

Abstract
N-Glycosylation is a common post-translational modification of monoclonal antibodies with a potential effect on the efficacy and safety of the drugs; detailed knowledge about this glycosylation is therefore crucial. We have developed a reversed-phase liquid chromatographic–mass spectrometric method, with different fluorescent labels, for analysis of N-glycosylation, and compared the sensitivity and selectivity of the methods. Our work demonstrates that anthranilic acid as fluorescent label in combination with reversed-phase liquid chromatography–mass spectrometry is an advantageous method for identification and quantification of neutral and acidic N-glycans. Our results show that mass spectrometry-based quantification correlates with quantification by fluorescence. Chromatographic discrimination between several structural glycan isomers was achieved. The sharp peaks of the eluting anthranilic acid-labeled N-glycans enabled on-line mass spectrometric analysis of even low-abundance glycan species. The method is broadly applicable to N-glycan analysis and is an orthogonal analytical method to the widely established hydrophilic-interaction liquid chromatography of 2-aminobenzamide-labeled N-glycans for characterization of N-glycans derived from biopharmaceuticals.
Keywords: Anthranilic acid, 2-aminobenzamide, Mass spectrometry, N-glycosylation, Reversed-phase chromatography, Monoclonal antibody
Introduction
Recombinant protein drugs belong to the most complex active pharmaceutical ingredients. Monoclonal antibodies (mAbs), glycoproteins with a molecular mass of approximately 150 kDa are one important class. Glycosylation has attracted interest because many investigations have shown that such modification may have an effect on the safety and efficacy of these therapeutic protein drugs [1–5]. In general, IgGs have one conserved N-glycosylation site on each heavy chain at their Fc part, usually at approximate position 297 of the heavy chain, and several mAbs carry a second N-glycosylation site in their variable region. The heterogeneity of the N-glycans attached to these sites is very high, far more than a dozen different glycans can be found [6]. Identification and quantification of the glycans in this mixture is difficult, and structural isomers of several glycans make discrimination even more challenging [7–9]. A comprehensive analytical approach is therefore required. N-Glycosylation can be studied, after enzymatic release of the glycans by use of peptide N-glycosidase F (PNGaseF), by liquid chromatography, mass spectrometry (MS) [10], or a combination of these [9–14]. Structural analysis of underivatized or labeled N-glycans is usually performed by MALDI-MS or by use of porous graphitized carbon (PGC) liquid chromatography in combination with on-line ESI MS2 [15–17].
Underivatized N-glycans have no light-absorbing properties. They can be derivatized with a fluorophore [18] for quantitative analysis by HPLC. Labeling with a fluorescent dye via reductive amination is widely used [12, 18]. This derivatization results in high sensitivity and, because every N-glycan carries only one label, irrespective of size or branching, quantitative information can be obtained from the intensity of the fluorescence signal. The tag not only enables UV or fluorescence detection—it also improves ionization and fragmentation of the labeled N-glycan in ESI–MS [19, 20].
For characterization of N-glycans derived from biopharmaceuticals, 2-aminobenzamide (2-AB) is routinely used as label [19, 21, 22], typically with separation and quantification by HILIC (hydrophilic interaction liquid chromatography) with fluorescence detection [23]. HILIC results in high resolution and selectivity for many glycan isomers [24]. However, the small injection volume of aqueous samples necessary, solvent conditions that are essential because of the solubility of the glycans, leads to reduced sensitivity in on-line MS detection, and the buffered mobile phase may lead to ion suppression.
In contrast with this, the mobile phases used for RP HPLC usually consist only of water, an organic solvent, and an acid; they are, therefore, highly MS-compatible. In addition, there are almost no limitations with regard to injection volumes of aqueous solutions, which results in high sensitivity. It has been shown that RP LC–MS of 2-AB or ANTS-derivatized oligosaccharides can be performed and used to characterize the N-glycosylation pattern of mAbs [13, 14, 25, 26]. However, 2-AB as fluorescent tag adds only weak hydrophobicity to the hydrophilic glycan, which necessitates use of a shallow and long chromatography gradient. Furthermore, labeled acidic glycans elute early and are poorly separated [13]. This limitation was solved by Melmer et al. [24] by addition of an ion-pairing reagent. However, use of an ion-pairing reagent again leads to ion suppression and thus reduced MS sensitivity. For analysis of negatively charged N-glycans containing sialic acids, labeling with a negatively charged tag, for example 2-aminobenzoic acid (2-AA) or 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) and MS detection in negative-ionization mode is frequently used [14, 27–29]. Prien et al. separated oligomannose structures (especially mannose 5 isomers) by RP LC after 2-AA derivatization and analyzed them by use of MS in negative mode [28]. The ability to separate complex N-glycan mixtures or isomers, for example G1F N-glycans with either 1,3 or 1,6 galactosylation, highly abundant in most mAbs, has not been reported for any N-glycan RP LC approach.
To provide solutions for the above mentioned limitations of current N-glycan analytical methods we have developed and optimized an RP LC–MS method with positive ionization for characterization of complex N-glycosylation profiles. The method was designed to characterize acidic and neutral N-glycans in a single LC–MS approach. We compared the sensitivity and selectivity of 2-AB and 2-AA as fluorescent labels and showed that our newly developed 2-AA RP LC–MS method has advantageous sensitivity and selectivity for a variety of structural isomers analysis of which has not yet been reported in the literature. By comparing results from quantitative MS and from FLD we also demonstrated that quantification of mAb glycans by these methods leads to very similar results. The method is versatile and can be used to address various questions in glycobiology or glycomics, from basic screening of N-glycan composition, because of grouping of the N-glycan types, to detailed analysis of low-abundance glycan species. Furthermore our results show that even complex and highly sialylated N-glycans can be investigated. This flexibility and versatility make the method broadly applicable to analysis of N-glycans and it can be applied with little effort to analysis of many (glyco)proteins, or in proteomics and/or glycomics laboratories, because the combination of RP LC and MS is a routine application in such laboratories.
