

Abstract
Hair bundles of the inner ear have a unique structure and protein composition that underlies their sensitivity to mechanical stimulation. Using mass spectrometry, we identified and quantified >1100 proteins, present from a few to 400,000 copies per stereocilium, from purified chick bundles; 336 of these were significantly enriched in bundles. Bundle proteins that we detected have been shown to regulate cytoskeleton structure and dynamics, energy metabolism, phospholipid synthesis, and cell signaling. Three-dimensional imaging using electron tomography allowed us to count the number of actin-actin crosslinkers and actin-membrane connectors; these values compared well to those obtained from mass spectrometry. Network analysis revealed several hub proteins, including RDX (radixin) and SLC9A3R2 (NHERF2), which interact with many bundle proteins and may perform functions essential for bundle structure and function. The quantitative mass spectrometry of bundle proteins reported here establishes a framework for future characterization of dynamic processes that shape bundle structure and function.
An outstanding example of a specialized organelle devoted to a single purpose, the vertebrate hair bundle transduces mechanical signals for the inner ear, converting sound and head movement to electrical signals that propagate to the central nervous system. Protruding from the apical surface of a sensory hair cell, a bundle typically consists of 50–100 actin-filled stereocilia and, at least during development, an axonemal kinocilium1. A bundle enlists ~100 transduction channels, which are mechanically gated by tip links as external forces oscillate the bundle; opening and closing of the channels in turn modulates the hair cell's membrane potential, controlling neurotransmitter release.
Because hair bundles have a reduced protein complement and carry out a specialized task, once we know which proteins are present—as well as their concentrations and interactions—understanding bundles' assembly and operation seems possible. While genetics studies have identified many proteins essential for bundle function2, others may have escaped detection because they are essential during development or, in some cases, can be compensated for by paralogs. To discover these additional proteins, biochemical strategies are essential; although bundles are scarce, quantitative mass spectrometry3 has the sensitivity and accuracy to detect and quantify the bundle's protein complement.
Our previous analysis of hair-bundle proteins using mass spectrometry detected 59 proteins, including several that are critical for bundle function4. Here, using a more sensitive mass spectrometer, we detected over 1100 proteins from chick vestibular bundles and identified those proteins selectively targeted to bundles. Many bundle-enriched proteins are expressed from deafness genes, confirming their essential role for the inner ear. We also imaged stereocilia using electron tomography and counted actin-actin crosslinkers and actin-membrane connectors; those counts compared favorably to mass-spectrometric estimates for crosslinker and connector proteins. To place the bundle's proteome into a network of functional and structural interactions, we assembled an interaction map that highlights the central roles in hair-bundle function played by actin, PI(4,5)P2, Ca2+, and CALM (calmodulin). Moreover, two other key hub proteins were identified: the ezrin-radixin-moesin (ERM) family member RDX (radixin), important in hair-bundle function5, and SLC9A3R2 (NHERF2; solute carrier family 9 member 3 regulator 2), a PDZ-domain adapter protein that couples RDX to many transmembrane proteins6. The comprehensive view offered by quantitative mass spectrometry reveals functional pathways in hair bundles and, based on the absence of key protein families, also rules out alternative mechanisms.
RESULTS
Mass spectrometry of purified hair bundles
Using liquid-chromatographic tandem mass spectrometry (LC-MS/MS), we identified proteins from hair bundles and epithelia of utricles (Supplementary Fig. 1), vestibular organs that detect linear acceleration, from embryonic day 20–21 (E20-E21) chicks; at this age, utricles are functional7. Bundles (BUN) were enriched 40-fold, to ~80% purity (see below), using the twist-off technique4,8. To obtain utricular epithelia, an eyelash was used to peel the hair-cell and supporting-cell layer from the underlying stroma layer (UTR). Supplementary Fig. 1b shows a cross-section of the utricle, showing the interface between the epithelium and stroma where the peel occurs. Four experiments each of BUN and UTR were analyzed.
We identified proteins using an Orbitrap mass spectrometer, analyzing data with the Andromeda search engine and MaxQuant9,10. Proteins that shared more than 20% of their detected peptides were combined into protein groups, which were denoted by their best scoring member. A total of 2944 proteins or protein groups were identified in the union of BUN and UTR. Increasing stringency by only considering proteins found in two or more experiments, we identified 1125 proteins from bundles (Fig. 1a; Supplementary Table 1); 728 proteins were identified in all four experiments. Only 20 proteins (<2%) were identified with a single unique peptide. In utricular epithelia, we identified 2753 proteins in two or more experiments, including 2147 in all four experiments.
Figure 1.
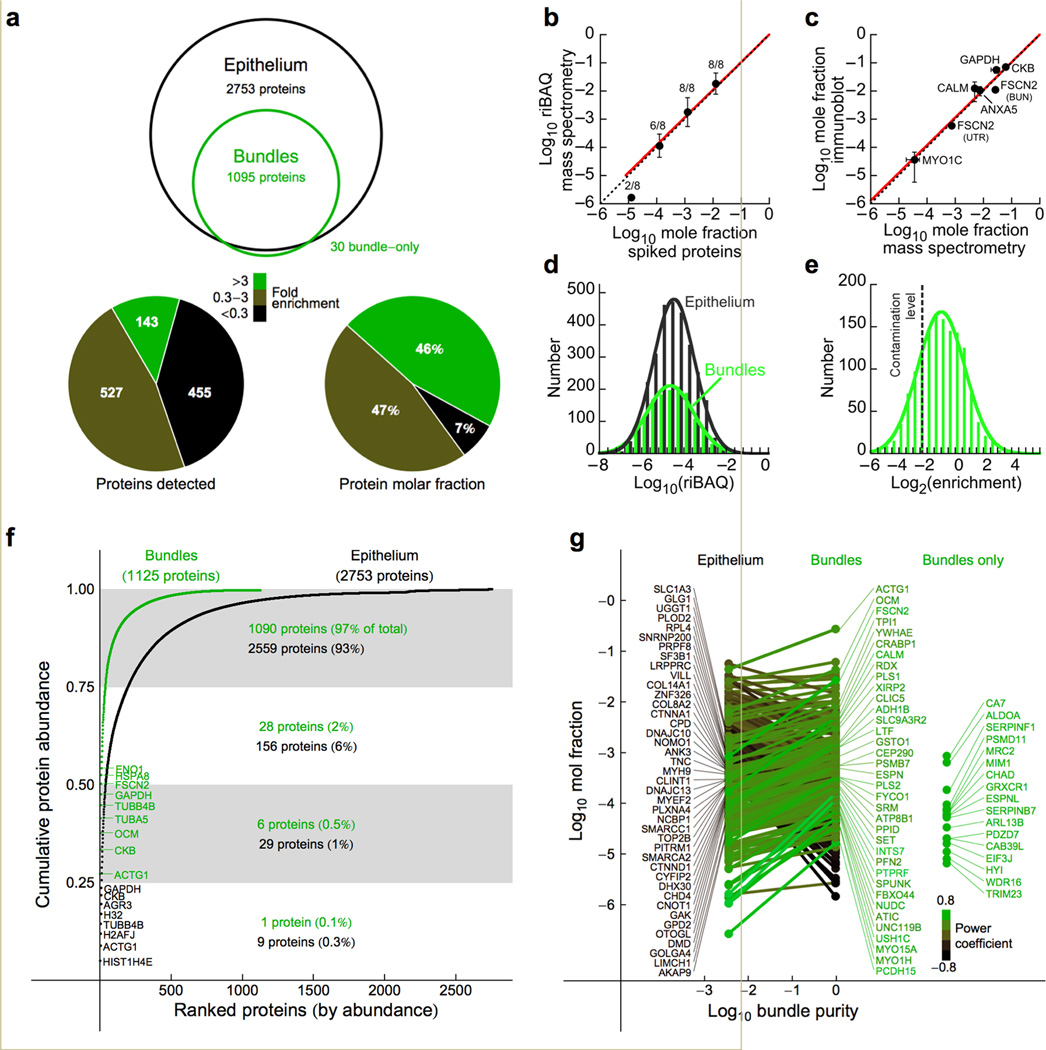
Quantitative analysis of chick hair-bundle proteins. (a) Top, proteins identified in bundles and epithelium (2 or more experiments). Bottom left, representation of bundle proteins as bundle-enriched, unenriched, and epithelium-enriched (by protein frequency). Bottom right, same as middle except the molar fractions of proteins in each class were summed. (b) Calibration curve relating mole fraction of human protein standards spiked into Escherichia coli extract to riBAQ value. The number of identified proteins is indicated for each data point (mean ± s.d.). The points corresponding to mole fractions of 10−2, 10−3, and 10−4 were fit with a line constrained through the 0,0 point (y = 1.02×; R = 0.999). (c) Calibration curve relating mole fraction determined from riBAQ values to mole fraction measured using quantitative immunoblots with purified proteins as standards; data points are mean ± s.e.m. and are fit by y = 0.98× (R = 0.97). Data for CKB and GAPDH were from ref. 4. Dotted lines in a and b correspond to the unity line. (d) Abundance distribution of bundle and epithelium proteins. Single Gaussian fits. (e) Enrichment distribution of proteins detected in bundles and epithelium; single Gaussian fit. (f) Cumulative protein molar abundance from highest to lowest abundance proteins. The most abundant 8–9 proteins in bundles and epithelium are indicated. (g) Mole fractions of proteins in epithelium (left) and bundle (right); the slope of the line connecting them represents bundle-to-epithelium enrichment. Proteins most highly enriched in the epithelium are indicated at left, while bundle-enriched proteins are at right. Hue represents relative enrichment (power coefficient of fit connecting points) for each protein. Far right, proteins detected only in bundles.
Quantitation using riBAQ
To quantify hair-bundle proteins, we used the iBAQ algorithm, which divides the sum of all precursor-peptide intensities by the number of theoretically observable peptides11. We normalized each protein's iBAQ value to the sum of all iBAQ values, generating a relative iBAQ (riBAQ) value for each protein. Although a previous report demonstrated the linearity of the iBAQ approach11, we sought a more rigorous validation: does riBAQ accurately report the mole fraction of each protein? We mimicked experiments with complex protein mixtures by detecting human proteins diluted in an Escherichia coli extract as a protein background. Only the more abundant human proteins were detected, demonstrating the limitations in detecting proteins at low mole fraction. The linear regression of Fig. 1b (log10 riBAQ = 1.02 ± 0.01 • log10 mole fraction) was carried out with the 10−2, 10−3, and 10−4 mole fraction data points. While the 10−5 data point does not fall upon the regression line, only two of eight proteins were detected. We conclude that the correspondence between riBAQ and mole fraction is nearly exact, at least down to a mole fraction of ~10−5 (Fig. 1b).
To independently verify our riBAQ calibration, we measured the concentrations of five hair-bundle proteins using quantitative immunoblotting (Fig. 1c). We only used proteins for which we had purified protein standards, which allowed us to generate accurate standard curves with known amounts of protein. The fit was very close to 1:1 (y = 0.98×; R = 0.97), confirming that riBAQ values reported mole fraction accurately.
