

Abstract
The controlled release of diflunisal and fluconazole from tablets made of novel polymers, poly(acrylic acid) (PAA) crosslinked with either β-cyclodextrin (βCD) or hydroxypropyl-βCD (HPβCD), was investigated and Carbopol 934P (Carbopol) was used as a highly crosslinked PAA for comparison. Diflunisal strongly associates with βCD-PAA and HPβCD-PAA polymers (Ka of 486 and 6,055 M−1 respectively); thus, it was physically mixed into the conjugates and also precomplexed to identify whether decomplexation has any influence on release kinetics. Fluconazole has poor complexing ability (Ka of 34 M−1 with HPβCD-PAA); thus, it was only tested as a physical mixture. Swelling and adhesion studies were conducted on all tablet combinations and adhesivity of the CD-PAA polymer tablets was maintained. Diflunisal release was much slower from HPβCD-PAA tablets than from βCD-PAA, suggesting that a higher degree of complexation retards release. The precomplexed diflunisal release was also slower than the physically mixed diflunisal of the corresponding conjugate. The release closely followed zero-order kinetics for HPβCD-PAA, but was more sigmoidal for βCD-PAA and especially Carbopol. Conversely, poorly associating fluconazole released in almost exactly the same way across both polymers and Carbopol, indicating that the release kinetics of poorly associating drugs are not influenced by the presence of cyclodextrins. In view of the varying profiles and release rates shown with diflunisal for the different polymers, the fluconazole data support the concept that adequate complexation can indeed modulate the release kinetics of drugs.
Electronic supplementary material
The online version of this article (doi:10.1208/s12249-012-9903-3) contains supplementary material, which is available to authorized users.
Key words: controlled release, cyclodextrin, diflunisal, fluconazole, poly(acrylic acid)
INTRODUCTION
Hydrogels are useful carriers for drug delivery due to their inertness and ability to modulate the release of pharmaceutical compounds (1,2). Another focus of intense research has been buccal administration of drugs because this delivery route avoids the issue of first pass effect and poor absorption in the gut thus improving bioavailability (3,4). In comparison to conventional per oral dosing, buccal administration has the advantage of low enzymatic activity and tolerance to potential sensitizers (5). Poly(acrylic acid) (PAA) is an excipient suitable for buccal drug delivery due to its mucoadhesivity and it is used to synthesize hydrogels that exhibit reversible swelling behavior in response to changes in the physiological medium (e.g., pH, temperature, and ionic strength). Cyclodextrins (CDs) are torus-shaped cyclic oligosaccharides that are able to complex a wide variety of organic molecules within their cavity thus modifying the molecules' physicochemical properties. Incorporating CDs into hydrogels can potentially enable controlled drug release via the dual functionality arising from the responsive swelling of hydrogels and complexation with CD.
CD-crosslinked PAA polymers (CD-PAA) (Fig. 1) formulated as hydrogels were recently shown to be useful controlled delivery platforms for the release of diflunisal and fluconazole (6). Depending on the degree of crosslinking of the polymers and the magnitude of the association constant (Ka) of the model drug, different rates of release were achieved. The companion work is extended here by developing and characterizing solid dosage forms of the CD-PAA polymers intended for buccal drug administration. The impact of these tablet dosage forms on the release of two different drugs, fluconazole and diflunisal, was evaluated in comparison to release from within a Carbopol 934P (Carbopol) matrix, a highly crosslinked PAA. The effect of precomplexation of diflunisal with free CD dispersed in a matrix of the linear PAA polymer was also evaluated.
Fig. 1.

Structure of the CD-PAA polymers and schematic representation of inclusion complexation and release of the drug from the matrix. CD is either βCD or HPβCD. The chemical composition and K a values are described in Table I. Release kinetics are described in Table II and Table III
In addition, the adhesion of CD-PAA dosage forms to a model hydrophobic elastic substrate made of polydimethylsiloxane (PDMS) was measured and evaluated in comparison to Carbopol adhesion; the adhesion of the solid dosage disk to the PDMS was simply the peak force following retraction of the disk from the contact. PDMS was chosen as the model surface because its hydrophobicity and elasticity are similar to that of the underlying tissue within the oral cavity (7). Unlike excised tissue, PDMS has a consistent surface chemistry and roughness, which can also be easily controlled. Our adhesion measurements followed procedures similar to those found in the literature (8,9). Relatively short times were used for pre-hydration and for pressing the tablet onto PDMS in order to simulate the real-life scenario of a patient applying a mucoadhesive tablet to the inside of their cheek.
MATERIALS AND METHODS
Materials and Sample Preparation
Diflunisal and Carbopol 934P (Carbopol) were purchased from Sigma-Aldrich (Sydney, Australia). Fluconazole was extracted from Fluconazole Sandoz capsules (Pyrmont, NSW, Australia) as described by Kutyla et al. (6). Disodium hydrogen orthophosphate dodecahydrate and potassium dihydrogen orthophosphate were of analytical grade and were used as received. Milli-Q water was used for all experiments.
Synthesis of Polymers
The polymers βCD-PAA and hydroxypropyl-βCD-PAA (HPβCD-PAA) were prepared from PAA (molecular weight (MW) 450,000 g/mol) and βCD or HPβCD (molar substitution of 0.65) according to the procedure of Kutyla et al. (6), using 24 h activation time and 1.2 g of CD. The chemical composition and Ka values are described in Table I.
Table I.
Chemical Composition of CD-PAA Polymers and Association Constants with Diflunisal and Fluconazole
| Polymer properties | βCD-PAA | HPβCD-PAA |
|---|---|---|
| CD | 33.7% w/w | 37.7% w/w |
| COOH | 34.2% w/w | 31.3% w/w |
| Calculated ester crosslinks to a CDa | 8.4 | 6.1 |
| K a diflunisalb | 486 M−1 | 6,055 M−1 |
| K a fluconazoleb | –c | 34 M−1 |
Preparation of Tablets
Tablets 100 mg in weight with 10% w/w drug component were manufactured as follows: polymer powders (including a previously prepared physical mixture of 30% w/w βCD and 70% w/w PAA) were gently mixed with the drug in a mortar and pestle using geometric progression and directly compressed with a tablet press (Manesty E2, Sussex, England) on a flat single 10 mm punch and die. In the case of CD-PAA polymer precomplexation with diflunisal, 0.9 g polymer and 0.1 g drug were dissolved in 100 mL 50% v/v ethanol and lyophilized for 48 h. For the physical mixture of βCD and PAA, precomplexation was achieved by dissolving diflunisal (0.1 g) in 50 mL of ethanol and adding this solution dropwise to 50 mL aqueous solution of PAA and βCD (0.9 g) under gentle stirring, followed by lyophilization. The powder was then ground in a mortar and pestle and compressed into tablets. The weight and thickness uniformity are presented in the Supplementary material.
In Vitro Tablet Characterization
Drug and CD Release
Drug release studies were conducted in triplicate. The tablets were placed in a basket dissolution apparatus, mesh size 40 (Varian, North Carolina, USA), then submerged in 100 mL 0.15 M phosphate buffer (PB), pH 7.0, maintained at 37°C, and gently stirred at 100 rpm. At predetermined time intervals, 2.0 mL samples were removed for UV analysis with reference to a standard curve (diflunisal λmax 252 nm, fluconazole λmax 261 nm). CD content was analyzed with the phenol-sulfuric acid assay (10) calculated by reference to a standard curve at 486 nm. Cumulative release was calculated with a correction for the respective dilutions resulting from replacement of the sample with an equal volume of fresh buffer. The studies were conducted over a period of 12 h.
