphous dispersions over a wider composition range than Soluplus® for the four sulfonamides tested. Moreover, amorphous dispersions made with PVP were homogeneous [single glass transition (Tg)], while amorphous dispersions made from Soluplus® were heterogeneous (two Tgs). This behaviour is consistent with the fact that all the sulfonamides tested presented a lower solubility in Soluplus® than in PVP, as evidenced by Flory–Huggins parameters determined. Amorphous dispersions of SDM with Soluplus® could be produced even though SDM does not amorphise alone upon milling and Soluplus® presents Tg at a lower temperature than SDM. Amorphous dispersions of SMZ could be prepared with a lower excipient concentration compared to STZ, SDM and SDZ, which may reflect the one-dimensional H-bonding network in SMZ compared to the 2D or 3D H-bonding network found in the other sulfonamides. Stability tests (60% RH/25°C) revealed that dispersions made with Soluplus® remained dry and powdery compared to those made with PVP that formed a sticky paste in less than 2 weeks, indicating a possible advantage of using Soluplus® in terms of increased physical stability under high humidity storage conditions.
KEY WORDS: amorphous dispersions, milling, polyvinylpyrrolidone, Soluplus, sulphonamide
INTRODUCTION
Due to a high Gibbs free energy, amorphous formulations represent a promising approach to overcome limited bioavailability of poorly soluble drugs (1–4). However, the amorphous state is unstable (5), and there is a risk that an amorphous material will recrystallise during the typical shelf-life of a medicine (about 2 years), limiting the benefits of such formulations (6). In order to improve the stability of an active pharmaceutical ingredient (API) in the amorphous state, a well-known strategy consists in co-processing it with a high glass transition (Tg) polymeric excipient to produce an amorphous dispersion (6,7). The higher the Tg compared to the storage temperature, the lower the likelihood of crystallisation (8). It is critical to carefully select the components to make an amorphous dispersion so as to optimise the solubility of the API in the polymer and thus reduce the risk of phase separation in the amorphous system (9–11). An important point to consider is that attainment of a one-phase system requires the two components to be thermodynamically miscible during processing (11). An understanding of drug-polymer thermodynamic phase diagrams which indicate not only the solubility of (crystalline) drug in the polymer but also the amorphous drug-polymer miscibility is important, due to the potential correlation of these parameters to the physical stability of the amorphous solid dispersion (11).
Several methods are traditionally used to produce amorphous dispersions such as melt quenching (6), hot melt extrusion (12,13), spray drying (10,14,15) and freeze drying (16,17). Milling is a technique widely used to reduce particle size (18) but is not commonly used for making amorphous dispersions, although it has been shown to be suitable to amorphise pharmaceutical compounds and to produce glassy solutions (10,19–22). Although milling presents some disadvantages compared to the more traditional techniques mentioned above, such as potential for material caking in the mill and challenges with scale-up, advantages associated with the process include the absence of thermal stress if the process is properly controlled (23) and the absence of solvent.
The aim of this article is to investigate the production and the physicochemical characteristics of binary amorphous dispersions sulfonamides/polymeric excipients by ball milling. The sulfonamides selected, which are all poorly soluble in aqueous media, are sulfathiazole (STZ), sulfadimidine (SDM), sulfamerazine (SMZ) and sulfadiazine (SDZ). Despite similar molecular structures, they present several differences. STZ and SMZ present several polymorphs (10,24) while SDM and SDZ present, so far, only one crystalline form (25,26). The excipients selected are polyvinylpyrolidone (PVP) and polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol graft co-polymer (commercially available as Soluplus®). PVP is an amorphous polymer which is widely used in the pharmaceutical industry, known to stabilise amorphous pharmaceutical systems (6,7) and to enhance their dissolution (10). Soluplus® is a new amorphous polymer specifically designed to produce amorphous dispersions by hot melt extrusion (27,28) (due to a low Tg of 72°C) and to enhance solubility and dissolution of poorly soluble drugs (27,28). The co-processed mixtures were characterised by powder X-ray diffraction (PXRD) and by standard and modulated differential scanning calorimetry (DSC and MDSC). Solubility of the sulfonamides in PVP or in Soluplus® was also investigated.
MATERIALS AND METHODS
Materials
STZ [molecular weight (Mw) = 255.32 g mol−1], SDM (Mw = 278.33 g mol−1), SMZ (Mw = 264.30 g mol−1), SDZ (Mw = 250.28 g mol−1) and PVP (Mw = 10,000 g mol−1 according to the supplier) were purchased from Sigma-Aldrich and used without further purification. Soluplus® (polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol graft co-polymer; 90,000 g mol−1 ≤ Mw ≤ 140,000 g mol−1 according to the supplier) was obtained as a gift from BASF and used without further purification.
Methods
Milling and Cryo-Milling
Ball milling at room temperature was performed with a PM 100 high energy planetary mill (Retsch, Germany) at room temperature. Zirconium oxide milling jars of 50 cm3 with three balls (Ø = 20 mm) of the same material were used. API/excipient (2.5 g total powder weight; PVP or Soluplus® as excipient) mixture was placed in the jar corresponding to a ball/sample weight ratio of 32:1. The rotation speed of the solar disk was set to 400 rpm. In order to avoid possible overheating of the container, pause periods of 10 min were made after every 20 min of milling. Total milling time was kept constant at 15 h corresponding to an effective milling time of 10 h. This duration of milling ensured that a stationary state [no more evolution of the physical state of the compounds upon milling (19)] was reached. The ambient relative humidity in the room where the mill was located was in the range 25–45% RH during milling.
Cryo-milling was performed in an oscillatory ball mill (Mixer Mill MM400, Retsch GmbH & Co., Germany). A fresh 1 g batch of powder was used for each milling. The powder samples were placed in a 25-mL volume stainless steel milling jar containing one stainless steel ball (Ø = 15 mm). The ball/sample weight ratio was 14:1. Milling jars were then sealed and immersed in liquid nitrogen for 3 min before milling at 25 Hz. Re-cooling of the milling chambers for 2 min with liquid nitrogen was performed every 7.5 min. The total milling time was 1 h for all the cryomilled samples. Solid-state analysis by DSC and PXRD, as detailed below, was undertaken immediately post-milling.
Melt Quenching and Determination of the Glass Transition Temperatures of the Sulfonamides
Melt quenching of STZ, SDM and SMZ was performed as previously described (10) by melting the initially crystalline powder, placed in an aluminium weighing boat, on a hot plate (IKA RCT basic, Germany) and quenching the obtained liquid by removing the aluminium weighing boat from the hot plate and allowing to it cool at room temperature. This enabled fully amorphous samples (as determined by powder X-ray diffraction and differential scanning calorimetry) to be prepared. It should be noted that, preliminary thermogravimetric analysis performed for this article revealed that SDZ undergoes extensive thermal degradation during melting preventing the production of amorphous SDZ by melt quenching.
