

Abstract
This study evaluated the effect of counterions on the physicochemical properties of prazosin salts. Salt forms of prazosin, namely, mesylate, besylate, tosylate, camsylate, oxalate, and maleate, were prepared and compared with the marketed anhydrous and polyhydrate forms of prazosin hydrochloride. Physicochemical characterization was performed in the order of crystallinity, hygroscopicity, solubility, and stability to select the optimal salt(s). Permeability study in Caco-2 cell lines and in vivo bioavailability study in rat model were investigated to ascertain their biopharmaceutical advantage. All salt forms were crystalline, nonhygroscopic (except the anhydrous hydrochloride salt), and had solubility in the range of 0.2 to 1.6 mg/ml. All salts were physically and chemically stable at 40°C/75% relative humidity, but degraded in UV-visible light, except the anhydrous hydrochloride salt. Prazosin mesylate was selected as the optimal salt, as it possessed higher solubility, permeability, and bioavailability, compared to the commercial hydrochloride salts. Hydrochloride salt is reported to have poor bioavailability that is partially attributed to its low solubility and extensive common-ion effect in the gastric region. Factors like hydrophilicity of the counterion, hydration state of the salt, and melting point of the salt contribute to the physicochemical properties of the salts. This study has implications in the selection of an optimal salt form for prazosin, which is suitable for further development.
KEY WORDS: prazosin, preformulation, salt form, salt screening
INTRODUCTION
Salt formation is an acid–base reaction, involving either a proton transfer or neutralization reaction, and affects a range of physicochemical and biopharmaceutical properties, including solubility, hygroscopicity, and stability (1–7). The salt selection process, therefore, is an integral part of the preformulation activities during early stages of drug development. A tiered approach is generally adopted for salt screening to enable phase studies, thus economizing on the time and compound requirements (8,9).
Prazosin (Fig. 1) is an α1 adrenergic blocker used in the treatment of hypertension. It is also useful in the treatment of Raynaud’s disease and benign prostatic hyperplasia (10,11). Commercially, anhydrous and polyhydrate forms of the hydrochloride salt, with a water content of 2% and 8–15%, respectively, are available (12). However, the polyhydrate form of prazosin hydrochloride has low solubility (1 mg/ml) and is photodegradable (13). Similarly, the anhydrous hydrochloride form converts to the more stable polyhydrate form in aqueous solution as well as at high humidity (14). This justifies the need to screen for alternate salt forms of prazosin.
Fig. 1.
Structure of prazosin. Protonation occurs at nitrogen of the quinazoline ring, at the para position to the exocyclic amino group
The present work evaluates the effect of counterions on the properties of prazosin. Correlation between the physicochemical properties of prazosin salts and the properties of corresponding counterions is also envisaged in this manuscript.
MATERIALS AND METHODS
Materials
Prazosin hydrochloride anhydrous (PRB HCl A) and prazosin hydrochloride polyhydrate (PRB HCl P) were purchased from Synthokem Laboratories, India and used as supplied. Methanesulfonic acid A.R. and toluenesulfonic acid A.R. were purchased from Acros Organics, USA. Camphorsulfonic acid (±) A.R. and oxalic acid A.R. were purchased from Aldrich, USA. Benzenesulfonic acid A.R. and maleic acid A.R. were obtained from Merck, Germany. All other chemicals used were of analytical grade.
Preparation of Prazosin Salts
Prazosin-free base (PRB) was first generated by basification of PRB HCl A. Briefly, PRB HCl A was dissolved in Ultrapure® water, followed by pH adjustment to 9–10 with 2.5 M sodium hydroxide. Prazosin, at this pH, exists as unionized species, with a low intrinsic aqueous solubility of 3.2 μg/ml at 25°C (15). Precipitates of PRB were collected by filtration. Conversion to PRB was confirmed by the negative chloride test in the precipitated solid. The identity of PRB was further confirmed by differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), 1H nuclear magnetic resonance (NMR), and elemental analysis.
PRB was dispersed in acetonitrile/water (4:1), followed by the addition of counterion (1:1.5, molar equivalent drug/counterion) dissolved in acetonitrile/water (4:1). In all cases, white precipitate of the corresponding salt ensued immediately, which was slurried overnight in Ultrapure® water to generate the most stable hydrate form. Slurry was filtered, air-dried, and characterized by elemental analysis, 1H NMR, powder X-ray diffraction (PXRD), and DSC/TGA to confirm salt formation.
Mass Spectrometry
Mass spectrographs of prazosin salts were obtained by LCQ Mass Spectrometer (Finnigan MAT, UK) in atmospheric pressure chemical ionization mode, with an inner temperature of 200°C. Samples were dissolved in methanol, filtered (0.45 μm membrane filter), and analyzed in the range of m/z 0–500. Data interpretation was performed using X’Calibur® software.
Head Space Gas Chromatography
Residual solvent was analyzed with a head space gas chromatograph (GC-17A, Shimadzu) equipped with class-GC software, utilizing a capillary column (Chrompack, The Netherlands). Experimental conditions were flame ionization detector (temperature, 300°C); column, G-300 (2 μm); column temperature, 100°C; injection temperature, 250°C; and carrier gas, helium (21 ml/s).