Materials and methods
Materials
PNGase F was from New England Biolabs (Frankfurt am Main, Germany). Acetonitrile (ACN) and acetic acid were from Merck (Darmstadt, Germany). Formic acid, DMSO, and sodium cyanoborohydride were from Fluka (Sigma, Munich, Germany). Sephadex G-10 columns were custom made by GE Healthcare (Vienna, Austria). Amicon Ultra 30-K filter devices were from Millipore (Schwalbach, Germany). The mAb glycan standard was prepared at Sandoz. Monoclonal antibodies 1–3 were obtained from in-house development at Sandoz. The acidic N-glycan standards were from Thera Proteins (Barcarena, Portugal).
Methods
Enzymatic N-glycan release by use of PNGaseF
Desalted mAb (1 mg) was used. The N-glycans (15 nmol) were released by incubating the samples with PNGaseF overnight (~17 h) at 37 °C. The N-glycans were separated from the proteins by use of Amicon 30K filter devices and were brought to dryness by use of a Speedvac.
Fluorescence labeling of released N-glycans or N-glycan standards
Na[BH3(CN)] and either 2-AA or 2-AB were dissolved in 70:30 (%, v/v) DMSO–acetic acid to furnish concentrations of 63 and 50 mg mL−1, respectively. Labeling solution (15 μL) and deionized water (10 μL) were added either to 15 nmol enzymatically released and dried glycans or to 250 pmol lyophilized N-glycan standard. The labeling reaction was performed at 37 °C for 17 h.
Excess label was removed by gel filtration on G-10 columns. Columns were conditioned with 10 mL H2O. Samples were diluted to 100 μL with deionized water then applied to the column. After rinsing the column with 700 μL H2O the purified fluorescence-labeled N-glycans were eluted with 600 μL H2O.
Reversed-phase HPLC of labeled N-glycans
Liquid chromatography was performed with an Agilent 1200 series chromatograph on a Waters Acquity UPLC BEH130 C18 (2.1 mm × 150 mm, 1.7-μm particle) column.
Analysis of 2-AA-labeled glycans was performed with a gradient prepared from 1.0 % formic acid in H2O (component A) and 50 % ACN in 1.0 % formic acid in H2O (component B). The column was equilibrated with 4 % B. After injection of up to 100 μL sample the mobile phase composition was held at 4 % B for 2 min. The proportion of B was then raised in four steps to 28 %, first to 10 % over 27 min, then to 11.5 % over 10 min, then to 14 % over 8 min, and finally to 28 % over 19 min. The column was regenerated by increasing to 90 % B over 4 min, followed by isocratic elution for 2 min. The column was then re-equilibrated at 8 % B for 5 min. Oven temperature was 50 °C and the flow-rate was 0.30 mL min−1. Fluorescence detection was performed with an excitation wavelength of 250 nm and an emission wavelength of 425 nm.
Analysis of 2-AB-labeled glycans was performed with a gradient prepared from 0.5 % formic acid in H2O (component A) and 0.5 % formic acid and 5 % ACN in H2O (component B). The column was equilibrated with 25 % B. After injection the mobile phase was held at 25 % B for 2 min. The proportion of B was then increased to 55 % over 60 min and then to 61 % over 24 min. This composition was held for 2 min then the initial conditions were restored in 2 min and held for an additional 5 min. Oven temperature was 40 °C and the flow rate was 0.3 mL min−1. Fluorescence detection was performed with an excitation wavelength of 250 nm and an emission wavelength of 428 nm.
Mass spectrometry
The HPLC was directly coupled to a 3D ion trap ESI–MS (Bruker AmaZon). The ion trap was operated in enhanced resolution mode with a capillary potential of 4 kV. The capillary temperature was set to 250 °C with a nebulizer pressure of 2 bar and a dry gas flow of 6 L min−1. MS2 spectra were generated by use of the Auto MS2 mode and collision induced dissociation (CID).
Results and discussion
LC–MS of 2-AB-labeled N-glycans
Our RP LC–MS method entails use of two structurally closely similar chemical labels, 2-AB and 2-AA. In a first step an RP LC–MS method for 2-AB-labeled N-glycans was developed. Because 2-AB-labeled N-glycans are weakly retained on C18 columns, a mobile phase gradient with a low organic solvent content was used. The separation was optimum under the mobile phase conditions described in the section “Reversed-phase HPLC of labeled N-glycans”. A fluorescence chromatogram obtained from the 2-AB RP LC–MS method for the mAb glycan standard is shown in Fig. 1. The glycans elute in groups. To reduce the run time the mobile phase composition was adapted. Use of formic acid instead of acetic acid improved retention and led to sharper peaks. A run time of 95 min was sufficient for analysis of the N-glycans of an mAb. A shorter gradient and the resulting reduction of run time led to loss of resolution, because of the low hydrophobicity of the labeled N-glycans.
Fig. 1.

(A) Fluorescence chromatogram obtained from separation of 2-AB N-glycans on a RP column. The magnified view (B) of the chromatogram shows the smaller peaks of less abundant N-glycans. The numbered peaks were identified by use of MS and MS2. Stacked numbering indicates co-elution of N-glycans. MS data of the identified peaks are listed in Table 1. The 2-AB glycans elute in different groups depending on their type. Oligomannose (green) glycans elute first, followed by acidic hybrid and complex type (pink) and neutral hybrid glycans (orange). Acidic hybrid with core fucose (pink) elute after complex glycans (blue). Neutral hybrid structures (orange) co-elute between approximately 57 and 86 min, with acidic complexes followed by complex 2-AB glycans (blue), with all three groups carrying a core fucose
The first compounds eluting between 16 and 30 min from the column are the high-mannose glycans, in the order high to low number of mannose residues (Fig. 1, green). The acidic hybrid and complex glycans (Fig. 1, pink) overlap with the oligomannose group from approximately 22–26 min. The next glycans to elute are the hybrid N-glycans lacking the core fucose at the terminal GlcNAc (Fig. 1, orange), eluting from 28–36 min. The complex bi-antennary 2-AB glycans elute in the middle of the chromatogram (Fig. 1, blue, 42–48 min) immediately before the acidic hybrid glycans with core fucose. Hybrid (Fig. 1, orange, 57–71 min) and acidic complex (Fig. 1, pink, 48–66 min) 2-AB glycans, both groups with a fucose residue attached to their core, co-elute. The group with the most abundant glycans in most mABs, the complex type glycans with core fucose, elute at the end of the chromatogram with retention times of 74–88 min. Sialic acid-containing 2-AB-labeled glycans elute as sharp peaks before their corresponding neutral glycans Fig. 2.