Swelling
Swelling was conducted in triplicate, concomitantly with dissolution studies. The baskets were removed from the dissolution medium and gently shaken to remove any excess moisture and the external and internal surfaces of the basket and tablet were blotted with tissue. The swollen tablets were not removed from the basket to prevent disturbing their integrity. The swelling index (SI) was calculated by the following formula:
 |
1 |
where mi is the initial dry tablet mass and mt is the swollen tablet mass at time t, obtained by subtracting the combined swollen tablet mass in the basket (Ct) from the dry basket mass (B):
 |
2 |
Adhesion
Adhesion of hydrated tablets was measured on a Haake MARS III stress-controlled rheometer (Thermo Scientific, Karlsruhe, Germany) with a 20 mm diameter titanium parallel plate (as the top surface) and a 35 mm diameter PDMS disc (as the bottom surface). The PDMS disc was formed in a custom-made cup attachment as follows. A 35 mm diameter (ID) hole was machined into an aluminum base to a depth of ~10 mm. The PDMS monomer and initiator were mixed according to manufacturer's instructions (10:1 w/v, monomer/initiator) (Sylgard 184, Dow Corning) in a plastic cup, degassed in a vacuum oven at −100 kPa for 30 min, poured into the custom-made attachment, and allowed to react while in position on the rheometer at 24°C. This last step ensured the PDMS disk formed parallel to the top surface. The PDMS was allowed to cure for over 24 h before use (Supplementary material).
There are three main stages to an adhesion test: (1) hydration of the tablet; (2) compression of the tablet onto the lower surface; and (3) controlled removal of the tablet in the direction normal to the lower surface. A review of the literature on similar studies reveals that the hydration time, compression force and time, and the separation speed vary substantially within the three stages, respectively (11–19): the hydration period ranges from seconds to several minutes; the compression time and compression force range from 1 to 10 min and 0.1 to 10 N, respectively; and the detachment speed ranges from 0.050 to 10 mm/s. The choice of any and all of these parameters will affect the measured adhesion. With that in mind, this study took a more pragmatic approach by asking the question, “What set of conditions best represent the clinical scenario of this dosage form application?” All experiments were performed at 37°C.
Stage 1
The hydrating solution was 0.15 M PB pH 7.0, used to simulate the buffering action of physiological solutions such as saliva. The tablet was attached to the top plate with double-sided tape (Nachi 745 Tissue Tape with acrylic adhesive, Stylus Tapes, Brisbane, Australia) and lowered into the hydrating solution in the reservoir (no PDMS surface contact) for a hydration time of either 2 or 30 s; a time of 2 s was chosen because it is representative of the end use of a patient licking the tablet prior to pressing onto the buccal mucosa. The tablet was then removed from the solution by winding up the upper surface, and the solution was removed from the custom-made attachment reservoir via capillary action using Kimtech Kimwipes (Kimberly-Clark, NSW Australia). The wet tablet was untouched and exposed to air for ~10 s between stages 1 and 2.
Stage 2
The tablet was pressed onto the PDMS surface under a compression force of 0.40 N for 30 s. There were two reasons for choosing this value. First, a minimal force is necessary to ensure the tablet and the lower surface come into intimate contact. Second, a hydrating tablet will spread under a sustained load, which increases contact between tablet and lower surface thus increasing the measured adhesive force. The practical use of a buccal tablet in the clinical setting will require very low compressive forces during application of the tablet and then minimal compressive force after application. Thirty seconds was chosen to mimic a typical end use situation of pressing a tablet against the mucosal surfaces.
Stage 3
Following compression, the tablet was removed at a constant speed of 0.167 mm/s (which is on the lower end of the values reported in the literature). The normal force, FN, was recorded throughout the process. Every tablet formulation was tested in triplicate and the averaged data were subsequently analyzed for FN, detachment, which was considered to be the adhesion force.
Rationale for Using PDMS Substrate and PB for Adhesion Studies
Historically, experiments that measure the mucoadhesion of a polymer tablet do so with glass or animal tissue as the substrate and mucin solutions to hydrate. Interestingly, there has been little correlation found between ex vivo/in vitro “mucoadhesion” or residence time and in vivo studies (13,20,21). The numerous methods of evaluating mucoadhesion in vitro range from spectroscopic characterization of molecular interactions via FTIR (22,23) or NMR (24,25), assessment of any rheological synergism between mucin and polymer gels (26,27), through evaluation of surface energies (28,29) to the most common that measure tensile detachment strength from mucin-covered surfaces or excised animal tissue (18,30). It should be emphasized that in mucoadhesion studies, animal tissue and mucin are treated and washed to be safe to use, which changes their physical properties and thereby limits their ability to mimic the in vivo environment (31). Regarding mucin, the reconstituted mucin solutions bear no physical resemblance to biofluids such as saliva; mucin solutions are relatively inelastic viscous liquids compared with saliva that is extremely viscoelastic and relatively low in viscosity (32). Mucin, and more importantly the salt accompanying mucin, has been shown to alter the rheological properties of a model polymer gel (Carbopol) (Fig. 13.8, page 344 of (33)). Finally, the presence of ions, different pH, or temperatures can affect the final material properties of a gelled network. These various approaches to the measurement of mucoadhesivity result in data that are difficult to compare and that will change depending on the variables in each technique and particularly from variations in mucin type (or the presence of impurities and salts) as well as the innate variation in excised animal tissues. There is also the question of the relevance of such results to real systems and the reproducibility of data obtained with biological substrates, substrates with physical properties that vary depending on preparation, and storage methods.
PDMS is not biological and can be made reproducibly with known surface chemistry. Although hydrophobic, PDMS is known to interact with substances through polar–polar associations or hydrogen bonds between its siloxane group and the H atom of the alcohol or acid of the substance (34). Furthermore, adhesion to biological tissue has been shown to encompass hydrophobic interactions (35). Whilst PDMS has not been previously used for evaluating bioadhesion per se, it is being used to understand and evaluate food–oral substrate interactions (36).
Simple hydrating solution was used to limit the effects of interactions with mucin, but also in the expectation that the magnitude of FN, detachment would be large enough that relative differences could be attributed to the presence of drugs or different crosslinkers. Therefore, PB was used to hydrate the polymer tablets.
RESULTS AND DISCUSSION
Effect of Drug Loading on Drug Release Characteristics
Diflunisal
Diflunisal was physically mixed with CD-PAA polymer or precomplexed into the CD-PAA polymer to evaluate any differences in release profiles with respect to drug loading and whether the difference in Ka between the two polymer types affects the rate of drug release. Carbopol 934P, a commercially available, highly crosslinked PAA (with sucrose allyl ether) (37), was used as a benchmark compound. It was chosen for this study because it is a standardized reference pharmaceutical excipient to compare with our CD-crosslinked PAA. It has wide applicability, ranging from drug delivery applications that include stabilization of emulsions and suspensions, sustained release formulations and localized drug delivery, to numerous uses in the cosmetics industry (38,39). Non-crosslinked PAA with or without physically incorporated βCD (the starting materials used for synthesis) was also used as a comparison, with diflunisal that was either physically mixed or precomplexed in the βCD. Linear PAA exhibited rapid diflunisal release (Fig. 2a, ~100% release in 200 min) compared with crosslinked PAA (Fig. 2b, c, ~100% release in >600 min).
Fig. 2.

