

Abstract
A hybrid electrolysis and Pd-catalytic oxidation process is evaluated for degradation of trichloroethylene (TCE) in groundwater. A three-electrode, one anode and two cathodes, column is employed to automatically develop a low pH condition in the Pd vicinity and a neutral effluent. Simulated groundwater containing up to 5 mM bicarbonate can be acidified to below pH 4 in the Pd vicinity using a total of 60 mA with 20 mA passing through the third electrode. By packing 2 g of Pd/Al2O3 pellets in the developed acidic region, the column efficiency for TCE oxidation in simulated groundwater (5.3 mg/L TCE) increases from 44 to 59 and 68% with increasing Fe(II) concentration from 0 to 5 and 10 mg/L, respectively. Different from Pd-catalytic hydrodechlorination under reducing conditions, this hybrid electrolysis and Pd-catalytic oxidation process is advantageous in controlling the fouling caused by reduced sulfur compounds (RSCs) because the in situ generated reactive oxidizing species, i.e., O2, H2O2 and •OH, can oxidize RSCs to some extent. In particular, sulfite at concentrations less than 1 mM even greatly increases TCE oxidation by the production of SO4•−, a strong oxidizing radical, and more •OH.
Keywords: Pd, electrolytic, TCE, groundwater remediation, reduced sulfur compounds
1. Introduction
Electrochemical methods have attracted great interests for water treatment, particularly for degradation of biorefractory pollutants (Brillas et al., 2009; Chen, 2004; Reddy and Cameselle, 2009). Contaminated groundwater has moderate electrical conductivity (500–1000 μΩ/cm) and low concentrations of pollutants. These features support the applicability of electrochemical processes for groundwater remediation. Compared with wastewater treatment, in situ groundwater remediation requires high stability electrodes because the remediation could be applied for a few years and electrode replacement is costly. Direct oxidation or reduction of contaminants on the surface of the anode or the cathode requires special electrocatalytic electrode materials, that will work at high overpotential conditions to obtain vigorous redox condition (Anglada et al., 2009; Chen et al., 2003; Carter and Farrell, 2009; Mishra et al., 2008; Vlyssides et al., 2004; Yang et al., 2007; Zhao et al., 2010). These electrodes, e.g., boron doped diamond (Anglada et al., 2009; Carter and Farrell, 2009; Mishra et al., 2008) and Pd supported materials (Chen et al., 2003; Yang et al., 2007), can achieve high degradation efficiency. However, they are costly and are difficult to implement for large-scale groundwater remediation.
The products of water electrolysis, H2, O2, H+ and OH− (1–2), using low-cost stable electrodes, such as Ti-based mixed metal oxide (Ti/MMO), provide opportunities for combining electrolysis with other processes for groundwater remediation. For example, O2 produced at the anode can be used for aerobic degradation (Franz et al., 2002; Lohner and Tiehm, 2009; Lohner et al., 2011), H2 produced at cathode can be delivered for anaerobic degradation (Lohner and Tiehm, 2009; Lohner et al., 2011; Weathers et al., 1997), and OH− produced at cathode can be utilized for contaminant hydrolysis (Gent et al., 2009).
Although catalytic hydrodechlorination by Pd and H2 is very effective in transformation of chlorinated solvents in groundwater (Chaplin et al., 2012; Hildebrand et al., 2009; Lowry and Reinhard, 2000, 2001; Nutt et al., 2005; Schueth et al., 2004), transportation, storage and in situ injection of gaseous H2 is hazardous and costly. As a solution, in situ generation of H2 at the cathode by water electrolysis (2) is proposed for Pd-catalytic hydrodechlorination of chlorinated organics (Mcnab and Ruiz, 1998; Zheng et al., 2012). Simultaneous generation of O2 at the anode (1) may compete for [H] and suppress contaminant hydrodechlorination (Lowry and Reinhard, 2000). Our recent work shows that anodic O2 may combine with cathodic H2 on Pd surface generating H2O2 (3) (Yuan et al., 2011), which can be decomposed to strong oxidizing •OH radicals (oxidation potential: 2.8 V vs SHE) by Pd and intrinsic Fe(II) in groundwater (4) (Yuan et al., 2012). In this hybrid electrolysis and Pd catalytic process, oxidation instead of hydrodechlorination is proven as the dominant pathway for trichloroethylene (TCE) degradation (Yuan et al., 2012). Weak acidity (pH≤4) is required to attain high degradation efficiency, which could be critical for actual application.