Powder X-ray Diffraction
PXRD measurements were performed on samples placed on a low background silicon sample holder (except for co-milled SDM/PVP where a standard glass sample holder was used), using a Rigaku Miniflex II desktop X-ray diffractometer (Rigaku, Tokyo, Japan) with the Bragg-Brentano geometry, as previously described (10). The PXRD patterns were recorded from 5° to 40° on the 2θ scale at a step of 0.05°/s. X-ray tube composed of Cu anode (λCuKα = 1.54 Å) was operated under a voltage of 30 kV and current of 15 mA. PXRD patterns of cryo-milled samples were recorded on an Inel Equinox 3000 powder diffractometer with 1,200 s data collection time and a Cu anode X-ray tube operated at 30 kV and 25 mA
Thermal Analysis
DSC experiments for STZ/PVP, SDM/PVP and SDZ/PVP co-milled systems were conducted using a Mettler Toledo 821e with a refrigerated cooling system (LabPlant RP-100), as previously described (10). Nitrogen was used as the purge gas. Aluminium pans (40 μL) with three vent holes were used throughout the study, and sample weights varied between 3 and 6 mg. The system was calibrated for temperature and cell constant using indium and zinc. A heating rate of 10°C/min was implemented in all DSC measurements. Analysis was carried out and monitored by Mettler Toledo STARe software (version 6.10) with a Windows NT operating system.
DSC experiments for STZ/Soluplus®, SDM/ Soluplus®, SMZ/Soluplus®, SDZ/Soluplus® and SMZ/PVP co-milled systems were conducted using a DSC Q200 (TA Instruments, UK) in hermetic pans with one pin hole, as previously described (10). The instrument was calibrated for temperature and cell constant using high purity indium. A heating rate of 10°C/min was implemented in all DSC measurements.
Measurements of heat capacity of amorphous APIs were conducted on a DSC Q200 (TA Instruments, UK) in modulation mode, as previously described (10). Sealed pans with one pin hole were used. The instrument was calibrated for temperature and cell constant using high purity indium. Heat capacity readings were calibrated using sapphire. The parameters used for these experiments were: heating rate, 1°C min−1; amplitude of modulation, 1°C; and period of modulation, 120 s.
The temperature of exothermic or endothermic events given in this paper was the onset temperature. Glass transition temperatures were measured at the midpoint of the Cp shift.
Thermogravimetric analysis (TGA) experiments were conducted on a TGA Q50 (TA Instruments, UK). Weighed samples (5–10 mg) were analysed in open aluminium pans placed. All samples were heated from 25°C to the melting point at a scanning rate of 10°C/min under nitrogen purge.
Determination of Solubility of Crystalline APIs in Polymer
The method developed by Tao et al. (29) to determine the solubility of a crystalline API in a polymer was used to determine the solubility of the four sulfonamides studied in PVP or Soluplus®. Physical mixtures of API with excipient were prepared by gravimetrically mixing the compounds in the required proportions. The mixtures were milled at room temperature with a mixer mill MM 200 (Retsch, Germany) for 30 min at 70% of maximum intensity of milling in order to improve the intimate contact between particles of API and particles of excipient in the physical mix (10). Attention was paid that crystalline API was still present at the end of milling (checked by PXRD). The obtained mixtures were then held at 100°C for 5 min in a DSC cell flushed with dry nitrogen to remove moisture, cooled back to room temperature and subjected to a DSC scan at 1°C min−1 to measure the temperature of the dissolution endpoint (Tend) of the drug in the excipient. The relationship between Tend and composition allowed the solubility of API in PVP as a function of temperature to be obtained (29).
Stability Testing of Amorphous Dispersions
The physical stability of the amorphous dispersions was investigated as a function of humidity and temperature using an Amebis stability testing system (Amebis, Ireland). Samples were placed in Amebis humidity devices at 60% RH and stored in a Gallenkamp incubator at 25°C. The samples were tested periodically by PXRD.
RESULTS
Production of Sulfonamides/PVP Composites by Co-milling
PXRD Analysis
Figure 1 presents the PXRD patterns of the unprocessed sulfonamides, unprocessed PVP, milled sulfonamides and co-milled sulfonamide/PVP binary systems with various PVP weight fractions (XPVP). PXRD patterns of unprocessed sulfonamides present sharp Bragg peaks characteristic of crystalline materials. The diffractogram of unprocessed STZ reveals that the polymorphic form is III (10), stable at room temperature and atmospheric pressure. The diffractogram of unprocessed SMZ is characteristic of form I (24), the metastable polymorph at room temperature. According to Zhang et al. (24), the kinetics of conversion of the metastable to stable form of SMZ in the solid state is so slow that the metastable form does not convert back to the stable one at room temperature. SDM and SDZ do not present polymorphism, and the PXRD patterns recorded for this article correspond to the unique PXRD patterns previously published (25). The PXRD pattern of PVP presents a diffuse halo characteristic of an amorphous material. After milling, SDM, STZ and SDZ diffractograms present a clear broadening and a loss of intensity of the Bragg peaks. The PXRD pattern of milled STZ reveals that form III is mainly present. Polymorph IV may also be present, but the broadening of the peaks in the PXRD prevents definitive conclusions from being made (10). The changes in PXRD patterns of these sulfonamides correlate with reduction in crystallite sizes, formation of crystallite strains and partial amorphisation (30–32). The PXRD pattern of milled SMZ is clearly different to that of the unprocessed material. There are Bragg peaks at 2θ = 15.7°, 19.2°, 21.3° and 26.6° that were not present in the pattern of the unmilled material. Comparison with results previously published by Zhang et al. (24) reveals that the PXRD pattern is characteristic of form II, the stable polymorph of SMZ at room temperature. This shows a remarkable counterintuitive example of stabilisation of material upon milling as the Gibbs free energy of the stable form II is lower than that of the metastable form I (24). Such polymorphic transformations upon milling from a metastable/unstable state towards a stable state have been observed in other systems such as indomethacin (33) for example. Overall, the PXRD patterns of co-milled sulfonamide/PVP systems present only a diffuse halo with no Bragg peaks, characteristic of a PXRD amorphous system, when a sufficient proportion of PVP is present. The minimum amount of PVP required for the complete disappearance of the Bragg peaks depends on the sulfonamide. For STZ, SDM, SMZ and SDZ, this amount is XPVP = 0.6, 0.7, 0.3 and 0.6, respectively. For PVP loads below these values, the X-ray patterns present residual Bragg peaks, characteristics of a crystalline phase.
Fig. 1.