Quantitation of hair-bundle proteins
With knowledge of total number of molecules per stereocilium, or of molecules per stereocilium of one accurately measured protein, mole fraction values can be used to estimate the number of molecules per stereocilium for any protein. Because actin monomers are present at minimal levels in stereocilia8 and because each stereocilium has 400,000 filamentous actin molecules (by electron tomography; see below), we used this estimate and actin's mole fraction value to convert mole fraction values for each hair bundle protein into molecules per stereocilium. These values are reported in Supplementary Table 1. The distribution of protein abundance values was similar for hair-bundle and epithelium proteins (Fig. 1d), indicating that low-abundance proteins were similarly detected in both preparations.
Proteins that are selectively targeted to hair bundles may be particularly important for function. Given that bundles constitute ~2% of the epithelium (Online Methods), targeted proteins could have a bundle-to-epithelium ratio (enrichment) as large as 50-fold. Because stereocilia are not closed compartments, however, diffusible cell-body proteins will also be present in bundles, with an enrichment of ~1. Finally, the bundle preparation will also contain cell-body contaminants. The broad histogram of binned enrichment values reflected the presence of all three types of proteins (Fig. 1e).
We determined the contamination level, the average BUN/UTR ratio for proteins known not to be in stereocilia, by measuring relative molar abundances of proteins from mitochondria and nuclei, which are absent from stereocilia12. We chose 81 nuclear and mitochondrial proteins detected in three or more utricle experiments; proteins that were not detected in bundles were assigned an enrichment value of 0. The contamination level estimated from these 81 proteins was 0.20 ± 0.25 (mean ± s.d.), which suggests that the BUN preparation contains ~80% hair bundles; bundles were thus purified approximately 40-fold.
To validate the estimated contamination level, we used quantitative immunoblotting (Supplementary Fig. 2a–b) to measure the presence in the BUN sample of five proteins known to be absent from hair bundles: ATP1A1 (ATPase, Na+/K+ transporting, alpha 1 polypeptide), found on the basolateral membranes of hair cells and supporting cells13; HSPA5 (heat shock protein 5; GRP78), an endoplasmic reticulum marker; MDH2 (malate dehydrogenase 2), a component of the mitochondrial citric acid cycle; PTPRJ (protein tyrosine phosphatase receptor J; also known as the supporting cell antigen), present on supporting cell apical surfaces14; and VIM (vimentin), an intermediate filament protein found in cell bodies of hair cells and supporting cells of the vestibular system15. Using immunocytochemistry, we confirmed that these proteins are absent from hair bundles (Supplementary Fig. 2c). As controls, we also examined actin and FSCN2 by immunoblotting (Supplementary Fig. 2a); each is concentrated in hair bundles (Supplementary Fig. 2c). In 14 measurements from 6 sets of BUN and UTR samples, we measured a contamination level of 0.30 ± 0.14 (mean ± s.d.), similar to that estimated by mass spectrometry.
We used riBAQ measurement errors, propagated in combination with the error in the calibration slope measurement, to estimate errors in protein enrichment and abundance reported in Supplementary Table 1. To determine the slope error, we plotted log10 riBAQ against log10 mole fraction (reversed axes from Fig. 1b) for all human proteins detected in the 10−2–10−4 mole fraction range. We used a linear mixed-effects model to generate a fit through the data, constrained through the (0,0) point; the slope of the calibration curve was 1.00 ± 0.03. Using the contamination level and the mole fraction of each protein in hair-bundle and epithelium samples, we corrected the abundance of each protein to reflect its actual concentration in bundles (Supplementary Table 1). Because of the substantial uncertainty in the contamination level, these corrected values are much more reliable for bundle-enriched proteins.
The most abundant proteins in hair bundles included ACTG1 (gamma actin, representing all actins), CKB (brain creatine kinase), OCM (parvalbumin CPV3), TUBA5 and TUBB4B (representing all alpha and beta tubulins), and FSCN2 (fascin 2) (Fig. 1f). Glycolytic enzymes were abundant, as were HSPA8 and HSP90AA1 (representing the 70 and 90 kD heat shock protein families). Only 7 proteins (0.6%) accounted for 50% of the total proteome molar abundance in bundles (Fig. 1f).
In Fig. 1g, we highlighted proteins with high bundle or epithelium enrichment, plotting mole fraction for all proteins detected in both samples against approximate bundle purity. Some proteins were only identified in bundles, for example PDZD7 (PDZ domain containing 7)16, presumably because their epithelium concentration is below the limit of detection. We may have underestimated the hair-bundle riBAQ values for high-molecular-mass proteins, however. Because strong cation exchange (SCX) purification was carried out only for BUN gel slices that were adjacent to sample wells (Online Methods), recovery of proteins from these slices may have been less efficient than from the corresponding UTR slices. Consequently, our estimates of concentrations and enrichment values for CDH23 (cadherin 23), GPR98 (VLGR1; G-protein coupled receptor 98), and USH2A (usherin), all of which are especially large, may be too low.
We used the contamination level to determine which proteins were reliably present in hair bundles. Of the 1095 proteins detected in two or more experiments each of bundles and epithelium, 336 had a bundle-to-epithelium enrichment level higher than the contamination level with a p-value of <0.05, adjusted for the false-discovery rate (FDR), the rate of incorrect assignments among enriched proteins17. Many actin-associated proteins were present at 100 or more copies per stereocilium (Fig. 2a; Table 1). Moreover, proteins known to be in stoichiometric complexes were at similar abundance (Fig. 2a), which independently corroborated our quantitation.
Figure 2.

Protein composition of chick hair bundles. (a) Hair bundle proteins ranked in order of abundance. Data-point color indicates protein class (key in panel b); symbol size represents bundle-to-epithelium enrichment. Red callouts indicate the most abundant actin-associated proteins; proteins significantly enriched over the contamination level are indicated by bold symbols. Blue and magenta callouts highlight proteins known to be in 1:1 stoichiometry. (b) Bundle proteins ranked in order of enrichment. Color indicates protein class, while symbol size indicates abundance. Proteins encoded by deafness genes are indicated; deafness proteins significantly enriched over the contamination level are indicated by bold symbols, those detected in 2 or fewer epithelium runs (hence not subject to statistical analysis) are indicated by italic symbols.
Table 1.
Quantitation of actin and actin-binding proteins in chick stereocilia.
| Identifier or protein group |
Description | Protein symbol |
Group members | Paralog in BUN? |
BUN/UTR ratio |
Corrected molecules per SC |
|---|---|---|---|---|---|---|
| ACT | Actin gamma 1 | ACTG1 | ACTG1; ACTA1; ACTA2; ACTB; ACTBL2; ACTC1; ACTG2 | Yes | 7 | 400,000 |
| NP_001171209 | Fascin 2 | FSCN2 | FSCN2 | Yes | 40 | 40,000 |
| ERM | Radixin | RDX | RDX; EZR; MSN | Yes | 9 | 6,800 |
| ENSGALP00000025573 | Myosin VI | MYO6 | MYO6 | No | 0.8 | 6,600 |
| ENSGALP00000004164 | Plastin 1 | PLS1 | PLS1 | Yes | 12 | 5,500 |
| ENSGALP00000017765 | Xin actin-binding repeat-containing protein 2 | XIRP2 | XIRP2 | Yes | 13 | 4,600 |
| ENSGALP00000030892 | Chloride intracellular channel 5 | CLIC5 | CLIC5 | Yes | 6 | 2,400 |
| ENSGALP00000008988 | Solute carrier family 9 member 3 regulator 2 | SLC9A3R2 | SLC9A3R2 | No | 17 | 2,000 |
| SPTAN1 | Spectrin, alpha, non-erythrocytic 1 | SPTAN1 | SPTAN1 | No | 0.5 | 1,400 |
| NP_001171603 | Fascin 1 | FSCN1 | FSCN1 | Yes | 4 | 1,300 |
| ENSGALP00000013109 | ARP1 actin-related protein 1 homolog A, centractin alpha | ACTR1A | ACTR1A | No | 1.7 | 1,300 |
| ENSGALP00000014097 | Destrin (actin depolymerizing factor) | DSTN | DSTN | No | 1.3 | 1,300 |
| ENSGALP00000013240 | Spectrin, beta, non-erythrocytic 1 | SPTBN1 | SPTBN1 | No | 0.4 | 1,300 |
| ENSGALP00000023085 | Actinin, alpha 4 | ACTN4 | ACTN4 | Yes | 1.4 | 1,100 |
| TWF2 | Twinfilin-2 | TWF2 | TWF2; TWF2/WDR82 | No | 4 | 950 |
| ENSGALP00000015053 | Shootin-1 | SHOOTIN1 | SHOOTIN1 | No | 5 | 910 |
| ENSGALP00000036480 | WD repeat domain 1 | WDR1 | WDR1 | No | 3 | 870 |
| ACTN1/2 | Actinin, alpha 1 | ACTN1 | ACTN1; ACTN2 | Yes | 0.9 | 790 |
| XP_417532.3 | Espin | ESPN | ESPN | Yes | 10 | 710 |
| ENSGALP00000006414 | Capping protein, beta 2 | CAPZB2 | CAPZB2; CAPZB1 | No | 2 | 690 |
| ENSGALP00000015277 | Capping protein, alpha 2 | CAPZA2 | CAPZA2 | Yes | 1.3 | 540 |
| ENSGALP00000007451 | Actin related protein 2/3 complex, subunit 1A, 41 kDa | ARPC1A | ARPC1A; ARPC1B | Yes | 1.2 | 470 |
| ENSGALP00000027391 | Plastin 2 | PLS2 | PLS2 | Yes | 21 | 460 |
| ENSGALP00000015632 | Myosin IIIB | MYO3B | MYO3B | Yes | 4 | 430 |
| ENSGALP00000038669 | ARP2 actin-related protein 2 homolog | ACTR2 | ACTR2 | Yes | 2 | 430 |
| ENSGALP00000009526 | Plastin 3 | PLS3 | PLS3 | Yes | 3 | 400 |
| ENSGALP00000019832 | ARP3 actin-related protein 3 homolog | ACTR3 | ACTR3; ACTR3B | Yes | 0.7 | 400 |
| TPM1/3 | Tropomyosin 1 | TPM1 | TPM1; TPM3 | Yes | 4 | 300 |
| ENSGALP00000039390 | Capping protein, alpha 1 | CAPZA1 | CAPZA1 | Yes | 2 | 260 |
| ENSGALP00000001044 | Myosin VIIA | MYO7A | MYO7A | No | 0.3 | 250 |
| ENSGALP00000010800 | Actin related protein 2/3 complex, subunit 4, 20 kDa | ARPC4 | ARPC4 | 1.1 | 210 | |
| ENSGALP00000002197 | Gelsolin | GSN | GSN | No | 0.4 | 190 |
| ENSGALP00000038130 | Profilin 2 | PFN2 | PFN2 | No | 5 | 180 |
| ENSGALP00000018652 | Actin related protein 2/3 complex, subunit 2, 34 kDa | ARPC2 | ARPC2 | No | 1.7 | 180 |
| ENSGALP00000039755 | EPS8-like 2 | EPS8L2 | EPS8L2 | No | 0.97 | 130 |
| RAC | Ras-related C3 botulinum toxin substrate 3 | RAC3 | RAC3; RAC1; RAC2; RHOG | No | 1.8 | 130 |
| ENSGALP00000001914 | Chloride intracellular channel 4 | CLIC4 | CLIC4; CLIC6 | Yes | 1.0 | 100 |
| ENSGALP00000001794 | Myosin, heavy chain 10, non-muscle | MYH10 | MYH10; MYH1 | Yes | 0.2 | 95 |
| ENSGALP00000010106 | Espin-like | ESPNL | ESPNL | Yes | BUN only | 91 |
| ENSGALP00000036676 | Glutaredoxin, cysteine rich 1 | GRXCR1 | GRXCR1 | No | BUN only | 81 |
| ENSGALP00000013036 | Coronin, actin binding protein, 2B | CORO2B | CORO2B | Yes | 0.9 | 69 |
| ENSGALP00000007931 | Synapsin I | SYN1 | SYN1 | Yes | 1.0 | 65 |
| ENSGALP00000007959 | Myosin XVA | MYO15A | MYO15A | No | 34 | 50 |
| ENSGALP00000008214 | Myosin IC | MYO1C | MYO1C | Yes | 6 | 47 |
| ENSGALP00000008212 | Myosin IH | MYO1H | MYO1H | Yes | 16 | 46 |
| ENSGALP00000001365 | Huntingtin interacting protein 1 | HIP1 | HIP1 | No | 0.4 | 36 |
| ENSGALP00000039629 | Vinculin | VCL | VCL | No | 0.4 | 31 |
| EBP41/L3 | Erythrocyte membrane protein band 4.1 | EPB41 | EPB41; EPB41L3 | Yes | 0.7 | 24 |
| ENSGALP00000022935 | Coronin, actin binding protein, 1C | CORO1C | CORO1C | Yes | 1.3 | 19 |
| ENSGALP00000019631 | Actin-related protein 10 homolog (ARP11) | ACTR10 | ACTR10 | Yes | 0.9 | 18 |
| ENSGALP00000017268 | Mediator of cell motility 1 | MEMO1 | MEMO1 | No | 0.5 | 9 |
| ENSGALP00000027304 | LIM domain 7 | LMO7 | LMO7 | No | 1.3 | 6 |
| ACTL6A | Actin-like 6A (ARP4) | ACTL6A | ACTL6A | No | 0.2 | 5 |
| ENSGALP00000012361 | Myosin IIIA | MYO3A | MYO3A | Yes | 2.7 | 4 |
| ENSGALP00000009946 | LIM domain and actin binding 1 | LIMA1 | LIMA1 | Yes | 0.3 | 3 |
| ENSGALP00000007644 | Myosin phosphatase Rho interacting protein | MPRIP | MPRIP | No | 0.2 | 1 |
Key: Identifier or protein group, Ensembl or NCBI identifier, or experimentally-assigned protein group name; Description, common name of the principal entry, the protein with most mass-spectrometric evidence; Protein symbol, official protein symbol (based on human genes); Group members, symbols for all proteins in bundles that are summed together in entry (alphabetical order after principal entry); Paralog in BUN, indicates whether a paralog is present in bundles; BUN/UTR ratio, bundle/utricle ratio (enrichment); Corrected molecules per SC, estimated molecules per stereocilium, determined with relative iBAQ quantitation, corrected for enrichment. Actin crosslinking proteins are indicated in bold, while actin-to-membrane connectors are indicated by bold italics.