Differential Scanning Calorimetry
Calorimetric response of the sample was measured using DSC (DSC Q2000, TA Instruments, USA), equipped with RCS90 cooling accessory. Prior to analysis, the instrument was calibrated for temperature and heat flow, using high-purity indium. Samples were heated at 20°C/min nitrogen purging (50 ml/min) in pin-holed aluminum pans. Analysis of data was performed with Universal® Analysis software (version 4.5A).
Thermogravimetric Analysis
TGA was performed using Mettler Toledo 851e TGA/SDTA, in pin-holed aluminum crucibles at 20°C/min under nitrogen purging (40 ml/min).
Karl Fischer Titrimetry
Water content was determined by Karl Fischer titration, using pyridine-free reagents and the dead-stop method (Karl Fischer titrator 794 Basic Titrino, automatic burette 794 for presentation of solvent, 703 Ti stand, Metrohm AG, CH-Herisau). Analytical grade disodium tartrate dihydrate (15.65% water content) was used as Karl Fischer standard. Sample sizes ranged from 50 to 150 mg.
Hygroscopicity Study
Prazosin salts were uniformly spread as a thin layer in a Petri dish and kept in sealed desiccators at different humidity conditions: 21% relative humidity (RH; silica gel) and 92% RH (saturated solution of potassium nitrate). Changes in the sample gross weight were recorded after 1 week. Withdrawn samples were analyzed by DSC, TGA, and PXRD to assess any associated solid-state changes.
Solid-State Stability
Physical and chemical stability was performed by keeping 100–200 mg sample at 40°C/75% RH in an open Petri dish for 1 month, followed by analysis using DSC, PXRD, TGA, and high-performance liquid chromatography (HPLC). Photodegradation of samples was also assessed in the photochamber as per the ICH guidelines, by exposing to UV light (200 Wh/m2) and fluorescent light (1.2 × 106 lx h) (16).
Solubility Study
Solubility of prazosin salts was determined by shake flask method. Briefly, excess salt was suspended in Ultrapure® water, followed by equilibration in shaker water bath for 24 h (37°C; 200 rpm). Supernatant was analyzed by previously mentioned HPLC method. Residual solids were assessed for associated solid-state transitions by DSC and PXRD.
Caco-2 Permeability
Caco-2 permeability was determined using an in-house developed protocol. Preliminary experimentation indicated a high efflux for prazosin, as reported (17). Therefore, a high concentration of prazosin (500 μM) was employed in the permeability experiments. Stock solution was prepared in dimethylformamide/water (4:1), which was later diluted with water to the desired concentration. Concentration of dimethylformamide was kept below 5% v/v. Permeability studies were conducted under pH gradient condition (apical, 6.8; basolateral, 7.4).
Bioavailability Study in the Rat Model
All animal experiments were performed in accordance with the Committee for the Purpose of Control and Supervision on Experiments on Animals guidelines. Experimental protocol was approved by the Institutional Animals Ethics Committee (IAEC/11/39). Male Sprague–Dawley rats (225–250 g body weight) were kept on fasting for 12 h before the start, with free access to water before and during the experimentation. Prazosin salts were suspended in double-distilled water and administered at a dose of 10 mg/kg of rat body weight via oral gavage. Drug suspension (0.5 ml) was administered to the rats, at zero time point. Initial particle size of salts was controlled by sieving to achieve comparative particle size distribution. Particle size in the final formulations was also comparable, in the range of 5–25 μ.
Prazosin salts were suspended in double-distilled water and administered at a dose of 10 mg/kg of rat body weight via oral gavage. Approximately 0.5 ml of blood samples were collected from retro-orbital plexus after 0.25, 0.5, 1, 1.5, 2, 3, 5, and 8 h in heparinized microcentrifuge tubes. Plasma was separated immediately by centrifugation at 10,000 rpm for 5 min at 4°C and stored at −20°C until it was processed and analyzed. Plasma samples were extracted with acetonitrile and quantified by a validated HPLC-RF method. Analysis was performed on Lichrospher® CN column (5 μm, 250 × 4.6 mm; Merck). The mobile phase consisted of a mixture of methanol/water/glacial acetic acid (43:57:0.5 v/v/v), adjusted to pH 5.0 with diethylamine. Flow rate was kept at 1 ml/min. Fluorescence detector was set at λex/λem of 246/389 nm. Column temperature was kept at 30°C and the injection volume was 20 μl. Prazosin was quantified by the ratio of the peak area of prazosin to that of carbamazepine (internal standard).
Mean plasma profiles were generated and the standard noncompartmental pharmacokinetic parameters were calculated using Kinetica® pharmacokinetics software. Statistical comparisons were performed using SigmaStat® for Windows Version 2.03 (SPSS Inc.). Statistical testing between two mean values, at the 5% level of significance, was performed using two-sided unpaired t test.