Release profiles of precomplexed or physically mixed diflunisal from (a) PAA and PAA physically mixed with βCD, (b) βCD-PAA polymer and from physically mixed Carbopol, and (c) HPβCD-PAA polymer and from physically mixed Carbopol (each point and error bar represent the mean ± SD of three experiments)
Overall, the precomplexed diflunisal exhibited slower release from the CD-PAA polymers and the PAA and βCD mixture than the corresponding physically incorporated diflunisal, but the difference was small (Fig. 2). The rationalization of this minimal difference is that the physically mixed diflunisal dissolves in the matrix as the tablet absorbs water and readily complexes with the available CD cavities.
Due to the comparable crosslink density to a CD for the two CD-PAA polymers (Table I), the convolution and mesh size were expected to be similar. Whilst the βCD-PAA polymer (Fig. 2b) released diflunisal faster than benchmark Carbopol, the HPβCD-PAA polymer (Fig. 2c) exhibited slower release. The differences observed in the rate of release may be due to the different Kas of diflunisal with the polymers, previously determined as 486 M−1 for βCD-PAA and 6,055 M−1 for HPβCD-PAA (6). When looking at the diflunisal release data in isolation and considering the added complexity of different swelling profiles (Fig. 3), it is difficult to ascertain which parameter has the predominant influence over drug kinetics. A global comparison with the fluconazole release data, however, enables for some important conclusions to be drawn: the weakly associating fluconazole exhibits a release profile that is very similar to the reference Carbopol matrix (Fig. 4), whereas diflunisal presents with a dissimilar profile (Fig. 2b, c). Diflunisal's rate of release and “type” of release profile are different between the βCD-PAA and HPβCD-PAA matrixes, an observation which is not apparent with fluconazole. This suggests that complexation with polymer bound CD does modulate release kinetics, despite the swelling rate variations seen between the matrixes prepared from the different CD-PAA polymer combinations.
Fig. 3.

Equilibrium swelling of (a) physically mixed diflunisal and fluconazole tablets (b) physically mixed vs complexed diflunisal in CD-PAA polymer tablets (each point and error bar represent the mean ± SD of three experiments)
Fig. 4.

Release profiles of physically mixed fluconazole from HPβCD-PAA, βCD-PAA and from Carbopol (each point and error bar represent the mean ± SD of three experiments)
The Weibull Function
The sigmoidal release profiles were modeled with the Weibull function (40):
 |
3 |
where Q is the amount of drug dissolved as a function of time t, Q0 is the total amount of drug released (often set to 100% (41)), T is the lag time as a result of the dissolution process, a is a scale parameter describing the time dependence (in the form a = (Td)b it can be used to represent the more useful dissolution time, Td, which is equal to the time when 63.2% of drug is dissolved or released (40)), and b is a shape parameter (for sigmoidal shapes b > 1, indicating a complex release mechanism (42)).
Whilst the use of this equation has been criticized for its non-kinetic basis and the lack of parameters related to the intrinsic dissolution rate (42), it has been widely used for modeling purposes (43) and has been subject to investigations to validate its use (44–46). Papadopoulou et al. (47) reason that b values characterize the release mechanism because of linear correlations seen with the n exponent of the power law, universally used to describe how a tablet's geometry and properties of its matrix influence release kinetics (48).
The results presented in Table II show that in all instances, the b parameter is much larger than 1, indicating that the rate of release increases to an inflection point, decreasing thereafter, giving a sigmoidal shape to the release curve. This occurrence has also been attributed to complex release mechanisms where the change in the rate of release is not uniform (47). The b parameters indicate that precomplexation influences the shape of the release profile (complex < physical mix). Surprisingly, the highly crosslinked Carbopol and linear PAA presented with the most variable parameters among all the results and shape parameters that were quite removed from the others (3.81 and 3.43, respectively, compared with 1.85–2.39). It is also apparent that out of polymers with physically incorporated vs precomplexed drug, it is the physical mixtures that present with greater variability in the release kinetic parameters. This indicates that the more uniform drug distribution in lyophilized formulations is superior to manual mixing in a mortar and pestle, although machine mixing may circumvent this problem.
Table II.
Parameter b of the Weibull Equation and Resultant Dissolution Time (n = 3, R 2 ≥ 0.993)
| Tablet | DIF | βCD | ||
|---|---|---|---|---|
| b | T d (min) | b | T d (min) | |
| CBPL and DIF | 3.81 ± 1.05 | 446 ± 106 | NA | NA |
| βCD-PAA and DIF phys | 2.39 ± 0.36 | 232 ± 26 | NA | NA |
| βCD-PAA and DIF cplx | 2.00 ± 0.48 | 266 ± 28 | NA | NA |
| PAA and DIF phys | 3.43 ± 0.86 | 112 ± 27 | NA | NA |
| PAA and βCD and DIF phys | 2.23 ± 0.13 | 88 ± 6.0 | 2.59 ± 0.34 | 100 ± 5.5 |
| PAA and βCD and DIF cplx | 1.85 ± 0.11 | 96 ± 2.9 | 1.83 ± 0.06 | 93 ± 2.3 |
DIF diflunisal, FLZ fluconazole, phys physical mixture, cplx complex
The calculated Td value for precomplexed diflunisal in βCD-PAA was approximately 1.15 larger than for the physically mixed diflunisal (Table II). For the physical mixture of PAA and βCD, the precomplexed diflunisal Td was 1.09 times larger than the physically mixed diflunisal. These results suggest that association of diflunisal with βCD modulates the release kinetics by increasing the time taken to release the drug.
The release of βCD from the mixture of βCD and PAA was also measured (Supplementary material). The Td values for βCD and diflunisal were similar for precomplexed diflunisal (96 cf. 93 min), showing that the drug and CD are released in unison. The physically mixed diflunisal showed more dissimilarity, with the Td of diflunisal occurring at 88 min, whereas βCD was at 100 min. Thus, even though in situ complexation occurs, it is reduced and has less effect than when the drug and CD are in precomplexed form. It is interesting to note that βCD has a “delayed” release compared with diflunisal, but it is a much larger molecule, thus its diffusion is hindered throughout the matrix when compared with diflunisal.
Zero-Order Release
The first 80% of physically mixed diflunisal released over 450 min from the HPβCD-PAA polymer very closely followed zero-order kinetics (r2 ≥ 0.994), whereas the complexed form exhibited zero-order release for almost the entire release profile, up to ~99% at 690 min (r2 ≥ 0.991).
Numerous explanations have been proposed as the rate-limiting component of zero-order release and include the synchronization of the swelling and dissolution/erosion fronts (49,50), synchronization of the erosion and drug diffusion fronts (51), solvent-induced polymer relaxation (52,53), or simply erosion of the polymer (54) although, in this case, zero-order release is considered to only occur if the drug is immobilized (55,56).
The front synchronization process, as proposed by Colombo et al. (51), has been used successfully to explain release independent of time for devices of constant release area with low drug incorporation. This allows one to disregard nonconformity with thermodynamic ideality, such as differing diffusion coefficients and drug/solvent or drug/polymer interactions (50).
Distinct fronts were observed in the cases presented here, but dissociation of the drug may also play a role in the zero-order kinetic profile observed. The slower release of diflunisal out of precomplexed rather than physically mixed system of HPβCD-PAA points towards decomplexation of the drug as regulating the release. A shift of the equilibrium towards dissociation upon drug depletion is expected to have kept the amount of diffusing drug constant. This would also have been anticipated in the βCD-PAA tablets, but the release of diflunisal deviated from linearity, which may suggest that a certain threshold Ka value is required before zero-order release occurs.