"Deafness proteins" are enriched in hair bundles
We ranked proteins by bundle enrichment and labeled those proteins encoded by deafness or vestibular-dysfunction genes (Fig. 2b). A list of 7112 Online Mendelian Inheritance in Man (OMIM) terms and their mapping to human genes and MGI marker accession IDs (downloaded 10/2012) was used to identify deafness proteins in the list of proteins identified from BUN and UTR samples, including redundant proteins present in protein groups (Supplementary Table 1). Two terms were used to search the OMIM data: "deafness" and "Usher syndrome."
Most "deafness proteins" detected were enriched in hair bundles; 4% of the 277 proteins enriched >2-fold were associated with deafness in the OMIM database18, compared to only 0.7% of 2667 proteins enriched <2-fold (p < 10−4, Fisher's exact test). The OMIM database has 163 entries annotated with "deafness" or "Usher syndrome," corresponding to 0.7% of the ~23,500 genes in the human Ensembl database. Proteins enriched >2-fold were also significantly (p < 10−2) associated with mouse deafness entries in the Mouse Genome Database (MGD)19. The list of proteins enriched in bundles over epithelium is thus a rich reference for proteins with demonstrated significance for auditory and vestibular function.
The OMIM and MGI databases do not include all genes associated with deafness or vestibular dysfunction that are expressed in stereocilia. Adding in additional known deafness proteins (Supplementary Table 1), including those arising from targeted mutagenesis, our mass spectrometry experiments detected 22 of 27 mouse deafness proteins known to be expressed in stereocilia2. These 22 proteins had an average bundle-to-epithelium enrichment of 29 ± 12 (mean ± s.e.m.), confirming that the enrichment analysis successfully identified functionally important proteins. Only DFNB31 (whirlin), CLRN1 (clarin 1), LHFPL5 (TMHS; lipoma HMGIC fusion partner-like 5), USH1G (Sans), and STRC (stereocilin) were not detected. Two of these proteins were not expected to be detected; USH1G transcripts were undetectable in E20–21 utricles20, accounting for the absence of the protein, and STRC is not present in the chick Ensembl database, so cannot be detected with our mass spectrometric approach. Thus only about 10% of known, detectable stereocilia deafness proteins (3/25) escaped observation by mass spectrometry, either because of their low abundance or because they are in auditory but not vestibular stereocilia.
Additional proteins enriched in hair bundles may be encoded by as-yet-undiscovered deafness genes. At least 1/3 of all deafness genes are expressed in bundles, and given ~400 human loci for non-syndromic deafness21, an additional 100 bundle proteins might plausibly be associated with deafness. By identifying human homologs for bundle-enriched proteins and correlating genomic map locations with identified but uncloned deafness loci, we identified 13 new candidates for 8 deafness loci (Table 2).
Table 2.
Candidates for mapped but uncloned deafness genes.
| Human deafness locus |
Chick protein identifier | Homologous human gene |
Chr | Description | Protein symbol |
BUN:UTR enrichment |
|---|---|---|---|---|---|---|
| AUNA1 | ENSGALP00000027391 | ENSG00000136167 | 13 | Plastin 2 | PLS2 | 21x |
| AUNA1 | ENSGALP00000027417 | ENSG00000102547 | 13 | Calcium binding protein 39-like | CAB39L | Bundle only |
| DFNA16 | ENSGALP00000015632 | ENSG00000071909 | 2 | Myosin IIIB | MYO3B | 3x |
| DFNA18 | ENSGALP00000004164 | ENSG00000120756 | 3 | Plastin 1 | PLS1 | 4x |
| DFNA32 | ENSGALP00000039755 | ENSG00000177106 | 11 | Epidermal growth factor receptor kinase substrate 8-like protein 2 | EPS8L2 | 1x |
| DFNB55 | ENSGALP00000022269 | ENSG00000128050 | 4 | Multifunctional protein ADE2 | PAICS | 1x |
| DFNB55 | ENSGALP00000022326 | ENSG00000109265 | 4 | Uncharacterized protein KIAA1211 | KIAA1211 | 3x |
| DFNB57 | ENSGALP00000013870 | ENSG00000108039 | 10 | Xaa-Pro aminopeptidase 1 | XPNPEP1 | 3x |
| DFNB57 | ENSGALP00000015053 | ENSG00000187164 | 10 | Shootin-1 | SHOOTIN1 | 5x |
| DFNB57 | ENSGALP00000032553 | ENSG00000186862 | 10 | PDZ domain containing 7 | PDZD7 | Bundle only |
| DFNB85 | ENSGALP00000007825 | ENSG00000175662 | 17 | TOM1-like protein 2 | TOM1L2 | 1x |
| DFNX5 | ENSGALP00000009526 | ENSG00000102024 | X | Plastin 3 | PLS3 | 3x |
| DFNX5 | ENSGALP00000031748 | ENSG00000165704 | X | Hypoxanthine-guanine phosphoribosyl-transferase | HPRT | 4x |
Key: Human deafness locus, unmapped human deafness locus identifier; Chick protein identifier, chicken Ensembl identifier for protein mapping to deafness gene; Homologous human gene, human gene to which chick protein maps; Chr, human chromosome number; Description, descriptive name for protein; Abbrv, abbreviation; Chick enrichment, bundle/utricle enrichment in chick.
Actin cytoskeleton structure from electron tomography
To further validate the mass-spectrometric data, we used electron tomography22 to count cytoskeletal structures in stereocilia from chick utricles (Fig. 3). We generated tomograms from stereocilia oriented longitudinally (Fig. 3a–c), transversely (Fig. 3f–h; Supplementary Fig. 3), and obliquely (Fig. 3i–k) to the plane of section. Each dataset has distinct advantages for quantitation. The longitudinal view allows us to follow individual actin filaments for long distances, but out-of-plane crosslinkers are less reliably detected due to limited tilt-related data anisotropy. Transverse views allow ready measurement of actin-actin distances in all orientations, but the number of actin-actin links that can be detected is relatively small because of the ultrathin sections. Oblique views allow more reliable detection of out-of-plane crosslinkers, but are complicated by the section plane jumping from one actin filament to another. To interpret density maps obtained by electron tomography, we used two density thresholds to build simple ball-and-stick models. The use of two thresholds, which generated maximum and minimum estimates of crosslinker numbers, addressed difficulties in objectively thresholding density maps, which was complicated by reconstruction noise and possible staining inhomogeneity. Out-of-plane crosslinkers were omitted in model building of our high-threshold (low estimate) rendered maps.
Figure 3.

Electron tomography of chick stereocilia. (a–c) Tomogram from a stereocilium oriented longitudinally with respect to the plane of section. (a) Two-dimensional 0° tilt projection image recorded for tomographic reconstruction. (b) Single ~0.8 nm slice of unfiltered three-dimensional reconstruction. (c) Single ~0.8 nm slice of bilaterally filtered density map. Scale bars in a–k are 100 nm. (d–e) Stereocilia model from longitudinal tomogram. Red lines represent actin, blue lines represent actin-actin crosslinkers, orange lines represent actin-membrane connectors, and membrane density is depicted in light green. (d) Overview of stereocilia model overlaid on the surface-rendered density map (6.4 nm thick). (e) Overview of model alone and segmented membrane density. (f–h) Transverse stereocilia tomogram. (f) Two-dimensional 0° tilt projection of transverse image recorded for tomographic reconstruction. (g) Single 0.8 nm slice of unfiltered three-dimensional reconstruction. (h) Single 0.8 nm slice of bilaterally filtered density map of transverse orientation, allowing precise measurements of actin-actin distances. (i–k) Oblique stereocilia tomogram. (i) Two-dimensional 0° tilt projection of oblique image used for tomographic reconstruction (stereocilia longitudinal axis is at an angle of ~18° with respect to sectioning plane). (j) Single 0.8 nm slice of unfiltered three-dimensional reconstruction. (k) Single 0.8 nm slice of bilaterally filtered three-dimensional density map. (l–m) Scaled views of density and model in mid-shaft region; from longitudinal stereocilia orientation. (l) Density map only. (m) Model overlaid on the density map. (n) Model alone. Scale bars in l–q are 20 nm. (o–q) Close-up views of density map in a region adjacent to the membrane. (o) Density map only. (p) Model overlaid on the density map. (q) Model alone. (r) Histogram showing distribution of actin-actin distances at sites of cross-bridges. The data were fit with the sum of three Gaussians (red), with equal σ (width) for each. Individual fits for 8, 11, and 15 nm peaks are shown in gray. (s) Close-up view of model. Scale bar is 50 nm.