Fig. 2.

RP chromatogram obtained from the mAb glycan standard showing the grouping of the eluting 2-AA N-glycans. (A) High-mannose structures elute first (green), followed by non-fucosylated hybrid (orange) and complex glycans (blue). Fucosylated hybrid (orange) and complex structures (blue) elute last in the chromatogram. Acidic glycans elute immediately before their appropriate neutral glycans (pink). (B) Magnified view of the region between 18 and 42 min showing the less abundant glycans. The identified glycans are numbered and the appropriate masses are listed in Table 1. Stacked numbering indicates co-elution of N-glycans. The glycan structures are depicted in Fig. 3
Labeled oligomannose and hybrid structures elute from high to low number of monosaccharides whereas complex type 2-AB glycans, including the acidic variants, elute from low to high number of monosaccharide units. Several 2-AB glycans elute with similar retention times and could not be separated. The 2-AB-labeled glycans were identified by MS and MS2 by use of the ion-trap mass spectrometer. Table 1 contains the MS data for the identified glycans, and the respective glycan structures are drawn in Fig. 3, with the appropriate nomenclature.
Table 1.
2-AA and 2-AB-labeled glycans from mAb glycan standard identified by ion-trap MS and MS2. Observed and theoretical mass are shown for each assigned glycan. For N-glycan isomers only one mass is shown. The peak numbers correspond to the appropriate peak numbering in the chromatograms and to the structures in Fig. 3
| Peak | 2-AA Glycans | 2-AB Glycans | N-Glycan | ||
|---|---|---|---|---|---|
| Observed mass | Theoretical mass | Observed mass | Theoretical mass | ||
| 5 | 1841.602 | 1841.645 | 1840.689 | 1840.661 | M8 |
| 6, 7, 8, 9 | 1679.541 | 1679.592 | 1678.604 | 1678.608 | M7 |
| 10 | 1031.406 | 1031.381 | 1030.425 | 1030.397 | M3 |
| 11 | 1234.470 | 1234.460 | 1233.489 | 1233.476 | M3G0 |
| 12 | 2027.716 | 2027.709 | 2026.739 | 2026.725 | SM5G1 |
| 13, 14 | 1517.592 | 1517.539 | 1516.573 | 1516.555 | M6 |
| 15 | 1396.550 | 1396.513 | 1395.54 | 1395.529 | M3G1 |
| 16 | 1855.655 | 1865.656 | 1865.678 | 1864.672 | SM4G1 |
| 17, 18, 19 | 1703.619 | 1703.603 | 1702.615 | 1702.619 | SM3G1 |
| 20, 21 | 1193.457 | 1193.433 | 1192.49 | 1192.449 | M4 |
| 22 | 1355.525 | 1355.486 | 1354.533 | 1354.502 | M5 |
| 23, 24 | 1558.617 | 1558.566 | 1557.582 | 1557.582 | M5G0 |
| 25 | 1720.647 | 1720.618 | 1719.643 | 1719.634 | M5G1 |
| 26, 27, 28, 29 | 1599.618 | 1599.592 | 1598.612 | 1598.608 | G1 |
| 30 | 1720.647 | 1720.618 | 1719.643 | 1719.634 | M6G0 |
| 31 | 1558.592 | 1558.566 | 1557.576 | 1557.582 | M4G1 |
| 32, 33 | 1437.586 | 1437.539 | 1436.565 | 1436.555 | G0 |
| 34 | 1687.579 | 1687.608 | – | 1686.624 | SM3G1 |
| 35, 36 | 1786.675 | 1786.677 | 1785.708 | 1785.693 | G0F + GN |
| 37 | 1583.710 | 1583.597 | 1582.648 | 1582.613 | G0F |
| 38 | 2173.726 | 2173.767 | 2172.78 | 2172.783 | SM5G1F |
| 39 | 2011.693 | 2011.714 | 2010.754 | 2010.73 | SM4G1F |
| 40 | 2376.763 | 2376.846 | 2375.787 | 2375.862 | SG3F |
| 41 | 1849.705 | 1849.661 | 1848.692 | 1848.677 | SM3G1F |
| 42, 43 | 2214.754 | 2214.793 | 2213.818 | 2213.809 | SG2F |
| 44, 45 | 2052.732 | 2052.740 | 2051.768 | 2051.756 | SG1F |
| 46 | 1948.741 | 1948.729 | 1947.804 | 1947.745 | G1F + NG |
| 47 | 2028.702 | 2028.729 | – | 2027.745 | M6G1F |
| 48, 49 | 1907.689 | 1907.703 | 1906.775 | 1906.719 | G2F |
| 50 | 1866.679 | 1866.676 | 1865.698 | 1865.692 | M5G1F |
| 51, 52 | 1704.609 | 1704.624 | 1703.644 | 1703.64 | M5G0F |
| 53 | 1866.650 | 1866.676 | 1865.668 | 1865.692 | M6G0F |
| 54 | 1583.533 | 1583.597 | 1582.608 | 1582.613 | M3G0F + NG |
| 55 | 1704.609 | 1704.624 | 1703.643 | 1703.64 | M4G1F |
| 56, 57 | 1542.566 | 1542.571 | 1541.582 | 1541.587 | M3G1F |
| 58 | 2069.748 | 2069.756 | – | 2068.772 | G3F |
| 59 | 1380.553 | 1380.518 | 1379.569 | 1379.534 | M3G0F |
| 60 | 1907.690 | 1907.703 | 1906.746 | 1906.719 | G2F |
| 61, 62 | 1745.610 | 1745.650 | 1745.655 | 1744.666 | G1F |
| 63 | 1583.709 | 1583.597 | 1582.607 | 1582.613 | G0F |
Fig. 3.