The slower release of precomplexed diflunisal compared with physically mixed diflunisal is in contrast with a number of studies in the literature conducted on hydroxypropyl methylcellulose (HPMC). Zugasti et al. (57) performed diflunisal release studies in matrixes with excess soluble βCD polymers or monomeric βCD. The matrix with diflunisal precomplexed in βCD exhibited faster release than the physical mixture. The authors also found that precomplexed diflunisal released approximately according to first-order kinetics, whereas physically mixed diflunisal was closer to zero-order release. Similar studies with carbamazepine carried out by Koester et al. (58) showed faster release for precomplexed than physically mixed drug. These dissimilarities with our results may be attributable to the different polymer and formulation type used and mobile CD within the matrix. On the other hand, Pose-Vilarnovo and others (59) found that the presence of excess HPβCD and βCD in HPMC/lactose blends decreased the release of diclofenac, a hydrophilic drug, whereas it increased the release of the hydrophobic sulfamethizole. The influence of decreased diffusion rate was speculated as being predominant for the former drug, whereas increased dissolution was the principal effect seen with the latter. These contrasting results show that release of a drug is not only based on the properties of the matrix, but also on the contributing factors of the drug (hydrophobicity, solubility, MW, etc.).
Salmaso et al. (60) found that high concentration of βCD-hexamethylene crosslinker in polyethylene glycol matrixes significantly decreased the release of hydrophobic β-estradiol due to extremely high affinity for the βCD (Ka 77,947 M−1 (61)). A similar effect was seen with quinine, although the slow release rate was offset by increased diffusivity seen in the matrix due to the small nature of this drug. Nielsen et al. (62) found retarded release of hydrophilic ibuprofen in studies conducted on βCD covalently bonded within poly(vinylpyrolidone)/poly(ethyleneglycol-dimethacrylate). This is aligned with the results of our studies, where a physically immobilized CD can hinder the release of drug through complexation.
Fluconazole
Fluconazole was tested only as a physical mixture because of its poor complexing ability (Ka of 39.7 and 132 M−1 for HPβCD and βCD, respectively) (6). The release profiles were very similar for the three different polymers (Fig. 4). There is no real distinction between these different polymers and fluconazole release, in contrast to the distinction seen with diflunisal. This signifies that complexation of a compound with high Ka does modulate release kinetics, regardless of the differences seen in swelling of the different CD-PAA polymers.
The release of fluconazole is likely to be governed by a partition mechanism by interaction with hydrophobic entities of the polymer, with zero-order kinetics prevailing. The n values of the power law, however, were all between 0.73 and 0.76 (data not shown) but values in excess of 0.66 are considered to predominantly exhibit zero-order (case II transport) (54). It was noted that for approximately the first 30% drug release, corresponding to 50 min of release for both Carbopol and HPβCD-PAA, the polymers exhibited a zero-order profile, whereas βCD-PAA released drug faster and displayed this behavior for the first 30 min, where the diffusional path length is short (r2 ≥ 0.994 and 0.998, respectively). Between ~30% and 70% drug release, the power law indicated a switch of kinetic mechanism to more diffusion controlled with n values of 0.60 for both CD polymers and a value of 0.43 for Carbopol (R2 ≥ 0.983 and 0.995, respectively).
Swelling of Tablets
Swelling is a factor in drug release kinetics, and conversely, the physicochemical properties of the drug used (solubility, pKa, etc.) influence the mechanism and extent of swelling (63,64). Swelling in this case occurs as water, an efficient plasticizer, penetrates the polymer forming a gel layer on the outside of the tablet, which mobilizes the polymer chains and the drug molecules.
The mechanism by which diflunisal and fluconazole exhibit their plasticizing effects on the polymers is through decreasing the within-polymer chain interactions thus increasing mobility and swelling. As pH increases, ionization of the COOH groups of the polymers causes electrostatic repulsion and increased swelling. Diflunisal is also expected to ionize over time, thus there is also electrostatic repulsion between it and the charged polymer. Fluconazole, on the other hand, is neutral and any interactions would be based on H-bonding or hydrophobic interactions. This suggests that greater swelling for the diflunisal and polymer is likely and may also promote the diffusional process of the diflunisal through the matrix (65). Nonetheless, differences in swelling barely start becoming apparent at the tail end of release for Carbopol and HPβCD-PAA only (Fig. 3a). A pH gradient is expected in the tablet, with the center maintaining a very acidic environment (66), therefore ionization of diflunisal in this situation may not occur and account for the lack of differences in swelling between the fluconazole and diflunisal tablets.
Precomplexed diflunisal with HPβCD and βCD has a lower interaction with the polymer and diffuses out through pores forming in the matrix when the tablet imbibes water. This results in a decreased plasticizing effect of the drug when compared with physically mixed diflunisal (Fig. 3b). Fluconazole has also shown similar plasticizing efficacy in these polymers, by exhibiting comparable swelling profiles and a similar equilibrium SI (Fig. 3a). Plain tablets of the CD-PAA conjugates were also tested and the SIs were similar with the said conjugates and complexed mixtures of drugs, although the swelling rates were slightly slower at first (Supplementary material). Unexpectedly, plain Carbopol tablets swelled faster and reached a comparable SI to Carbopol physically mixed with fluconazole or diflunisal. Due to the high SI and highly crosslinked nature (resulting in the formation of discrete microgels with many interstitial spaces), this polymer is not expected to be greatly influenced by any packing disruption/osmotic activity of either of these drugs at the low weight percent present.
Mesh size has been shown to be inversely proportional to equilibrium swelling degree (67) and this indicates that HPβCD-PAA has the lowest mesh size, followed by βCD-PAA (Fig. 3). Carbopol, therefore, may be considered to have the highest mesh size, but it is highly crosslinked; thus, these assumptions do not apply. Observations revealed distinct particles sloughing off from the tablet that remained in the basket, unlike the slow dissolution that occurred with the CD-PAA tablets. For drugs such as diflunisal that are expected to diffuse out of the polymers, the different mesh sizes may have an influence on retardation of release, with Carbopol expected to exhibit least resistance in the low microviscosity interstitial spaces. Whilst HPβCD-PAA retarded release more than βCD-PAA, Carbopol demonstrated intermediate release. Hydrogen bonding and electrostatic interactions between diflunisal and the acrylic acid residue COOH moieties may have influenced the release due to a higher free COOH percentage in Carbopol compared with the βCD-PAA polymer.
The swelling data are quite variable for the tablets made of a mixture of PAA and βCD (Supplementary material). This is due to the fragility of the tablets and difficulty with which to dry them without adsorbing dissolved polymer on the tissue. However, it appears that the swelling and erosion process are quite similar across the three different PAA tablet types.
Water uptake data were analyzed using the following model (68,69), although originally it was intended for application to systems that swell less than 25% of their original volume (70):
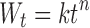 |
4 |
where Wt is the amount of water sorbed at time t, k is a swelling constant, and n represents the mechanism of water uptake.
Table III shows that incorporation of fluconazole in the matrixes of HPβCD-PAA and βCD-PAA increases k, but n stays relatively the same, indicating an increase in the rate of water uptake but no real effect on the mechanism. Water uptake is almost Fickian for HPβCD-PAA with or without fluconazole, whereas for βCD-PAA, anomalous behavior is seen. The physical incorporation of diflunisal, however, shows a decrease in k and an increase in n, indicating a lowering of polymer chain relaxation involvement. Precomplexed diflunisal shows that water uptake is governed mostly by solvent diffusion. Lyophilization has been shown to result in a highly porous structure with high surface area of the material, which expedites uptake of water (71) and results in faster swelling and higher swelling capacity (72). This seems in contrast to the results obtained for precomplexed (lyophilized) CD-PAA with diflunisal compared with physically mixed diflunisal. The lower swelling indexes seen for the polymers with complexed diflunisal may be due to their amorphous state post-lyophilization (73,74) and thus rapid disintegration (30). Of course, the CD-PAA polymers are lyophilized after synthesis, thus the additional cycle of lyophilization may not change these properties to any significant degree than what is seen already. Evidently, it may be that release of diflunisal from matrixes with physically incorporated drug results in the formation of cavities, causing a more porous structure that seems to be confirmed in the results.