We measured in several independent subvolumes the total actin-filament length, actin-actin spacing, number of actin-actin crosslinkers, and number of actin-membrane connectors (Fig. 3d–e, l–s). A prototypical chick utricle stereocilium visualized by fluorescence and transmission electron microscopy was ~250 nm in diameter, ~5 µm in length, and hence ~0.2 fl in volume. Electron tomography indicated that there were ~210 actin filaments in a stereocilium of that diameter, or ~400,000 actin monomers in filaments (~3 mM).
Using the low threshold (upper limit) and averaging the data from six subvolumes per tomogram, we estimated 62,000 ± 1,000 (longitudinal-orientation tomogram) and 91,000 ± 2,000 (oblique-orientation tomogram) crosslinkers per prototypical stereocilium (mean ± s.e.m.). The conservative lower-limit estimates with a high-density threshold, which also do not consider out-of-plane crosslinkers, were 30,000 ± 1,000 and 36,000 ± 2,000. Assuming three crosslinkers for every 36 nm of actin filament23, the theoretical maximum is ~87,000.
We also counted actin-to-membrane connectors with electron tomography. The prototypical stereocilium has ~52 actin filaments adjacent to the plasma membrane; since a binding site should appear every 36 nm along each actin filament, each stereocilium could contain as many as 7200 actin-membrane connectors. We counted 7300 ± 1100 connectors per stereocilium with the low-density threshold and 5800 ± 900 using the high threshold (Fig. 3g–i).
Stereociliary protein network
Focusing on the actin cytoskeleton, we identified potential interactions between hair-bundle proteins and depicted them using graph theory24 with spring-electrical modeling25, generating an undirected, mathematically-defined graph that illustrates these relationships. Most of the major cytoskeletal proteins that were significantly enriched in hair bundles (Table 1) were chosen to seed the network. Some interactions for these proteins were identified by searching the STRING (http://string-db.org) and BioGRID (http://thebiogrid.org) databases; however, not all known interactions are in these or other protein-protein interaction databases. We therefore manually curated our protein interaction list (Supplementary Table 2) by searching PubMed for each protein in the network, allowing us to both validate interactions and identify additional ones. All interactions identified are cited in Supplementary Table 3.
The network figure (Fig. 4) highlights important hair-bundle proteins, as well as signaling molecules and ions. In most molecular networks, most nodes have only a few links but others—highly-connected hubs—have a very large number of links, which hold the sparsely-linked nodes together.26 For the 69 nodes with at least two interactions, the protein-interaction distribution data were fit well with a power law26, with P(k) ~ k −1.3, R = 0.79, and p < 10−3. The average clustering coefficient Ci = 2ni / ki (ki − 1), where ni is the number of links connecting the ki neighbors of node i to each other, represents how nodes interconnect26. Ci = 0.24, measured for our network, indicates strong clustering27. Nodes with the largest numbers of links were actin (33 interactions), PI(4,5)P2 (20), SLC9A3R2 (12), CALM (9), RDX (8), and Ca2+ (8).
Figure 4.

Interaction network for hair-bundle proteins. Symbols (nodes) represent bundle proteins or second messengers; only nodes with two or more interactions are plotted, with exceptions of OCM and CALB2. Underline labels indicate deafness proteins. Node colors indicate functional classification (same key as in Fig. 2b), while node symbol size represents protein abundance in bundles. Ca2+, PI(4,5)P2, and cAMP are indicated by diamond node symbols. Solid links (lines between nodes) represent interactions validated with literature citations; Supplementary Table 3 lists all interactions and evidence. Dotted links correspond to interactions involving paralogs of bundle proteins; dashed links represent hypothetical interactions (e.g., SLC9A3R2 interactions from Table 3). The layout of the plot is controlled by the density of links between nearby nodes. The distribution of nodes and links in the plot is fit well by a power law, which indicates that the plot contains a few highly connected nodes (hubs) and many other less-connected nodes. Supplementary Fig. 5 reproduces this figure with each link hyperlinked to a PubMed reference supporting the interaction.
RDX and SLC9A3R2
RDX and SLC9A3R2, identified as hubs in the hair-bundle protein network, were each detected in chick utricle bundles by immunoblotting (Fig. 5a). We also found that SLC9A3R2 expressed in tissue-culture cells immunoprecipitated with RDX; SLC9A3R2 binds to activated ERM proteins28, and binding was indeed stronger to RDX with a threonine-to-aspartate mutation that mimics the activating phosphorylation (Fig. 5b). In bullfrog stereocilia, RDX is activated by PI(4,5)P2 and phosphorylation is found in a narrow band above basal tapers, at the site of the ankle links5. Similarly, SLC9A3R2 and total RDX were concentrated in the bottom half of each stereocilium (Fig. 5c,d), and phosphorylated total ERM protein (pERM) was only found above stereociliary tapers (Fig. 5c). Likewise, pERM is concentrated in the upper half of supporting-cell microvilli (Fig. 5c, inset). Although MYO7A also appears in a band above basal tapers,29 its distribution was distinct from those of RDX and SLC9A3R2 (Fig. 5e). Finally, we detected in hair bundles the Rho-family GTPases RHOA, RAC3, and CDC42, which control the actin cytoskeleton (Supplementary Table 1; Fig. 5a,f).
Figure 5.

Identification of RDX and SLC9A3R2 in hair bundles. (a) Protein immunoblotting with purified hair bundles. Lanes from left to right for each blot: Hair bundles, bundles from 40 chick ears (~0.6 µg total protein); Agarose, agarose equivalent to that in the bundles sample; Epithelium, utricle sensory epithelium from 4 chick ears (~10 µg protein). Antibodies used are indicated at left. Both anti-RDX and anti-pERM (which recognizes phosphorylated ezrin, radixin, and moesin) detected bands both at the expected size (~70 kD) and at ~80 kD (asterisk). (b) RDX-SLC9A3R2 interaction. Epitope-tagged chick WT RDX, T564D-RDX, and SLC9A3R2 were expressed in HEK cells in the indicated combinations. Tagged SLC9A3R2 was immunoprecipitated, and associated RDX was detected by immunoblotting. (c) RDX and pERM immunocytochemistry. RDX and pERM co-localize except in the taper region (double arrows in bottom panel). Inset, magnification of apical surfaces of supporting and hair cells. Note that pERM immunoreactivity is absent from bases of microvilli (MV), as in stereocilia (SC). Scale bar in panel c is 10 µm and applies to panels c-f. (d) RDX and SLC9A3R2 immunocytochemistry. RDX and SLC9A3R2 overlap throughout the bundle, but SLC9A3R2 is more concentrated towards stereociliary bases (double arrows in bottom panel), including the tapers, than is RDX. (e) MYO7A immunocytochemistry. MYO7A is concentrated towards stereociliary tips and in a band above the taper region (asterisks). (f) RHOA immunocytochemistry. Staining is seen in stereocilia (arrow) and the kinocilium or the tallest stereocilia of the bundle (arrowheads). RHOA is also substantially enriched in hair cells over supporting cells.
When bound to RDX, the PDZ domains of SLC9A3R2 are available for binding; based on consensus sequences for SLC9A3R2 ligands30, we identified 24 hair-bundle proteins as candidate binding partners, including CDH23, PCDH15, USH1C, USH2A, and GPR98 (Table 3).
Table 3.
Candidate SLC9A3R2-binding proteins.
| Identifier | Last 4 residues |
Description | Protein symbol |
BUN:UTR enrichment |
|---|---|---|---|---|
| ENSGALP00000004315 | STAL | Protocadherin-15 | PCDH15 | 35 |
| ENSGALP00000038711 | MTFF | Harmonin (Usher syndrome 1C) | USH1C | 22 |
| ENSGALP00000000916 | ETKL | Espin | ESPN | 10 |
| ENSGALP00000007402 | ITEL | Cadherin-23 | CDH23 | 3 |
| ENSGALP00000004504 | CTVF | Ras-related C3 botulinum toxin substrate 3 | RAC3 | 2 |
| ENSGALP00000015700 | DTHL | Usherin (Usher syndrome 2A) | USH2A | 2 |
| ENSGALP00000023576 | DTHL | G protein-coupled receptor 98 | GPR98 | 1.3 |
| ENSGALP00000018350 | ETSL | ATPase, Ca2+ transporting, plasma membrane 1* | ATP2B1 | 1 |
| ENSGALP00000015409 | ESDL | Actinin, alpha 1† | ACTN1 | 0.9 |
| ENSGALP00000019939 | LTLL | Cold inducible RNA binding protein | CIRBP | 0.9 |
| ENSGALP00000040234 | PTGF | Casein kinase 1, alpha 1 | CSNK1A1 | 0.9 |
| ENSGALP00000003482 | ATVL | Dmx-like 1 | DMXL1 | 0.8 |
| ENSGALP00000009572 | STAL | Rho GTPase activating protein 17 | ARHGAP17 | 0.4 |
| ENSGALP00000015964 | NTFF | Ribosomal protein L18a | RPL18A | 0.3 |
| ENSGALP00000009665 | KTSL | Coatomer protein complex, subunit beta 1 | COPB1 | 0.2 |
| ENSGALP00000024331 | DTEL | Tight junction protein 2 (zona occludens 2) | TJP2 | 0.2 |
| ENSGALP00000007487 | VTLL | CSE1 chromosome segregation 1-like | CSE1L | 0.1 |
| ENSGALP00000039286 | QTEF | Family with sequence similarity 129, member B | FAM129B | 0.1 |
| ENSGALP00000019402 | DTDL | Catenin (cadherin-associated protein), beta 1, 88 kDa | CTNNB1 | 0.08 |
| ENSGALP00000017577 | QTEL | Anterior gradient 3 homolog | AGR3 | 0.05 |
| ENSGALP00000023016 | PTTL | LIM and calponin homology domains 1 | LIMCH1 | 0.04 |
| ENSGALP00000019992 | GTSL | Golgin A4 | GOLGA4 | 0.02 |
| ENSGALP00000012817 | STCL | Laminin, beta 1 | LAMB1 | 0.01 |
| ENSGALP00000023309 | ESDL | Actinin, alpha 2† | ACTN2 | n/a |
| ENSGALP00000005687 | ETSL | ATPase, Ca2+ transporting, plasma membrane 2* | ATP2B2 | n/a |
ATP2B1 and ATP2B2 are in the same protein group. Only the "b" splice forms of these proteins bind PDZ domains; the splice form of ATP2B2 in hair bundles is "a".
ACTN1 and ACTN2 are in the same protein group.