Symbol structures of the identified N-glycans. Numbering is in accordance with the peak numbering for the N-glycans in the fluorescence chromatograms and in Table 1. For structures with multiple peak assignments only one possible isomer is drawn. Symbols: inverted red triangles, l-fucose; blue squares, N-acetyl-d-glucosamine; green circles, d-mannose; yellow circles, d-galactose; blue diamonds, N-glycoylneuraminic acid; purple diamonds, N-acetyl-neuraminic acid
Separation of 2-AA-labeled N-glycans
As an alternative to 2-AB-labeled glycans, 2-AA-labeled glycans were also tested using the developed 2-AB method. Most 2-AA glycans remained on the column after the 95-min gradient, only the oligomannose structures eluted late. Because of this stronger retention of the 2-AA-labeled glycans the mobile phase had to be adapted to enable optimum separation. In contrast with 2-AB, 2-AA is negatively charged at neutral pH. Mobile phase pH was therefore reduced to provide sufficient protons for efficient ionization in positive MS mode. Use of 1 % formic acid and an ACN content of component B increased by a factor of 10 compared with the 2-AB method, to 50 %, enabled optimum separation and good ionization. Higher signal intensity was observed with positive-ionization MS compared with negative-ionization mode. The resulting run-time was 78 min. As observed for the 2-AB method a shorter gradient resulted in substantial loss of resolution.
Figure 2 shows a fluorescence chromatogram obtained from the 2-AA-labeled mAb glycan standard; it is similar to that from the 2-AB method (Fig. 1). The labeled glycans elute in different groups, highlighted with different colors in Fig. 2. High-mannose structures are the first group eluting from high number to small number of monosaccharide residues, for example from M9 to M5 (green, 19–27 min). Sialic acid containing non-fucosylated hybrid and complex variants (pink, 23–30 min) are then followed by the neutral non-fucosylated hybrid variants (orange, 28–31 min). Complex structures lacking the core fucose (blue, 30–34 min) elute before sialic acid-containing hybrid glycoforms and sialic acid-containing complex forms (pink, 34–40 min), both with core fucosylation. Bisecting structures co-elute with the latter. Neutral core-fucosylated hybrid glycans (orange, 39–42 min) elute before the most abundant core fucosylated complex variants in mAbs, the bi-antennary N-glycans (blue, 42–51 min). This grouping is similar to that of our 2-AB approach and to those of previous investigations [13]. Sialic acid-containing non-fucosylated hybrid and complex variants elute after the high-mannose and before the neutral non-fucosylated hybrid group. The same order is observed for the core fucosylated hybrid and complex glycoforms. Again, the same sharp peak shape was obtained for the eluting sialic acid-containing 2-AA glycans as for neutral glycans, without the need for an ion-pairing reagent. Compared with the 2-AB method, the peaks were sharper (e.g. the peak width of peak 63 is 0.72 min (2-AB) compared with 0.25 min (2-AA)) and more condensed. Because of the greater number of separated glycan structures more partially resolved peaks are present in the chromatogram.
Identification of N-glycans by use of positive mode ESI–MS and MS2
So far, 2-AA has been used almost exclusively with negative-ionization MS [19, 20], because of the negative charge of the acid group. The acidic mobile phase used in this investigation favors proton adducts and resulted in good ionization efficiency in positive-ionization mode. We observed higher signal intensities for positive ionization than for negative ionization. The high formic acid content of the mobile phase also led to formation of formic acid clusters. 2-AA N-glycans were identified from their mass derived from mass spectra and from the mass of appropriate fragments generated by CID. All 2-AB and 2-AA N-glycans identified in the mAb glycan standard, with their observed and calculated masses, are listed in Table 1. Some glycan structures occur several times in course of the chromatogram. These may be structural isomers or bisecting or tri-antennary variants with the same mass that cannot be distinguished because no linkage information is obtained by use of this LC–MS approach.
