

Abstract
Mining gene expression profiles has proven valuable for identifying signatures serving as surrogates of cancer phenotypes. However, the similarities of such signatures across different cancer types have not been strong enough to conclude that they represent a universal biological mechanism shared among multiple cancer types. Here we present a computational method for generating signatures using an iterative process that converges to one of several precise attractors defining signatures representing biomolecular events, such as cell transdifferentiation or the presence of an amplicon. By analyzing rich gene expression datasets from different cancer types, we identified several such biomolecular events, some of which are universally present in all tested cancer types in nearly identical form. Although the method is unsupervised, we show that it often leads to attractors with strong phenotypic associations. We present several such multi-cancer attractors, focusing on three that are prominent and sharply defined in all cases: a mesenchymal transition attractor strongly associated with tumor stage, a mitotic chromosomal instability attractor strongly associated with tumor grade, and a lymphocyte-specific attractor.
Author Summary
Cancer is known to be characterized by several unifying biological capabilities or “hallmarks.” However, attempts to computationally identify patterns, such as gene expression signatures, shared across many different cancer types have been largely unsuccessful. A typical approach has been to classify samples into mutually exclusive subtypes, each of which is characterized by a particular gene signature. Although occasional similarities of such signatures in different cancer types exist, these similarities have not been sufficiently strong to conclude that they reflect the same biological event. By contrast, we have developed a computational methodology that has identified some signatures of co-expressed genes exhibiting remarkable similarity across many different cancer types. These signatures appear as stable “attractors” of an iterative computational procedure that tends to collect mutually associated genes, so that its convergence can point to the core (“heart”) of the underlying biological co-expression mechanism. One of these “pan-cancer” attractors corresponds to a transdifferentiation of cancer cells empowering them with invasiveness and motility. Another represents a mitotic chromosomal instability of cancer cells. A third attractor is lymphocyte-specific.
Introduction
Despite their type-specific features, cancers share some common traits, or “hallmarks,” related to, e.g., the abilities of some cancer cells to divide uncontrollably or to invade surrounding tissues [1]. Furthermore, it has been recognized that gene expression signatures resulting from analysis of cancer datasets can serve as surrogates of cancer phenotypes [2]. Therefore, it is reasonable to hypothesize that computational analysis of rich biomolecular cancer datasets may reveal signatures that are shared across many cancer types and are associated with specific cancer phenotypes. Such rich datasets become publicly available at an increasing rate from many sources, such as The Cancer Genome Atlas (TCGA). However, attempts to identify any such robust “bioinformatic hallmarks” of cancer have so far been largely unsuccessful.
Gene signatures may occasionally be found to exhibit similarities across multiple cancer types. However, to our knowledge no algorithm has ever produced a set of nearly identical signatures after independently and separately analyzing datasets from different cancer types.
There are various ways by which modules of co-expressed genes can be identified from rich datasets, some of which may be within the context of regulatory network discovery [3]. Clustering approaches can classify a selected set of genes into subsets each of which contains mutually related genes. Related techniques can also be used to classify samples into cancer subtypes [4]–[6], each characterized by a set of characteristic genes. One of the most powerful computational approaches for this task has been nonnegative matrix factorization (NMF) [7] combined with consensus clustering [8], resulting in successful subtype identification in several types of cancer.
The main objective addressed by techniques such as NMF is to reduce dimensionality by identifying a number of metagenes jointly representing the gene expression dataset as accurately as possible, in lieu of the whole set of individual genes. Each metagene in NMF is defined as a positive linear combination of the individual genes, so that its expression level is an accordingly weighted average of the expression levels of the individual genes. The identity of each resulting metagene is influenced by the presence of other metagenes within the objective of overall dimensionality reduction achieved by joint optimization.
By contrast, if the aim is exclusively to identify metagenes as surrogates of biomolecular events, then a fully unconstrained algorithm should be devised, without any effort to achieve dimensionality reduction, classification, mutual exclusivity, orthogonality, regulatory interaction inference, etc.
We can consider, for example, a hypothetical case in which we have found a cluster consisting of a number of co-expressed genes in a rich gene expression dataset. We may wish to scrutinize and “sharpen” this co-expression trying to identify the “heart” (core) of the genes that are most strongly co-expressed in that case. In the absence of a defining phenotype, we can continue applying an unsupervised methodology, as follows: We can first define a consensus metagene from the average expression levels of all genes in the cluster, and rank all the individual genes in terms of their association (defined numerically by some form of correlation) with that metagene. We can then replace the member genes of the cluster with an equal number of the top-ranked genes. Some of the original genes may naturally remain as members of the cluster, but some may be replaced, as this process will “attract” some other genes that are more strongly correlated with the cluster. We can now define a new metagene defined by the average expression levels of the genes in the newly defined cluster, and re-rank all the individual genes in terms of their association with that new metagene; and so on. It is intuitively reasonable to expect that this iterative process will eventually converge to a cluster that contains precisely the genes that are most associated with the metagene of the same cluster, so that any other individual genes will be less strongly associated with the metagene. We can think of this particular cluster defined by the convergence of this iterative process as an “attractor,” i.e., a module of co-expressed genes to which many other gene sets with close but not identical membership will converge using the same computational methodology.
The above description represents a simplified conceptual introduction of the computational methodology presented in this paper. Rather than using the average of the expression values in gene clusters of a particular size, the “attractors” are metagenes defined as weighted averages of all genes where each individual gene has a nonnegative weight, just like the metagenes defined using NMF [7]. We found that, given a rich (loosely defined as containing at least 200 samples) dataset represented by a gene expression matrix, such metagenes can be naturally identified as stable and precise attractors using a simple iterative approach. We experimentally verified these nice convergence properties without any exception after trying numerous times the method described in this paper on such rich datasets.
This methodology is totally unsupervised, as it does not make use of any phenotypic association. As we show in this paper, however, once identified, a metagene attractor is likely to be found associated with a phenotype.
We found that several attractor metagenes are present in nearly identical form in multiple cancer types. This provides an additional opportunity to combine the powers of a large number of rich datasets to focus, at an even sharper level, on the core genes of the underlying mechanism. For example, this methodology can precisely point to the causal (driver) oncogenes within amplicons to be among very few candidate genes. Importantly, this can be done from rich gene expression data, which already exist in abundance, without making any use of sequencing data.
We identified several attractors, each of which has the potential to lead to corresponding testable biological hypotheses after scrutinizing their top-ranked genes and finding a putative underlying mechanism. For the purposes of this paper we present the general methodology for the benefit of the research community together with a listing of the attractors in six datasets from three cancer types (ovarian, colon, breast).
Here, we focus on a few interesting cancer-associated attractors that we found present in multiple cancer types. Particular emphasis is given to what we consider to be three key “bioinformatic hallmarks” of cancer, related to the ability of cancer cells to invade surrounding tissues; to divide uncontrollably; and the ability of the organism to recruit the immune system to fight cancer: a tumor stage-associated mesenchymal transition attractor, a tumor grade-associated mitotic chromosomal instability (mitotic CIN) attractor, and a lymphocyte-specific attractor.
Results
Derivation of Attractor Metagenes
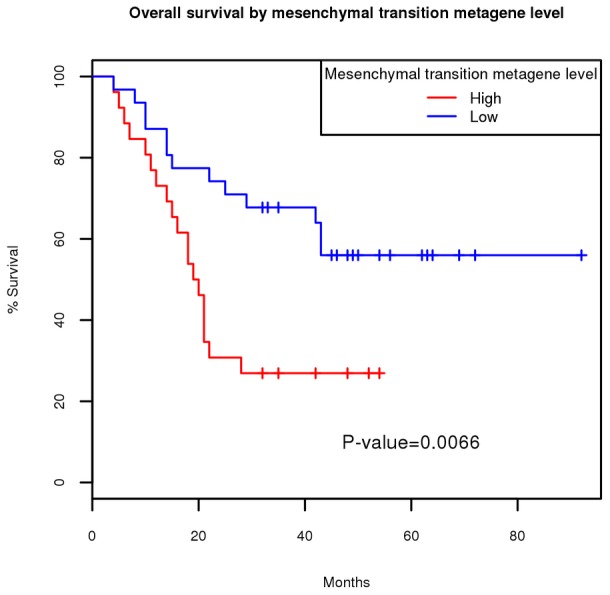
Given a nonnegative measure  of pairwise association between genes
of pairwise association between genes  and
and  , we define an attractor metagene
, we define an attractor metagene  to be a linear combination of the individual genes with weights
to be a linear combination of the individual genes with weights  . The association measure
. The association measure 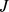 is assumed to have minimum possible value 0 and maximum possible value 1, so the same is true for the weights. It is also assumed to be scale-invariant, therefore it is not necessary for the weights to be normalized so that they add to 1, and the metagenes can still be thought of as expressing a normalized weighted average of the expression levels of the individual genes. See Materials and Methods for the choice of the measure
is assumed to have minimum possible value 0 and maximum possible value 1, so the same is true for the weights. It is also assumed to be scale-invariant, therefore it is not necessary for the weights to be normalized so that they add to 1, and the metagenes can still be thought of as expressing a normalized weighted average of the expression levels of the individual genes. See Materials and Methods for the choice of the measure  .
.
According to this definition, the genes with the highest weights in an attractor metagene will have the highest association with the metagene (and, by implication, they will tend to be highly associated among themselves) and so they will often represent a biomolecular event reflected by the co-expression of these top genes. This can happen, e.g., when a biological mechanism is activated, or when a copy number variation (CNV), such as an amplicon, is present, in some of the samples included in the expression matrix. In the following we use the term “attractor” for simplicity to refer to an attractor metagene, and the term “top genes” to refer to the genes with the highest weights in the attractor. The definition of an attractor metagene can readily be generalized to include features other than gene expression, such as methylation values. It can also be used in datasets of any objects (not necessarily genes) characterized by any type of feature vectors, with applications in other disciplines, such as social and economic sciences.