The C-terminal four amino acids of all chick bundle proteins were searched for instances of XTXL (X is any amino acid), XTXF, GVGL, ESDL, STHM, TLGA, all which bind to SLC9A3R230.
DISCUSSION
Here we used quantitative mass spectrometry to establish an extensive compendium of the proteins of vestibular hair bundles, available for browsing on the SHIELD website of inner-ear gene expression datasets (https://shield.hms.harvard.edu). These data will allow us to systematically address two crucial topics for the cell biology of hair cells, construction of the bundle cytoskeleton during development, as well as composition and assembly of the transduction complex. By determining functional relationships between bundle proteins, investigating static and dynamic protein localization, measuring protein abundance, and cataloging protein-protein interactions, we will gain a comprehensive understanding of how bundle proteins cooperate to make the bundle and carry out transduction.
Actin cytoskeleton
As expected given the structure of the stereocilium cytoskeleton, actin and actin-associated proteins were abundant in the mass spectrometry data. We compared the tomography estimates to abundances of known actin-actin crosslinkers and actin-membrane connectors measured by mass spectrometry using the riBAQ method (Table 1). We detected three classes of crosslinkers: fascins (40,000 molecules per stereocilium of FSCN2, 1300 FSCN1), plastins (5500 PLS1, also known as fimbrin, 460 PLS2, 400 PLS3), and espins (710 ESPN and 90 ESPNL), similar to what we found previously31. Mass spectrometry thus estimates that each stereocilium has ~48,000 crosslinkers, in good agreement with the tomography estimates (33,000–77,000).
The ERM family, which crosslinks membrane-protein complexes to actin at ~36 nm intervals, likely contributes most of the actin-membrane connectors. By mass spectrometry, EZR (ezrin), RDX, and MSN (moesin) together totaled 6800 molecules per stereocilium (Table 1), with RDX accounting for the majority; this value is within the range estimated by tomography (5800–7300).
Remaining actin-to-membrane connectors may be members of the myosin superfamily1. Mass-spectrometric quantitation gave estimates of myosin abundance that corresponded well to independent measurements. By quantitative immunoblotting, bullfrog bundles have >700 molecules/stereocilium of MYO6, >400 of MYO7A, and 100 of MYO1C29; mass spectrometry estimated 6600 chick MYO6, 250 MYO7A, and ~50 each of MYO1C and the closely related MYO1H.
Actin polymerization in stereocilia, which is dynamic at least through late development32, is controlled by the myosin-III and -XV families1. We detected 430 myosin-III molecules per stereocilum, nearly all of which was MYO3B. Notably, the concentration of MYO3B was very close to that of its binding partner ESPN and that of PFN2, the profilin paralog that binds to ESPN.
We detected 50 molecules per stereocilium of MYO15A. Although the MYO15A binding partner DFNB31 was not detected, we found 130 molecules of EPS8L2 (EPS8-like 2); because its paralog EPS8 (epidermal growth factor receptor pathway substrate 8) binds the MYO15A-DFNB31 complex, EPS8L2 might partner with MYO15A in the vestibular system. Altogether we counted ~7500 myosin molecules per stereocilium, which could account for remaining actin-to-membrane connectors.
Several other proteins also control polymerization of actin networks33. We detected five of seven subunits of the ARP2/3 complex, which mediates polymerization of branched actin networks; at 3 µM (~340 molecules per stereocilum), the ARP2/3 complex is present at concentration similar to that in human neutrophils34. Because we did not detect any activating WASP/WAVE family members, however, the ARP2/3 complex likely is inactive in stereocilia. We did not detect any Ena/VASP family members, and while we detected one formin (DIAPH2 in one bundle experiment), its enrichment value suggested it is a contaminant. Together these results suggest that control of actin-filament polymerization in late development involves only myosin-mediated mechanisms.
Although actin may not treadmill from tip to taper35, the stability of stereociliary actin filaments suggests that their barbed ends, at stereociliary tips, are capped. If one barbed-end capper is present per filament, and if filaments run the length of the stereocilium, there should be ~210 cappers per stereocilium. We detected two major cappers: ~700 CAPZ (capping protein) heterodimers per stereocilium and 950 TWF2 (twinfilin-2) molecules. The excess of cappers over actin filaments suggests that they compete for filament ends, which could occur differentially in longer and shorter stereocilia.
Pointed ends of stereociliary actin filaments progressively terminate to form stereociliary tapers, suggesting a systematic capping or depolymerization there. We did not detect tropomodulin, the best-characterized pointed-end capper36, nor did we detect taperin, a candidate pointed-end capper37. The pointed ends of stereociliary actin filaments either terminate on the taper membrane23 or gather into the central rootlet material38; if the former, MYO6 could anchor the pointed ends to the membrane protein PTPRQ (protein tyrosine phosphatase, receptor type, Q)39, present at 1500 copies per stereocilum, or if the latter, the rootlet component TRIOBP (TRIO and F-actin binding protein)40 (detected in one experiment only) could crosslink filament ends to the rootlet.
At 4600 molecules/stereocilium, XIRP2 (Xin-related protein 2) is the most abundant protein in hair bundles without an obvious role. Although most species' XIRP2 proteins contain >30 actin-binding Xin repeats, chick and rat bundle XIRP2 do not contain these domains (Supplementary Fig. 4b). However, XIRP2 is a paralog of the actin-binding protein LIMA141, also known as EPLIN, suggesting that XIRP2 may nevertheless bind actin.
Network analysis highlighted the role of several other cytoskeletal proteins. The largest hair-bundle hub was actin, with 33 interactions; PI(4,5)P2 had 20 interactions, which was expected given its prominence in stereocilia5 and roles in actin polymerization42 and membrane targeting43. As is clear from co-clustering of the two hubs in the network map (Fig. 4), many bundle proteins bind both actin and PI(4,5)P2; moreover, several bundle proteins not known to interact with actin do bind to PI(4,5)P2, suggesting that the phospholipid may concentrate some membrane-associated proteins in stereocilia. Also prominent were CALM and Ca2+, with respectively 9 and 8 interactions; Ca2+ entering stereocilia through transduction channels couples mechanotransduction to CALM-dependent enzyme activity.
RDX and SLC9A3R2 (2000 molecules/stereocilium) had respectively 8 and 12 interactions, including a direct interaction between them that is facilitated by RDX phosphorylation. A major binding partner for RDX is thought to be CLIC5 (chloride intracellular channel 5)44, which can interconvert between cytosolic and transmembrane forms. At 2400 molecules/stereocilium, CLIC5 could bind RDX molecules not bound by SLC9A3R2. RHOA, detected in stereocilia, induces translocation of CLIC4, a paralog of CLIC5, to the plasma membrane45; moreover, RHOA can activate RDX after preactivation by PI(4,5)P246. RHOA may therefore both activate RDX and target its receptor to stereociliary membranes, providing a scaffold for other protein-protein interactions.
Other membrane proteins will interact with RDX via SLC9A3R2. Given the critical role for the paralog SLC9A3R1 in assembling microvilli47, we propose that SLC9A3R2 is necessary for assembling stereocilia. SLC9A3R1 is highly dynamic in microvilli48, suggesting that SLC9A3R2 complexes may be dynamic in stereocilia. Moreover, likely ligands for SLC9A3R2 are of known importance for bundle structure and function. USH2A and GPR98 are located at the ankle links1; RDX may anchor ankle links there through SLC9A3R2. SLC9A3R2 and RDX might also anchor the CDH23- and PCDH15-containing transient lateral links required for forming a cohesive bundle1.
Other hair bundle proteins
Mechanotransduction molecules are rare in hair bundles; there is only one tip link and two active transduction channels per stereocilium1. Nevertheless, we detected ~20 molecules/stereocilium each of the tip link cadherins, CDH23 and PCDH15 (protocadherin 15), as well as 60 USH1C (harmonin) molecules, which cluster to anchor the upper end of a tip link1. While the tip link of a single stereocilium should only contain two each of the cadherins, they are also present in the lateral links of developing bundles and in kinocilial links (Supplementary Fig. 4)1.
Our inability to detect other transduction proteins—like the elusive transduction channel—likely arises from the limited dynamic range of mass spectrometry, rather than lack of sensitivity. In each mass spectrometry run, we matched ~104 spectra to chick peptides; because only 1–10 molecules of the transduction channel may be present for every 106 bundle molecules, substantial enrichment may be required to detect it above the background of actin and other cytoskeletal proteins.
Axonemal kinocilia are present in the hair-bundle preparation4; besides tubulin, we detected the axonemal dyneins DNAH5 and DNAH9, radial spoke head molecules RSHL1, RSPH6A, and RSPH9, the intraflagellar transport molecule IFT172, and the axonemal small GTPase ARL13B.
Diffusible Ca2+ buffers were prominent; we estimated 63,000 OCM, 8000 CALB2 (calretinin), and 7300 CALM (calmodulin) per stereocilium, together corresponding to a total of ~2 mM Ca2+ binding sites. The estimated CALM concentration (~60 µM) is nearly identical to the 70 µM estimated for bullfrog hair bundles by quantitative immunoblotting49. The membrane area of the prototypical stereocilium is ~4 µm2; if the density of the Ca2+ pump in chick bundles is the same as in bullfrog (2000 molecules µm−2; ref. 50), each stereocilium would have 8000 Ca2+ pumps. Mass spectrometry estimated significantly fewer, ~1700 (mostly ATP2B2), perhaps because transmembrane peptides are not well detected in LC-MS/MS experiments.
Conclusions
Many proteins enriched in hair bundles are encoded by deafness genes, suggesting that other bundle-enriched proteins may be linked to deafness in the future. Mass spectrometry allows us to identify functionally important bundle proteins that have not yet been identified by genetics, such as proteins that carry out multiple roles in an organism and could have an embryonic lethal phenotype. Indeed, genetic screens for deafness likely miss ubiquitously expressed proteins with developmental roles; by contrast, the mass spectrometric approach can in principle identify any protein that contributes to maintenance and function of the hair bundle.
ONLINE METHODS
Animals
Animal experiments reported here were approved by the Oregon Health & Science University Institutional Animal Care and Use Committee (IACUC); the approval number was A684. All experiments began with euthanasia of the animal, which was carried out using methods approved by American Veterinary Medical Association Panel on Euthanasia.
Preparation of samples for mass spectrometry
Utricle hair bundles were purified from E20–21 chicks using the twist-off method4,8. We estimated the fraction of epithelium protein accounted for by bundles using two independent methods. In the first, we divided the amount of bundle protein per utricle (16 ng; ref. 4), measured with a fluorescence protein assay, by the estimated recovery (~40%); this value was then divided by the protein per utricle (estimated here at 2.4 ± 0.2 µg). This approach suggested bundle protein was 1.7% of the utricle's total protein. In the second method, we estimated the areas taken up by bundles and cell bodies in images of plastic sections of fixed, osmium-stained utricles. Using Fiji (http://fiji.sc) to measure regions of interest, this method estimated that bundles make up 1.8 ± 0.6% of the total protein in the utricle. Given the uncertainties in each, the methods suggested that bundles make up ~2% of the total protein in the utricle.