RESULTS AND DISCUSSIONS
Physicochemical Properties of PRB and PRB HCl
Table I shows the physicochemical properties of PRB and PRB HCl salts. PRB, PRB HCl A, and PRB HCl P were all crystalline in nature. PRB and PRB HCl A showed a melting onset at 264°C and 284°C, respectively (Fig. 2; Table II). In contrast, PRB HCl P showed an initial dehydration endotherm at 60–160°C, followed by recrystallization and finally the melting of stable anhydrate form at 272°C (onset).
Table I.
Physicochemical Characterization of PRB and PRB HCl Salts
| Property | PRB | PRB HCl A | PRB HCl P |
|---|---|---|---|
| Microscopy (optical-polarized) | Crystalline | Crystalline | Crystalline |
| Melting point (onset °C) | 264 | 284 (with degradation) | 272 (with degradation) |
| Hygroscopicity (21, 92% RH; 1 week) | Nonhygroscopic at 21, 92% RH (<1% weight gain) | Nonhygroscopic at 21% RH (<1% weight gain); converts to PRB HCl P at 92% RH | Nonhygroscopic at 92% RH (<1% weight gain); becomes dihydrate at 21% RH |
| Solid form | Anhydrous form; no other forms reported/detected | Anhydrous alpha and polyhydrate forms marketed. Eight other polymorphic forms reported (14,22–25) | |
| Aqueous solubility (μg/ml)a | 6 | 990 | 930 |
| pH (aqueous saturated solution, 37°C) | 6.75–6.80 | 3.75–3.80 | 3.75–3.80 |
| Photostability; color after photostability | 93.96 (0.07); orange | 99.55 (0.15); creamy white | 91.04 (0.06); orange |
aAqueous solubility determined after 24 h (parentheses indicate the polymorphic form of the solid obtained after solubility study)
Fig. 2.
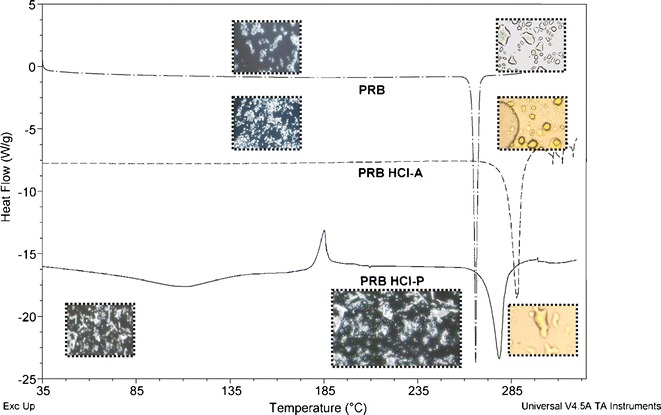
DSC traces of PRB, PRB HCl A, and PRB HCl P
Table II.
Characterization of Salt Forms of Prazosin
| Compound | Melting point (onset °C) | TGA (% weight loss) | Karl Fischer (% water) | Mass spectrometry (M+ + 1)a | Residual solventb | Elemental analysisc | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| C | H | N | S | O | ||||||
| PRB | 264 | 0.2 (anhydrous) | 0.6 | 384.4 | Not detected | 59.5 (59.4) | 5.5 (5.5) | 18.3 (19.4) | – | 16.7 (15.8) |
| PRB HCl A | 284 | 0.2 (anhydrous) | 0.4 | 384.4 | Not detected | 54.3 (54.3) | 5.2 (5.3) | 16.7 (17.8) | – | 15.2 (14.1) |
| PRB HCl P | 272 | 12.3 (hydrate) | 13.6 | 384.4 | Not detected | 49.2 (49.5) | 5.8 (6.0) | 15.1 (15.7) | – | 22.1 (21.5) |
| PRB BSA | 316 | 4.2 (hydrate) | 5.5 | 384.4 | Not detected | 53.6 (52.6) | 5.2 (5.0) | 12.5 (13.2) | 5.7 (5.5) | 22.9 (23.7) |
| PRB CSA | 332 | 0.2 (anhydrous) | 0.6 | 384.4 | Not detected | 56.5 (56.3) | 6.0 (6.1) | 11.4 (12.1) | 5.2 (5.4) | 20.8 (20.0) |
| PRB MEA | 237 | 4.3 (hydrate) | 5.2 | 384.4 | Not detected | 51.6 (53.3) | 5.4 (5.2) | 13.1 (13.5) | – | 29.9 (28.0) |
| PRB MES | 267 | 5.0 (hydrate) | 5.2 | 384.4 | Not detected | 48.3 (47.2) | 5.4 (5.3) | 14.1 (14.8) | 6.4 (6.3) | 25.7 (26.4) |
| PRB OA | 254 | 7.4 (hydrate) | 8.3 | 384.4 | Not detected | 49.5 (47.3) | 5.3 (5.4) | 13.7 (12.9) | – | 31.4 (34.5) |
| PRB TSA | 300 | 3.6 (hydrate) | 4.2 | 384.4 | Not detected | 52.8 (53.4) | 5.6 (5.5) | 11.8 (12.8) | 5.4 (5.3) | 24.3 (23.0) |
aAtmospheric pressure chemical ionization mode of mass spectrometry analysis
bResidual solvent analysis by head space gas chromatography
cExpected (observed) for elemental analysis. Stoichiometry of salts was 1:1, as determined by elemental and high-performance liquid chromatography analysis
PRB was nonhygroscopic at 21–92% RH. PRB HCl A converted to PRB HCl P at 92% RH. Conversion of PRB HCl A to PRB HCl P was further confirmed by slurrying with water, wherein the irregular crystals of PRB HCl A converted to needles of PRB HCl P arranged in a lamellar fashion (Fig. 3), as reported by Bianco (18). In contrast, PRB HCl P was nonhygroscopic at high RH, but converted to the corresponding PRB HCl dihydrate at 21% RH. We recently demonstrated that PRB HCl P is a nonstoichiometric hydrate, which reversibly exchanges water to convert from PRB HCl dihydrate to PRB HCl P (19).