Table III.
Mean Swelling k and n Values with SD (n = 3)
| Tablet | k | n | R 2a |
|---|---|---|---|
| CBPL and DIF phys | 1.01 ± 0.53 | 0.63 ± 0.10 | 0.995 |
| CBPL and FLZ phys | 1.25 ± 0.30 | 0.58 ± 0.04 | 0.992 |
| CBPL | 0.38 ± 0.02 | 0.81 ± 0.01 | 0.993 |
| HPβCD-PAA and DIF cplx | 1.00 ± 0.47 | 0.48 ± 0.07 | 0.991 |
| HPβCD-PAA and DIF phys | 0.65 ± 0.20 | 0.60 ± 0.05 | 0.987 |
| HPβCD-PAA and FLZ phys | 0.97 ± 0.31 | 0.52 ± 0.05 | 0.989 |
| HPβCD-PAA | 0.86 ± 0.17 | 0.51 ± 0.03 | 0.984 |
| βCD-PAA and DIF cplx | 1.50 ± 0.22 | 0.48 ± 0.02 | 0.981 |
| βCD-PAA and DIF phys | 0.86 ± 0.20 | 0.65 ± 0.05 | 0.987 |
| βCD-PAA and FLZ phys | 1.01 ± 0.04 | 0.61 ± 0.01 | 0.979 |
| βCD-PAA | 0.72 ± 0.13 | 0.62 ± 0.03 | 0.993 |
DIF diflunisal, FLZ fluconazole, phys physical mixture, cplx complex, k has units of t −n
aReported as the lowest obtained for the three experiments
In Vitro Adhesion
The Effect of Hydration of Carbopol and Adhesion to PDMS
Figure 5a displays the results for Carbopol detachment from PDMS after hydration with buffer. Two hydration/compression conditions were used and found to result in similar values of FN, detachment. As such, the practical conditions of 2 s hydration with 30 s compression were used for all tests.
Fig. 5.

a F N, detachment, for Carbopol on PDMS hydrated with PB solution. The hydration time (H) and compression time (C) are listed alongside. b βCD-PAA F N, detachment with and without drug present. (c) HPβCD-PAA F N, detachment with and without drug present (at 2 s H and 30 s C). Each graph represents the mean ± SD of three experiments
For completeness, long-time hydration and high-compressive force data were collected for Carbopol on PDMS in PB (data not shown). The measured detachment forces were in the range of 3 to 7 N. The major difference is that, at very long hydration times (e.g. 2 h), a longer time was necessary to fully detach the tablet, and in some cases, a thin film remained attached to the PDMS plate and the upper plate.
Adhesion to PDMS Using PB to Hydrate
Each CD-PAA polymer/drug combination was tested using PB to hydrate for 2 s, with 30 s of compression before detachment. Figure 5b, c shows FN, detachment for βCD-PAA and HPβCD-PAA, respectively, with and without diflunisal and fluconazole present. There is adhesion between the hydrated tablet and the PDMS surface. However, within each set of experiments involving either βCD-PAA or HPβCD-PAA, there is no significant difference between CD-PAA polymer with or without drug or with how they are incorporated into the tablet matrix (physically mixed or precomplexed) (compared via a one-way ANOVA). Some drugs have been shown to be efficient plasticizers that disrupt attractive forces between polymer macromolecules (75), but the lack of effect implies that either diflunisal or fluconazole does not act as plasticizers or that the quantity of added drug is insufficient to impact the adhesion (76). If more replicates were conducted, these differences may have become more apparent, as indicated in Fig. 6a.
Fig. 6.

a Interaction plots between the possible predictor variables showing that differences may exist between polymers depending on drug. b F N, detachment for each polymer without drug present (mean ± SD of three experiments). The hydration time was 2 s and the compression time was 30 s. The F N, detachment for HPβCD-PAA was significantly different from βCD-PAA (p value = 0.007143) and significantly different from Carbopol (p value = 0.07557). The Carbopol and βCD-PAA were not significantly different
Figure 6b, however, shows that the adhesion between each polymer tablet (βCD-PAA or HPβCD-PAA or Carbopol) is significantly different; a one-way ANOVA showed that polymer has a significant effect on adhesion (p value = 0.07158). Two-way and three-way interaction terms were tested for and found to be nonsignificant. Post hoc multiple comparison testing, with Bonferroni adjustment to give familywise error rate of 5%, revealed that Carbopol and HPβCD-PAA are significantly different, and that βCD-PAA and HPβCD-PAA are significantly different. The difference in adhesion between βCD-PAA, HPβCD-PAA, and Carbopol is likely to be due to a number of factors such as rate and extent of hydration. The highest initial rate of hydration has been shown to attain the highest adhesive strength in studies conducted on PAA (77). The initial swelling rates in the equilibrium swelling studies conducted (Supplementary Material Figs. S2 and S3) show that Carbopol swells faster than βCD-PAA, which swells faster than HPβCD-PAA. This follows the strength of adhesion forces.
Adhesive strength increases with molecular weight, but chains that are too long are unfavorable to producing an interpenetrating layer and entanglements thus reducing the possibility of formation of a bio/mucoadhesive bond (78). Both CD-PAA polymers are less crosslinked than Carbopol; therefore, their longer chain conformation may be responsible for the lower adhesion. A study by Warren and Kellaway that looked at crosslink density, swelling, and mucoadhesion of sucrose crosslinked PAA found that increased crosslink density resulted in increased detachment forces (69). This was attributed to the increasing density of polymer chains per unit surface area of the polymer and it was argued by the authors that the decreased mesh size and lower mobility of polymer chains were still able to sufficiently swell to enable physical entanglement between the two substrates (69).
Finally, physical factors such as particle size and rate of hydration of polymers affect bioadhesion (79) with dialysis and lyophilization altering the physical characteristics of polymers and having been shown to cause a decrease in adhesion (80). This may also serve to explain Carbopol's highest adhesion, the only polymer that was not lyophilized. Carbopol also has the most COOH moieties by weight available for H-bonding, the predominating interaction in bioadhesion (31,81).
Conclusions
A new method of utilizing CD to both crosslink PAA and molecularly encapsulate and release drugs was evaluated. The variations in swelling between the βCD-PAA and HPβCD-PAA polymers rendered it more difficult to establish the role of complexation in drug release. However, in view of fluconazole with its poor associating capabilities and highly similar rates of release between the different polymers, as well as the change in the type of release profile for diflunisal (zero-order for HPβCD-PAA with a Ka of 6,055 M−1, sigmoidal for βCD-PAA 486 M−1), it can be concluded that release kinetics are influenced by complexation of drug with CD bound to PAA. This conclusion is also based upon the change in the type of release kinetics seen for diflunisal among the two different CD-PAA polymers. CD-PAA tablets were generally as adhesive as Carbopol tablets and the HPβCD-PAA polymer evaluated in this study has great potential as a buccal dosage form because it retains adhesivity whilst exhibiting zero-order release for diflunisal. Future work will focus on studying the behavior of these dosage forms in the in vivo environment.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(DOCX 134 kb)
Acknowledgments
Mr. Ron West of the School of Pharmacy is thanked for assistance with tablet manufacturing. We also thank Dr. Geraldine Elliot for helpful discussions. Financial support from the School of Pharmacy is gratefully acknowledged.