Separation of proteins by a short SDS-PAGE run prior to reduction, alkylation, and trypsin digestion substantially increased sensitivity and reproducibility of detection compared to other methods31, in part because we could remove interfering polymers from the agarose used for bundle isolation. NuPAGE LDS sample buffer (Invitrogen) with 50 mM dithiothreitol was added to a combined final volume of 80 µl per 100 utricles' worth of bundles; samples were heated to 70°C for 15 min. Epithelial proteins were also solubilized with NuPAGE LDS sample buffer. Proteins were separated by running ~1 cm into a NuPAGE 4–12% Bis-Tris gel (1.5 mm × 10 well; one or two lanes per bundle sample); gels were rinsed with water, then stained with Imperial Protein Stain (Thermo Scientific). The 1 cm of gel with separated proteins was manually sliced into six pieces.
Gel pieces were transferred to siliconized tubes and washed and destained in 200 µl 50% methanol overnight. The gel pieces were dehydrated in acetonitrile, rehydrated in 30 µl of 10 mM dithiothreitol in 0.1 M ammonium bicarbonate and reduced at room temperature for 0.5 h. The DTT solution was removed and the sample alkylated at room temperature for 0.5 h with 30 µl of 50 mM iodoacetamide in 0.1 M ammonium bicarbonate. The reagent was removed and the gel pieces dehydrated in 100 µl acetonitrile. The acetonitrile was removed and the gel pieces rehydrated in 100 µl of 0.1 M ammonium bicarbonate. The pieces were dehydrated in 100 µl acetonitrile, the acetonitrile removed and the pieces completely dried by vacuum centrifugation. The gel pieces were rehydrated in 20 ng/µl trypsin (Sigma-Aldrich T6567 proteomics grade, from porcine pancreas, dimethylated) in 50 mM ammonium bicarbonate on ice for 10 min. Any excess enzyme solution was removed and 20 µl of 50 mM ammonium bicarbonate added. The sample was digested overnight at 37°C and the peptides formed extracted from the polyacrylamide in two 30 µl aliquots of 50% acetonitrile/5% formic acid. These extracts were combined and evaporated to 15 µl for MS analysis. For the gel slice immediately adjacent to the agarose in the sample well, peptides were first purified away from interfering polymers on a SCX CapTrap from Bruker-Michrom (TR1/25109/35; size 0.5 × 2 mm, bed volume 0.5 µl). The CapTrap was washed with 50 µl of 1% acetic acid (void volume collected) and then eluted with 25 µl 1 M ammonium acetate into an separate Eppendorf tube. The eluate was vacuum dried, then the sample then reconstituted with 15 µl of 3% acetic acid. A single experiment's worth of hair bundles or epithelium was analyzed by six LC-MS/MS runs, corresponding to the six gel pieces.
Mass spectrometry data acquisition and analysis
The LC-MS/MS system consisted of a Thermo Electron Orbitrap Velos ETD mass spectrometer system; the exceptional mass accuracy of the Orbitrap instrument allows for high resolution of peptide peak m/z (mass-to-charge ratio), which leads to increased numbers of confident protein assignments. Peptides were introduced into the mass spectrometer with a Protana nanospray ion source, which was interfaced to a reversed-phase capillary column of 8 cm length × 75 µm internal diameter, self-packed with Phenomenex C18 Jupiter of 10 µm particle size. An extract aliquot (7.5 µl) was injected and peptides eluted from the column by an acetonitrile/0.1 M acetic acid gradient at a flow rate of 0.5 µl/min over 1.2 hr. The nanospray ion source was operated at 2.5 kV. The digest was analyzed using the data-dependent capability of the instrument, acquiring—in sequential scans—a single full scan mass spectrum in the Orbitrap detector at 60,000 resolution to determine peptide molecular weights, and 20 product-ion spectra in the ion trap to determine amino acid sequence.
MaxQuant version 1.2.2.5 software was used for protein identification and quantitation10. The default contaminants file associated with the MaxQuant download was edited to remove entries known to be present in hair bundles (e.g., actin) and to add additional impurities that entered the bundle-purification workflow (keratins, hemoglobins, egg white components). Nevertheless, alpha and beta hemoglobins, which appear in the preparation due to contamination from red blood cells4, are expressed in chick utricle20, suggesting that they should not be fully dismissed as components of hair cells. Mass spectrometry data were searched against Ensembl version 66 (released February, 2012) using Andromeda9; the Ensembl FASTA file was edited by replacing several sequences with longer or full-length sequences, including actin gamma 1 (NP_001007825.1), actin beta (NP_990849.1), fascin 1 (NP_001171603), fascin 2 (NP_001171209), ATP synthase beta (NP_001026562.1), peptidylprolyl isomerase A (NP_001159798.1), calbindin 2 (NP_990647.1), PDZD7 (XP_003641537.1), espin (XP_417532.3), and CACNA2D2 (XP_427707.3).
Protein identifications were reported with an FDR of 1%. Proteins identified with a single unique peptide are flagged in Supplementary Table 1. If a set of peptides for a protein was identical to or completely contained within that of another protein, MaxQuant groups those proteins together ("redundant groups"); the entry with the largest number of peptides was used to identify the redundant group. Redundant groups that shared more than 20% of their identified peptides were further grouped in our analysis ("shared-peptide groups"); the entry with the greatest intensity associated with it was used to identify the shared-peptide group. All mass spectrometry proteomics data, including raw data, MaxQuant output files, and modified Ensembl FASTA database, have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository51 with the dataset identifier PXD000104.
Annotation of the chicken genome is incomplete and occasionally wrong. For all proteins identified in the BUN preparation, we manually examined annotations of the chicken Ensembl entry and Ensembl-identified orthologs of other species, particularly mouse and human, to determine an appropriate description and symbol. Whenever possible, we chose the human ortholog's gene name—as determined by the Human Genome Organization—for a protein's symbol.
Gene Ontology annotation of the chicken genome is poor and, in many cases, was not useful for annotation of bundle proteins. Accordingly, we chose a simple, consistent set of ontology annotations to apply to all bundle proteins (see Fig. 2b). All proteins in the BUN preparation had one (and only one) of these ontologies assigned to it (Supplementary Table 1).
Paralog identification
Using data downloaded during 10/2012 from the Chicken Ensembl database (http://www.ensembl.org/Gallus_gallus/Info/Index) BioMart tool, we also identified all paralogs in the chicken genome for each protein entry or group, calculating the average sequence identity for all paralogs matching to a protein or protein group. We also determined which paralogs for a protein or protein group were identified in the combined BUN and UTR datasets, as well as which were present in the group of all proteins that were significantly enriched over the contamination level. These data are reported in Supplementary Table 1.
Intensity-based mass-spectrometric quantitation
To quantify proteins, we used a label-free method that relies each detected peptide's ion-current signal in the mass spectrometer. As peptides elute from the liquid chromatograph, undergo ionization, and are delivered to the mass spectrometer, the Orbitrap instrument we used measures their intensities, as well as their mass-to-charge ratio with high-resolution. Intensity depends both on the charge and amount of peptide delivered to the detector, although the efficiency of delivery varies widely from peptide to peptide because of variable recovery following liquid chromatography and differing degrees of ionization. Thus the relationship between intensity measured in the mass spectrometer and the amount of a peptide injected on to the liquid chromatograph also varies, which limits quantitation accuracy when standards for each protein are lacking. For example, hydrophobic peptides derived from transmembrane segments of integral membrane proteins are particularly poorly recovered, leading to reduced detection of this class of proteins52. Moreover, mass spectrometers like the one we used are tuned to optimally detect peptides in a relatively narrow mass range, typically 6–30 amino acids, so proteins with an overabundance of short or long tryptic peptides are quantified less accurately than an average peptide. Nevertheless, measurement of protein abundance uses the sum of many peptide measurements, so with larger proteins—which generate many peptides—inter-peptide variability is averaged out and quantitation accuracy is improved.
In the iBAQ algorithm11, the intensities of the precursor peptides that map to each protein are summed together and divided by the number of theoretically observable peptides, which is considered to be all tryptic peptides between 6 and 30 amino acids in length. This operation converts a measure that is proportional to mass (intensity) into one that is proportional to molar amount (iBAQ). The release of the MaxQuant10 we used (version 1.2.2.5) reports for each identified protein both its summed intensity and its iBAQ value.
To determine absolute amounts of each protein in stereocilia, we generated a normalized measure of molar abundance (relative iBAQ). We first removed from the analysis all contaminant proteins that entered our sample-preparation workflow, including include keratins (from human skin), hemoglobins (from blood), egg white proteins (e.g., ovalbumin), and trypsin. We then divided each remaining protein's iBAQ value by the sum of all non-contaminant iBAQ values, generating an riBAQ value for each protein:
| (1) |
For iBAQ validation experiments, we spiked 1/5 of a vial of UPS2 standard proteins (Universal Proteomics Standard 2; Sigma-Aldrich) into 25 µg (~500 pmol total) of the E. coli extract. Each experiment thus included 48 human proteins, eight each at 10 pmol, 1 pmol, 100 fmol, 10 fmol, 1 fmol, and 100 amol. We carried out four independent experiments using methods identical to those for hair bundles and utricular epithelia, including a 1-cm SDS-PAGE separation that was followed by slicing the gel into six pieces; reduction, alkylation, and trypsin proteolysis; LC-MS/MS using the Orbitrap mass spectrometer; and MaxQuant analysis with riBAQ determination.
For quantitative immunoblot validation of riBAQ values, BUN and UTR samples were loaded into 10- or 15-well Novex NuPAGE Bis-Tris 4–12% gels, along with dilutions of purified protein standards, and separated by SDS-PAGE. Protein standards were: CALM (bovine brain; EMD Millipore), ANXA5 (recombinant, from chicken; ImmunoTools GmbH), MYO1C (recombinant, from rat; gift of Lynne Coluccio, Boston Biomedical Research Institute), and FSCN2 (recombinant, from human; USCN Life Sciences). Five dilutions between 1 and 200 ng were used for CALM and ANXA5 standard curves, four dilutions between 0.01 and 1 ng were used for MYO1C, and four dilutions between 1.25 and 10 ng were used for the FSCN2. SDS-PAGE and immunoblotting were carried out essentially as described previously4,31. Band intensities were measured using Fiji imaging software and linear regression analysis was carried to determine the amount of each protein (ng/ear) within the bundle and epithelium samples. Three experiments were carried out for each protein. To estimate the mole fraction of each protein, the estimate of ng/ear was converted to mol/ear, then was divided by the number of moles of total protein per ear. We estimated moles of protein per ear by dividing the amount of protein per ear's worth of bundles or epithelium, then dividing by the average molecular mass for all proteins in each sample, which was weighted by the mole fraction estimated by mass spectrometry (46 kD for bundles, 56 kD for epithelium).
Statistical analysis
Fisher's exact test was used to determine the significance of the increased numbers of deafness proteins in the hair bundle samples. Permutation tests were employed to test whether the mean values of log2(BUN riBAQ/UTR riBAQ) were significantly greater than the corresponding contamination level of log2(0.20). Exact p-values were computed and adjusted for multiple test corrections by the false discovery rate17. All computations were done by using the exactRankTests package in the R statistical computing environment.