The charge states of the labeled glycans increase with increasing mass. Glycans with a mass < 1200 Da occur almost exclusively as [M + H]1+ ions, whereas glycans with mass >1200 Da are doubly charged [M + 2H]2+; singly charged [M + H]1+ ions, also, are present for glycans of mass <1500 Da. For masses >1800 Da, the N-glycans begin to ionize as triply charged [M + 3H]3+ ions, with the exception of oligomannose-type glycans which usually furnish doubly charged ions. Adduct ions are present for all charge states and the oligomannose glycans tend to have the highest affinity for adduct formation. Doubly and triply charged ions occur as mixed adducts also. [M + Na]1+, [M + K]1+, [M + H + Na]2+, [M + 2Na]2+, and [M + H + K]2+ are the most abundant adduct ions. On-line MS detection provides more information than is required for identification of N-glycans solely on the basis of their mass and fragments, because co-eluting 2-AA N-glycans can be identified and quantified by use of the extracted ion chromatograms (EICs) of the appropriate m/z values. Figures 4 and 5 show the MS2 spectra obtained after fragmentation of different N-glycoylneuraminic acid-containing 2-AA glycans. The fragments were labeled in accordance with the nomenclature of Domon and Costello [30]. The spectra were acquired on-line by use of positive ionization ESI–MS and CID fragmentation. Each of the SG1F 2-AA glycan fragments (Fig. 4) can be explained by the dissociation of a single bond, as expected from use of CID in ion-traps. The MS2 spectrum at m/z 1027.9 is dominated by B and Y ions. The fragmentation pattern of the sialic acid-containing SM5G1F (Fig. 5) is slightly different. In the MS2 spectrum of the [M + 2H]2+ ion at m/z 1088.4 only B ions are observed derived from the GlcNAc-containing branch, but not from the mannose containing-branch of the hybrid structure. The fragments B3α and Y6α with m/z 528 and 366, respectively, can be explained by loss of the terminal sialylation. Figure 6 shows the MS2 spectrum at m/z 1035.9 of the rare G3F glycan, which accounts for less than 0.01 % of the glycans. B ions are observed for both branches of the glycan, because both contain a GlcNAc. This 2-AA glycan co-elutes with the two overlapping and more abundant M3G0F and G2F peaks, but it can be identified and quantified by use of on-line MS detection. To check the performance of the method for more complex sialic acid glycans we labeled and analyzed six acidic glycan standards (Fig. 8 and also the section “Selectivity of the two approaches”). The bi-antennary glycans ionized as described above. For the tri and the tetra-antennary glycans we observed mainly [M + 3H]3+ and [M + 4H]4+ ions. Loss of terminal sialic acids was minimal and we observed almost no in-source fragmentation. Only loss of an antenna was monitored for the tetra-antennary N-glycan.
Fig. 4.

MS2 spectrum of the 2-AA-labeled SG1F glycan from mAb3 at m/z 1027.9. The dissociated bonds of the [M + 2H]2+ ion are depicted and the assigned B and Y ions are labeled in the spectrum. Dissociation of two bonds is indicated by a slash
Fig. 5.

MS2 spectrum of the 2-AA-labeled SM5G1F glycan from mAb3 at m/z 1088.4. Singly charged B and Y ions resulting from the [M + 2H]2+ ion are shown. B ions were exclusively from the α-branch containing a GlcNAc that is able to carry a charge. Dissociation of two bonds is indicated by a slash
Fig. 6.

MS2 spectrum of the 2-AA-labeled G3F N-glycan from mAb3. The dissociated bonds of the [M + 2H]2+ ion are depicted and the assigned B and Y ions are labeled in the spectrum. The glycan accounts for <0.01 % of the glycans of mAb3. Dissociation of two bonds is indicated by a slash
Fig. 8.
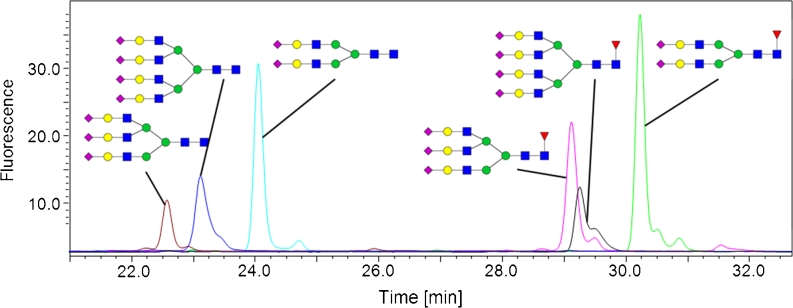
Overlay of fluorescence chromatograms derived from six different 2-AA-labeled sialic N-glycan standards. The appropriate structures are depicted. The grouping into non-fucosylated (three peaks on the left) and fucosylated (three peaks on the right) glycans is obvious
Selectivity of the two approaches
As mentioned in the “Introduction”, the complexity of glycosylation is not only because of the multitude of different N-glycan variants with different monosaccharide composition. It is also because of the existence of structural isomers with different linkage types. To obtain a glycan-map as comprehensive as possible it is important to separate these isomers. Separation of oligomannose isomers, for example, has been investigated and is described in several publications [6, 9, 27].
Figure 7 shows the EIC of the M7 isomers of mAb2 for the 2-AB (Fig. 7A) and 2-AA (Fig. 7B)-labeled glycans. Four different isomers are observed for this high-mannose glycan. The linkage could not be deduced with the reducing end derivatization used. The selectivity of the two methods is identical for the high-mannose structures; by comparison of the areas of peaks 3 and 4, however, we deduced that the order of elution might have changed because the ratio of the peak areas was inverted for the two labels (approx. 1.6 for 2-AA peaks 3:4 and approx. 1.6 for 2-AB peaks 4:3).
Fig. 7.

EICs of four structural M7 isomers (A, B) and G1F isomers (C, D) from mAb2. (A) EIC of 2-AB-labeled N-glycans. (B) EIC for the 2-AA-labeled glycans. Although selectivity is identical for the M7 isomers in both approaches, comparison of the peak areas reveals the order of elution changed for peaks 3 and 4. Selectivity is different for the G1F isomers. The 2-AB-labeled G1F elutes as one peak (C) whereas the 2-AA-labeled glycans (D) are separated into the two isomers. The terminal galactose residue (E) can be linked either to the α1,6 or the α1,3 branch of the bi-antennary N-glycan
G1F isomers are usually highly abundant structural isomers on mAbs and IgG (Fig. 7E) with the terminal galactose residue at the α1,3 or α1,6 branch [6]. So far, separation of these two isomers has been achieved by use of HILIC and porous graphitized carbon liquid chromatography only [24]. In Fig. 7 it is clearly apparent that the combination of 2-AA as label and the RP chromatography conditions chosen is capable of separating these isomers whereas the 2-AB RP method is not. Figure 7C shows the EIC of the G1F 2-AB glycan, which elutes as one peak because separation could not be achieved during method development. Figure 7D illustrates the selectivity of the 2-AA method toward the complex N-glycans. From quantitative data obtained by HILIC chromatography we could deduce that the first, larger peak is that of the α1,6 isomer and the second, smaller peak is that of the α1,3 isomer.