Contributor Information
Marguerite J. Kutyła, Phone: +61-7-33461900, FAX: +61-7-33461999, Email: m.kutyla@uqconnect.edu.au, Email: b.ross1@uq.edu.au
Benjamin P. Ross, Email: b.ross1@uq.edu.au
References
- 1.Miro A, Rondinone A, Nappi A, Ungaro F, Quaglia F, La Rotonda MI. Modulation of release rate and barrier transport of Diclofenac incorporated in hydrophilic matrices: role of cyclodextrins and implications in oral drug delivery. Eur J Pharm Biopharm. 2009;72(1):76–82. doi: 10.1016/j.ejpb.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 2.Peppas NA, Bures P, Leobandung W, Ichikawa H. Hydrogels in pharmaceutical formulations. Eur J Pharm Biopharm. 2000;50(1):27–46. doi: 10.1016/S0939-6411(00)00090-4. [DOI] [PubMed] [Google Scholar]
- 3.Cui F, He C, He M, Tang C, Yin L, Qian F, et al. Preparation and evaluation of chitosan-ethylenediaminetetraacetic acid hydrogel films for the mucoadhesive transbuccal delivery of insulin. J Biomed Mater Res. 2009;89A(4):1063–71. doi: 10.1002/jbm.a.32071. [DOI] [PubMed] [Google Scholar]
- 4.Sudhakar Y, Kuotsu K, Bandyopadhyay AK. Buccal bioadhesive drug delivery—a promising option for orally less efficient drugs. J Contr Release. 2006;114(1):15–40. doi: 10.1016/j.jconrel.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 5.Senel S, Kremer M, Nagy K, Squier C. Delivery of bioactive peptides and proteins across oral (buccal) mucosa. Curr Pharm Biotechnol. 2001;2(2):175–86. doi: 10.2174/1389201013378734. [DOI] [PubMed] [Google Scholar]
- 6.Kutyla MJ, Lambert LK, Davies NM, McGeary RP, Shaw PN, Ross BP. Cyclodextrin-crosslinked poly(acrylic acid): synthesis, physicochemical characterization and controlled release of diflunisal and fluconazole from hydrogels. Int J Pharm. doi:10.1016/j.ijpharm.2013.01.005 [DOI] [PubMed]
- 7.Bongaerts J, Rossetti D, Stokes J. The lubricating properties of human whole saliva. Tribol Lett. 2007;27(3):277–87. doi: 10.1007/s11249-007-9232-y. [DOI] [Google Scholar]
- 8.Mortazavi SA, Smart JD. An investigation of some factors influencing the in vitro assessment of mucoadhesion. Int J Pharm. 1995;116:223–30. doi: 10.1016/0378-5173(94)00299-K. [DOI] [Google Scholar]
- 9.Maggi L, Carena ML, Torre ML, Giunchedi P, Conte U. In vitro/ex vivo methods for the evaluation of bioadhesive polymers. A preliminary study. STP Pharma. 1994;4:343–8. [Google Scholar]
- 10.Koh GL, Tucker IG. Variability in the phenol-sulphuric acid assay for sodium carboxymethylcellulose. Int J Pharm. 1986;34:183–4. doi: 10.1016/0378-5173(86)90029-3. [DOI] [Google Scholar]
- 11.Jones DS, Muldoon BCO, Woolfson AD, Sanderson FD. An examination of the rheological and mucoadhesive properties of poly(acrylic acid) organogels designed as platforms for local drug delivery to the oral cavity. J Pharm Sci. 2007;96(10):2632–46. doi: 10.1002/jps.20771. [DOI] [PubMed] [Google Scholar]
- 12.Munasur AP, Govender T, Mackraj I. Using an experimental design to identify and quantify the effects of environment related test parameters on the in vitro mucoadhesivity testing of a propanolol buccal tablet. Drug Dev Ind Pharm. 2007;33(7):709–16. doi: 10.1080/03639040701199282. [DOI] [PubMed] [Google Scholar]
- 13.Shanker G, Kumar C, Gonugunta C, Kumar B, Veerareddy P. Formulation and evaluation of bioadhesive buccal drug delivery of tizanidine hydrochloride tablets. AAPS PharmSciTech. 2009;10(2):530–9. doi: 10.1208/s12249-009-9241-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel VM, Prajapati BG, Patel MM. Effect of hydrophilic polymers on buccoadhesive Eudragit patches of propranolol hydrochloride using factorial design. AAPS PharmSciTech. 2007;8(2):E1–7. doi: 10.1208/pt0802027. [DOI] [PubMed] [Google Scholar]
- 15.Bonacucina G, Cespi M, Misici-Falzi M, Palmieri GF. Rheological, adhesive and release characterisation of semisolid Carbopol/tetraglycol systems. Int J Pharm. 2006;307:129–40. doi: 10.1016/j.ijpharm.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 16.Bonacucina G, Martelli S, Palmieri GF. Rheological, mucoadhesive and release properties of Carbopol gels in hydrophilic cosolvents. Int J Pharm. 2004;282:115–30. doi: 10.1016/j.ijpharm.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 17.Tamburic S, Craig DQM. A comparison of different in vitro methods for measuring mucoadhesive performance. Eur J Pharm Biopharm. 1997;44:159–67. doi: 10.1016/S0939-6411(97)00073-8. [DOI] [Google Scholar]
- 18.Bromberg L, Temchenko M, Alakhov V, Hatton TA. Bioadhesive properties and rheology of polyether-modified poly(acrylic acid) hydrogels. Int J Pharm. 2004;282:45–60. doi: 10.1016/j.ijpharm.2004.05.030. [DOI] [PubMed] [Google Scholar]
- 19.Anlar S, Capan Y, Guven O, Gogus A, Dalkara T, Hincal AA. Formulation and in vitro-in vivo evaluation of buccoadhesive morphine sulfate tablets. Pharm Res. 1994;11(2):231–6. doi: 10.1023/A:1018951323522. [DOI] [PubMed] [Google Scholar]
- 20.Bouckaert S, Lefebvre RA, Remon JP. In vitro/in vivo correlation of the bioadhesive properties of a buccal bioadhesive miconazole slow-release tablet. Pharm Res. 1993;10(6):853–6. doi: 10.1023/A:1018957126809. [DOI] [PubMed] [Google Scholar]
- 21.Laulicht B, Cheifetz P, Tripathi A, Mathiowitz E. Are in vivo gastric bioadhesive forces accurately reflected by in vitro experiments? J Contr Release. 2009;134(2):103–10. doi: 10.1016/j.jconrel.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 22.Jabbari E, Wisniewski N, Peppas NA. Evidence of mucoadhesion by chain interpenetration at a poly(acrylic acid)/mucin interface using ATR-FTIR spectroscopy. J Contr Release. 1993;26(2):99–108. doi: 10.1016/0168-3659(93)90109-I. [DOI] [Google Scholar]
- 23.Saiano F, Pitarresi G, Cavallaro G, Licciardi M, Giammona G. Evaluation of mucoadhesive properties of α,β-poly(N-hydroxyethyl)-dl-aspartamide and α,β-poly(aspartylhydrazide) using ATR-FTIR spectroscopy. Polymer. 2002;43(23):6281–6. doi: 10.1016/S0032-3861(02)00504-9. [DOI] [Google Scholar]
- 24.Mortazavi SA. An in vitro assessment of mucus/mucoadhesive interactions. Int J Pharm. 1995;124(2):173–82. doi: 10.1016/0378-5173(95)00073-R. [DOI] [Google Scholar]
- 25.Patel MM, Smart JD, Nevell TG, Ewen RJ, Eaton PJ, Tsibouklis J. Mucin/poly(acrylic acid) interactions: a spectroscopic investigation of mucoadhesion. Biomacromolecules. 2003;4(5):1184–90. doi: 10.1021/bm034028p. [DOI] [PubMed] [Google Scholar]