Electron microscopy sample preparation and tomography
We used high-pressure freezing and freeze substitution53 to optimally preserve osmotically sensitive samples. Chick utricle epithelia were dissected in chick saline (155 mM NaCl, 5 mM KCl, 5 mM D-glucose, and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)) containing 0.1 mM CaCl2. The tissue was fixed for 2 hr at room temperature in 3% glutaraldehyde (Electron Microscopy Sciences), 0.2% tannic acid, 100 mM KCl, 5 mM MgCl2, and 20 mM 3-(N-morpholino) propanesulfonic acid (MOPS) at pH 6.8, then washed 3× for 5 min each at room temperature in the same solution without glutaraldehyde and tannic acid.
Samples were stained with 1% OsO4 in 0.1 M sodium phosphate buffer for 1 hr on ice, followed by three rinses with 0.1 M sodium phosphate buffer and three additional rinses with deionized water. Subsequent staining was carried out with 2% aqueous uranyl acetate at room temperature for 1 hr, followed immediately by three rinses with deionized water. Samples were coated with a 20% glycerol cryoprotectant and high-pressure frozen using a Bal-Tec HPM 010 high pressure freezer. Thereafter, freeze substitution was performed using a freeze substitution mix containing 0.1% uranyl acetate in 100% acetone. Epon-araldite resin embedding and infiltration was carried out as described22. Sections were cut at 70 or 120 nm for screening or for tomography, respectively. Thin sections, nominally 120 nm in thickness, were placed on 2 × 1 mm oval hole copper/rhodium grids with 0.6% Formvar coating for imaging and decorated with 10 nm or 15 nm gold fiducial markers for tomography. For additional stability upon beam exposure and to minimize charging, a thin film of carbon was deposited on grids containing sections using a Denton Vacuum system (DV-502). Initial screening and tomography was primarily done with JEOL1200-EX (TEM only), Philips CM200 FEG, and FEI Tecnai F20 electron microscopes, whereas analyzed tilt series were collected using an FEI Tecnai T12 LaB6 electron microscope operated at 120 kV.
Images were recorded with a Gatan MegaScan Model 794/20 2K CCD (JEOL1200-EX), a Gatan First Light 4K CCD (CM200), a Tietz F415 4K CDD (F20), or a Gatan 1000 2048 × 2048-pixel CCD camera (T12). Tomograms for quantification were collected using ~0.8 µm underfocus at a nominal 13,500× magnification, corresponding to a pixel size of 0.8 nm at the specimen level. Dual-axis tilt series were recorded at 1° intervals for angles of up to ±65°. Projections were aligned and reconstructed into a three-dimensional volume with the software package IMOD54. Resulting maps were processed either with three iterations of a bilateral filter using PRIISM55 or with successive rounds of smoothing, diffusion, or median filtering using the Clip program in IMOD. Segmentation and interactive simplified model building was done using UCSF Chimera56. The simplified model displaying actin filaments, crosslinkers, and connectors deliberately used cylinders smaller in diameter than their respective structures to allow adequate display of the crosslinkers and connectors.
Stereociliary protein network
The protein interaction data in Supplementary Table 2 can be represented in the form of a graph G = ⎨N, L⎬ where the set of nodes N correspond to bundle proteins and links L correspond to specific protein interactions. Two vertices n and m form an edge of the graph if ⎨n, m⎬ ∈ L; because in our data, ⎨n, m⎬ ∈ L implies ⎨m, n⎬ ∈ L, the graph is undirected and is drawn with line segments rather than arrows. To visualize the interrelationships between these interactions, they are displayed in a drawing (a mathematically-defined "graph"). To represent the interactions aesthetically, link crossings were minimized and spacing between nodes was kept relatively even. We used a straight-edge drawing algorithm, spring-electrical embedding25, which minimizes the energy of a physical model of the graph in two dimensions. Spring-electrical embedding uses two forces. The "attractive force" fa = dij2 / K between adjacent nodes is proportional to the Euclidian distance between them (dij); K is a spring-like constant that maintains the optimal distance between nodes. The "electrical force" (repulsive) is global and inversely proportional to the Euclidian distance between nodes: fr = K2 / dij. The layout of the graph vertices is then determined by minimizing the energy function described by these two forces25. These modeling rules draw together nodes with similar interactions and disperse unrelated ones, thus clustering related proteins, which may carry out similar roles in bundles. The spring-electrical embedding algorithm is implemented in Mathematica 8 (Wolfram Research; http://www.wolfram.com/mathematica/), which we used to generate the network figure.
Antibody methods
Primary antibodies were: for ANXA5, polyclonal anti-chick ANXA5 from Guy Richardson (University of Sussex); for alpha-tubulin, Sigma-Aldrich DMA1; for ATP1A1, Developmental Studies Hybridoma Bank a5; for ATP2B2, F2a (ref. 50); for ATP5A1, BD Transduction 612516; for CALM, Millipore 05-173; for CDC42, Cell Signaling #2466 (11A11); for CDH23, C2367 goat anti-CDH23 EC15/16 junction (CATRPAPPDRERQ); for CKB, rabbit anti-chBCK from Theo Wallimann (ETH Zürich); for CTNNB1, BD Transduction Labs #610154; for DSTN, anti-ADF/cofilin from James Bamberg (Colorado State University); for ENO1, Santa Cruz H-300; for ESPN, anti-espin from Stefan Heller (Stanford University) and pan-espin from Bechara Kachar (NIH); for FSCN2, anti-CYTLEFKAGKLAFKD (ref. 31); for GAPDH, Chemicon MAB374; for HSPA5 (GRP78), Abcam ab32618; for KIAA1211, rabbit anti-CVSTEPAWLALAKRKAKAWSD; for MDH2, Sigma-Aldrich HPA019714; for MYO1C, antibody 2652 (ref. 57); for MYO1H, rabbit G5991 anti-chick MYO1H C-terminal 15 kD; for MYO3A, QHF anti-Xenopus laevis MYO3A C-terminal 22 amino acids (from Beth Burnside, University of California, Berkeley); for MYO3B, rabbit anti-GDWIRKPLYGLFQYNSSMIGLESLC; for MYO7A, 138-1 (Developmental Studies Hybridoma Bank); for PCDH15, G19 anti-tip link antigen58; for PRKAR2A, Santa Cruz M-20; for PTPRJ, mAb D37 (ref. 14); for RDX, Abnova #H00005962-M06 (1D9); pERM, Cell Signaling #3149 (41A3); RHOA, Cell Signaling #2117 (67B9); and for talin, Sigma-Aldrich 8D4. We raised polyclonal sera against a mixture of two chicken SLC9A3R2 peptides (CHSDLQSPGKESEDGDSEK and CQRHSHSFSSHSSRKDLNGQKE). Antibodies against MYO6 were obtained from John Kendrick-Jones (MRC Laboratory of Molecular Biology). We also used anti-SLC9A3R2 (anti-NHERF2) 2331B (from Mark Donowitz; Johns Hopkins University) for immunoblotting.
For immunoblotting, utricles were dissected from E20-E21 chick embryos in cold, oxygenated chicken saline containing 4 mM CaCl2, and ototlithic membranes were removed without protease treatment. SDS-PAGE and immunoblotting were carried out essentially as described previously4,31. Primary antibodies were used at 1:1000, except anti-CALM (1:500). All secondary antibodies were diluted 1:10,000.
For immunoprecipitation, RDX and SLC9A3R2 were cloned from chicken utricle cDNA into expression vectors with respectively Myc and HA epitope tags. Proteins were expressed for 24 hr in HEK293T cells using Effectene (Qiagen) transfection. Cells were lysed using a probe sonicator with PBS containing 1% Triton X-100, 0.5% NP40, and protease inhibitors; the extract was spun at 16,000 g for 15 min. The supernatant fluid was removed and incubated with anti-HA-agarose (Sigma) overnight at 4°C. Immunoprecipitates were washed and proteins were eluted with SDS sample buffer. SDS-PAGE and immunoblotting was carried out essentially as above.
For immunocytochemistry, dissected utricles were fixed for 25 min in 4% formaldehyde (Electron Microscopy Sciences) in chicken saline. Organs were rinsed in PBS, permeabilized for 10 min in 0.5% Triton X-100 in PBS, and blocked for 2 hr in 2% bovine serum albumin/5% normal goat serum in PBS. Organs were incubated overnight at 4°C with primary antibodies diluted in blocking solution (1:250 dilution for all primary antibodies except anti-SLC9A3R2 and anti-RDX, both at 1:500), then rinsed 3× for 10 min each. Organs were then incubated for 3–4 hr in blocking solution with 1:1000 Alexa Fluor secondary antibodies and 0.4 U/ml Alexa Fluor 488 Phalloidin (Molecular Probes, Invitrogen). Organs were then rinsed 3× for 20 min each and mounted on slides in Vectashield (Vector Laboratories) using one Secure-Seal spacer (8 wells, 0.12 mm deep, Invitrogen).
Images were acquired on an Olympus Fluoview FV1000 laser scanning confocal microscope system and AF10-ASW 3.0 acquisition software, using a 60× 1.42 NA Plan Apo objective with 3× zoom and 0.2 µm z-steps. Confocal images were deconvolved with the optical transfer function optimized for that objective using an iterative algorithm of 10 iterations. The histogram was adjusted for the most positive image and applied to all the other images for consistency before saving the images as 24 bit merged TIFF. All z-stacks were processed using Fiji (fiji.sc); the Reslice function was used to generate an x–z slab of the stack.
Supplementary Material
ACKNOWLEDGEMENTS
Mass spectrometry was carried out by the W.M. Keck Biomedical Mass Spectrometry Laboratory and The University of Virginia Biomedical Research Facility. We thank Kent McDonald, Reena Zalpuri, and Guangwei Min of the UC Berkeley Electron Microscopy Laboratory for assistance with high-pressure freezing and imaging; Danielle Jorgens provided mentoring in high-pressure freezing. We would like to thank Anchi Cheng, Bridget Carragher, and Clint Potter for help with EM data collection at the National Resource for Automated Molecular Microscopy (NRAMM), supported by NIH NCRR grant RR017573. For technical assistance, we acknowledge Aurelie Snyder, of the Advanced Light Microscopy Core (ALM) at The Jungers Center (OHSU), supported by shared instrumentation grants S10 RR023432 and S10 RR025440 from the National Center for Research Resources (NIH). Work described here was supported by NIH grants K99/R00 DC009412 (JBS), R01 DC002368 (PGG), R01 DC011034 (PGG), P30 DC005983 (PGG), R01 EY007755 (LLD), P30 EY10572 (LLD), and P01 GM051487 (MA).
Footnotes
DATABASE ACCESSION NUMBERS
The mass spectrometry data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD000104.