We also evaluated the performance of our 2-AA method with more highly branched N-glycans with additional sialic acids. Six N-glycan standards with two, three, and four antennae were labeled. Each glycoform is present with and without a core fucose. All antennae carry one terminal N-acetylsialic acid. Overlays of the chromatograms, with the appropriate glycan structure, are shown in Fig. 8. The acidic N-glycans elute in accordance with the overall grouping (Fig. 2) and the separation between fucosylated and non-fucosylated glycans is obvious.
High sensitivity as a result of large injection volume
The possibility of using large injection volumes of aqueous samples in RP LC enables detection and identification, by fluorescence detection or mass spectrometry, of N-glycan variants which occur only at very low levels. Compared with HILIC, in which only few microliters of an aqueous sample can be injected, this circumstance is of huge advantage because the labeled and purified N-glycans are eluted in 600 μL H2O in the last step of our sample-preparation procedure. The sample can be injected without any additional concentration steps. N-Glycans accounting for less than 0.01 % of total mAb N-glycans, for example G3F (shown in Fig. 6) can be easily detected and identified by ion-trap MS, resulting in a high-sensitivity glycan map of the analyzed mAb.
Analysis of different glycosylated mAbs
We analyzed the glycosylation pattern of three different mAbs by use of the 2-AA RP LC–MS method to demonstrate the flexibility and versatility of the method for mAb N-glycan characterization. The fluorescence chromatograms are shown in Figs. 9, 10 and 11. These mAbs were chosen because of their different glycan content. The N-glycosylation pattern of mAb1 (Fig. 9) is indicative of the smallest amount of high-mannose structures but the largest amount of non-fucosylated complex glycans. Moreover it is the only mAb with both N-acetylneuraminic acid and N-glycoylneuraminic acid. It consists of 2 % high-mannose structures and <1 % hybrid structures without core fucose, and 7 % are complex structures lacking the core fucose. Hybrid and complex N-glycans with α1,6 core fucose account for <1 % and 90 %, respectively. Approximately 2 % of the N-glycans carry a terminal sialic acid.
Fig. 9.

RP FLD chromatogram obtained from mAb1 N-glycans after labeling with 2-AA. Peaks identified by MS and MS2 are numbered, and are listed in Table 1. The appropriate 2-AA glycans are shown in Fig. 3. The magnified view (small window) of the chromatogram shows the smaller peaks
Fig. 10.

RP FLD chromatogram obtained from mAb2 N-glycans after labeling with 2-AA. Peaks identified by MS and MS2 are numbered, and are listed in Table 1. The appropriate 2-AA glycans are shown in Fig. 3. The magnified view (small window) of the chromatogram shows the smaller peaks. Stacked numbering indicates co-elution of N-glycans. Quantitative data obtained from FLD and MS for mAb2 are listed in Table 2
Fig. 11.

RP FLD chromatogram obtained from mAb3 N-glycans after labeling with 2-AA. Peaks identified by MS and MS2 are numbered, and are listed in Table 1. The appropriate 2-AA glycans are shown in Fig. 3. The magnified view (small window) of the chromatogram shows the smaller peaks. Stacked numbering indicates co-elution of N-glycans
mAb2 (Fig. 10) has the highest amount of different oligomannose glycans and carries no sialylation. High-mannose structures account for 7 % of all glycans. Non-fucosylated hybrids account for <1 % and non-fucosylated complexes account for 2 % of the glycans. Hybrid and complex glycoforms with core fucose account for 1 % and 90 %, respectively. Relative amounts of glycans obtained from either fluorescence or MS results are listed, as examples, in Table 2. FLD data were obtained by integration of the peaks. Quantification by MS was performed by use of extracted ion chromatograms for the appropriate glycans. In general, the comparison shows there is good correlation between FLD and MS results, demonstrating the accuracy of the methods. Co-eluting structures 50, 52, 59, and 60 could be quantified individually by MS. The differences between MS and FLD detection for peaks 62 and 63 can be explained by the overlapping of the two peaks. For FLD quantification the peaks were split, resulting in a higher peak area for peak 62, whereas for MS detection the peaks could be quantified individually by use of their EICs. In the sum the peak areas are equal. There are larger differences between the EIC and FLD values for some glycan species of minor abundance; these result from larger relative integration errors of the EIC and FLD signals.
Table 2.
Comparison of fluorescence and MS data of 2-AA-labeled mAb2. Relative amounts of glycans (%) derived from the fluorescence signal (upper panel) and the MS data (lower panel) are listed for the appropriate peak number
| Peak# | 5 | 6 | 10 | 7 | 13 | 11 | 8 | 9 | 20 | 22 | 14 | 25 | 28 | 29 |
| FLD | 0.07 | 0.22 | 0.02 | 0.17 | 1.29 | 0.15 | 0.02 | 0.05 | 0.22 | 4.08 | 0.11 | 0.02 | 0.72 | 0.06 |
| MS | 0.08 | 0.22 | 0.03 | 0.15 | 1.12 | 0.26 | 0.07 | 0.08 | 0.27 | 4.37 | 0.14 | 0.03 | 0.66 | 0.17 |
| Peak# | 33 | 37 | 46 | 48 | 54 | 53 | 61 | 62 | 63 | 36 | 50 | 52 | 59 | 60 |
| FLD | 0.83 | 0.01 | 0.23 | 0.20 | 0.96 | 0.12 | 12.96 | 2.73 | 70.06 | 0.32 | 4.27 | |||
| MS | 1.20 | 0.10 | 0.05 | 0.08 | 0.79 | 0.08 | 12.95 | 5.29 | 67.43 | 0.26 | 0.15 | 0.18 | 2.42 | 1.37 |
mAb3 (Fig. 11) has the most complex N-glycosylation pattern of the antibodies analyzed. It is characterized by a large amount of hybrid structures and by oligomannose and several sialic acid moieties carrying glycans. The glycosylation consists of 5 % oligomannose structures and 5 % non-fucosylated hybrid structures. Complex variants account for 4 % of all glycans. Fucosylated hybrid glycans account for 5 %. Complex fucosylated glycans account for 85 % and the sialylation level is rather high at 5 % for this mAb. The relative glycan composition of the three mAbs was calculated by using the peak area from the fluorescence chromatogram of the RP LC runs; for unresolved or coeluting glycans the EIC was used for integration. The N-glycan structures assigned to each numbered peak are illustrated in Fig. 3.