The computational problem of identifying attractor metagenes given an expression matrix can be addressed heuristically using a simple iterative process: Starting from a particular seed (or “attractee”) metagene  , a new metagene is defined in which the new weights are
, a new metagene is defined in which the new weights are  . The same process is then repeated in the next iteration resulting in a new set of weights, and so forth. In all gene expression datasets that we tried we found that this process converges to a limited number of stable attractors. Each attractor is defined by a precise set of weights, which are reached with high accuracy typically within 10 or 20 iterations.
. The same process is then repeated in the next iteration resulting in a new set of weights, and so forth. In all gene expression datasets that we tried we found that this process converges to a limited number of stable attractors. Each attractor is defined by a precise set of weights, which are reached with high accuracy typically within 10 or 20 iterations.
This algorithmic behavior with nice convergence properties is not surprising, because if a metagene represents co-expressed genes, then the next iteration will naturally “attract” other similarly co-expressed genes, and so forth, until there are no other genes more associated with the top genes than those genes themselves. Furthermore, the set of the few genes with the highest weight are likely to represent the “heart” (core) of the underlying biomolecular event. In support of this concept, the association of any of the top-ranked individual genes with the attractor metagene is consistently and significantly higher than the pairwise association between any of these genes, suggesting that the set of these top genes jointly comprise a proxy representing a biomolecular event better than each of the individual genes would.
Indeed, related versions of the signatures identified by attractors in this paper have been previously identified in various contexts in individual cancer types, often intermingled with additional genes. However, the contribution of our work is that these signatures are found as pan-cancer biomolecular events, sharply pointing to the underlying mechanism. Therefore the top genes of the attractors will be appropriate for being used as biomarkers or for understanding the underlying biology. For example, one of the attractors that we identified (the “mitotic chromosomal instability” attractor, described below) has previously been found in approximate forms among sets of genes described generally [9] as “proliferation” or “cell cycle related” markers, while the actual attractor points much more sharply to particular elements in the structure of the kinetochore-microtubule interface.
A reasonable implementation of an “exhaustive” search of attractor metagenes is to start from each individual gene as a seed (“attractee”) assigning a weight of 1 to that gene, and 0 to all the other genes. Each gene participating in a particular co-expression event will then lead to the same attractor when used as attractee. The computational implementation of the algorithm is described in Materials and Methods. We note that a dual method can be used to identify attractor “metasamples” as representatives of subtypes, and we can also combine such metasamples with the attractor metagenes in various ways to achieve biclustering, but this topic is not examined in this paper.
We analyzed six datasets, two from ovarian cancer, two from breast cancer and two from colon cancer (Supplementary Text S1). In each case, we identified general (Supplementary Table S1) and genomically localized (Supplementary Table S2) attractors and we found that many among them appear in similar forms in all six datasets using particular merging and ranking criteria in each case (Materials and Methods and Supplementary Text S1). Following are descriptions of some of our results, starting with the three strongest multi-cancer attractors.
Mesenchymal Transition Attractor Metagene
This attractor contains mostly epithelial-mesenchymal transition (EMT)-associated genes. Table 1 provides a listing of the top 100 genes based on their average mutual information (Materials and Methods) with their corresponding attractor metagenes.
Table 1. Top 100 genes of the mesenchymal transition attractor based on six datasets.
| Rank | Gene Symbol | Avg MI | Rank | Gene Symbol | Avg MI |
| 1 | COL5A2 | 0.814 | 51 | SULF1 | 0.505 |
| 2 | VCAN | 0.775 | 52 | LOXL1 | 0.502 |
| 3 | SPARC | 0.766 | 53 | PRRX1 | 0.502 |
| 4 | THBS2 | 0.758 | 54 | PPAPDC1A | 0.499 |
| 5 | FBN1 | 0.749 | 55 | COL10A1 | 0.498 |
| 6 | COL1A2 | 0.749 | 56 | ITGA11 | 0.495 |
| 7 | COL5A1 | 0.747 | 57 | NTM | 0.494 |
| 8 | FAP | 0.734 | 58 | MXRA8 | 0.494 |
| 9 | AEBP1 | 0.711 | 59 | FIBIN | 0.493 |
| 10 | CTSK | 0.709 | 60 | WISP1 | 0.483 |
| 11 | COL3A1 | 0.688 | 61 | RCN3 | 0.483 |
| 12 | COL1A1 | 0.683 | 62 | TNFAIP6 | 0.481 |
| 13 | SERPINF1 | 0.674 | 63 | ECM2 | 0.480 |
| 14 | COL6A3 | 0.669 | 64 | HTRA1 | 0.480 |
| 15 | CDH11 | 0.663 | 65 | EFEMP2 | 0.478 |
| 16 | GLT8D2 | 0.658 | 66 | MXRA5 | 0.474 |
| 17 | LUM | 0.654 | 67 | ACTA2 | 0.472 |
| 18 | MMP2 | 0.654 | 68 | LOX | 0.470 |
| 19 | DCN | 0.650 | 69 | ITGBL1 | 0.466 |
| 20 | CCDC80 | 0.637 | 70 | PMP22 | 0.465 |
| 21 | POSTN | 0.631 | 71 | P4HA3 | 0.464 |
| 22 | CTHRC1 | 0.616 | 72 | PTRF | 0.463 |
| 23 | ADAM12 | 0.613 | 73 | CALD1 | 0.460 |
| 24 | COL6A2 | 0.608 | 74 | HEG1 | 0.458 |
| 25 | MSRB3 | 0.608 | 75 | NEXN | 0.455 |
| 26 | OLFML2B | 0.607 | 76 | NID2 | 0.455 |
| 27 | INHBA | 0.600 | 77 | TAGLN | 0.455 |
| 28 | FSTL1 | 0.600 | 78 | FAM26E | 0.452 |
| 29 | SFRP2 | 0.596 | 79 | ZNF521 | 0.452 |
| 30 | SNAI2 | 0.577 | 80 | SFRP4 | 0.451 |
| 31 | CRISPLD2 | 0.574 | 81 | PALLD | 0.450 |
| 32 | PCOLCE | 0.571 | 82 | OLFML1 | 0.447 |
| 33 | PDGFRB | 0.567 | 83 | FILIP1L | 0.447 |
| 34 | BGN | 0.565 | 84 | TIMP3 | 0.445 |
| 35 | COL12A1 | 0.560 | 85 | SPON2 | 0.443 |
| 36 | ANGPTL2 | 0.555 | 86 | SPOCK1 | 0.443 |
| 37 | COPZ2 | 0.553 | 87 | COL8A2 | 0.441 |
| 38 | CMTM3 | 0.549 | 88 | GPC6 | 0.438 |
| 39 | ASPN | 0.547 | 89 | PDPN | 0.437 |
| 40 | FN1 | 0.545 | 90 | GFPT2 | 0.436 |
| 41 | CNRIP1 | 0.540 | 91 | LHFP | 0.436 |
| 42 | FNDC1 | 0.538 | 92 | GREM1 | 0.436 |
| 43 | LRRC15 | 0.533 | 93 | TGFB1I1 | 0.435 |
| 44 | COL11A1 | 0.529 | 94 | C1S | 0.433 |
| 45 | ANTXR1 | 0.528 | 95 | EDNRA | 0.432 |
| 46 | RAB31 | 0.527 | 96 | GAS1 | 0.431 |
| 47 | FRMD6 | 0.524 | 97 | NOX4 | 0.431 |
| 48 | TSHZ3 | 0.520 | 98 | FBLN2 | 0.428 |
| 49 | THY1 | 0.519 | 99 | TCF4 | 0.428 |
| 50 | NNMT | 0.519 | 100 | NUAK1 | 0.427 |
The consistency of the attractor is established by the fact (Supplementary Table S1) that there are many genes (COL5A2, COL1A2, SPARC, CTSK, FBN1, VCAN, AEBP1, SERPINF1) that are among the top 50 genes in the attractors of all six datasets. The corresponding P value is less than 10−7 by permutation test (Materials and Methods). Similar results are found in other solid cancer types in all cases that we tried.
This is a stage-associated attractor, in which the signature is significantly present only when a particular level of invasive stage, specific to each cancer type, has been reached. Supplementary Table S3 demonstrates this phenomenon in three cancer datasets from different types (breast, ovarian and colon) that were annotated with clinical staging information, by providing a listing of differentially expressed genes, ranked by fold change, when ductal carcinoma in situ (DCIS) progresses to invasive ductal carcinoma; ovarian cancer progresses to stage III; and colon cancer progresses to stage II. In all three cases, the attractor is highly enriched among the top genes. Specifically, among the top 100 differentially expressed genes, the number of attractor genes included in Table 1 is 47 in breast cancer, 42 in ovarian cancer and 37 in colon cancer. The corresponding P values are 2×10−93, 4×10−80 and 8×10−78, respectively.