- 26.Hassan EE, Gallo JM. A simple rheological method for the in vitro assessment of mucin-polymer bioadhesive bond strength. Pharm Res. 1990;7(5):491–5. [DOI] [PubMed]
- 27.Rossi S, Bonferoni MC, Lippoli G, Bertoni M, Ferrari F, Caramella C, et al. Influence of mucin type on polymer-mucin rheological interactions. Biomaterials. 1995;16(14):1073–9. doi: 10.1016/0142-9612(95)98903-R. [DOI] [PubMed] [Google Scholar]
- 28.Lehr C-M, Boddé HE, Bouwstra JA, Junginger HE. A surface energy analysis of mucoadhesion II. Prediction of mucoadhesive performance by spreading coefficients. Eur J Pharm Sci. 1993;1(1):19–30. doi: 10.1016/0928-0987(93)90014-2. [DOI] [Google Scholar]
- 29.Rillosi M, Buckton G. Modelling mucoadhesion by use of surface energy terms obtained from the Lewis Acid—Lewis Base Approach. II. Studies on anionic, cationic, and unionisable polymers. Pharm Res. 1995;12(5):669–75. doi: 10.1023/A:1016299223369. [DOI] [PubMed] [Google Scholar]
- 30.Grabovac V, Guggi D, Bernkop-Schnürch A. Comparison of the mucoadhesive properties of various polymers. Adv Drug Deliv Rev. 2005;57(11):1713–23. doi: 10.1016/j.addr.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 31.Gu JM, Robinson JR, Leung SH. Binding of acrylic polymers to mucin/epithelial surfaces: structure-property relationships. Crit Rev Ther Drug Carrier Syst. 1988;5(1):21–67. [PubMed] [Google Scholar]
- 32.Stokes JR, Davies GA. Viscoelasticity of human whole saliva collected after acid and mechanical stimulation. Biorheology. 2007;44(3):141–60. [PubMed] [Google Scholar]
- 33.Stokes JR. Rheology of industrially relevant microgels. In: Fernandez-Nieves A, Wyss HM, Mattsson J, Weitz DA, editors. Microgel suspensions: fundamentals and applications. Germany: Wiley-VCH Verlag GmbH & Co. KGaA; 2011. p 327–53.
- 34.Boscaini E, Alexander M, Prazeller P, Mark T. Investigation of fundamental physical properties of a polydimethylsiloxane (PDMS) membrane using a proton transfer reaction mass spectrometer (PTRMS) Int J Mass Spectrom. 2004;239(2–3):179–86. [Google Scholar]
- 35.Wang PY. Evidence of hydrophobic interaction in adhesion to tissue. Nature. 1974;249:367–8. doi: 10.1038/249367a0. [DOI] [PubMed] [Google Scholar]
- 36.de Vicente J, Stokes JR, Spikes HA. Soft lubrication of model hydrocolloids. Food Hydrocolloids. 2006;20(4):483–91. doi: 10.1016/j.foodhyd.2005.04.005. [DOI] [Google Scholar]
- 37.Lubrizol. Lubrizol pharmaceutical polymers for controlled release tablets and capsules. Lubrizol Advanced Materials I. Cleveland, Ohio; 2010. p 1–9.
- 38.Elkheshen S. Interaction of verapamil hydrochloride with Carbopol 934P and its effect on the release rate of the drug and the water uptake of the polymer matrix. Drug Dev Ind Pharm. 2001;27(9):925. doi: 10.1081/DDC-100107673. [DOI] [PubMed] [Google Scholar]
- 39.Flick EW. Cosmetic and toiletry formulations, volume 7. 2. Norwich: William Andrew Publishing/Noyes; 1999. [Google Scholar]
- 40.Langenbucher F. Linearization of dissolution rate curves by the Weibull distribution. J Pharm Pharmacol. 1972;24(12):979–81. doi: 10.1111/j.2042-7158.1972.tb08930.x. [DOI] [PubMed] [Google Scholar]
- 41.Adams E, Coomans D, Smeyers-Verbeke J, Massart DL. Non-linear mixed effects models for the evaluation of dissolution profiles. Int J Pharm. 2002;240(1–2):37–53. doi: 10.1016/S0378-5173(02)00127-8. [DOI] [PubMed] [Google Scholar]
- 42.Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–33. doi: 10.1016/S0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 43.Koester LS, Ortega GG, Mayorga P, Bassani VL. Mathematical evaluation of in vitro release profiles of hydroxypropylmethylcellulose matrix tablets containing carbamazepine associated to β-cyclodextrin. Eur J Pharm Biopharm. 2004;58(1):177–9. doi: 10.1016/j.ejpb.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 44.Macheras P, Dokoumetzidis A. On the heterogeneity of drug dissolution and release. Pharm Res. 2000;17(2):108–12. doi: 10.1023/A:1007596709657. [DOI] [PubMed] [Google Scholar]
- 45.Lánský P, Weiss M. Classification of dissolution profiles in terms of fractional dissolution rate and a novel measure of heterogeneity. J Pharm Sci. 2003;92(8):1632–47. doi: 10.1002/jps.10419. [DOI] [PubMed] [Google Scholar]
- 46.Elkoshi Z. On the variability of dissolution data. Pharm Res. 1997;14(10):1355–62. doi: 10.1023/A:1012108402682. [DOI] [PubMed] [Google Scholar]
- 47.Papadopoulou V, Kosmidis K, Vlachou M, Macheras P. On the use of the Weibull function for the discernment of drug release mechanisms. Int J Pharm. 2006;309(1–2):44–50. doi: 10.1016/j.ijpharm.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 48.Ritger PL, Peppas NA. A simple equation for description of solute release I. Fickian and non-Fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Contr Release. 1987;5(1):23–36. doi: 10.1016/0168-3659(87)90034-4. [DOI] [PubMed] [Google Scholar]
- 49.Colombo P, Bettini R, Santi P, De Ascentiis A, Peppas NA. Analysis of the swelling and release mechanisms from drug delivery systems with emphasis on drug solubility and water transport. J Contr Release. 1996;39(2–3):231–7. doi: 10.1016/0168-3659(95)00158-1. [DOI] [Google Scholar]
- 50.Harland RS, Gazzaniga A, Sangalli ME, Colombo P, Peppas NA. Drug/polymer matrix swelling and dissolution. Pharm Res. 1988;5(8):488–94. doi: 10.1023/A:1015913207052. [DOI] [PubMed] [Google Scholar]
- 51.Colombo P, Bettini R, Massimo G, Catellani PL, Santi P, Peppas NA. Drug diffusion front movement is important in drug release control from swellable matrix tablets. J Pharm Sci. 1995;84(8):991–7. doi: 10.1002/jps.2600840816. [DOI] [PubMed] [Google Scholar]
- 52.Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15(1):25–35. doi: 10.1016/0378-5173(83)90064-9. [DOI] [PubMed] [Google Scholar]
- 53.Catellani P, Vaona G, Plazzi P, Colombo P. Compressed matrices: formulation and drug release kinetics. Acta Pharmaceutica Technologica. 1988;34:38–41. [Google Scholar]
- 54.Möckel JE, Lippold BC. Zero-order drug release from hydrocolloid matrices. Pharm Res. 1993;10(7):1066–70. doi: 10.1023/A:1018931210396. [DOI] [PubMed] [Google Scholar]
- 55.Fischel-Ghodsian F, Newton JM. Analysis of drug release kinetics from degradable polymeric devices. J Drug Target. 1993;1(1):51–7. doi: 10.3109/10611869308998764. [DOI] [PubMed] [Google Scholar]
- 56.Hopfenberg HB. Controlled release from erodible slabs, cylinders, and spheres. In: Controlled release polymeric formulations: American Chemical Society; 1976. vol 33 p. 26–32.