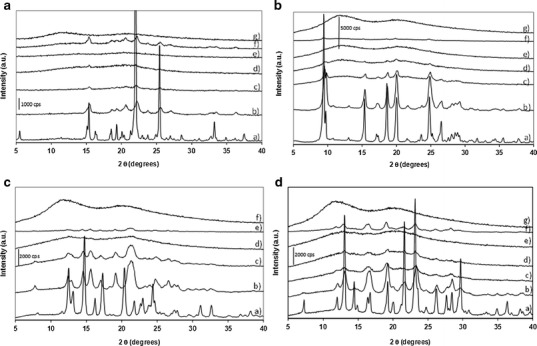
PXRD patterns of sulfonamide/PVP systems. (X PVP = weight fraction of PVP, milled systems milled for 10 h at 400 rpm). a STZ/PVP systems. a Unprocessed STZ; b milled STZ (X PVP = 0); c co-milled STZ/PVP with X PVP = 0.3; d co-milled STZ/PVP with X PVP = 0.5; e co-milled STZ/PVP with X PVP = 0.6; f physical mix of separately milled STZ and PVP with X PVP = 0.6 and g) milled PVP, X PVP = 1. b SDM/PVP systems. a Unprocessed SDM; b milled SDM (X PVP = 0); c co-milled SDM/PVP with X PVP = 0.3; d co-milled SDM/PVP with X PVP = 0.6; e co-milled SDM/PVP with X PVP = 0.7; f physical mix of separately milled SDM and PVP with X PVP = 0.7; g milled PVP, X PVP = 1. c SMZ/PVP systems. a Unprocessed SMZ; b milled SMZ (X PVP = 0); c co-milled SMZ/PVP with X PVP = 0.2; d co-milled SMZ/PVP with X PVP = 0.3; e physical mix of separately milled SMZ and PVP with X PVP = 0.7; f milled PVP, X PVP = 1. d SDZ/PVP systems. a Unprocessed SDZ; b milled SDZ (X PVP = 0); c co-milled SDZ/PVP with X PVP = 0.3; d co-milled SDZ/PVP with X PVP = 0.5; e co-milled SDZ/PVP with X PVP = 0.6; f physical mix of separately milled SDZ and PVP with X PVP = 0.6; g milled PVP, X PVP = 1
Thermal Behaviour
Figure 2 presents the DSC scans of the unprocessed sulfonamides, of milled sulfonamides and of co-milled sulfonamides/PVP binary systems with XPVP equal to the minimum PVP load necessary to obtain X-ray amorphous dispersions. The thermograms of the unprocessed SDM, SMZ and SDZ reveal only an endotherm attributed to the melting of the crystalline phase at 196.7°C, 237.1°C and 259.9°C, respectively. Moreover, TGA reveals that SDZ loses more than 25% of its initial mass during the melting process which is characteristic of severe thermal decomposition (data not shown). This decomposition is also confirmed by visual examination of SDZ samples, which, after melting, have the appearance of black foamed solids. The thermogram of unprocessed STZ reveals a first endotherm at 168.6°C attributed to the transition from form III to form I (34) followed by a second endotherm at 201.7°C attributed to the melting of form I.
Fig. 2.
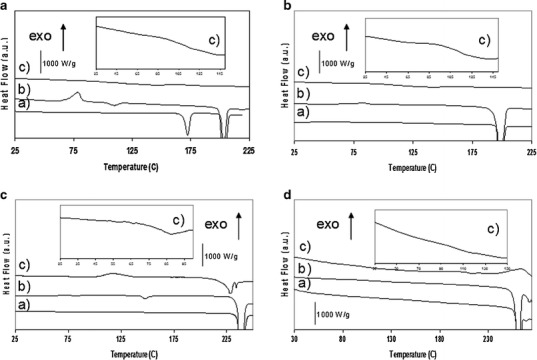
DSC scans of sulfonamide/PVP systems. (X PVP = weight fraction of PVP, milled systems milled for 10 h at 400 rpm). a STZ/PVP systems. a Unprocessed STZ; b milled STZ (X PVP = 0); c co-milled STZ/PVP with X PVP = 0.6; insert: glass transition zone for X PVP = 0.6. b SDM/PVP systems. a Unprocessed SDM; b milled SDM (X PVP = 0); c co-milled SDM/PVP with X PVP = 0.7 insert: glass transition zone for X PVP = 0.7. c SMZ/PVP systems. a Unprocessed SMZ; b milled SMZ (X PVP = 0); c co-milled SMZ/PVP with X PVP = 0.3 insert: glass transition zone for X PVP = 0.3. d SDZ/PVP systems. a Unprocessed SDZ; b milled SDZ (X PVP = 0); c co-milled SDZ/PVP with X PVP = 0.6 insert: glass transition zone for X PVP = 0.6
Melt quenched STZ, SDM and SMZ presented a glass transition (Tg) at 58°C, 74°C and 62°C, respectively. Due to thermal degradation, it was not possible to melt quench SDZ. Its glass transition was therefore evaluated as 2/3 of the value of the temperature of melting in Kelvin (35). This leads to an estimated Tg of ~84°C for SDZ. The DSC scans of co-milled systems with XPVP greater than or equal to the quantity used to make X-ray amorphous samples reveal that these systems present a single glass transition (Tg) located at a temperature between the Tg of the pure API and the PVP. This single Tg shows that comilling induced formation of glass solutions in which the API and the PVP are mixed at a molecular level.
Production of Sulfonamides/Soluplus® Composites by Co-milling
PXRD Analysis
Figure 3 presents the PXRD patterns of unmilled Soluplus® and of co-milled sulfonamide/Soluplus® composites. The diffractogram of unmilled Soluplus® presents a diffuse halo characteristic of an amorphous material. The patterns of co-milled systems reveal that it is possible to form amorphous dispersions with Soluplus® for a minimum load which is XSoluplus® (weight fraction of Soluplus®) = 0.7, 0.8, 0.5 and 0.9 for STZ/Soluplus®, SDM/Soluplus®, SMZ/Soluplus® and SDZ/Soluplus®, respectively. Compared to the sulfonamide/PVP systems described in the “Production of Sulfonamides/PVP Composites by Co-milling” section, the composition range in which it is possible to prepare sulfonamide/Soluplus® amorphous dispersions by milling is narrower.
Fig. 3.