This attractor has been previously identified with remarkable accuracy as representing a particular kind of mesenchymal transition of cancer cells present in all types of solid cancers tested leading to a published list of top 64 genes [10], [11]. This list was generated using a supervised algorithm using association with tumor stage. Indeed 52 of these top 64 genes also appear in Table 1 (P<10−114), and furthermore all top 19 genes of Table 1 are among the 64. We found that most of the genes of the signature were expressed by the cancer cells themselves, and not by the surrounding stroma, at least in a neuroblastoma xenograft model that we tried [11]. We also found that the signature is associated with prolonged time to recurrence in glioblastoma [12]. Related versions of the same signature were previously found to be associated with resistance to neoadjuvant therapy in breast cancer [13]. These results are consistent with the finding that EMT induces cancer cells to acquire stem cell properties [14]. It has been hypothesized that EMT is a key mechanism for cancer cell invasiveness and motility [15]–[17]. The attractor, however, appears to represent a more general phenomenon of transdifferentiation present even in nonepithelial cancers such as neuroblastoma, glioblastoma and Ewing's sarcoma.
Although similar signatures are often labeled as “stromal,” because they contain many stromal markers such as α-SMA and fibroblast activation protein, the fact that most of the genes of the signature were expressed by xenografted cancer cells [11], and not by mouse stromal cells, suggests that this particular attractor of coordinately expressed genes represents cancer cells having undergone a mesenchymal transition. The signature may indicate a non-fibroblastic transition, as occurs in glioblastoma, in which case collagen COL11A1 is not co-expressed with the other genes of the attractor. We have hypothesized that a full fibroblastic transition of the cancer cells occurs when cancer cells encounter adipocytes [11], in which case they may well assume the duties of cancer-associated fibroblasts (CAFs) in some tumors [1]. In that case, the best proxy of the signature [10] is COL11A1 and the strongly co-expressed genes THBS2 and INHBA. Indeed, the 64 genes of the previously identified signature were found from multi-cancer analysis [10] as the genes whose expression is consistently most associated with that of COL11A1.
The only EMT-inducing transcription factor found upregulated in the xenograft model [11] is SNAI2 (Slug), and it is also the one most associated with the signature in publicly available datasets. We also found that the microRNAs most highly associated with this attractor are miR-214, miR-199a, and miR-199b. Interestingly, miR-214 and miR-199a were found to be jointly regulated by another EMT-inducing transcription factor, TWIST1 [18].
The expression of the mesenchymal transition attractor indicates that the tumor is actively invasive at the specific sample site, so its prognostic value is cancer type and stage specific. As an example, we analyzed an oral squamous cell carcinoma dataset deposited in the Gene Expression Omnibus (GEO) under accession number GSE25104. The corresponding Kaplan-Meier survival curve (P = 0.0066) is shown in Figure 1.
Figure 1. Kaplan-Meier curves of mesenchymal transition attractor metagene in oral squamous cell carcinoma dataset.
Gene expression data from 57 patients (GSE25104) were divided into two groups: high mesenchymal transition metagene level and low mesenchymal transition metagene level depending on whether the metagene expression value exceeding the mean of the 57 patients. The P value of the association was determined by log-rank test.
Mitotic CIN Attractor Metagene
This attractor contains mostly kinetochore-associated genes. Table 2 provides a listing of the top 100 genes based on their average mutual information (Materials and Methods) with their corresponding attractor metagenes, starting from CENPA, which encodes for a histone H3-like centromeric protein.
Table 2. Top 100 genes of the mitotic CIN attractor based on six datasets.
| Rank | Gene Symbol | Avg MI | Rank | Gene Symbol | Avg MI |
| 1 | CENPA | 0.720 | 51 | CDCA8 | 0.532 |
| 2 | DLGAP5 | 0.693 | 52 | CDC45 | 0.528 |
| 3 | MELK | 0.684 | 53 | KIF18A | 0.524 |
| 4 | BUB1 | 0.674 | 54 | HMMR | 0.506 |
| 5 | KIF2C | 0.660 | 55 | TOP2A | 0.505 |
| 6 | KIF20A | 0.658 | 56 | CENPF | 0.503 |
| 7 | KIF4A | 0.656 | 57 | ZWINT | 0.503 |
| 8 | CCNA2 | 0.654 | 58 | PLK1 | 0.501 |
| 9 | CCNB2 | 0.652 | 59 | RAD51AP1 | 0.501 |
| 10 | NCAPG | 0.649 | 60 | FAM83D | 0.498 |
| 11 | TTK | 0.642 | 61 | E2F8 | 0.497 |
| 12 | CEP55 | 0.638 | 62 | CENPE | 0.497 |
| 13 | CCNB1 | 0.632 | 63 | MKI67 | 0.492 |
| 14 | CDK1 | 0.629 | 64 | CENPN | 0.491 |
| 15 | HJURP | 0.626 | 65 | MAD2L1 | 0.489 |
| 16 | CDC20 | 0.624 | 66 | CHEK1 | 0.486 |
| 17 | CDCA5 | 0.615 | 67 | GTSE1 | 0.477 |
| 18 | NCAPH | 0.615 | 68 | RAD51 | 0.475 |
| 19 | BUB1B | 0.609 | 69 | SGOL2 | 0.474 |
| 20 | KIF23 | 0.592 | 70 | PARPBP | 0.469 |
| 21 | KIF11 | 0.591 | 71 | TRIP13 | 0.467 |
| 22 | BIRC5 | 0.589 | 72 | SHCBP1 | 0.465 |
| 23 | NUF2 | 0.587 | 73 | DTL | 0.465 |
| 24 | TPX2 | 0.586 | 74 | CENPL | 0.462 |
| 25 | AURKB | 0.582 | 75 | FEN1 | 0.461 |
| 26 | RACGAP1 | 0.580 | 76 | FANCI | 0.461 |
| 27 | NUSAP1 | 0.580 | 77 | FBXO5 | 0.459 |
| 28 | ASPM | 0.579 | 78 | ECT2 | 0.457 |
| 29 | MCM10 | 0.579 | 79 | MND1 | 0.456 |
| 30 | PRC1 | 0.576 | 80 | CDC25C | 0.456 |
| 31 | DEPDC1B | 0.572 | 81 | PBK | 0.456 |
| 32 | UBE2C | 0.569 | 82 | KPNA2 | 0.452 |
| 33 | UBE2T | 0.567 | 83 | RAD54L | 0.452 |
| 34 | NEK2 | 0.566 | 84 | ESPL1 | 0.447 |
| 35 | FOXM1 | 0.565 | 85 | CDCA2 | 0.446 |
| 36 | NDC80 | 0.556 | 86 | FAM64A | 0.440 |
| 37 | CDCA3 | 0.556 | 87 | CENPK | 0.436 |
| 38 | FAM54A | 0.553 | 88 | MYBL2 | 0.435 |
| 39 | ANLN | 0.551 | 89 | SPAG5 | 0.434 |
| 40 | KIF15 | 0.548 | 90 | EZH2 | 0.431 |
| 41 | STIL | 0.547 | 91 | SMC4 | 0.430 |
| 42 | EXO1 | 0.542 | 92 | TACC3 | 0.428 |
| 43 | AURKA | 0.540 | 93 | C11orf82 | 0.427 |
| 44 | PTTG1 | 0.539 | 94 | MASTL | 0.426 |
| 45 | OIP5 | 0.539 | 95 | ASF1B | 0.426 |
| 46 | RRM2 | 0.539 | 96 | PTTG3P | 0.425 |
| 47 | DEPDC1 | 0.539 | 97 | CENPW | 0.424 |
| 48 | CDKN3 | 0.538 | 98 | ORC1 | 0.424 |
| 49 | KIF14 | 0.537 | 99 | HELLS | 0.422 |
| 50 | SPC25 | 0.534 | 100 | TK1 | 0.421 |
The consistency of the attractor is established by the fact (Supplementary Table S1) that there are many genes (CENPA, DLGAP5, KIF2C, CCNB2, MELK, CCNA2, KIF20A, HJURP, NUSAP1, BUB1, TTK, KIF11, NCAPH) that are among the top 50 genes in the attractors of all six datasets. The corresponding P value is less than 10−7 by permutation test (Materials and Methods). Similar results are found in other solid cancer types in all cases that we tried.
Contrary to the stage-associated mesenchymal transition attractor, this is a grade-associated attractor, in which the signature is significantly present only when an intermediate level of tumor grade is reached. Supplementary Table S4 demonstrates this phenomenon in three cancer datasets from different types (breast, ovarian and bladder) that were annotated with tumor grade information, by providing a listing of differentially expressed genes, ranked by fold change, when grade G2 is reached. In all three cases, the attractor is highly enriched among the top genes. Specifically, among the top 100 differentially expressed genes, the number of attractor genes included Table 2 is 40 in breast cancer, 38 in ovarian cancer and 27 in colon cancer. The corresponding P values are 4×10−74, 3×10−69 and 3×10−49, respectively. Consistently, a similar “gene expression grade index” signature [19] was previously found differentially expressed between histologic grade 3 and histologic grade 1 breast cancer samples. Furthermore, that same signature [19] was found capable of reclassifying patients with histologic grade 2 tumors into two groups with high versus low risks of recurrence.
This attractor is associated with chromosomal instability (CIN), as evidenced from the fact that another similar gene set comprising a “signature of chromosomal instability” [20] was previously derived from multiple cancer datasets purely by identifying the genes that are most correlated with a measure of aneuploidy in tumor samples. This led to a 70-gene signature referred to as “CIN70.” Indeed 31 of these 70 genes appear in Table 2 (P<10−53). However, several top genes of the attractor, such as CENPA, DLGAP5, KIF2C, BUB1 and CCNA2 are not present in the CIN70 list. Mitotic CIN is increasingly recognized [21] as a widespread multi-cancer phenomenon.