Fig. 3.
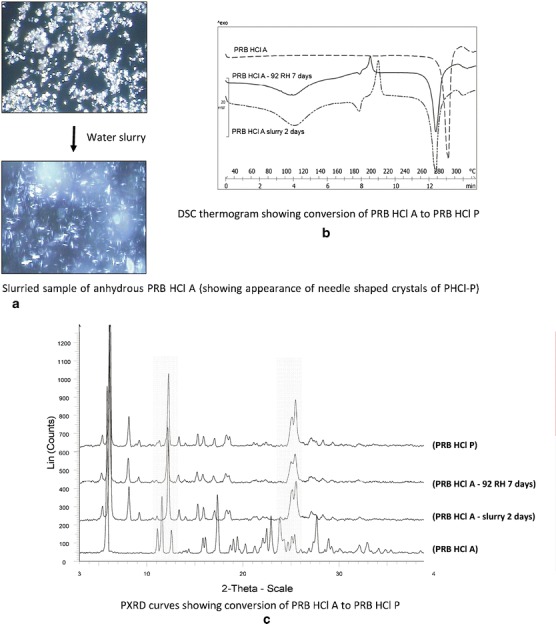
Conversion of PRB HCl A to PRB HCl P, as shown by a microscopy, b DSC, and c PXRD
Solubility of PRB, PRB HCl A, and PRB HCl P followed the order: PRB HCl A > PRB HCl P > PRB. Higher solubility of PRB HCl salts, compared to PRB, is attributed to the contribution of ionization of the drug. Solubility of PRB HCl A is initially higher, but reduces to the level of PRB HCl P due to conversion to the corresponding hydrate salt.
PRB, as well as PRB HCl P, showed photodegradation (Table III), unlike PRB HCl A, which remained unaffected by UV-visible light, as reported by Bianco (18).
Table III.
Physicochemical Characterization of Salt Forms of Prazosin
| Property | PRB MES | PRB BSA | PRB CSA | PRB TSA | PRB OA | PRB MEA |
|---|---|---|---|---|---|---|
| Microscopy (optically polarized) | Crystalline | Crystalline | Crystalline | Crystalline | Crystalline | Crystalline |
| Melting point (onset °C) | 267 | 316 | 332 | 300 | 254 | 237 |
| Solid form | Monohydrate | Monohydrate | Anhydrous | Monohydrate | Dihydrate | Monohydrate |
| Hygroscopic behavior (92% RH) | <1% weight gain (nonhygroscopic) | <1% weight gain (nonhygroscopic) | <1% weight gain (nonhygroscopic) | <1% weight gain (nonhygroscopic) | <1% weight gain (nonhygroscopic) | Converts to dihydrate |
| Aqueous solubility (mg/ml)a | 1.57 (hydrate) | 0.41 (hydrate) | 0.22 (anhydrous) | 0.28 (hydrate) | 0.16 (hydrate) | 0.14 (hydrate) |
| pH (aqueous saturated solution, 37°C) | 4.45–4.50 | 5.55–5.60 | 5.55–5.60 | 5.50–5.55 | 3.30–3.35 | 5.90–5.95 |
| Photostability; color after photostability | 92.6 (2.76); orange | 78.39 (4.31); orange | 94.25 (0.08); creamy white | 87.11 (0.05); orange | 76.84 (0.02); orange | 85.30 (3.04); orange |
aAqueous solubility determined after 24 h (parentheses indicate the polymorphic form of the solid obtained after the solubility study)
Preparation and Characterization of Salts of Prazosin
Prazosin is a weakly basic drug, with a reported aqueous pKa of 6.8 (14,20). Speciation profile and the established rule of 2–3 pKa difference between the drug and the counterion (21) were considered for salt formation. Acidic counterions having pKa below 4, like methanesulfonic acid (pKa, −1.2), benzenesulfonic acid (pKa, 0.7), toluenesulfonic acid (pKa, −1.34), and camphorsulfonic acid (pKa, 2.0) were selected for salt formation. Aliphatic organic carboxylic acid counterions, namely, maleic acid (pKa, 1.92; 6.23) and oxalic acid (pKa, 1.2) were also evaluated to assess the effect of organic counterions on the physicochemical properties of prazosin.