- 57.Zugasti ME, Zornoza A, del Mar Goni M, Isasi JR, Velaz I, Martin C, et al. Influence of soluble and insoluble cyclodextrin polymers on drug release from hydroxypropyl methylcellulose tablets. Drug Dev Ind Pharm. 2009;35(10):1264–70. doi: 10.1080/03639040902882306. [DOI] [PubMed] [Google Scholar]
- 58.Koester LS, Xavier CR, Mayorga P, Bassani VL. Influence of β-cyclodextrin complexation on carbamazepine release from hydroxypropyl methylcellulose matrix tablets. Eur J Pharm Biopharm. 2003;55(1):85–91. doi: 10.1016/S0939-6411(02)00127-3. [DOI] [PubMed] [Google Scholar]
- 59.Pose-Vilarnovo B, Rodriguez-Tenreiro C, dos Santos JFR, Vazquez-Doval J, Concheiro A, Alvarez-Lorenzo C, et al. Modulating drug release with cyclodextrins in hydroxypropyl methylcellulose gels and tablets. J Contr Release. 2004;94(2–3):351–63. doi: 10.1016/j.jconrel.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 60.Salmaso S, Semenzato A, Bersani S, Matricardi P, Rossi F, Caliceti P. Cyclodextrin/PEG based hydrogels for multi-drug delivery. Int J Pharm. 2007;345(1–2):42–50. doi: 10.1016/j.ijpharm.2007.05.035. [DOI] [PubMed] [Google Scholar]
- 61.Caliceti P, Salmaso S, Semenzato A, Carofiglio T, Fornasier R, Fermeglia M, et al. Synthesis and physicochemical characterization of folate—cyclodextrin bioconjugate for active drug delivery. Bioconjug Chem. 2003;14(5):899–908. doi: 10.1021/bc034080i. [DOI] [PubMed] [Google Scholar]
- 62.Nielsen AL, Madsen F, Larsen KL. Cyclodextrin modified hydrogels of PVP/PEG for sustained drug release. Drug Deliv. 2009;16(2):92–101. doi: 10.1080/10717540802605129. [DOI] [PubMed] [Google Scholar]
- 63.Mitchell K, Ford JL, Armstrong DJ, Elliott PNC, Hogan JE, Rostron C. The influence of drugs on the properties of gels and swelling characteristics of matrices containing methylcellulose or hydroxypropylmethylcellulose. Int J Pharm. 1993;100(1–3):165–73. doi: 10.1016/0378-5173(93)90087-V. [DOI] [Google Scholar]
- 64.Dahlberg C, Fureby A, Schuleit M, Dvinskikh SV, Furó I. Polymer mobilization and drug release during tablet swelling. A 1H NMR and NMR microimaging study. J Contr Release. 2007;122(2):199–205. doi: 10.1016/j.jconrel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 65.Peppas NA, Wright SL. Solute diffusion in poly(vinyl alcohol)/poly(acrylic acid) interpenetrating networks. Macromolecules. 1996;29(27):8798–804. doi: 10.1021/ma9613392. [DOI] [Google Scholar]
- 66.Smart JD, Mortazavi SA. An investigation of the pH within the gel layer of a hydrating poly(acrylic acid) compact. J Pharm Pharmacol. 1995;47:1099. [Google Scholar]
- 67.Canal T, Peppas NA. Correlation between mesh size and equilibrium degree of swelling of polymeric networks. J Biomed Mater Res. 1989;23(10):1183–93. doi: 10.1002/jbm.820231007. [DOI] [PubMed] [Google Scholar]
- 68.Vergnaud JM. Liquid transport controlled release processes in polymeric materials: applications to oral dosage forms. Int J Pharm. 1993;90(2):89–94. doi: 10.1016/0378-5173(93)90145-6. [DOI] [Google Scholar]
- 69.Warren SJ, Kellaway IW. The synthesis and in vitro characterization of the mucoadhesion and swelling of poly(acrylic acid) hydrogels. Pharm Dev Technol. 1998;3(2):199–208. doi: 10.3109/10837459809028496. [DOI] [PubMed] [Google Scholar]
- 70.Ritger PL, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Contr Release. 1987;5(1):37–42. doi: 10.1016/0168-3659(87)90035-6. [DOI] [PubMed] [Google Scholar]
- 71.Fakes MG, Dali MV, Haby TA, Morris KR, Varia SA, Serajuddin ATM. Moisture sorption behavior of selected bulking agents used in lyophilized products. PDA J Pharm Sci Technol. 2000;54(2):144–9. [PubMed] [Google Scholar]
- 72.Llabot JM, Manzo RH, Allemandi DA. Drug release from carbomer:carbomer sodium salt matrices with potential use as mucoadhesive drug delivery system. Int J Pharm. 2004;276(1–2):59–66. doi: 10.1016/j.ijpharm.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 73.Bernkop-Schnürch A, Humenberger C, Valenta C. Basic studies on bioadhesive delivery systems for peptide and protein drugs. Int J Pharm. 1998;165(2):217–25. doi: 10.1016/S0378-5173(98)00017-9. [DOI] [Google Scholar]
- 74.Bashaiwoldu AB, Podczeck F, Newton JM. A study on the effect of drying techniques on the mechanical properties of pellets and compacted pellets. Eur J Pharm Sci. 2004;21(2–3):119–29. doi: 10.1016/j.ejps.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 75.Siepmann F, Le Brun V, Siepmann J. Drugs acting as plasticizers in polymeric systems: a quantitative treatment. J Contr Release. 2006;115(3):298–306. doi: 10.1016/j.jconrel.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 76.Siepmann J, Lecomte F, Bodmeier R. Diffusion-controlled drug delivery systems: calculation of the required composition to achieve desired release profiles. J Contr Release. 1999;60(2–3):379–89. doi: 10.1016/S0168-3659(99)00093-0. [DOI] [PubMed] [Google Scholar]
- 77.Jacques Y, Buri P. An investigation of the physical behaviour of moisture-activated mucoadhesive hydrogels upon contact with biological and non-biological substrates. Pharm Acta Helv. 1997;72(4):225–32. doi: 10.1016/S0031-6865(97)00017-4. [DOI] [PubMed] [Google Scholar]
- 78.Peppas NA, Buri P. Surface, interfacial and molecular aspects of polymer bioadhesion on soft tissues. J Contr Release. 1985;2:257–75. doi: 10.1016/0168-3659(85)90050-1. [DOI] [Google Scholar]
- 79.Park H, Robinson JR. Physico-chemical properties of water insoluble polymers important to mucin/epithelial adhesion. J Contr Release. 1985;2:41–57. doi: 10.1016/0168-3659(85)90032-X. [DOI] [Google Scholar]
- 80.Henriksen I, Green KL, Smart JD, Smistad G, Karlsen J. Bioadhesion of hydrated chitosans: an in vitro and in vivo study. Int J Pharm. 1996;145(1–2):231–40. doi: 10.1016/S0378-5173(96)04776-X. [DOI] [Google Scholar]
- 81.Smart JD. The basics and underlying mechanisms of mucoadhesion. Adv Drug Deliv Rev. 2005;57(11):1556–68. doi: 10.1016/j.addr.2005.07.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 134 kb)