The attractor is characterized by overexpression of kinetochore-associated genes, which is known [22] to induce CIN. Overexpression of several of the genes of the attractor, such as the top gene CENPA [23], as well as MAD2L1 [24] and TPX2 [25], has also been independently previously found associated with CIN. Included in the mitotic CIN attractor are key components of mitotic checkpoint signaling [26], such as BUB1B, MAD2L1 (aka MAD2), CDC20, and TTK (aka MSP1). Also among the genes in the attractor is MKI67 (aka Ki-67), which has been widely used as a proliferation rate marker in cancer.
Among transcription factors, we found MYBL2 (aka B-Myb) and FOXM1 to be strongly associated with the attractor. They are already known to be sequentially recruited to promote late cell cycle gene expression [27] to prepare for mitosis.
Inactivation of the retinoblastoma (RB) tumor suppressor promotes CIN [28] and the expression of the attractor signature. Indeed, a similar expression of a “proliferation gene cluster [29]” was found strongly associated with the human papillomavirus E7 oncogene, which abrogates RB protein function and activates E2F-regulated genes. Consistently, many among the genes of the attractor correspond to E2F pathway genes controlling cell division or proliferation. Among the E2F transcription factors, we found that E2F8 and E2F7 are most strongly associated with the attractor.
Lymphocyte-Specific Attractor Metagene
This attractor consists mainly of lymphocyte-specific genes with prominent presence of CD53, PTPRC, LAPTM5, DOCK2, LCP2 and IL10RA. It is strongly associated with the expression of microRNA miR-142 as well as with particular hypermethylated and hypomethylated gene signatures [30]. There is also significant overlap between the sets of hypomethylated and overexpressed genes, suggesting that their expression is triggered by hypomethylation. Gene set enrichment analysis reveals that the attractor is found enriched in genes known to be preferentially expressed in differentiation into lymphocytes [31]. Table 3 provides a listing of the top 100 genes of the lymphocyte-specific attractor based on their average mutual information (Materials and Methods) with their corresponding attractor metagenes.
Table 3. Top 100 genes of the lymphocyte-specific attractor based on six datasets.
| Rank | Gene Symbol | Avg MI | Rank | Gene Symbol | Avg MI |
| 1 | PTPRC | 0.782 | 51 | NCF1 | 0.560 |
| 2 | CD53 | 0.768 | 52 | CCL5 | 0.557 |
| 3 | LCP2 | 0.739 | 53 | LST1 | 0.557 |
| 4 | LAPTM5 | 0.708 | 54 | CD3D | 0.553 |
| 5 | DOCK2 | 0.699 | 55 | RCSD1 | 0.548 |
| 6 | IL10RA | 0.699 | 56 | FGL2 | 0.538 |
| 7 | CYBB | 0.698 | 57 | HCST | 0.538 |
| 8 | CD48 | 0.691 | 58 | MARCH1 | 0.538 |
| 9 | ITGB2 | 0.679 | 59 | FERMT3 | 0.536 |
| 10 | EVI2B | 0.675 | 60 | FCGR2B | 0.533 |
| 11 | MS4A6A | 0.673 | 61 | GIMAP5 | 0.530 |
| 12 | TFEC | 0.659 | 62 | MYO1F | 0.530 |
| 13 | SLA | 0.657 | 63 | KLHL6 | 0.530 |
| 14 | TNFSF13B | 0.657 | 64 | GIMAP1 | 0.527 |
| 15 | C1orf162 | 0.656 | 65 | CD163 | 0.524 |
| 16 | SAMSN1 | 0.652 | 66 | CLEC7A | 0.522 |
| 17 | PLEK | 0.649 | 67 | CCR1 | 0.518 |
| 18 | GMFG | 0.647 | 68 | GBP5 | 0.517 |
| 19 | GIMAP4 | 0.647 | 69 | NCF2 | 0.516 |
| 20 | SASH3 | 0.645 | 70 | HLA-DPA1 | 0.516 |
| 21 | EVI2A | 0.638 | 71 | RNASE6 | 0.515 |
| 22 | SRGN | 0.638 | 72 | CD14 | 0.515 |
| 23 | AIF1 | 0.636 | 73 | FAM26F | 0.511 |
| 24 | LAIR1 | 0.627 | 74 | CD4 | 0.510 |
| 25 | FYB | 0.625 | 75 | FCGR1A | 0.506 |
| 26 | FCER1G | 0.623 | 76 | GZMA | 0.506 |
| 27 | MPEG1 | 0.621 | 77 | GPR183 | 0.505 |
| 28 | CD86 | 0.621 | 78 | CD84 | 0.505 |
| 29 | C3AR1 | 0.611 | 79 | NKG7 | 0.504 |
| 30 | C1QB | 0.608 | 80 | C1QA | 0.502 |
| 31 | CD2 | 0.606 | 81 | CD300LF | 0.500 |
| 32 | HCLS1 | 0.599 | 82 | FPR3 | 0.499 |
| 33 | HCK | 0.592 | 83 | PARVG | 0.496 |
| 34 | MNDA | 0.587 | 84 | TRAF3IP3 | 0.494 |
| 35 | CD37 | 0.587 | 85 | TYROBP | 0.492 |
| 36 | LY96 | 0.585 | 86 | LPXN | 0.492 |
| 37 | CCR5 | 0.585 | 87 | GIMAP8 | 0.492 |
| 38 | ARHGAP9 | 0.580 | 88 | MS4A7 | 0.490 |
| 39 | CD52 | 0.580 | 89 | IL2RB | 0.489 |
| 40 | GPR65 | 0.580 | 90 | CD300A | 0.488 |
| 41 | GIMAP6 | 0.578 | 91 | IGSF6 | 0.488 |
| 42 | SLAMF8 | 0.577 | 92 | SELPLG | 0.488 |
| 43 | WIPF1 | 0.577 | 93 | FCGR2A | 0.487 |
| 44 | MS4A4A | 0.574 | 94 | NCKAP1L | 0.483 |
| 45 | ARHGAP15 | 0.573 | 95 | DOK2 | 0.483 |
| 46 | HAVCR2 | 0.567 | 96 | CD247 | 0.481 |
| 47 | ARHGAP30 | 0.566 | 97 | SELL | 0.480 |
| 48 | CLEC4A | 0.566 | 98 | GZMK | 0.479 |
| 49 | TAGAP | 0.564 | 99 | CCR2 | 0.479 |
| 50 | CYTIP | 0.563 | 100 | LY86 | 0.479 |
The gene membership of the attractor provides hints about the underlying immune mechanism, which could be valuable towards generating hypotheses for potential immunotherapies such as adoptive transfer of lymphocytes. For example, the presence of the signal-transducing LCP2 (aka SLP-76) gene, together with the adaptor FYB (aka ADAP), suggests the formation of the SLP-76-ADAP adaptor module, which is known to regulate lymphocyte co-stimulation mediated by integrin ITGB2 (aka LFA-1), another prominent gene in the attractor [32].
Association of the Three Main Attractor Metagenes with Prognosis in Breast Cancer
We found that each of the above three main attractors under particular conditions is highly prognostic in breast cancer by analysing the METABRIC discovery breast cancer dataset [33] which includes both expression as well as survival data.
Mesenchymal transition attractor
In breast cancer, the mesenchymal transition attractor is expressed very early, as cancer becomes invasive. The presence of the attractor in a particular sample of high-stage tumor in not as informative, because of heterogeneity. On the other hand we found that the presence of the attractor in early-stage tumors is highly prognostic, consistent with the hypothesis that it indicates increased invasiveness. As shown in Figure 2, the Kaplan-Meier 15-year survival curves of the mesenchymal transition attractor using all samples does not show any significance. However, when we restrict the samples to early stage patients, defined as having no positive lymph nodes and tumor size less than 30 mm, the association between the attractor and survival became significant (P = 0.032).
Figure 2. Kaplan-Meier curve of mesenchymal transition attractor metagenes in breast cancer dataset.

The mesenchymal transition attractor metagene is most prominent in the early stage of breast cancer. The survival curve of the full dataset is insignificant (left). However, when the samples are restricted to only those at early stage (with no positive lymph nodes and tumor size less than 30 mm), the association between the mesenchymal transition attractor and the survival becomes significant (right), with P = 0.032.
Mitotic CIN attractor
The expression of the mitotic CIN attractor indicates that the tumor is dividing uncontrollably and therefore, in all cases, the attractor is highly prognostic for survival. The corresponding Kaplan-Meier 15-year survival curve (P<2×10−8) is shown in Figure 3. Furthermore, we ranked all the genes in terms of the concordance index [34] between the survival and the individual gene's expression value from the same rich dataset. Table 4 shows the top 100 genes, within which 47 (indicated by underline and boldface), including the top six, are also among the genes shown in Table 2 (P = 2×10−98 by Fisher's exact test). This extraordinary enrichment (also note that eight among the top ten genes, including the top three, are among the genes of Table 2) demonstrates that the members of the mitotic CIN attractor are, among all genes, the most prognostic ones, at least in breast cancer.
Figure 3. Kaplan-Meier curve of mitotic CIN attractor metagene in breast cancer dataset.

To evaluate the association between the mitotic CIN metagene expression and the 15-year survival, patients were divided into two groups: high mitotic CIN and low mitotic CIN. This binary expression level was determined by whether the mitotic CIN metagene expression value exceeding the mean of the patients. The P value of the association based on log-rank test is 1.8×10−8.