AUTHOR CONTRIBUTIONS
J.B.S. and P.G.G. designed the overall approach and analysis. J.F.K. carried out immunoblotting and immunocytochemistry experiments of Figs. 2 and 5, as well as Supplementary Figs. 2 and 4; she also carried out quantitative immunoblots of Fig. 1. J.B.S. and J.M.P. prepared hair-bundle and epithelium samples for mass spectrometric analysis. A.H., Z.M., A.N.T., and M.A. carried out electron tomography and analyzed tomography data. N.E.S. and E.D.J. carried out mass spectrometry experiments. K.J.S. carried out immunocytochemistry experiments of Supplementary Fig. 4. H.Z. carried out immunoprecipitation experiments of Fig. 5. D.C. carried out the statistical analyses. P.G.G. carried out mass spectrometry data analysis, with assistance from P.A.W. and L.L.D. The manuscript was written by P.G.G.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
MAIN-TEXT REFERENCES
- 1.Schwander M, Kachar B, Müller U. The cell biology of hearing. J Cell Biol. 2010;190:9–20. doi: 10.1083/jcb.201001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peng AW, Salles FT, Pan B, Ricci AJ. Integrating the biophysical and molecular mechanisms of auditory hair cell mechanotransduction. Nat Commun. 2011;2:523. doi: 10.1038/ncomms1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Domon B, Aebersold R. Options and considerations when selecting a quantitative proteomics strategy. Nat Biotechnol. 2010;28:710–721. doi: 10.1038/nbt.1661. [DOI] [PubMed] [Google Scholar]
- 4.Shin JB, et al. Hair bundles are specialized for ATP delivery via creatine kinase. Neuron. 2007;53:371–386. doi: 10.1016/j.neuron.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao H, Williams DE, Shin JB, Brugger B, Gillespie PG. Large membrane domains in hair bundles specify spatially constricted radixin activation. J Neurosci. 2012;32:4600–4609. doi: 10.1523/JNEUROSCI.6184-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donowitz M, et al. NHERF family and NHE3 regulation. J Physiol. 2005;567:3–11. doi: 10.1113/jphysiol.2005.090399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones SM, Jones TA. Ontogeny of vestibular compound action potentials in the domestic chicken. J Assoc Res Otolaryngol. 2000;1:232–242. doi: 10.1007/s101620010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillespie PG, Hudspeth AJ. High-purity isolation of bullfrog hair bundles and subcellular and topological localization of constituent proteins. J. Cell Biol. 1991;112:625–640. doi: 10.1083/jcb.112.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cox J, et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 10.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 11.Schwanhäusser B, et al. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka K, Smith CA. Structure of the chicken's inner ear: SEM and TEM study. Am J Anat. 1978;153:251–271. doi: 10.1002/aja.1001530206. [DOI] [PubMed] [Google Scholar]
- 13.Burnham JA, Stirling CE. Quantitative localization of Na-K pump sites in the frog sacculus. J Neurocytol. 1984;13:617–638. doi: 10.1007/BF01148082. [DOI] [PubMed] [Google Scholar]
- 14.Kruger RP, et al. The supporting-cell antigen: a receptor-like protein tyrosine phosphatase expressed in the sensory epithelia of the avian inner ear. J Neurosci. 1999;19:4815–4827. doi: 10.1523/JNEUROSCI.19-12-04815.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schulte BA, Adams JC. Immunohistochemical localization of vimentin in the gerbil inner ear. J Histochem Cytochem. 1989;37:1787–1797. doi: 10.1177/37.12.2685109. [DOI] [PubMed] [Google Scholar]
- 16.Grati M, et al. Localization of PDZD7 to the stereocilia ankle-link associates this scaffolding protein with the Usher syndrome protein network. J Neurosci. 2012;32:14288–14293. doi: 10.1523/JNEUROSCI.3071-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Statist. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 18.McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet. 2007;80:588–604. doi: 10.1086/514346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eppig JT, et al. The Mouse Genome Database (MGD): from genes to mice--a community resource for mouse biology. Nucleic Acids Res. 2005;33:D471–D475. doi: 10.1093/nar/gki113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spinelli KJ, et al. Distinct energy metabolism of auditory and vestibular sensory epithelia revealed by quantitative mass spectrometry using MS2 intensity. Proc Natl Acad Sci U S A. 2012;109:E268–E277. doi: 10.1073/pnas.1115866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steel KP. In: Genes and Common Diseases: Genetics in Modern Medicine. Wright AF, Hastie ND, editors. Cambridge: Cambridge University Press; 2007. pp. 505–515. [Google Scholar]
- 22.Auer M, et al. Three-dimensional architecture of hair-bundle linkages revealed by electron-microscopic tomography. J Assoc Res Otolaryngol. 2008;9:215–224. doi: 10.1007/s10162-008-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tilney LG, Tilney MS, DeRosier DJ. Actin filaments, stereocilia, and hair cells: how cells count and measure. Ann. Rev. Cell Biol. 1992;8:257–274. doi: 10.1146/annurev.cb.08.110192.001353. [DOI] [PubMed] [Google Scholar]
- 24.Ma'ayan A. Insights into the organization of biochemical regulatory networks using graph theory analyses. J Biol Chem. 2009;284:5451–5455. doi: 10.1074/jbc.R800056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fruchterman TMJ, Reingold EM. Graph drawing by force-directed placement. Software: Practice and Experience. 1991;21:1129–1164. [Google Scholar]
- 26.Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 27.Watts DJ, Strogatz SH. Collective dynamics of 'small-world' networks. Nature. 1998;393:440–442. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- 28.Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010;11:276–287. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasson T, et al. Unconventional myosins in inner-ear sensory epithelia. J. Cell Biol. 1997;137:1287–1307. doi: 10.1083/jcb.137.6.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tonikian R, et al. A specificity map for the PDZ domain family. PLoS Biol. 2008;6:e239. doi: 10.1371/journal.pbio.0060239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin JB, et al. The R109H variant of fascin-2, a developmentally regulated actin crosslinker in hair-cell stereocilia, underlies early-onset hearing loss of DBA/2J mice. J Neurosci. 2010;30:9683–9694. doi: 10.1523/JNEUROSCI.1541-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rzadzinska AK, Schneider ME, Davies C, Riordan GP, Kachar B. An actin molecular treadmill and myosins maintain stereocilia functional architecture and self-renewal. J. Cell Biol. 2004;164:887–897. doi: 10.1083/jcb.200310055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chhabra ES, Higgs HN. The many faces of actin: matching assembly factors with cellular structures. Nat Cell Biol. 2007;9:1110–1121. doi: 10.1038/ncb1007-1110. [DOI] [PubMed] [Google Scholar]
- 34.Higgs HN, Blanchoin L, Pollard TD. Influence of the C terminus of Wiskott-Aldrich syndrome protein (WASp) and the Arp2/3 complex on actin polymerization. Biochemistry. 1999;38:15212–15222. doi: 10.1021/bi991843+. [DOI] [PubMed] [Google Scholar]
- 35.Zhang DS, et al. Multi-isotope imaging mass spectrometry reveals slow protein turnover in hair-cell stereocilia. Nature. 2012;481:520–524. doi: 10.1038/nature10745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fischer RS, Fowler VM. Tropomodulins: life at the slow end. Trends Cell Biol. 2003;13:593–601. doi: 10.1016/j.tcb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 37.Rehman AU, et al. Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79. Am J Hum Genet. 2010;86:378–388. doi: 10.1016/j.ajhg.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Furness DN, Mahendrasingam S, Ohashi M, Fettiplace R, Hackney CM. The dimensions and composition of stereociliary rootlets in mammalian cochlear hair cells: comparison between high- and low-frequency cells and evidence for a connection to the lateral membrane. J Neurosci. 2008;28:6342–6353. doi: 10.1523/JNEUROSCI.1154-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakaguchi H, et al. Dynamic compartmentalization of protein tyrosine phosphatase receptor Q at the proximal end of stereocilia: implication of myosin VI-based transport. Cell Motil Cytoskeleton. 2008;65:528–538. doi: 10.1002/cm.20275. [DOI] [PubMed] [Google Scholar]
- 40.Kitajiri S, et al. Actin-bundling protein TRIOBP forms resilient rootlets of hair cell stereocilia essential for hearing. Cell. 2010;141:786–798. doi: 10.1016/j.cell.2010.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maul RS, et al. EPLIN regulates actin dynamics by cross-linking and stabilizing filaments. J Cell Biol. 2003;160:399–407. doi: 10.1083/jcb.200212057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yin HL, Janmey PA. Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol. 2003;65:761–789. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- 43.Heo WD, et al. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314:1458–1461. doi: 10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gagnon LH, et al. The chloride intracellular channel protein CLIC5 is expressed at high levels in hair cell stereocilia and is essential for normal inner ear function. J Neurosci. 2006;26:10188–10198. doi: 10.1523/JNEUROSCI.2166-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ponsioen B, et al. Spatiotemporal regulation of chloride intracellular channel protein CLIC4 by RhoA. Mol Biol Cell. 2009;20:4664–4672. doi: 10.1091/mbc.E09-06-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaw RJ, Henry M, Solomon F, Jacks T. RhoA-dependent phosphorylation and relocalization of ERM proteins into apical membrane/actin protrusions in fibroblasts. Mol Biol Cell. 1998;9:403–419. doi: 10.1091/mbc.9.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garbett D, LaLonde DP, Bretscher A. The scaffolding protein EBP50 regulates microvillar assembly in a phosphorylation-dependent manner. J Cell Biol. 2010;191:397–413. doi: 10.1083/jcb.201004115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garbett D, Bretscher A. PDZ interactions regulate rapid turnover of the scaffolding protein EBP50 in microvilli. J Cell Biol. 2012 doi: 10.1083/jcb.201204008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walker RG, Hudspeth AJ, Gillespie PG. Calmodulin and calmodulin-binding proteins in hair bundles. Proc. Natl. Acad. Sci. USA. 1993;90:2807–2811. doi: 10.1073/pnas.90.7.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dumont RA, et al. Plasma membrane Ca2+-ATPase isoform 2a is the PMCA of hair bundles. J. Neurosci. 2001;21:5066–5078. doi: 10.1523/JNEUROSCI.21-14-05066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
ONLINE-METHODS REFERENCES
- 51.Vizcaino JA, et al. The Proteomics Identifications database: 2010 update. Nucleic Acids Res. 2010;38:D736–D742. doi: 10.1093/nar/gkp964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barrera NP, Robinson CV. Advances in the mass spectrometry of membrane proteins: from individual proteins to intact complexes. Annu Rev Biochem. 2011;80:247–271. doi: 10.1146/annurev-biochem-062309-093307. [DOI] [PubMed] [Google Scholar]
- 53.Sosinsky GE, et al. The combination of chemical fixation procedures with high pressure freezing and freeze substitution preserves highly labile tissue ultrastructure for electron tomography applications. J Struct Biol. 2008;161:359–371. doi: 10.1016/j.jsb.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 55.Chen H, Clyborne WK, Sedat JW, Agard DA. Priism: an integrated system for display and analysis of 3-D microscope images. Proceedings of SPIE. 1992;1660:784–790. [Google Scholar]
- 56.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 57.Dumont RA, Zhao YD, Holt JR, Bahler M, Gillespie PG. Myosin-I isozymes in neonatal rodent auditory and vestibular epithelia. J. Assoc. Res. Otolaryngol. 2002;3:375–389. doi: 10.1007/s101620020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goodyear RJ, Richardson GP. A novel antigen sensitive to calcium chelation that is associated with the tip links and kinocilial links of sensory hair bundles. J Neurosci. 2003;23:4878–4887. doi: 10.1523/JNEUROSCI.23-12-04878.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.