Qualification of the method
The method was qualified to demonstrate its robustness and reliability. Sample preparation, including deglycosylation by use of PNGaseF, N-glycan separation by ultrafiltration, N-glycan labeling using 2-AB, and the gel filtration step to remove excess label is a standard company procedure which has been reported elsewhere [23]. Reproducibility of fluorescence labeling by 2-AA was monitored by performing the two labeling reactions in parallel and by comparing the relative amounts of glycans. Robustness and linearity were assessed by testing different column batches and by analyzing dilutions of analyte (e.g. mAb1 total peak area R2 = 0.9992), respectively. Intra-assay precision was monitored by repeated sample preparation and analysis by the same operator. For example, for relative quantification of mAb2 glycans by fluorescence coefficient of variation (%CV) values were calculated (e.g. peaks 22 (1.24 %) and 63 (1.17 %), and total glycan area (5.42 %)).All HPLC and MS experiments were conducted with qualified instruments.
Conclusion
This glycan-mapping method using RP HPLC of 2-AB or 2-AA-labeled N-glycans in combination with fluorescence detection and on-line ion-trap mass spectrometry enables high-sensitivity and high-resolution N-glycan analysis. The 2-AA method enables analysis of neutral and acidic N-glycans with positive ESI–MS. Compared with the RP LC–MS of 2-AB-labeled N-glycans more structural isomers can be separated in a shorter analysis time. In particular, separation of the highly abundant and isobaric G1F isomers was achieved. The 2-AA chromatogram obtained from the mAb glycan standard (Fig. 2) also contains more peaks and partially resolved peaks, because of the greater selectivity and resolution of the method. This selectivity of the reversed-phase chromatography in combination with 2-AA labeling enables separation of a variety of structural isomers of different types of N-glycan and, more important, separates the N-glycans into seven different groups: oligomannose, hybrid, and complex without core fucosylation, hybrid and complex with core fucosylation, and two sialic acid-containing groups again with and without the core fucose residues; this enables rapid screening of the glycan composition. The ability to separate N-glycans with multiple sialic acids and up to four antennae shows the versatility of the method. Retention of 2-AA on the reversed phase is better than that of 2-AB; this may result from the greater hydrophobicity of the protonated carboxyl group of the 2-AA label in the acidic mobile phase. The better retention and separation results in sharper peaks and, because of the greater sensitivity in fluorescence detection of 2-AA [19, 31], analysis of rare glycan species becomes possible. In addition, focusing of the analyte on the column leads to a more concentrated sample entering the ionization chamber of the mass spectrometer and enables efficient ionization and more sensitive MS detection. Fragmentation of the, mostly, [M + 2H]2+ ions by CID produces the mainly occurring B and Y ions, providing information about the N-glycan structure. We observed good ionization of the 2-AA-labeled N-glycans in positive ESI–MS, and even for sialylated structures the MS signal was intense and on-line MS2 data were obtained. Because of the high resolution of the liquid chromatography and the sensitive MS detection, the high complexity of mAb N-glycosylation can be investigated in detail. The large injection volume even enables detection and quantification N-glycans of very low abundance. Analysis of three mAbs with different glycosylation patterns showed the flexibility of the method for mAb N-glycan characterization. Furthermore, our data show that relative quantification with FLD data is comparable with quantification by MS for the labeled mAb N-glycans; this was somewhat expected, because the molecular composition of the glycans is similar and the masses are distributed over a relatively small range. The low hydrophobicity of the labeled glycans, which enables good separation, also makes MS quantification more accurate, because the ACN content changes slowly and conditions in the ionization chamber of the mass spectrometer during analyte elution are almost identical. Co-eluting glycans could also be quantified individually by use of MS data. For samples containing N-glycans with a larger size distribution, however, comparability must be evaluated individually.
To summarize, our 2-AA RP LC–MS approach can be used as a robust method which is orthogonal to the widely established HILIC of 2-AB glycans or MALDI MS of labeled neutral and acidic N-glycans derived from mAbs and other glycoproteins. In this work we demonstrated the strength and versatility of RP LC with on-line MS detection for analysis of N-glycosylation.