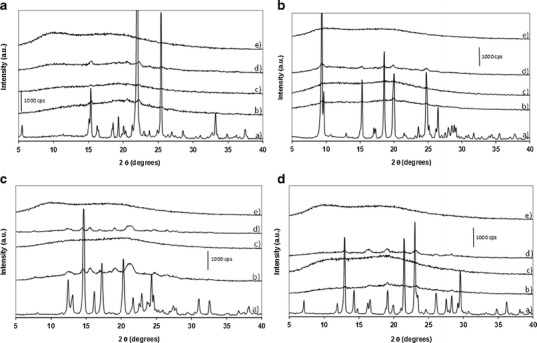
PXRD patterns of sulfonamide/Soluplus® systems. (X Soluplus® = weight fraction of Soluplus®, milled systems milled for 10 h at 400 rpm). a STZ/Soluplus® systems. a Unprocessed STZ; b co-milled STZ/Soluplus® with X Soluplus® = 0.6; c co-milled STZ/Soluplus® with X Soluplus® = 0.7; d physical mix of separately milled STZ and Soluplus® with X Soluplus® = 0.7; e milled Soluplus®, X Soluplus® = 1. b SDM/Soluplus® systems. a Unprocessed SDM; b co-milled SDM/Soluplus® with X Soluplus® = 0.7; c co-milled SDM/Soluplus® with X Soluplus® = 0.8; d physical mix of separately milled SDM and Soluplus® with X Soluplus® = 0.8; e milled Soluplus®, X Soluplus® = 1. c SMZ/Soluplus® systems. a Unprocessed SMZ; b co-milled SMZ/Soluplus® with X Soluplus® = 0.4; c co-milled SMZ/Soluplus® with X Soluplus® = 0.5; d physical mix of separately milled SMZ and Soluplus® with X Soluplus® = 0.5; e milled Soluplus®, X Soluplus® = 1. d SDZ/Soluplus® systems. a Unprocessed SDZ; b co-milled SDZ/Soluplus® with X Soluplus® = 0.8; c co-milled SDZ/Soluplus® with X Soluplus® = 0.9; d physical mix of separately milled SDZ and Soluplus® with X Soluplus® = 0.9; e milled Soluplus®, X Soluplus® = 1
Thermal Behaviour
Figure 4 presents the thermograms of unprocessed Soluplus® and of co-milled sulfonamide/Soluplus® systems. The thermogram of unprocessed Soluplus® reveals a glass transition at 72°C, consistent with the glass transition temperature reported by the supplier (around 70°C). The Cp shift of the baseline due to the glass transition spans over 100°C which is consistent with the polydispersity (90,000 g mol−1 ≤ Mw ≤ 140,000 g mol−1) reported by the supplier. As a consequence of the broadness of the Cp shift of the glass transition of Soluplus® and of the proximity of the Tg of Soluplus® to the Tg of the sulfonamides studied here, it is not possible to determine accurately if the amorphous dispersions present a single Tg or two overlapped Tgs although, as can be seen in the inserts of Fig. 4, it seems that these amorphous systems may present two Tgs. Therefore, in contrast to sulfonamide/PVP systems, the sulfonamide/Soluplus® amorphous dispersions appear to be heterogeneous.
Fig. 4.
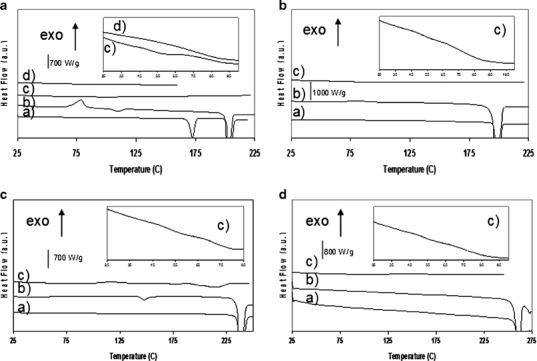
DSC scans of sulfonamide/Soluplus® systems. (X Soluplus® = weight fraction of Soluplus®, milled systems milled for 10 h at 400 rpm). a STZ/Soluplus® systems. a Unprocessed STZ; b milled STZ (X Soluplus® = 0); c co-milled STZ/Soluplus® with X Soluplus® = 0.7; d milled Soluplus®; insert: glass transition zone for co-milled system with X Soluplus® = 0.7 and X Soluplus® =1. b SDM/Soluplus® systems. a Unprocessed SDM; b milled SDM (X Soluplus® = 0); c co-milled SDM/Soluplus® with X Soluplus® = 0.8; insert: glass transition zone for co-milled system with X Soluplus® = 0.8. c SMZ/Soluplus® systems. a Unprocessed SMZ; b milled SMZ (XSoluplus® = 0); c co-milled SMZ/Soluplus® with X Soluplus® = 0.5; insert: glass transition zone for co-milled system with X Soluplus® = 0.5. d SDZ/Soluplus® systems. a Unprocessed SDZ; b milled SDZ (X Soluplus® = 0); c co-milled SDZ/Soluplus® with X Soluplus® = 0.9; insert: glass transition zone for X Soluplus® = 0.9
Solubility of Sulfonamides in PVP and Soluplus®
Table I presents the relationship between Tend, defined as the melting temperature endset (10,11), and the weight fraction of PVP for the sulfonamide/PVP systems. Table II presents the relationship between Tend and the weight fraction of Soluplus® for the sulfonamide/Soluplus® systems. For excipient fractions above the ones presented in the tables, the high viscosity of the systems and/or the weak intensity of the melting event prevented an accurate determination of Tend. In the case of the STZ, the polymorphic transformation III → I followed by the melting of form I during heating makes the analysis more complicated (10). As previously detailed (10), the endset of temperature of melting of form I is the one reported as Tend because above this temperature, the only stable form is the liquid (Form I is the polymorph with the highest melting point). However, for XSTZ ≤ 0.6 for STZ/PVP systems and for XSTZ ≤ 0.4 for STZ/Soluplus®, the DSC scans reveal only one endotherm. The nature of this endotherm is not clear. It appears in the temperature range of the transition III → I of STZ and the reason why we see it for these compositions is not clear. It could be because the temperature of transition III → I and the temperature of melting of form I overlapped or because the excipient prevented the conversion of form III toward form I (i.e. for these compositions, form III melts, but the resulting liquid cannot recrystallise to form I, possibly due to the presence of excipient). In order to circumvent this problem, we produced STZ form I by spray drying using the method described previously (10) and used it to make physical mixtures of STZ I/excipient. This enabled Tend for XSTZ = 0.6 and XSTZ = 0.4 for STZ/PVP and STZ/Soluplus® systems, respectively, to be determined. For XSTZ below these compositions, the high viscosity of the mixture at Tend and the weak intensity of the melting peak do not allow an accurate determination of the solution endpoints.
Table I.
Dissolution Endpoints (T end) Measured by DSC for STZ/PVP, SDM/PVP, SMZ/PVP and SDZ/PVP Systems
| X PVP | T end (°C) STZ/PVP | T end (°C) SDM/PVP | T end (°C) SMZ/PVP | T end (°C) SDZ/PVP |
|---|---|---|---|---|
| 0.0 | 202.5 ± 0.4 | 198.9 ± 0.1 | 237.5 ± 0.1 | 252.7 ± 0.1 |
| 0.1 | 199.9 ± 0.3 | 196.7 ± 0.0 | 232.7 ± 0.1 | 248.2 ± 0.2 |
| 0.2 | 195.4 ± 0.2 | 193.8 ± 0.0 | 230.6 ± 0.1 | 244.3 ± 0.0 |
| 0.3 | 186.4 ± 1.2 | 189.2 ± 0.2 | 228.2 ± 0.2 | 241.2 ± 0.2 |
| 0.4 | 173.2 ± 1.7 | 179.8 ± 1.5 | 224.3 ± 0.3 | 238.3 ± 0.0 |
| 0.5 | – | 165.1 ± 1.9 | 209.9 ± 4.0 | 233.6 ± 0.7 |
| 0.6 | – | 146.7 ± 2.8 | – | 217.9 ± 1.4 |
Each value is the average of two measurements. The value after ± is the standard deviation
X pvp weight fraction of PVP®
Table II.