Solid-state characterization of prazosin salts is shown in Table II. All salt forms were obtained as hydrates, except PRB CSA which was obtained in anhydrous form. All hydrate salts showed a characteristic endotherm in the 60–160°C range due to water loss, as determined by TGA and Karl Fischer analysis. Hydrate salts (Fig. 4) underwent recrystallization after initial water loss, followed by melting of the stable anhydrate form along with degradation. In contrast, PRB OA showed polymorphic transformation, as indicated by two melting events observed in the DSC thermogram.
Fig. 4.
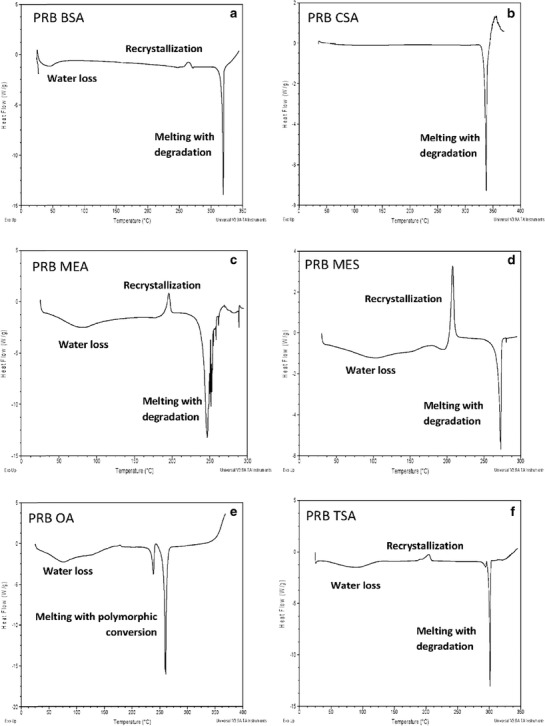
DSC traces of prazosin salts: a PRB BSA, b PRB CSA, c PRB MEA, d PRB MES, e PRB OA, and f PRB TSA
Physicochemical Evaluation of Prazosin Salts
Prepared prazosin salts were evaluated for the physicochemical properties of crystallinity, hygroscopicity, solubility, and stability in a tiered manner (Table III). All salt forms were crystalline in nature. Moreover, all prazosin salts were physically stable at higher humidity, except PRB MEA, which converted to the dihydrate form. Solubility of prazosin salts varied in the range of 0.2–1.6 mg/ml. Among the salts, PRB MES had the most optimal properties, in terms of being nonhygroscopic and with the highest solubility. All salt forms were physically and chemically stable at accelerated stability condition of 40°C/75% RH. However, all salts showed significant photodegradation, except for PRB HCl A (Table III).
Comparative Physicochemical Profiling of the Optimal Salt
On the basis of preliminary physicochemical characterization, PRB MES was selected as the optimal salts. Polymorph screening of PRB MES by methods, including solvent recrystallization and reaction crystallization method (data not shown), established the monohydrate form of PRB MES as the most stable form.
Figure 5a, b shows the pH solubility profile and solubility as a function of time, respectively, in comparison to PRB HCl A and PRB HCl P. It could be observed that PRB MES showed higher solubility, compared to PRB and PRB HCl A. This could be advantageous in terms of the resulting higher bioavailability for PRB MES. The intrinsic dissolution rate of PRB MES was also evaluated and was observed to be higher, compared to the hydrochloride salt (1.01 vs. 0.88 mg/min/cm2).
Fig. 5.
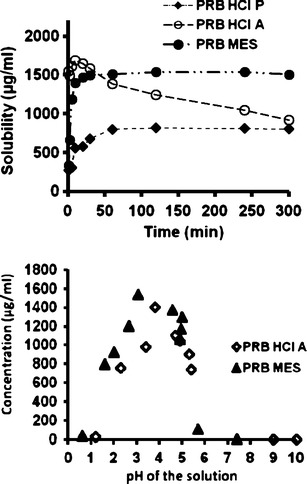
Solubility profile of the optimal salts: a solubility as a function of time and b solubility as a function of pH
Permeability coefficients of prazosin salts were further determined from the slope of cumulative concentration vs. time curve. The permeation rate of prazosin salts through the Caco-2 monolayer was linear over the period of study (120 min). PRB MES depicted higher permeability, compared to other salts, correlating with the trend in their solubility as: PRB MES > PRB HCl P > PRB CSA. However, no statistically significant difference in the permeability of prazosin salts was observed across the apical to basolateral side (Table IV).
Table IV.
Permeability of Prazosin Salts (n = 3)
| Salt | Permeability (cm/sec) ×10−6 (standard deviation) | Efflux ratio | |
|---|---|---|---|
| (A → B) | (B → A) | ||
| PRB HCl P | 3.7 (0.61) | 38.2 (3.9) | 10.41 |
| PRB MES | 4.3 (0.43) | 42.2 (2.0) | 9.70 |
A → B apical to basolateral, B → A basolateral to apical
The basolateral to apical transport of salts was statistically significantly (P < 0.001; Student’s t test) higher for all the salts, compared to the apical to basolateral transport. This may be attributed to the active efflux of prazosin by P-glycoproteins, thus reducing the permeability from the apical to the basolateral side (22).