Abbreviations
- 2-AA
2-Aminobenzoic acid
- 2-AB
2-Aminobenzamide
- ACN
Acetonitrile
- ANTS
8-Aminonaphthalene-1,3,6-trisulfonic acid
- DMSO
Dimethyl sulfoxide
- EIC
Extracted ion chromatogram
- ESI
Electrospray ionization
- FLD
Fluorescence detector
- GlcNAc
N-Acetyl-d-glucosamine
- HILIC
Hydrophilic interaction liquid chromatography
- HPLC
High-performance liquid chromatography
- LC
Liquid chromatography
- mAb
Monoclonal antibody
- MALDI
Matrix-assisted laser desorption ionization
- MS
Mass spectrometry
- PNGaseF
Peptide N-glycosidase F
- PGC
Porous graphitized carbon
- RP
Reversed phase
References
- 1.Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, Murphy BA, Satinover SM, Hosen J, Mauro D, Slebos RJ, Zhou Q, Gold D, Hatley T, Hicklin DJ, Platts-Mills TAE. N Engl J Med. 2008;358:1109–1117. doi: 10.1056/NEJMoa074943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morell AG, Gregoriadis G, Scheinberg IH, Hickman J, Ashwell G. J Biol Chem. 1971;246:1461–1467. [PubMed] [Google Scholar]
- 3.Shields RL, Lai J, Keck R, O'Connell LY, Hong K, Meng YG, Weikert SH, Presta LG. J Biol Chem. 2002;277:26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 4.Jefferis R. Nat Rev Drug Discov. 2009;8:226–234. doi: 10.1038/nrd2804. [DOI] [PubMed] [Google Scholar]
- 5.Liu C, Dong S, Xu XJ, Yin Y, Shriver Z, Capila I, Myette J, Venkataraman G. J Pharm Biomed Anal. 2011;54:27–36. doi: 10.1016/j.jpba.2010.07.044. [DOI] [PubMed] [Google Scholar]
- 6.Flynn GC, Chen X, Liu YD, Shah B, Zhang Z. Mol Immunol. 2010;47:2074–2082. doi: 10.1016/j.molimm.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Ashline DJ, Lapadula AJ, Liu Y, Lin M, Grace M, Pramanik B, Reinhold VN. Anal Chem. 2007;79:3830–3842. doi: 10.1021/ac062383a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prien JM, Ashline DJ, Lapadula AJ, Zhang H, Reinhold VN. J Am Soc Mass Spectrom. 2009;20:539–556. doi: 10.1016/j.jasms.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maslen S, Sadowski P, Adam A, Lilley K, Stephens E. Anal Chem. 2006;78:8491–8498. doi: 10.1021/ac0614137. [DOI] [PubMed] [Google Scholar]
- 10.Harvey DJ. Proteomics. 2005;5:1774–1786. doi: 10.1002/pmic.200401248. [DOI] [PubMed] [Google Scholar]
- 11.Huhn C, Selman MHJ, Ruhaak LR, Deelder AM, Wuhrer M. Proteomics. 2009;9:882–913. doi: 10.1002/pmic.200800715. [DOI] [PubMed] [Google Scholar]
- 12.Anumula KR. Anal Biochem. 1994;220:275–283. doi: 10.1006/abio.1994.1338. [DOI] [PubMed] [Google Scholar]
- 13.Chen X, Flynn GC. Anal Biochem. 2007;370:147–161. doi: 10.1016/j.ab.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 14.Gennaro L, Harvey DJ, Vouros P. Rapid Commun Mass Spectrom: RCM. 2003;17:1528–1534. doi: 10.1002/rcm.1079. [DOI] [PubMed] [Google Scholar]
- 15.Harvey DJ. J Mass Spectrom: JMS. 2000;35:1178–1190. doi: 10.1002/1096-9888(200010)35:10<1178::AID-JMS46>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 16.Pabst M, Grass J, Toegel S, Liebminger E, Strasser R, Altmann F. Glycobiology. 2012;22:389–399. doi: 10.1093/glycob/cwr138. [DOI] [PubMed] [Google Scholar]
- 17.Harvey DJ, Royle L, Radcliffe CM, Rudd PM, Dwek R. Anal Biochem. 2008;376:44–60. doi: 10.1016/j.ab.2008.01.025. [DOI] [PubMed] [Google Scholar]
- 18.Bigge JC, Patel TP, Bruce JA, Goulding PN, Charles SM, Parekh RB. Anal Biochem. 1995;230:229–238. doi: 10.1006/abio.1995.1468. [DOI] [PubMed] [Google Scholar]
- 19.Harvey DJ. J Chromatogr B Anal Technol Biomed Life Sci. 2011;879:1196–1225. doi: 10.1016/j.jchromb.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 20.Lattova E, Snovida S, Perreault HÃ, Krokhin O. J Am Soc Mass Spectrom. 2005;16:683–696. doi: 10.1016/j.jasms.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 21.Pabst M, Kolarich D, Pöltl G, Dalik T, Lubec G, Hofinger A, Altmann F. Anal Biochem. 2009;384:263–273. doi: 10.1016/j.ab.2008.09.041. [DOI] [PubMed] [Google Scholar]
- 22.Ruhaak LR, Zauner G, Huhn C, Bruggink C, Deelder AM, Wuhrer M. Anal Bioanal Chem. 2010;397:3457–3481. doi: 10.1007/s00216-010-3532-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melmer M, Stangler T, Schiefermeier M, Brunner W, Toll H, Rupprechter A, Lindner W, Premstaller A. Anal Bioanal Chem. 2010;398:905–914. doi: 10.1007/s00216-010-3988-x. [DOI] [PubMed] [Google Scholar]
- 24.Melmer M, Stangler T, Premstaller A, Lindner W. J Chromatogr A. 2011;1218:118–123. doi: 10.1016/j.chroma.2010.10.122. [DOI] [PubMed] [Google Scholar]
- 25.Morelle W, Page A, Michalski JC. Rapid Commun Mass Spectrom: RCM. 2005;19:1145–1158. doi: 10.1002/rcm.1900. [DOI] [PubMed] [Google Scholar]
- 26.Prater BD, Connelly HM, Qin Q, Cockrill SL. Anal Biochem. 2009;385:69–79. doi: 10.1016/j.ab.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 27.Prien JM, Prater BD, Qin Q, Cockrill SL. Anal Chem. 2010;82:1498–1508. doi: 10.1021/ac902617t. [DOI] [PubMed] [Google Scholar]
- 28.Prien JM, Prater BD, Cockrill SL. Glycobiology. 2010;20:629–647. doi: 10.1093/glycob/cwq012. [DOI] [PubMed] [Google Scholar]
- 29.Harvey DJ. J Mass Spectrom: JMS. 2005;40:642–653. doi: 10.1002/jms.836. [DOI] [PubMed] [Google Scholar]
- 30.Domon B, Costello CE. Glycoconj J. 1988;5:397–409. doi: 10.1007/BF01049915. [DOI] [Google Scholar]
- 31.Anumula KR, Dhume ST. Glycobiology. 1998;8:685–694. doi: 10.1093/glycob/8.7.685. [DOI] [PubMed] [Google Scholar]