Table 4. List of top-ranked genes in terms of survival concordance index of the METABRIC discovery dataset demonstrating enrichment of the mitotic CIN attractor.
| Rank | Gene Symbol | Concordance Index | Rank | Gene Symbol | Concordance Index |
| 1 | CDCA5 | 0.670 | 51 | PRR11 | 0.639 |
| 2 | AURKA | 0.663 | 52 | LOC651816 | 0.638 |
| 3 | KIF20A | 0.662 | 53 | KRT80 | 0.638 |
| 4 | TROAP | 0.661 | 54 | C15orf42 | 0.637 |
| 5 | UBE2C | 0.659 | 55 | SGOL1 | 0.637 |
| 6 | AURKA | 0.658 | 56 | GPI | 0.637 |
| 7 | FAM83D | 0.657 | 57 | CEP55 | 0.637 |
| 8 | SHMT2 | 0.655 | 58 | MCM10 | 0.636 |
| 9 | UBE2C | 0.655 | 59 | PKMYT1 | 0.635 |
| 10 | CCNB2 | 0.653 | 60 | CENPN | 0.635 |
| 11 | TPX2 | 0.653 | 61 | C20orf24 | 0.635 |
| 12 | EXO1 | 0.653 | 62 | SPC24 | 0.635 |
| 13 | ORC6 | 0.653 | 63 | RIPK4 | 0.635 |
| 14 | CENPA | 0.653 | 64 | TOMM40 | 0.634 |
| 15 | C1orf106 | 0.652 | 65 | ANLN | 0.634 |
| 16 | GTSE1 | 0.652 | 66 | ADRM1 | 0.634 |
| 17 | MELK | 0.651 | 67 | NCAPG | 0.633 |
| 18 | STIP1 | 0.651 | 68 | CDCA8 | 0.633 |
| 19 | SPC25 | 0.649 | 69 | AIF1L | 0.633 |
| 20 | CENPA | 0.649 | 70 | MRPS5 | 0.633 |
| 21 | GARS | 0.649 | 71 | GPR56 | 0.633 |
| 22 | MELK | 0.649 | 72 | PEX13 | 0.633 |
| 23 | UCK2 | 0.648 | 73 | ENO1 | 0.633 |
| 24 | HJURP | 0.648 | 74 | NUTF2 | 0.633 |
| 25 | PTTG1 | 0.647 | 75 | MEMO1 | 0.632 |
| 26 | CBX2 | 0.646 | 76 | TXNRD1 | 0.632 |
| 27 | CCNE1 | 0.646 | 77 | SLC7A5 | 0.631 |
| 28 | PLK1 | 0.646 | 78 | FOXM1 | 0.631 |
| 29 | KIF2C | 0.645 | 79 | KIF14 | 0.631 |
| 30 | CCNA2 | 0.645 | 80 | PPP1R14B | 0.631 |
| 31 | GMPSP1 | 0.645 | 81 | FAM54A | 0.630 |
| 32 | AURKB | 0.645 | 82 | C20orf24 | 0.630 |
| 33 | BUB1 | 0.644 | 83 | SGOL1 | 0.630 |
| 34 | TRIP13 | 0.643 | 84 | NUP93 | 0.630 |
| 35 | FOXM1 | 0.643 | 85 | ZNF695 | 0.630 |
| 36 | CDC20 | 0.643 | 86 | BIRC5 | 0.630 |
| 37 | LOC731049 | 0.642 | 87 | CENPL | 0.630 |
| 38 | POLQ | 0.642 | 88 | SOX11 | 0.630 |
| 39 | GSK3B | 0.642 | 89 | KIF23 | 0.629 |
| 40 | CCNE1 | 0.642 | 90 | SLC52A2 | 0.629 |
| 41 | KIF4A | 0.641 | 91 | AIF1L | 0.629 |
| 42 | PRC1 | 0.641 | 92 | FEN1 | 0.629 |
| 43 | LAD1 | 0.641 | 93 | CDC25A | 0.629 |
| 44 | FAM64A | 0.641 | 94 | CDCA3 | 0.628 |
| 45 | SAPCD2 | 0.641 | 95 | TMEM132A | 0.628 |
| 46 | RACGAP1 | 0.641 | 96 | CENPE | 0.628 |
| 47 | POLR2D | 0.641 | 97 | NACC2 | 0.628 |
| 48 | CKAP2L | 0.640 | 98 | TTK | 0.628 |
| 49 | PTTG1 | 0.640 | 99 | SNRPA1 | 0.628 |
| 50 | ECE2 | 0.639 | 100 | MMP15 | 0.628 |
The 47 underlined genes are also among the top 100 genes of the mitotic CIN attractor (Table 2).
Lymphocyte-specific attractor
We found the attractor to be strongly protective in ER-negative breast cancers. As shown in Figure 4, the Kaplan-Meier 15-year survival curve restricted to ER-negative reveals association with longer survival (P = 0.004). Although the precise underlying biological mechanisms are unclear, it appears that this effect is due to some type of immune system recruitment to fight cancer. Interestingly, however, this effect is reversed in the presence of multiple positive lymph nodes. Indeed, the corresponding Kaplan-Meier curve shown on the right side of Figure 4 when restricted to patients with more than five positive lymph nodes demonstrates that the presence of the signature is associated with bad prognosis. This reversal may be explained by the fact that the presence of the lymphocyte-specific signature when lymph nodes are already affected implies that the cancer has obtained a (devastating for the patient) tolerance to this type of immune system recruitment.
Figure 4. Kaplan-Meier curve of lymphocyte-specific attractor metagenes in breast cancer dataset.

For ER-negative patients, the expression of the attractor is highly protective (high expression implies longer survival, left). However, when multiple lymph nodes are already affected, the expression of the attractor has a reversed effect on survival. When we restrict the samples to those with more than five positive lymph nodes, higher expression of the lymphocyte-specific attractor implies shorter survival (right), although the association is not significant due to the limited number of samples (76).
Other Attractors
Chr8q24.3 amplicon attractor
Amplification in chr8q24 is often considered to be associated with cancer because of the presence of the MYC (aka c-Myc) oncogene at location 8q24.21. Indeed, MYC is one of 157 genes in “amplicon 8q23-q24” previously identified [35] in an extensive study of the breast cancer “amplicome” derived from 191 samples.
We found, however, that the core of the amplified genes occurs at location 8q24.3 and this is, in fact, our most prominent multi-cancer amplicon attractor. Core genes of the attractor are PUF60 (aka FIR), EXOSC4, SHARPIN, HSF1, BOP1, SLC52A2. It is known that PUF60 can repress c-Myc via its far upstream element (FUSE), although a particular isoform was found to have the opposite effect [36]. The other genes may also play important roles. For example, HSF1 (heat shock transcription factor 1) has been associated with cancer in various ways [37]. It was found [38] that HSF1 can induce genomic instability through direct interaction with CDC20, a key gene of the mitotic CIN attractor mentioned above (listed in Table 2). Furthermore, HSF1 was found [39] required for the cell transformation and tumorigenesis induced by the ERBB2 (aka HER2) oncogene (see subsequent discussion of HER2 amplicon) responsible for aggressive breast tumors.
The top ten genes of the chr8q24.3 attractor, ranked by the average of the highest five values of mutual information (Materials and Methods), are shown in Table 5. Interestingly, as shown in one of the attractors in Supplementary Table S1, an aneuploidy attractor involving a whole arm amplification of chr8q is also occasionally present in multiple cancer types, and this 8q whole arm amplification is the most prominent such aneuploidy attractor among all chromosomes.
Table 5. List of top ten genes in the chr8q24.3 and HER2 amplicons.
| chr8q24.3 | HER2 | ||||
| Rank | Gene Symbol | Avg MI of Top 4 Datasets | Rank | Gene Symbol | Avg MI of Top 4 Datasets |
| 1 | EXOSC4 | 0.716 | 1 | PGAP3 | 0.794 |
| 2 | PUF60 | 0.659 | 2 | ERBB2 | 0.793 |
| 3 | BOP1 | 0.653 | 3 | STARD3 | 0.768 |
| 4 | SLC52A2 | 0.639 | 4 | MIEN1 | 0.764 |
| 5 | SHARPIN | 0.634 | 5 | GRB7 | 0.718 |
| 6 | HSF1 | 0.616 | 6 | PSMD3 | 0.602 |
| 7 | FBXL6 | 0.615 | 7 | GSDMB | 0.539 |
| 8 | CYC1 | 0.608 | 8 | ORMDL3 | 0.498 |
| 9 | SCRIB | 0.552 | 9 | MED24 | 0.414 |
| 10 | GPAA1 | 0.551 | 10 | MED1 | 0.400 |
Furthermore, prognostic associations involving the 8q24.3 amplicon have recently been recognized in various cancers [40], [41].
Chr17q12 HER2 amplicon attractor
This amplicon is prominent in breast cancer [42] and we also found it present in some samples of ovarian cancer, but not as much in colon cancer. So we initially used the four datasets of breast and ovarian cancer for deriving the attractor. We found that ERBB2 (aka HER2), STAR3, GRB7 and PGAP3 were the top-ranked genes, consistent with their known presence in the amplicon. We also found that gene MIEN1 (aka C17orf37) was very highly ranked in the two datasets in which its probe set was present. MIEN1 has recently been identified as an important gene within the 17q12 amplicon in various cancers including prostate cancer [43]. Therefore, we augmented the choice of datasets to the following seven, of which MIEN1 is included in five: breast GSE2034, breast GSE32646, breast GSE36771, breast TCGA, ovarian GSE9891, ovarian GSE26193, ovarian TCGA. Table 5 shows the top ten genes ranked by the average of the top five scores of mutual information (Materials and Methods) in the seven datasets for each gene. The results suggest that the above-mentioned five genes, including MIEN1, are consistently strongly co-expressed, and therefore are likely “driver” genes in the amplicon.