Figure 6 and Table V depict the pharmacokinetics data for PRB HCl P and PRB MES MH. PRB MES MH showed greater area under the curve, compared to PRB HCl P (P < 0.001). This could be attributed to the higher solubility of PRB MES MH, compared to PRB HCl P.
Fig. 6.
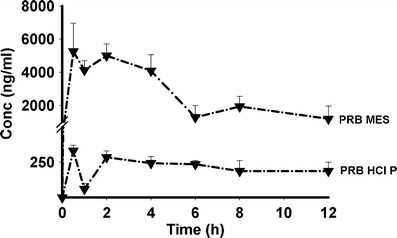
Pharmacokinetic profile of selected prazosin salts
Table V.
Pharmacokinetic Parameters of Prazosin Salts
| Solid form | Pharmacokinetic parameters | |
|---|---|---|
| C max (ng/ml) | AUC0 − t (ng h/ml) | |
| PRB HCl P | 292.8 (30.7)a | 1,538.2 (152.4) |
| PRB MES | 6,152.5 (946.5)b | 33,614.6 (7,753.8)b |
aParentheses indicate the standard error of the mean
bDifferences are significant at P < 0.001 level of significance
Common-ion effect is reported to reduce the bioavailability of hydrochloride salts of active pharmaceutical ingredients (23–25). It has also been reported that the nonhydrochloride salts may show higher solubility and thus higher bioavailability in the chloride-rich physiological media. Bogardus and Blackwood reported that the dissolution rate of doxycycline hydrochloride in 0.1 M HCl was reduced due to the common-ion effect, whereas the dissolution rate of nonhydrochloride doxycycline salt remains unaffected (23). Engel et al. observed that the mesylate salt of basic drugs showed higher bioavailability, compared to the hydrochloride salts (24). Prazosin hydrochloride salt shows extreme chloride ion dependence, with its solubility reducing from 1.4 to 0.037 mg/ml, in 0.1 M HCl (5). The higher bioavailability of PRB MES, compared to PRB HCl P, is thus attributed to higher solubility of the former, as well as the absence of the common-ion effect.
Effect of Counterion on the Properties of Prazosin Salts
Effect of Counterion on Melting Point of Prazosin
Melting point (in degrees Celsius; onset) of prazosin salts followed the trend: PRB MEA (237) < PRB OA (254) < PRB MES (267) ∼ PRB HCl P (272) < PRB HCl A (284) < PRB TSA (300) < PRB BSA (316) < PRB CSA (332). The melting point of stronger sulfonate and hydrochloride salt is higher, compared to the weaker organic maleate (PRB MEA) and oxalate salts (PRB OA). Sulfonate counterions are likely to show greater intramolecular as well as intermolecular interaction due to their stronger capability to form hydrogen bonding, electrostatic interactions, or steric effects. Thus, sulfonates are likely to show stronger interactions in the crystal lattice, resulting in higher melting points (26). A similar reasoning applies to PRB HCl, which has strong electrostatic interactions as well as hydrogen bonding capability. In contrast, the flexible aliphatic acid counterions like maleic and oxalic acid (PRB OA and PRB MEA salts) would have relatively weaker interactions in the crystal lattice.
Effect of Counterion on Solubility of Prazosin
Tables I and III show the solubility of prazosin salts. The solubility of prazosin salts varied from 0.2 to 1.6 mg/ml. PRB MES showed higher solubility, compared to commercial PRB HCl A and PRB HCl P. The solubility of salts followed the order: PRB MEA ∼ PRB OA < PRB CSA < PRB TSA < PRB BSA < PRB HCl P ∼ PRB HCl A < PRB MES.
Generally, a decrease in salt solubility accompanies an increase in melting point, which is partially attributed to higher crystal lattice energy for salt with higher melting point. Agharkar et al. studied the solubility of salts for the antimalarial drug α-(2-piperidyl)-3,6-bis(trifluoromethyl)-9-phenanthrene methanol. They attributed the lower solubility of hydrochloride and sulfate salts partly to their higher melting point, compared to lactate and 2-hydroxyethane-1-sulfonate salts (27). Gould also demonstrated a linear relationship between log10 solubility and a reciprocal relationship of absolute melting temperature (5). In contrast, Gu and Strickley found no correlation between the melting point and solubility of sodium and tris(hydroxymethyl)aminomethane salts of four analgesic/anti-inflammatory agents (28). Anderson and Conradi evaluated the solubility of six ammonium salts of flurbiprofen, differing in the hydrophobicity of counterion. Solubility correlated with the melting point of salts, but the most hydrophilic tromethamine salt ranked third in terms of solubility (29). Similarly, Chowhan evaluated sodium, potassium, magnesium, and calcium salts of four carboxylic acids and observed that no correlation between the melting point and solubility of the salt could be established (30).
Figure 7 shows the solubility of prazosin salts as a function of their melting points. No general correlation between the solubility of salts and their melting points, however, could be deduced, possibly due to the simultaneous involvement of the number of variables, like hydration energy and hydration state of the solid form. This behavior is similar to the previously reported studies, wherein the establishment of correlation between the solubility and melting point of the salt has been only partly successful (5,27–30). However, in stronger prazosin sulfonate salts like prazosin mesylate, prazosin besylate, and prazosin camsylate, however, solubility generally decreased with an increase in melting point of the salt.