Dissolution Endpoints (T end) Measured by DSC for STZ/Soluplus®, SDM/Soluplus®, SMZ/Soluplus® and SDZ/Soluplus® Systems
| X Soluplus® | T end (°C) STZ/Soluplus® | T end (°C) SDM/Soluplus® | T end (°C) SMZ/Soluplus® | T end (°C) SDZ/Soluplus® |
|---|---|---|---|---|
| 0.0 | 202.5 ± 0.4 | 198.9 ± 0.1 | 237.5 ± 0.1 | 252.7 ± 0.1 |
| 0.1 | 200.7 ± 0.1 | 197.8 ± 0.2 | 233.8 ± 0.1 | 247.2 ± 0.4 |
| 0.2 | 200.0 ± 0.1 | 196.2 ± 0.0 | 231.9 ± 0.1 | 244.0 ± 0.1 |
| 0.3 | 195.8 ± 0.0 | 193.6 ± 0.0 | 230.2 ± 0.1 | 242.0 ± 0.1 |
| 0.4 | 191.4 ± 0.1 | 189.7 ± 0.0 | 228.2 ± 0.1 | 240.0 ± 0.0 |
| 0.5 | 183.8 ± 0.0 | 183.7 ± 0.0 | 224.2 ± 0.0 | 237.7 ± 0.0 |
| 0.6 | 170.1 ± 0.0 | 173.8 ± 0.0 | 214.6 ± 0.2 | 234.1 ± 0.1 |
| 0.7 | – | 165.4 ± 0.4 | 201.8 ± 0.6 | 227.1 ± 0.0 |
X Soluplus® weight fraction of Soluplus®
Each value is the average of two measurements. The value after ± is the standard deviation
Stability Studies
The physical stability of amorphous dispersions made with SDM and Xexcipient (PVP or Soluplus) = 0.8 was tested at 60% RH and 25°C. The rationale behind this choice of composition was to select the same lowest concentration of excipient that would give an amorphous dispersion with PVP or Soluplus®. Results showed that, after 2 weeks of storage, both systems were still amorphous by PXRD, but SDM/PVP formed a sticky paste while SDM/Soluplus® was still powdery. TGA analysis showed a mass loss at 150°C of 3.4% for SDM/Soluplus® and of 11.6% for SDM/PVP (data not shown). This mass loss along with the visual inspection of the sample confirmed the higher hygroscopicity of SDM/PVP amorphous dispersions compared to SDM/Soluplus® amorphous dispersions.
DISCUSSION
Temperature-Composition Diagrams
An amorphous state will show a tendency to crystallise to lower its Gibbs free energy (5,6). It is therefore necessary to anticipate the instability of the amorphous dispersions produced to design amorphous formulations (7). Important factors to consider when probing the instability of amorphous dispersions of small molecules in polymers are the solubility of the API in the polymer and the position of Tg relative to the storage temperature. Based on the solubility of the API in the polymer and on the changes of Tg relative to the composition of the amorphous dispersions, it is possible to build temperature-composition diagrams in which the zones of stability and instability are defined (9–11). The solubility of the sulfonamides in PVP and Soluplus® has been determined from the Tend measurement based on the work of Yu’s group (29,36) and the Flory–Huggins (FH) theory (37). Tend represents the experimental solubility of the API in the polymer at a given composition and temperature (Tend). It is possible to determine the FH interaction parameter (which gives an indication of the solubility of drugs in polymeric excipients) by determining the activity of the API in the excipient using Eq. 1 and fitting this activity with Eq. 2 (36):
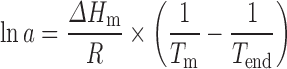 |
1 |
where a is the activity of the API in the polymer, ΔHm is the melting enthalpy (joules per mole) of the pure API, R is the gas constant (joules per degree Kelvin per mole), Tm is the melting point (Kelvin) of the pure API and Tend (Kelvin) is the melting point of the drug in a drug/polymer mixture.
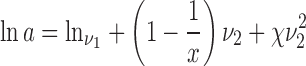 |
2 |
where ν1 is the volume fraction of the API, ν2 is the volume fraction of the excipient, x is the molar volume ratio of the excipient and the API and χ is the FH parameter.
The Tgs were determined for all sulfonamide/PVP mixtures and fitted with the Gordon-Taylor equation (38) using the Simha–Boyer rule (39). The fact that the experimental changes in Tg follow Gordon–Taylor indicates that there are no strong interactions between the APIs and the polymer (22). Table III gives the data necessary to determine the FH parameter from Eqs. 1 and 2 and the changes of Tg versus composition according to the Gordon–Taylor equation. Table IV gives the FH parameters determined for all the sulfonamide/excipients studied in this paper. For all the systems studied, the FH parameters were less than or equal to zero meaning that all the systems should be miscible according to the FH theory. It is worth noting that STZ and SDM on one hand and that SMZ and SDZ on the other hand have similar solubilities in the excipients. The FH parameters’ values reveal also that SMZ and SDZ should mix less easily with PVP or Soluplus® than STZ and SDM. These parameters reveal also that STZ, SDM, SMZ and SDZ are less soluble in Soluplus® than in PVP. This might explain why amorphous dispersions made from Soluplus® are less homogeneous (two Tgs) than the ones made from PVP (single Tg) and why, for a given sulfonamide, the concentration range in which it is possible to produce amorphous dispersions with Soluplus® is narrower than the concentration range in which it is possible to produce amorphous dispersions with PVP. An alternative explanation for the presence of two Tgs for amorphous dispersions made from Soluplus® is the high polydispersity of this polymer.
Table III.
Thermal Characteristics of STZ, SDM, SMZ, SDZ, PVP and Soluplus®
| Compounds/properties | T m (K) | ΔH m (kJ/mol) | Tg (K) | Density (g cm−3) | ΔCp (J K−1 g−1) |
|---|---|---|---|---|---|
| Sulfathiazole | 473.8 ± 0.1 (n = 3) | 28.7 ± 0.2 (n = 3) | 331.1 ± 0.8 (n = 7) | 1.53a | 0.44 ±0.02 (n = 3) |
| Sulfadimidine | 470.6 ± 0.1 (n = 3) | 36.0 ± 0.3 (n = 3) | 347.3 ± 0.6 (n = 7) | 1.34 ± 0.00 (n = 5) | 0.51 ±0.01 (n = 3) |
| Sulfamerazine | 510.5 ± 0.1 (n = 3) | 36.7 ± 1.0 (n = 3) | 335.5 ± 2.3 (n = 7) | 1.35 ± 0.00 (n = 5) | 0.79 ± 0.20 (n = 2) |
| Sulfadiazine | 535.3 ± 0.5 (n = 3) | 39.9 ± 0.5 (n = 3) | 356.9 (estimated as 2/3 of T m) | 1.50 ± 0.00 (n = 5) (measured on crystalline sample) | ND |
| PVP | – | – | 403.4 ± 5.1 (n = 3) | 1.23 ± 0.00 (n = 5) | 0.22b |
| Soluplus® | – | – | 344.8 ± 2.0 (n = 3) | 1.16 ± 0.00 (n = 5) | 0.29 ± 0.06 (n = 3) |
Table IV.