In addition to the narrow HER2 amplicon, it is known that sometimes a large amplicon extends to more than a million bases containing both HER2 as well as TOP2A (one of the genes of the mitotic CIN attractor) at 17q21 [44]. We have observed that TOP2A indeed appears among the top 50 genes in terms of its association with the attractor in breast cancer. HER2/TOP2A co-amplification has been linked with better clinical response to therapy.
Estrogen receptor breast cancer attractor
We found this attractor clearly present only in breast cancer, and therefore we derived it using six breast cancer datasets (GSE2034, GSE3494, GSE31448, GSE32646, GSE36771, breast TCGA). Table 6 shows the top 50 genes ranked by the average mutual information (Materials and Methods) in these datasets, revealing that genes AGR3, CA12, AGR2, GATA3, FOXA1, MLPH and TBC1D9 are strongly co-expressed with the estrogen receptor ESR1 in the attractor. However, this co-expression is not as uniform as in the other attractors. For example, the progesterone receptor PGR appear in the list, but in reality it can be underexpressed even if the estrogen receptor ESR1 is not.
Table 6. Top 50 genes of the estrogen receptor breast cancer attractor.
| Rank | Gene Symbol | Avg MI | Rank | Gene Symbol | Avg MI |
| 1 | AGR3 | 0.847 | 26 | ERBB4 | 0.393 |
| 2 | CA12 | 0.616 | 27 | AR | 0.383 |
| 3 | FOXA1 | 0.613 | 28 | P4HTM | 0.383 |
| 4 | GATA3 | 0.585 | 29 | SLC44A4 | 0.380 |
| 5 | MLPH | 0.580 | 30 | KDM4B | 0.375 |
| 6 | AGR2 | 0.570 | 31 | GFRA1 | 0.374 |
| 7 | ESR1 | 0.543 | 32 | MAPT | 0.370 |
| 8 | TBC1D9 | 0.540 | 33 | MYB | 0.364 |
| 9 | XBP1 | 0.460 | 34 | DACH1 | 0.359 |
| 10 | ANXA9 | 0.456 | 35 | SLC7A8 | 0.359 |
| 11 | PRR15 | 0.452 | 36 | MAGED2 | 0.358 |
| 12 | SCUBE2 | 0.444 | 37 | FBP1 | 0.357 |
| 13 | FSIP1 | 0.438 | 38 | SLC22A5 | 0.355 |
| 14 | TFF3 | 0.429 | 39 | CMBL | 0.346 |
| 15 | SPDEF | 0.429 | 40 | DYNLRB2 | 0.346 |
| 16 | NAT1 | 0.428 | 41 | C6orf211 | 0.342 |
| 17 | ABAT | 0.423 | 42 | GREB1 | 0.342 |
| 18 | CCDC170 | 0.422 | 43 | SIDT1 | 0.338 |
| 19 | DNALI1 | 0.418 | 44 | TTC39A | 0.330 |
| 20 | DEGS2 | 0.415 | 45 | FAM214A | 0.326 |
| 21 | DNAJC12 | 0.411 | 46 | IL6ST | 0.324 |
| 22 | SLC39A6 | 0.406 | 47 | CXXC5 | 0.323 |
| 23 | CAPN8 | 0.399 | 48 | ACADSB | 0.323 |
| 24 | TFF1 | 0.397 | 49 | CELSR1 | 0.322 |
| 25 | THSD4 | 0.395 | 50 | CLSTN2 | 0.322 |
Comparison with Other Unsupervised Algorithms
The scope of the algorithm identifying attractor metagenes is different from that of other unsupervised methods, which are usually aimed at identifying subtypes or mutually exclusive clusters. Nevertheless, it is interesting to find to what extent other algorithms can produce multiple cancer signatures each of which appears in nearly identical form across different types. We applied three widely used methods, k-means clustering, principal component analysis and hierarchical clustering on the six cancer datasets used in this paper. In all cases, we listed the top fifty genes in each cluster and applied the same clustering algorithm as in the main text to find common genes among them and group them together. The results are shown in Supplementary Text S2 and Supplementary Tables S5, S6, S7. We found that, in all cases, these well-established methods cannot identify multiple universal metagenes common in all six tested datasets.
Using Attractor Metagenes as Proxies of Biomolecular Events
A biomolecular event, whether it is present in multiple cancer types or it is cancer specific, can be represented by a “consensus attractor metagene” after analyzing multiple datasets. To generate such consensus attractors, we use genes that were profiled by at least three of the six datasets, then rank individual genes in terms of their average mutual information (Materials and Methods) with the corresponding attractor metagenes across all datasets in which the gene was profiled.
For example, Figure 5 contains scatter plots from four different rich breast cancer datasets connecting the mitotic CIN and estrogen receptor attractors. It has previously been reported [45] that breast tumors with high chromosomal instability are predominantly of the estrogen receptor negative phenotype. Although these scatter plots cannot be used for precise conclusions, they do suggest in all cases that ER-negative tumors have high mitotic chromosomal instability (or equivalently that low chromosomal instability implies that the tumor is ER-positive). The reverse relationship, however, is not as clear.
Figure 5. Scatter plots demonstrating the relationship between mitotic CIN attractor and estrogen receptor attractor in breast cancer.

The two metagenes were defined to be “consensus attractors” after ranking individual genes in terms of their average mutual information with the corresponding attractor metagenes, across all datasets, and selecting the genes having average mutual information greater than 0.5. These criteria led to 59 genes in the consensus mitotic CIN attractor (the top 59 genes in Table 2), and AGR3, ESR1, CA12, AGR2, GATA3, FOXA1, MLPH and TBC1D9 (the top eight genes in Table 6) in the consensus estrogen receptor breast cancer attractor. These scatter plots reveal that ER-negative breast tumors have high mitotic chromosomal instability, but not necessarily vice versa.
Discussion
Gene expression analysis has resulted in several cancer types being further classified into subtypes labeled, e.g. as “mesenchymal” or “proliferative.” Such characterizations, however, may sometimes simply reflect the presence of the mesenchymal transition attractor or the mitotic chromosomal instability attractor, respectively, in some of the analyzed samples. Similar subtype characterizations across cancer types often share several common genes, but the consistency of these similarities has not been significantly high.
By contrast, using an unconstrained algorithm independent of subtype classification or dimensionality reduction, we identified several attractors exhibiting remarkable consistency across many cancer types, suggesting that each of them represents a precise biological phenomenon present in multiple cancers.
We found that the mesenchymal transition attractor is significantly present only in samples whose stage designation has exceeded a threshold, but not in all of such samples. On the other hand, the absence of the mesenchymal transition attractor in a profiled high-stage sample (or the absence of the mitotic chromosomal instability attractor in a profiled high-grade sample) does not necessarily mean that the attractor is not present in other locations of the same tumor. Indeed, it is increasingly appreciated [46] that tumors are highly heterogeneous. Therefore it is possible for the same tumor to contain components, in which, e.g., some are migratory having undergone mesenchymal transition, some other ones are highly proliferative, etc. If so, attempts for subtype classification based on one particular site in a sample may be confusing.
Existing molecular marker products make use of multigene assays that have been derived from phenotypic associations in particular cancer types. For breast cancer, biomarkers such as Oncotype DX [47] and Mammaprint [48] contain several genes highly ranked in our attractors. For example, most of the genes used for the Oncotype DX breast cancer recurrence score directly converge to one of our identified attractors: MMP11 to the mesenchymal transition attractor; MKI67 (aka Ki-67), AURKA (aka STK15), BIRC5 (aka Survivin), CCNB1, and MYBL2 to the mitotic CIN attractor; CD68 to the lymphocyte-specific attractor; ERBB2 and GRB7 to the HER2 amplicon attractor; and ESR1, SCUBE2, PGR to the estrogen receptor attractor.
We envision, instead, a multi-cancer biomarker product that will include detection of the level of expression of each of the key attractor metagenes. These levels would need to be combined in different ways in different cancer types, but each of the metagenes would indicate the same attribute and the contributions of each component will be cleanly distinguished. Even though molecular marker genes in some existing products are already separated into groups that are related to our attractor designation, any improvement in diagnostic, prognostic, or predictive accuracy resulting from better such group designation and better choice of genes in each group would be highly desirable. We hope that the identification of the attractors of cancer, as presented here, will be valuable in that regard.
Materials and Methods
The full code of the attractor finding algorithm is publicly available in the Sage Bionetworks Synapse platform at https://synapse.sagebase.org/#Synapse:syn1446295. In addition, we provide a pseudo-code in Supplementary Text S3.
General Attractor Finding Algorithm
We chose the association measure  between genes to be a power function with exponent a of a normalized estimated information theoretic measure of the mutual information [49]
between genes to be a power function with exponent a of a normalized estimated information theoretic measure of the mutual information [49]
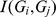 with minimum value 0 and maximum value 1 (see “Mutual information estimation” below; more sophisticated related association measures [50] can also be used, but computational complexity will be prohibitive). In other words,
with minimum value 0 and maximum value 1 (see “Mutual information estimation” below; more sophisticated related association measures [50] can also be used, but computational complexity will be prohibitive). In other words,  , in which the exponent
, in which the exponent  can be any nonnegative number. The value of
can be any nonnegative number. The value of  is set to zero if the Pearson correlation between the two genes is negative. Each iteration defines a new metagene in which the weight
is set to zero if the Pearson correlation between the two genes is negative. Each iteration defines a new metagene in which the weight for gene
for gene  is equal to
is equal to 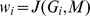 where
where  is the immediately preceding metagene. The process is repeated until the magnitude of the difference between two consecutive weight vectors is less than a threshold, which we chose to be equal to 10−7.
is the immediately preceding metagene. The process is repeated until the magnitude of the difference between two consecutive weight vectors is less than a threshold, which we chose to be equal to 10−7.