Fig. 7.
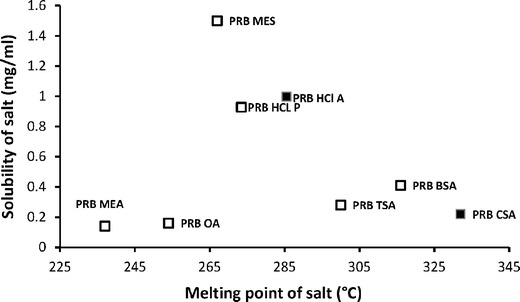
Correlation of solubility with the melting point of prazosin salt. Filled squares anhydrous compounds, open squares hydrates
Apart from the melting point of the salt, the hydration potential of the counterion is an important factor governing salt solubility. This behavior explains the overall trend in the solubility of sulfonate salts of prazosin, which follows the solubility (in milligrams per milliliter) behavior of corresponding counterions: MES (5,000) > BSA (930) > TSA (670) > CSA (100). On a similar basis, the solubility of PRB BSA, PRB CSA, and PRB TSA is lower, compared to the more hydrophilic mesylate counterion in PRB MES. Introduction of bulkier hydrophobic group in the structure reduces their interaction with water. This is in agreement with a previously reported study, wherein sulfonic acid salts with a moderate number of hydrocarbons, namely, bisulfate, mesylate, and besylate had favorable interaction with water. In contrast, counterions with larger, more hydrophobic structure, e.g., besylate and tosylate, had limited interaction with water (31).
The hydration state of the salt in solid state also affects its interaction with water and thus the corresponding solubility. Rubino evaluated the sodium salt of a range of drugs and observed that the salts forming crystal hydrates have lower solubility, compared to their anhydrous counterparts (32). The lower solubility of PRB HCl P compared to PRB HCl A could, thus, be attributed to this phenomenon, in spite of the higher melting point of the latter.
CONCLUSION
The present study evaluated an effect of counterions on the physicochemical properties of prazosin salts. Mesylate and camsylate salts of prazosin showed optimal properties, compared to other prazosin salts. Bioavailability study, using the rat model, indicated mesylate salt to have a higher bioavailability, compared to the marketed hydrochloride salts. Correlation of the solubility of salts with their structural attributes depicted an influence of parameters, including lattice energy, hydration energy, and solid-state hydration, on the solubility of salt.
ACKNOWLEDGMENTS
Lokesh Kumar acknowledges the Department of Science and Technology of the Government of India and the Ranbaxy Science Foundation for providing the research fellowship to carry out this work. Services provided by the Central Instrumentation Laboratory at NIPER Mohali are also gratefully acknowledged.
Declaration of Interest
The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- 1.Berge SM, Bighley LM, Monkhouse DC. Pharmaceutical salts. J Pharm Sci. 1977;66:1–19. doi: 10.1002/jps.2600660104. [DOI] [PubMed] [Google Scholar]
- 2.Black SN, Collier EA, Davey RJ, Roberts RJ. Structure, solubility, screening, and synthesis of molecular salts. J Pharm Sci. 2007;96:1053–68. doi: 10.1002/jps.20927. [DOI] [PubMed] [Google Scholar]
- 3.Bowker MJ. A procedure for salt selection and optimization. In: Stahl PH, Wermuth CG, editors. Handbook of pharmaceutical salts: properties, selection and use. Weinheim: Wiley-VCH Inc.; 2002. pp. 161–89. [Google Scholar]
- 4.Davies G. Changing the salt, changing the drug. Pharm J. 2001;266:322–3. [Google Scholar]
- 5.Gould PL. Salt selection for basic drugs. Int J Pharm. 1986;33:201–17. doi: 10.1016/0378-5173(86)90055-4. [DOI] [Google Scholar]
- 6.Serajuddin ATM. Salt formation to improve drug solubility. Adv Drug Deliv Rev. 2007;59:603–16. doi: 10.1016/j.addr.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 7.Stahl PH, Nakano M. Pharmaceutical aspects of the drug salt form. In: Stahl PH, Wermuth CG, editors. Handbook of pharmaceutical salts: properties, selection and use. Weinheim: Wiley-VCH Inc.; 2002. pp. 83–116. [Google Scholar]
- 8.Kumar L, Amin A, Bansal AK. Preparation and characterization of salt forms of enalapril. Pharm Dev Technol. 2008;13:345–57. doi: 10.1080/10837450802244686. [DOI] [PubMed] [Google Scholar]
- 9.Morris KR, Fakes MG, Thakur AB, Newman AW, Singh AK, Venit JJ, et al. An integrated approach to the selection of optimal salt form for a new drug candidate. Int J Pharm. 1994;105:209–17. doi: 10.1016/0378-5173(94)90104-X. [DOI] [Google Scholar]
- 10.Gilman AG, Goodman LS, Gilman A. Goodman and Gilman’s the pharmacological basis of therapeutics. 6. New York: McMillan; 1980. pp. 806–7. [Google Scholar]
- 11.Hofmann BB. Adrenergic receptor blocking agents. In: Katzung BG, editor. Basic and clinical pharmacology. Los Altos: Lange Medical; 1984. pp. 97–107. [Google Scholar]
- 12.Wu S. Polar and nonpolar interactions in adhesion. J Adhes. 1973;5:39–55. doi: 10.1080/00218467308078437. [DOI] [Google Scholar]
- 13.Bianco EJ. Novel crystalline forms of prazosin hydrochloride. US Patent 4092315; 1978.