Flory–Huggins Parameter for the Sulfonamide/Polymer Systems Studied in this Paper
| STZ | SDM | SMZ | SDZ | |
|---|---|---|---|---|
| PVP | −1.5 (R 2 = 0.99) | −1.8 (R 2 = 0.99) | −0.7 (R 2 = 0.73) | −0.5 (R 2 = 0.76) |
| Soluplus® | −0.2 (R 2 = 0.99) | −0.3 (R 2 = 0.99) | −0.1 (R 2 = 0.71) | 0.0 (R 2 = 0.40) |
Figure 5a represents the zones of stability and instability on composition-temperature diagrams for sulfonamide/PVP systems. As previously described (10), these diagrams can be split into four zones. Zone I is a zone where the API forms an undersaturated solution with the polymer that has a Tg above the zone temperature range making it therefore stable from a thermodynamic and kinetic point of view. Zone II may still be kinetically hindered but to a lesser extent than Zone I. Zone II is a zone where the API forms an undersaturated solution with the polymer that has a Tg below the zone temperature range making it stable from a thermodynamic point of view but in which the high molecular mobility might trigger some problems (such as crystallisation and chemical degradation, for example). Zone III is a zone where the API forms a supersaturated solution with the polymer that has a Tg below the zone temperature range making it unstable from a thermodynamic point of view and in which the high molecular mobility will allow phase separation, recrystallisation and/or chemical degradations. Zone IV is a zone where the API forms a supersaturated solution with the polymer that has a Tg above the zone temperature range making it unstable from a thermodynamic point of view but in which the low molecular mobility might prevent the likelihood of phase separation, recrystallisation and/or chemical degradations.
Fig. 5.
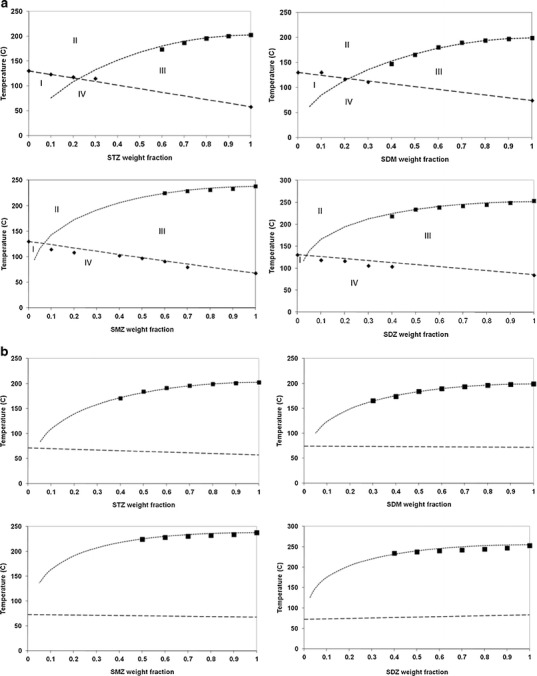
T end (dissolution endpoint) and Tg (glass transition temperature) as a function of API concentration. Diamonds represent experimental Tg, squares represent experimental T end, dashed line represents the Gordon–Taylor law, dotted line represents T end calculated from Eqs. 1 and 2. a API/PVP systems. b API/Soluplus® systems
Figure 5B represents the zones of stability and instability on composition-temperature diagrams for sulfonamide/Soluplus® systems. As the FH curve does not cross the Tg curve, it is not possible to split the diagrams in four zones like for the API/PVP systems. It is not clear from our results why co-milling SDM with Soluplus® produces amorphous dispersions while SDM does not amorphise when milled alone even though the Tg of SDM is higher than the Tg of Soluplus®. It seems reasonable to hypothesise that, upon milling, the mixing of the polymeric excipient with the API must prevent the sulfonamide molecules from rearranging themselves to form a crystal lattice.
Differing Behaviour of SMZ Compared to Other Sulfonamides
As shown by Figs. 1 and 3, SMZ is able to form amorphous dispersions with PVP or Soluplus® over a wider concentration range than STZ, SDM or SDZ (i.e. with a lower amount of excipient). It is clear from the FH parameters’ values that the greater ability of SMZ (compared to STZ, SDM and SDZ) to form amorphous dispersions with PVP or Soluplus® is not due to a greater miscibility with these excipients compared to the other sulfonamides studied in this article.
It has been shown that, the greater the glass transition temperature, the easier the amorphisation of a compound upon milling at a given temperature (19,21,22,32,40,41). However, this cannot explain the amorphisation of SMZ with lower excipient concentration compared to SDM as the Tg of SDM is higher than the Tg of SMZ, and therefore, one might expect that SDM/excipient systems should form amorphous dispersions over a wider concentration range than SMZ/excipient systems.
A striking feature of SMZ that might give an insight into its ability to form an amorphous dispersion with a lower excipient content compared the other sulfonamides studied here is the polymorphic transformation (from the metastable form I to the stable form II at room temperature) it undergoes upon milling. As a polymorphic transformation requires the rearrangement of the molecules in a different three-dimensional order, it is necessary that the system goes through a transitory amorphous phase. This transitory amorphous phase may not be observed at room temperature because, at this temperature, the molecular mobility is high enough to trigger the recrystallisation before the amorphous phase can be observed. However, milling at liquid nitrogen temperature allows the formation of an amorphous phase to be observed as shown in the PXRD patterns of Fig. 6. This amorphous form recrystallises towards the stable form II upon storage at room temperature. Form II is also the form obtained after milling SMZ I at room temperature. Therefore, this experiment supports the hypothesis of the formation of a transitory amorphous state during milling before conversion towards the stable form II. It seems therefore reasonable to hypothesise that SMZ is transitionally amorphised during milling at RT. For low content of excipient, the molecular mobility is high enough to prevent the direct observation of this amorphous form, but for XPVP ≥ 0.3 and XSoluplus® ≥ 0.5, the amorphous system obtained is stable enough to be observed. Therefore, a tentative explanation for the formation of amorphous dispersions with SMZ at lower excipient content compared to STZ, SDM and SDZ would be the formation of a transient amorphous state. The hypothesis of a transient amorphization upon milling of SMZ compared to STZ, SDM and SDZ is also supported by previous work reporting that SMZ I presents slip planes conferring plasticity on this crystalline form (42) and by the analysis of the crystalline networks of these sulfonamides (42) that revealed that SMZ I present a one-dimensional network of hydrogen bondings, while the other crystalline sulfonamides have a two or three-dimensional hydrogen bonding networks, making them less sensitive to milling. Therefore, the greater plasticity of form I and its one-dimensional network of hydrogen bonding facilitate the transient amorphisation upon milling. The fact that PVP or Soluplus® prevents the recrystallisation of the transitory amorphous form of SMZ more efficiently than for the other sulfonamides may be due to the one-dimensional nature of the SMZ H-bond network, allowing greater capacity for interaction with the stabilising excipients than the other sulfa compounds.