At one extreme, if  is sufficiently large then each of the seeds will create its own single-gene attractor because all other genes will always have near-zero weights. In that case, the total number of attractors will be equal to the number of genes. At the other extreme, if
is sufficiently large then each of the seeds will create its own single-gene attractor because all other genes will always have near-zero weights. In that case, the total number of attractors will be equal to the number of genes. At the other extreme, if  is zero then all weights will remain equal to each other representing the average of all genes, so there will only be one attractor. The higher the value of
is zero then all weights will remain equal to each other representing the average of all genes, so there will only be one attractor. The higher the value of  , the “sharper” (more focused on its top gene) each attractor will be and the higher the total number of attractors will be. As the value of
, the “sharper” (more focused on its top gene) each attractor will be and the higher the total number of attractors will be. As the value of  is gradually decreased, the attractor from a particular seed will transform itself, occasionally in a discontinuous manner, thus providing insight into potential related biological mechanisms.
is gradually decreased, the attractor from a particular seed will transform itself, occasionally in a discontinuous manner, thus providing insight into potential related biological mechanisms.
We empirically found that an appropriate choice of  (in the sense of maximizing the strength of the attractor, as defined below) for general attractors is around 5, in which case there will typically be approximately 50 to 150 resulting attractors, each resulting from many attractee genes. An alternative to the power function can be a sigmoid function with varying steepness, but we found that the consistency of the resulting attractors was worse in that case.
(in the sense of maximizing the strength of the attractor, as defined below) for general attractors is around 5, in which case there will typically be approximately 50 to 150 resulting attractors, each resulting from many attractee genes. An alternative to the power function can be a sigmoid function with varying steepness, but we found that the consistency of the resulting attractors was worse in that case.
As mentioned in the Introduction, an attractor metagene can also be interpreted as a set of the top genes of the attractor, i.e., a gene set that includes only the genes that are significantly associated with the attractor. One empirical choice for such a gene set would be to include only the genes whose mutual information (or the z-score thereof) with the attractor metagene exceeds a particular threshold. In fact, the attractor finding algorithm itself can be designed to discover “attractor gene sets,” without assigning weights to genes. In that case, metagenes are defined as simple averages of the genes in a gene set, and each iteration leads to a new gene set consisting of the new set of top-ranked genes in terms of their association with the previous metagene (gene set sizes can be constant or adaptively changing in various ways). We found, however, that such a method has the disadvantage of occasionally leading to attractors with significant overlap, which requires additional post-processing steps.
Identified attractors can be ranked in various ways. The “strength of an attractor” can be defined as the mutual information between the n th top gene of the attractor and the attractor metagene. Indeed, if this measure is high, this implies that at least the top n genes of the attractor are strongly co-expressed. We selected n = 10 as a reasonable choice, not too large, but sufficiently so to represent a real complex biological phenomenon of co-expression of at least ten genes. For amplicons, n = 5 is sufficient to ensure that the oncogenes are included in the co-expression). We use these choices when referring to the strength of an attractor.
The top genes of many among the found attractors are genomically localized. In that case the biomolecular event that they represent is often the presence of a particular copy number variation. In the cancer datasets that we tried, this phenomenon almost always corresponds to a local amplification event known as an amplicon. We therefore also devised a related amplicon-finding algorithm, custom-designed to identify localized amplicon-representing attractor metagenes, described below.
Genomically Localized Attractor Finding Algorithm
To identify genomically localized attractors – almost always amplicons – we use the same algorithm but for each seed gene we restrict the set of candidate attractor genes to only include those in the local genomic neighbourhood of the gene, and we optimize the exponent a so that the strength of the attractor is maximized. Specifically, we sort the genes in each chromosome in terms of their genomic location and we only consider the genes within a window of size 51, i.e., with 25 genes on each side of the seed gene. We further optimize the choice of the exponent  for each seed, by allowing
for each seed, by allowing  to range from 1.0 to 6.0 with step size of 0.5 and selecting the attractor with the highest strength.
to range from 1.0 to 6.0 with step size of 0.5 and selecting the attractor with the highest strength.
Because the set of allowed genes is different for each seed, the attractors will be different from each other, but “neighbouring” attractors will usually be very similar to each other. Therefore, following exhaustive attractor finding from each seed gene in a chromosome, we apply a filtering algorithm to only select the highest-strength attractor in each local genomic region, as follows: For each attractor, we rank all the genes in terms of their mutual information with the corresponding attractor metagene and we define the range of the attractor to be the chromosomal range of its top 15 genes. If there is any other attractor with overlapping range and higher strength, then the former attractor is filtered out. This filtering is done in parallel, so elimination of attractors occurs simultaneously.
Mutual Information Estimation
Assuming that the continuous expression levels of two genes  and
and  are governed by a joint probability density
are governed by a joint probability density  with corresponding marginal
with corresponding marginal  and
and  , the mutual information
, the mutual information 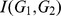 is defined as the expected value of
is defined as the expected value of  . It is a non-negative quantity representing the information that each one of the variables provides about the other. The pairwise mutual information has successfully been used as a general measure of the correlation between two random variables. We compute mutual information with a spline-based estimator [51] using six bins in each dimension. This method divides the observation space into equally spaced bins and blurs the boundaries between the bins with spline basis functions using third-order B-splines. We further normalize the estimated mutual information by dividing by the maximum of the estimated
. It is a non-negative quantity representing the information that each one of the variables provides about the other. The pairwise mutual information has successfully been used as a general measure of the correlation between two random variables. We compute mutual information with a spline-based estimator [51] using six bins in each dimension. This method divides the observation space into equally spaced bins and blurs the boundaries between the bins with spline basis functions using third-order B-splines. We further normalize the estimated mutual information by dividing by the maximum of the estimated  and
and  , so the maximum possible value of
, so the maximum possible value of  is 1.
is 1.
Pre-processing Gene Expression Datasets
We used Level 3 data when directly available, and imputed missing values using a k-nearest-neighbour algorithm with k = 10, as implemented in R [52]. We normalized the other datasets on the Affymetrix platform using the RMA algorithm as implemented in the affy package in Bioconductor [53]. To avoid biasing attractor convergence with multiple correlated probe sets of the same gene, we summarized the probe set-level expression values into the gene-level expression values by taking the mean of the expression values of probe sets for the same genes. We used the annotations for the probe sets given in the jetset package [54].
To investigate the associations between the attractor metagene expression and the tumor stage and grade, we used the following annotated gene expression datasets. For stage association: Breast (GSE3893), TCGA Ovarian, Colon (GSE14333). For grade association: Breast (GSE3494), TCGA Ovarian, Bladder (GSE13507). For Breast GSE3494 we used only the samples profiled by U133A arrays. For Breast GSE3893 we combined two platforms by taking the intersections of the probes in the U133A and the U133Plus 2.0 arrays. For datasets profiled by Affymetrix platforms all the datasets were normalized using the RMA algorithm. For Bladder GSE13507 normalization was done as provided in the GEO.
P Value Evaluation
P values for gene set enrichment were evaluated with the cumulative hypergeometric distribution using the total number of genes in each dataset.
The significance of the consistency of the mesenchymal transition and mitotic CIN attractors was evaluated as follows: Supplementary Table S1 contains 210 gene sets from six cancer datasets. Each of the gene sets contains 50 genes. The mesenchymal transition metagene has eight genes (COL5A2, COL1A2, SPARC, CTSK, FBN1, VCAN, AEBP1, SERPINF1) common across all six datasets. The mitotic CIN metagene has 13 common genes (CENPA, DLGAP5, KIF2C, CCNB2, MELK, CCNA2, KIF20A, HJURP, NUSAP1, BUB1, TTK, KIF11, NCAPH) across all six datasets. To evaluate the significance of the consistency across the six datasets, we randomly generated 210 gene sets with the same sizes as those in the Table. In other words, we randomly selected 50 genes out of the 11,395 common genes to generate a random gene set. We created 210 such random gene sets, and then assigned them to six different datasets according to the settings in the Table. We then performed the clustering algorithm described in Materials and Methods. Each time, we counted the maximum number of genes common in all six datasets, and we repeated this process ten million times. This constitutes a conservative way of evaluating consistency, in the sense that for each random experiment we only record the maximum number of common genes in the gene set cluster, and we created random gene sets using only the common genes in the six datasets. In these ten million experiments, it never occurred that more than one gene was common in all six datasets. Therefore, the corresponding P value for both the mesenchymal transition metagene as well as the mitotic CIN metagene is less than 10−7, and is in fact much lower than that given the large number (8 and 13 respectively) of the common genes.
Supporting Information
General attractors identified from the six datasets.
(XLS)
Genomically localized attractors identified from the six datasets.
(XLS)
Association of mesenchymal transition attractor with tumor stage.
(XLS)
Association of mitotic CIN attractor with tumor grade.
(XLS)
Common clusters from the six datasets using k-means.
(XLS)
Common clusters from the six datasets using principal component analysis.
(XLS)
Common clusters from the six datasets using hierarchical clustering.
(XLS)
Comparison with other unsupervised algorithms.
(DOCX)
Pseudo-code for attractor metagene finding algorithm.
(DOCX)
Acknowledgments
We thank the Chang Gung Memorial Hospital-Linkou and Chang Gung University, Taoyuan, Taiwan, R.O.C. and in particular their Head and Neck Oncology Group and Dr. Tzu-Chen Yen who served as our contact point, for providing us with survival data for dataset GSE25104. This study makes use of data generated by the Molecular Taxonomy of Breast Cancer International Consortium. Funding for that project was provided by Cancer Research UK and the British Columbia Cancer Agency Branch. Our results using these data (the METABRIC 996-sample discovery dataset) were produced during our participation in the Sage Bionetworks/DREAM breast cancer prognosis Challenge.