- 14.Kostek LJ. Prazosin hydrochloride. In: Brittain HG, editor. Analytical profile of drug substances and excipients, vol. 18. Amsterdam: Elsevier; 1989. p. 351–81.
- 15.Bergström CAS, Norinder U, Luthman K, Artursson P. Experimental and computational screening models for prediction of aqueous drug solubility. Pharm Res. 2002;19:182–8. doi: 10.1023/A:1014224900524. [DOI] [PubMed] [Google Scholar]
- 16.Kostek LJ. Prazosin hydrochloride. Analytical Profile of Drug Substances and Excipients. 1989;18:351–81.
- 17.Prazosin permeability. http://www.cyprotex.com/cloescreen/in-vitro-permeability/mdr1-mdck-permeability/. Accessed 1 May 2012.
- 18.Bianco EJ. Novel crystalline forms of prazosin hydrochloride. US Patent 4092315, Pfizer Inc.; 1978.
- 19.Kumar L, Bansal AK. Effect of humidity on the hydration behaviour of prazosin hydrochloride polyhydrate: thermal, sorption and crystallographic study. Thermochim Acta. 2012;525:206–10. doi: 10.1016/j.tca.2011.08.006. [DOI] [Google Scholar]
- 20.Volgyi G, Ruiz R, Box K, Comerc J, Bosch E, Takacs-Novak K. Potentiometric and spectrophotometric pKa determination of water-insoluble compounds: validation study in a new cosolvent system. Anal Chim Acta. 2007;583:418–28. doi: 10.1016/j.aca.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 21.Tong W. Salt screening and selection: new challenges and considerations in the modern pharmaceutical R&D paradigm. http://www.pharmacy.utah.edu/pharmaceutics/pdf/Salt.pdf. Accessed 5 May 2012.
- 22.MDR1–MDCK permeability (P-glycoprotein substrate identification). http://www.cyprotex.com/cloescreen/in-vitro-permeability/mdr1-mdck-permeability/. Accessed 1 May 2012.
- 23.Bogardus JB. Common ion equilibria of hydrochloride salts and the Setschenow equation. J Pharm Sci. 1982;71:588–90. doi: 10.1002/jps.2600710527. [DOI] [PubMed] [Google Scholar]
- 24.Engel GL, Farid NA, Faul MM, Richardson LA, Winneroski LL. Salt form selection and characterization of LY333531 mesylate monohydrate. Int J Pharm. 2000;198:239–47. doi: 10.1016/S0378-5173(00)00350-1. [DOI] [PubMed] [Google Scholar]
- 25.Miyazaki S, Oshiba M, Nadai T. Precaution on use of hydrochloride salts in pharmaceutical formulation. J Pharm Sci. 1981;70:594–6. doi: 10.1002/jps.2600700604. [DOI] [PubMed] [Google Scholar]
- 26.Bowker MJ, Stahl PH. Preparation of water-soluble compounds through salt formation. In: Wermuth CG, editor. The practice of medicinal chemistry. London: Academic; 2008. pp. 749–66. [Google Scholar]
- 27.Agharkar S, Lindenbaum S, Higuchi T. Enhancement of solubility of drug salts by hydrophilic counterions: properties of organic salts of an antimalarial drug. J Pharm Sci. 1976;65:747–9. doi: 10.1002/jps.2600650533. [DOI] [PubMed] [Google Scholar]
- 28.Gu L, Strickley RL. Preformulation salt selection—physical property comparison of the tris(hydroxymethyl)aminomethane salts of four analgesic/antiinflammatory agents with the sodium salts and the free acids. Pharm Res. 1987;4:255–7. doi: 10.1023/A:1016420514689. [DOI] [PubMed] [Google Scholar]
- 29.Anderson BD, Conradi RA. Predictive relationships in the water solubility of salts of a nonsteroidal anti-inflammatory drug. J Pharm Sci. 1985;74:815–20. doi: 10.1002/jps.2600740803. [DOI] [PubMed] [Google Scholar]
- 30.Chowhan JT. pH–solubility profiles of organic carboxylic acids and their salts. J Pharm Sci. 1978;67:1257–60. doi: 10.1002/jps.2600670918. [DOI] [PubMed] [Google Scholar]
- 31.Guerrieri P, Rumondor ACF, Li T, Taylor LS. Analysis of relationships between solid-state properties, counterion, and developability of pharmaceutical salts. AAPS PharmSciTech. 2010;11:1212–22. doi: 10.1208/s12249-010-9499-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rubino JT. Solubility and solid state properties of the sodium salts of drugs. J Pharm Sci. 1989;78:485–9. doi: 10.1002/jps.2600780614. [DOI] [PubMed] [Google Scholar]