Fig. 6.
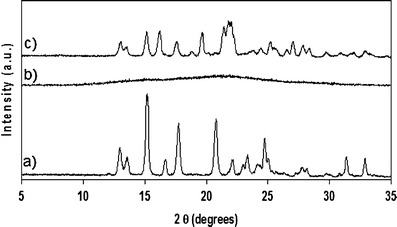
PXRD pattern of cryomilled SMZ and of cryomilled SMZ after storage at room temperature (RT) and humidity. a Unprocessed SMZ, b SMZ cryomilled for 1 h, c SMZ after 24 h of storage at RT post-cryomilling for 1 h
CONCLUSIONS
In this paper, we have shown that ball milling is a viable process to produce amorphous dispersions of small molecule API with amorphous polymeric excipients. The possibility of forming amorphous dispersions of SDM/Soluplus® while SDM milled alone does not amorphise highlighted the fact that using an excipient of higher Tg than the API was not a necessary condition to produce an amorphous system.
The possibility of forming amorphous dispersions of SMZ with a lower excipient concentration compared to STZ, SDM and SDZ seems consistent with the 1D H-bond network in the crystalline form I that induces a greater plasticity and facilitates the amorphisation of SMZ. This transient amorphous state was not observed during milling at RT, probably due to recrystallisation kinetics being too fast, but was evident in cryomilling experiments. The recrystallisation of the amorphous state obtained by cryomilling towards the same crystalline form as the one that is obtained after the polymorphic transformation observed upon milling at RT supports that hypothesis. Therefore, a tentative conclusion concerning the formation of amorphous dispersions of SMZ/excipient at lower excipient concentration compared to the other sulfonamides studied in this article would be that PVP or Soluplus® prevents the recrystallisation of the transitory amorphous form of SMZ more efficiently than for the other sulfonamides and that amorphisation is facilitated by the one-dimensional nature of the H-bond network. The stability diagrams established for these amorphous dispersions might help in the selection of the appropriate composition of the amorphous dispersions in order to control the stability at a given temperature.
ACKNOWLEDGMENTS
This paper is based upon works supported by the Science Foundation Ireland under grant no. [07/SRC/B1158] as part of the Solid State Pharmaceutical Cluster (SSPC).
REFERENCES
- 1.Murdande SB, Pikal MJ, Shanker RM, Bogner RH. Solubility advantage of amorphous pharmaceuticals: I. A thermodynamic analysis. J Pharm Sci. 2010;99(3):1254–64. doi: 10.1002/jps.21903. [DOI] [PubMed] [Google Scholar]
- 2.Patterson JE, James MB, Forster AH, Lancaster RW, Butler JM, Rades T. Preparation of glass solutions of three poorly water soluble drugs by spray drying, melt extrusion and ball milling. Int J Pharm. 2007;336(1):22–34. doi: 10.1016/j.ijpharm.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 3.Greco K, Bogner R. Crystallization of amorphous indomethacin during dissolution: effect of processing and annealing. Mol Pharm. 2010;7(5):1406–18. doi: 10.1021/mp1000197. [DOI] [PubMed] [Google Scholar]
- 4.Bogner RH, Murdande SB, Pikal MJ, Shanker RM. Solubility advantage of amorphous pharmaceuticals: II. Application of quantitative thermodynamic relationships for prediction of solubility enhancement in structurally diverse insoluble pharmaceuticals. Pharm Res. 2010;27(12):2704–14. doi: 10.1007/s11095-010-0269-5. [DOI] [PubMed] [Google Scholar]
- 5.Bhugra C, Pikal MJ. Role of thermodynamic, molecular, and kinetic factors in crystallization from the amorphous state. J Pharm Sci. 2008;97(4):1329–49. doi: 10.1002/jps.21138. [DOI] [PubMed] [Google Scholar]
- 6.Caron V, Bhugra C, Pikal MJ. Prediction of onset of crystallization in amorphous pharmaceutical systems: phenobarbital, nifedipine/PVP, and phenobarbital/PVP. J Pharm Sci. 2010;99(9):3887–900. doi: 10.1002/jps.22232. [DOI] [PubMed] [Google Scholar]
- 7.Tajber L, Corrigan OI, Healy AM. Physicochemical evaluation of PVP-thiazide diuretic interactions induced by processing—analysis of glass transition composition relationships. Eur J Pharm Sci. 2005;24(5):553–63. doi: 10.1016/j.ejps.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 8.Hancock BC, Shamblin SL, Zografi G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharm Res. 1995;12(6):799–806. doi: 10.1023/A:1016292416526. [DOI] [PubMed] [Google Scholar]
- 9.Qian F, Huang J, Hussain MA. Drug–polymer solubility and miscibility: stability consideration and practical challenges in amorphous solid dispersion development. J Pharm Sci. 2010;99(7):2941–7. doi: 10.1002/jps.22074. [DOI] [PubMed] [Google Scholar]
- 10.Caron V, Tajber L, Corrigan OI, Healy AM. A comparison of spray drying and milling in the production of amorphous dispersions of sulfathiazole/polyvinylpyrrolidone and sulfadimidine/polyvinylpyrrolidone. Mol Pharm. 2011;8(2):532–42. doi: 10.1021/mp1003674. [DOI] [PubMed] [Google Scholar]
- 11.Tian Y, Booth J, Meehan E, Jones DS, Li S, Andrews GP. Construction of drug–polymer thermodynamic phase diagrams using Flory–Huggins interaction theory: identifying the relevance of temperature and drug weight fraction to phase separation within solid dispersions. Mol Pharm. 2012. doi:10.1021/mp300386v. [DOI] [PubMed]
- 12.Andrews GP, Jones DS, Abu-Diak O, Margetson DN, McAllister MS. Hot-melt extrusion: an emerging drug delivery technology. Pharm Technol Eur. 2009;21(1):18–23. [Google Scholar]
- 13.Andrews GP, Abu-Diak O, Kusmanto F, Hornsby P, Hui Z, Jones DS. Physicochemical characterization and drug-release properties of celecoxib hot-melt extruded glass solutions. J Pharm Pharmacol. 2010;62(11):1580–90. doi: 10.1111/j.2042-7158.2010.01177.x. [DOI] [PubMed] [Google Scholar]
- 14.Corrigan OI, Holohan EM. Amorphous spray-dried hydroflumethiazide-polyvinylpyrrolidone systems: physicochemical properties. J Pharm Pharmacol. 1984;36(4):217–21. doi: 10.1111/j.2042-7158.1984.tb04353.x. [DOI] [PubMed] [Google Scholar]