Funding Statement
This work was funded by Columbia University's unrestricted-purpose allocation of inventor's research of royalties resulting from intellectual property totally unrelated to the work described in this paper. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 2. Nevins JR, Potti A (2007) Mining gene expression profiles: expression signatures as cancer phenotypes. Nat Rev Genet 8: 601–609. [DOI] [PubMed] [Google Scholar]
- 3. Segal E, Friedman N, Kaminski N, Regev A, Koller D (2005) From signatures to models: understanding cancer using microarrays. Nat Genet 37 Suppl: S38–45. [DOI] [PubMed] [Google Scholar]
- 4. Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, et al. (2011) Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 17: 500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, et al. (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cancer Genome Atlas Research N (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474: 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brunet JP, Tamayo P, Golub TR, Mesirov JP (2004) Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci U S A 101: 4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Monti S, Tamayo P, Mesirov J, Golub T (2003) Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Machine Learning 52: 91–118. [Google Scholar]
- 9. Whitfield ML, George LK, Grant GD, Perou CM (2006) Common markers of proliferation. Nat Rev Cancer 6: 99–106. [DOI] [PubMed] [Google Scholar]
- 10. Kim H, Watkinson J, Varadan V, Anastassiou D (2010) Multi-cancer computational analysis reveals invasion-associated variant of desmoplastic reaction involving INHBA, THBS2 and COL11A1. BMC Med Genomics 3: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anastassiou D, Rumjantseva V, Cheng W, Huang J, Canoll PD, et al. (2011) Human cancer cells express Slug-based epithelial-mesenchymal transition gene expression signature obtained in vivo. BMC Cancer 11: 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheng WY, Kandel JJ, Yamashiro DJ, Canoll P, Anastassiou D (2012) A multi-cancer mesenchymal transition gene expression signature is associated with prolonged time to recurrence in glioblastoma. PLoS One 7: e34705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, et al. (2009) A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med 15: 68–74. [DOI] [PubMed] [Google Scholar]
- 14. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, et al. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133: 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hay ED (1995) An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 154: 8–20. [DOI] [PubMed] [Google Scholar]
- 16. Thiery JP (2002) Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2: 442–454. [DOI] [PubMed] [Google Scholar]
- 17. Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119: 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yin G, Chen R, Alvero AB, Fu HH, Holmberg J, et al. (2010) TWISTing stemness, inflammation and proliferation of epithelial ovarian cancer cells through MIR199A2/214. Oncogene 29: 3545–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, et al. (2006) Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst 98: 262–272. [DOI] [PubMed] [Google Scholar]
- 20. Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z (2006) A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet 38: 1043–1048. [DOI] [PubMed] [Google Scholar]
- 21. Schvartzman JM, Sotillo R, Benezra R (2010) Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer 10: 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuen KW, Montpetit B, Hieter P (2005) The kinetochore and cancer: what's the connection? Curr Opin Cell Biol 17: 576–582. [DOI] [PubMed] [Google Scholar]
- 23. Amato A, Schillaci T, Lentini L, Di Leonardo A (2009) CENPA overexpression promotes genome instability in pRb-depleted human cells. Mol Cancer 8: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sotillo R, Schvartzman JM, Socci ND, Benezra R (2010) Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature 464: 436–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heidebrecht HJ, Adam-Klages S, Szczepanowski M, Pollmann M, Buck F, et al. (2003) repp86: A human protein associated in the progression of mitosis. Mol Cancer Res 1: 271–279. [PubMed] [Google Scholar]
- 26. Orr-Weaver TL, Weinberg RA (1998) A checkpoint on the road to cancer. Nature 392: 223–224. [DOI] [PubMed] [Google Scholar]
- 27. Sadasivam S, Duan S, DeCaprio JA (2012) The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev 26: 474–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Manning AL, Dyson NJ (2012) RB: mitotic implications of a tumour suppressor. Nat Rev Cancer 12: 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rosty C, Sheffer M, Tsafrir D, Stransky N, Tsafrir I, et al. (2005) Identification of a proliferation gene cluster associated with HPV E6/E7 expression level and viral DNA load in invasive cervical carcinoma. Oncogene 24: 7094–7104. [DOI] [PubMed] [Google Scholar]
- 30. Andreopoulos B, Anastassiou D (2012) Integrated Analysis Reveals hsa-miR-142 as a Representative of a Lymphocyte-Specific Gene Expression and Methylation Signature. Cancer Inform 11: 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee MS, Hanspers K, Barker CS, Korn AP, McCune JM (2004) Gene expression profiles during human CD4+ T cell differentiation. Int Immunol 16: 1109–1124. [DOI] [PubMed] [Google Scholar]
- 32. Wang H, Wei B, Bismuth G, Rudd CE (2009) SLP-76-ADAP adaptor module regulates LFA-1 mediated costimulation and T cell motility. Proc Natl Acad Sci U S A 106: 12436–12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, et al. (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486: 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pencina MJ, D'Agostino RB (2004) Overall C as a measure of discrimination in survival analysis: model specific population value and confidence interval estimation. Stat Med 23: 2109–2123. [DOI] [PubMed] [Google Scholar]
- 35. Nikolsky Y, Sviridov E, Yao J, Dosymbekov D, Ustyansky V, et al. (2008) Genome-wide functional synergy between amplified and mutated genes in human breast cancer. Cancer Res 68: 9532–9540. [DOI] [PubMed] [Google Scholar]
- 36. Matsushita K, Tomonaga T, Shimada H, Shioya A, Higashi M, et al. (2006) An essential role of alternative splicing of c-myc suppressor FUSE-binding protein-interacting repressor in carcinogenesis. Cancer Res 66: 1409–1417. [DOI] [PubMed] [Google Scholar]
- 37. Dai C, Whitesell L, Rogers AB, Lindquist S (2007) Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130: 1005–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee YJ, Lee HJ, Lee JS, Jeoung D, Kang CM, et al. (2008) A novel function for HSF1-induced mitotic exit failure and genomic instability through direct interaction between HSF1 and Cdc20. Oncogene 27: 2999–3009. [DOI] [PubMed] [Google Scholar]
- 39. Meng L, Gabai VL, Sherman MY (2010) Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene 29: 5204–5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bilal E, Vassallo K, Toppmeyer D, Barnard N, Rye IH, et al. (2012) Amplified Loci on Chromosomes 8 and 17 Predict Early Relapse in ER-Positive Breast Cancers. PLoS One 7: e38575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peng CH, Liao CT, Peng SC, Chen YJ, Cheng AJ, et al. (2011) A novel molecular signature identified by systems genetics approach predicts prognosis in oral squamous cell carcinoma. PLoS One 6: e23452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Theillet C (2010) What do we learn from HER2-positive breast cancer genomic profiles? Breast Cancer Res 12: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dasgupta S, Wasson LM, Rauniyar N, Prokai L, Borejdo J, et al. (2009) Novel gene C17orf37 in 17q12 amplicon promotes migration and invasion of prostate cancer cells. Oncogene 28: 2860–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arriola E, Marchio C, Tan DS, Drury SC, Lambros MB, et al. (2008) Genomic analysis of the HER2/TOP2A amplicon in breast cancer and breast cancer cell lines. Lab Invest 88: 491–503. [DOI] [PubMed] [Google Scholar]
- 45. Birkbak NJ, Eklund AC, Li Q, McClelland SE, Endesfelder D, et al. (2011) Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res 71: 3447–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, et al. (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Paik S, Shak S, Tang G, Kim C, Baker J, et al. (2004) A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 351: 2817–2826. [DOI] [PubMed] [Google Scholar]
- 48. van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, et al. (2002) Gene expression profiling predicts clinical outcome of breast cancer. Nature 415: 530–536. [DOI] [PubMed] [Google Scholar]
- 49.Cover TM, Thomas JA (2006) Elements of information theory. Hoboken, N.J.: Wiley-Interscience. xxiii, 748 p. [Google Scholar]
- 50. Reshef DN, Reshef YA, Finucane HK, Grossman SR, McVean G, et al. (2011) Detecting novel associations in large data sets. Science 334: 1518–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Daub CO, Steuer R, Selbig J, Kloska S (2004) Estimating mutual information using B-spline functions–an improved similarity measure for analysing gene expression data. BMC Bioinformatics 5: 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, et al. (2001) Missing value estimation methods for DNA microarrays. Bioinformatics 17: 520–525. [DOI] [PubMed] [Google Scholar]
- 53. Gautier L, Cope L, Bolstad BM, Irizarry RA (2004) affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20: 307–315. [DOI] [PubMed] [Google Scholar]
- 54. Li Q, Birkbak NJ, Gyorffy B, Szallasi Z, Eklund AC (2011) Jetset: selecting the optimal microarray probe set to represent a gene. BMC Bioinformatics 12: 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General attractors identified from the six datasets.
(XLS)
Genomically localized attractors identified from the six datasets.
(XLS)
Association of mesenchymal transition attractor with tumor stage.
(XLS)
Association of mitotic CIN attractor with tumor grade.
(XLS)
Common clusters from the six datasets using k-means.
(XLS)
Common clusters from the six datasets using principal component analysis.
(XLS)
Common clusters from the six datasets using hierarchical clustering.
(XLS)
Comparison with other unsupervised algorithms.
(DOCX)
Pseudo-code for attractor metagene finding algorithm.
(DOCX)