- 15.Corrigan DO, Corrigan OI, Healy AM. Predicting the physical state of spray dried composites: salbutamol sulphate/lactose and salbutamol sulphate/polyethylene glycol co-spray dried systems. Int J Pharm. 2004;273(1–2):171–82. doi: 10.1016/j.ijpharm.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Lu X, Pikal MJ. Freeze-drying of mannitol-trehalose-sodium chloride-based formulations: the impact of annealing on dry layer resistance to mass transfer and cake structure. Pharm Dev Technol. 2004;9(1):85–95. doi: 10.1081/PDT-120027421. [DOI] [PubMed] [Google Scholar]
- 17.Luthra SA, Hodge IM, Pikal MJ. Investigation of the impact of annealing on global molecular mobility in glasses: optimization for stabilization of amorphous pharmaceuticals. J Pharm Sci. 2008;97(9):3865–82. doi: 10.1002/jps.21255. [DOI] [PubMed] [Google Scholar]
- 18.Brittain HG. Effects of mechanical processing on phase composition. J Pharm Sci. 2002;91(7):1573–80. doi: 10.1002/jps.10115. [DOI] [PubMed] [Google Scholar]
- 19.Willart JF, Descamps M. Solid state amorphization of pharmaceuticals. Mol Pharm. 2008;5(6):905–20. doi: 10.1021/mp800092t. [DOI] [PubMed] [Google Scholar]
- 20.Willart JF, Caron V, Descamps M. Transformations of crystalline sugars upon milling. J Therm Anal Calorim. 2007;90(1):125–30. doi: 10.1007/s10973-007-8485-x. [DOI] [Google Scholar]
- 21.Willart JF, Descamps N, Caron V, Capet F, Danede F, Descamps M. Formation of lactose-mannitol molecular alloys by solid state vitrification. Solid State Commun. 2006;138(4):194–9. doi: 10.1016/j.ssc.2006.02.034. [DOI] [Google Scholar]
- 22.Caron V, Willart JF, Danede F, Descamps M. The implication of the glass transition in the formation of trehalose/mannitol molecular alloys by ball milling. Solid State Commun. 2007;144(7–8):288–92. doi: 10.1016/j.ssc.2007.09.006. [DOI] [Google Scholar]
- 23.Willart JF, Caron V, Lefort R, Danede F, Prevost D, Descamps M. Athermal character of the solid state amorphization of lactose induced by ball milling. Solid State Commun. 2004;132(10):693–6. doi: 10.1016/j.ssc.2004.09.007. [DOI] [Google Scholar]
- 24.Zhang GGZ, Gu C, Zell MT, Todd Burkhardt R, Munson EJ, Grant DJW. Crystallization and transitions of sulfamerazine polymorphs. J Pharm Sci. 2002;91(4):1089–100. doi: 10.1002/jps.10100. [DOI] [PubMed] [Google Scholar]
- 25.Yang SS, Guillory JK. Polymorphism in sulfonamides. J Pharm Sci. 1972;61:26–40. doi: 10.1002/jps.2600610104. [DOI] [PubMed] [Google Scholar]
- 26.Mesley RJ, Houghton EE. Infrared identification of pharmaceutically important sulphonamides with particular reference to the occurrence of polymorphism. J Pharm Pharmacol. 1967;19:295–304. doi: 10.1111/j.2042-7158.1967.tb08090.x. [DOI] [PubMed] [Google Scholar]
- 27.Occhipinti V, Djuric D. A new excipient for hot-melt extrusion. Manuf Chem. 2010;81–82:36–8. [Google Scholar]
- 28.Hardung H, Djuric D, Ali S. Combining HME & solubilization: Soluplus®—the solid solution. Drug Deliv Technol. 2010;10(3):20–7. [Google Scholar]
- 29.Tao J, Sun Y, Zhang GGZ, Yu L. Solubility of small-molecule crystals in polymers: d -mannitol in PVP, indomethacin in PVP/VA, and nifedipine in PVP/VA. Pharm Res. 2009;26(4):855–64. doi: 10.1007/s11095-008-9784-z. [DOI] [PubMed] [Google Scholar]
- 30.Langford JI, Louer D. Powder diffraction. Rep Prog Phys. 1996;59(2):131–234. doi: 10.1088/0034-4885/59/2/002. [DOI] [Google Scholar]
- 31.Louer D. Advances in powder diffraction analysis. Acta Crystallogr A. 1998;54(6):922–33. doi: 10.1107/S0108767398007363. [DOI] [Google Scholar]
- 32.Dujardin N, Willart JF, Dudognon E, Hédoux A, Guinet Y, Paccou L, et al. Solid state vitrification of crystalline α and β-D-glucose by mechanical milling. Solid State Commun. 2008;148(1–2):78–82. doi: 10.1016/j.ssc.2008.07.002. [DOI] [Google Scholar]
- 33.Desprez S, Descamps M. Transformations of glassy indomethacin induced by ball-milling. J Non-Cryst Solids. 2006;352(42–49 SPEC. ISS):4480–5. doi: 10.1016/j.jnoncrysol.2006.02.130. [DOI] [Google Scholar]
- 34.Zeitler JA, Newnham DA, Taday PF, Threlfall TL, Lancaster RW, Berg RW, et al. Characterization of temperature-induced phase transitions in five polymorphic forms of sulfathiazole by terahertz pulsed spectroscopy and differential scanning calorimetry. J Pharm Sci. 2006;95(11):2486–98. doi: 10.1002/jps.20719. [DOI] [PubMed] [Google Scholar]
- 35.Debenedetti PG, Stillinger FH. Supercooled liquids and the glass transition. Nature. 2001;410(6825):259–67. doi: 10.1038/35065704. [DOI] [PubMed] [Google Scholar]
- 36.Sun Y, Tao J, Zhang GGZ, Yu L. Solubilities of crystalline drugs in polymers: an improved analytical method and comparison of solubilities of indomethacin and nifedipine in PVP, PVP/VA, and PVAc. J Pharm Sci. 2010;99(9):4023–31. doi: 10.1002/jps.22251. [DOI] [PubMed] [Google Scholar]
- 37.Flory PJ. Principle of polymer chemistry. Ithaca: Cornell University Press; 1953. [Google Scholar]
- 38.Gordon M, Taylor JS. Ideal copolymers and the second order transitions of synthetic rubbers I. Non-crystalline copolymers. J Appl Chem. 1952;2:493–500. doi: 10.1002/jctb.5010020901. [DOI] [Google Scholar]
- 39.Simha R, Boyer RF. On a general relation involving glass temperature and coefficient of expansion of polymers. J Chem Phys. 1962;37(5):1003–7. doi: 10.1063/1.1733201. [DOI] [Google Scholar]
- 40.Descamps M, Willart JF, Dudognon E, Caron V. Transformation of pharmaceutical compounds upon milling and comilling: the role of Tg. J Pharm Sci. 2007;96(5):1398–407. doi: 10.1002/jps.20939. [DOI] [PubMed] [Google Scholar]
- 41.Willart JF, Dujardin N, Dudognon E, Danede F, Descamps M. Amorphization of sugar hydrates upon milling. Carbohydr Res. 2010;345(11):1613–6. doi: 10.1016/j.carres.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 42.Sun C, Grant DJW. Influence of crystal structure on the tableting properties of sulfamerazine polymorphs. Pharm Res. 2001;18(3):274–80. doi: 10.1023/A:1011038526805. [DOI] [PubMed] [Google Scholar]