| (1) |
| (2) |
| (3) |
| (4) |
It is well recognized that reduced sulfur compounds (RSCs), which are produced from anaerobic bacterial respiration in aquifers, dramatically deactivate Pd during hydrodechlorination (Chaplin et al., 2012; Davie et al., 2008; Lowry and Reinhard, 2000; Schueth et al., 2004). Oxidizing reagents, such as hypochlorite (Chaplin et al., 2007; Davie et al., 2008; Lowry and Reinhard, 2000), hydrogen peroxide (Schueth et al., 2004), and permanganate (Angeles-Wedler et al., 2008), can regenerate sulfur-fouled Pd catalyst to some extent by oxidizing RSCs, and co-existence of MnO4−, an oxidant, can also prevent Pd deactivation (Angeles-Wedler et al., 2008). When water electrolysis is combined with Pd catalytic oxidation in the presence of Fe(II), the in situ generated reactive oxidizing species, i.e., O2, H2O2 and •OH, are capable of oxidizing RCSs simultaneously while degrading contaminants, thereby preventing Pd deactivation. Our recent preliminary results in batch setups show that sulfite significantly enhances TCE oxidation whereas sulfide slightly inhibits the oxidation under electro-generated oxidizing conditions (Yuan et al., 2012). However, the mechanism for the distinct influence has not been elucidated. In particular, the unexpected degradation enhancement in the presence of sulfite, as well the transformation kinetics under flow conditions are interesting and requires investigation.
In this study, a Pd-containing electrolytic column using three electrodes, two cathodes and one anode, is developed to achieve automatic pH-regulation, that is, weak acidity in Pd vicinity and neutral effluent. The column performance is evaluated for transformation of TCE, a common contaminant at many sites in the United States, in simulated groundwater. The reactive transport of contaminants is simulated, and the influence of sulfite and sulfide on degradation under flow conditions is evaluated. Ultimately, the main objectives are to evaluate contaminant degradation with automatic electrochemical pH-regulation, and to reveal the mechanism for the influence of RCSs on TCE degradation by the process.
2. Materials and methods
2.1. Chemicals
TCE (99.5%) and cis-dichloroethylene (cis-DCE, 97%) were purchased from Sigma-Aldrich. H2O2 (30%) and Na2SO3 (98.1%) were purchased from Fisher Sci. Na2S •9H2O (98%) and tert-bytyl alcohol (TBA, 99%) were obtained from Acros. 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) was provided by Cayman Chemical Company (USA). Excess TCE was dissolved into 18.2 mΩ·cm high-purity water to form a TCE saturated solution (1.07 mg/mL at 20 °C), which was used as stock solution for preparing aqueous TCE solutions. Palladium on alumina powder (1% wt. Pd, Sigma-Aldrich) with average particle size of 6 μm was used as catalyst in batch experiments. Palladium on alumina pellet (0.5% wt. Pd, Sigma-Aldrich) at much larger average size of 3.2 mm (Fig. S1 in Supporting Information) was used in column experiments. Deionized water (18.0 mΩ·cm) obtained from a Millipore Milli-Q system was used in all the experiments. All chemicals used in this study were above analytical grade.
2.2. Three-electrode column experiments
A vertical acrylic column (3.175 cm inner diameter × 30 cm length) (Fig. 1) was used for flow-through experiments. Three pieces of MMO mesh were installed in sequence as Anode, Cathodes 1 and 2. Preliminary results indicate that Fe(II) was precipitated prior to Pd vicinity when Cathode 1 was placed at the bottom, so the aforementioned electrode sequence is employed. 2 g of Pd/Al2O3 pellets were supported by Cathode 1 reaching a monolayer of pellet bed. This dosage of catalyst is packed because moderate removals of contaminants can be achieved to facilitate comparisons. By adjusting the rheostat, small fraction of current will pass through Cathode 2 so that an automatic pH-regulation can be achieved. No fillings were packed between the Anode and Cathode 1 in order to reduce the electric resistance. All the remaining space in the column was packed with 4-mm glass bead with a porosity of 0.65. The total and pore volume (PV) of the column are 245 and 160 mL, respectively. Simulated TCE-contaminated groundwater (5.3 mg/L) was prepared by dissolving TCE-saturated solution in 3 mM Na2SO4 and 0.5 mM CaSO4 deoxygenated solution (~ 800 μS/cm). The simulated contaminated groundwater was stored in gas-tight collapsing bad. Prior to electrolysis, the column was flushed by 2 PVs of contaminated groundwater. Adsorption of TCE on Pd/Al2O3 pellets and glass beads was measured to be insignificant. The flow rate was maintained at 2 mL/min (0.25 cm/min) using a peristaltic pump (Cole Parmer). A total current of 60 mA, with voltage varying from 22 to 30 V, was sustained with 40 and 20 mA passing through Cathodes 1 and 2, respectively. At regular time intervals, 1 mL of aqueous solution was sampled from 6 ports (see Fig. 1 for locations) and immediately mixed with 1 mL of methanol for analysis.
Fig. 1.
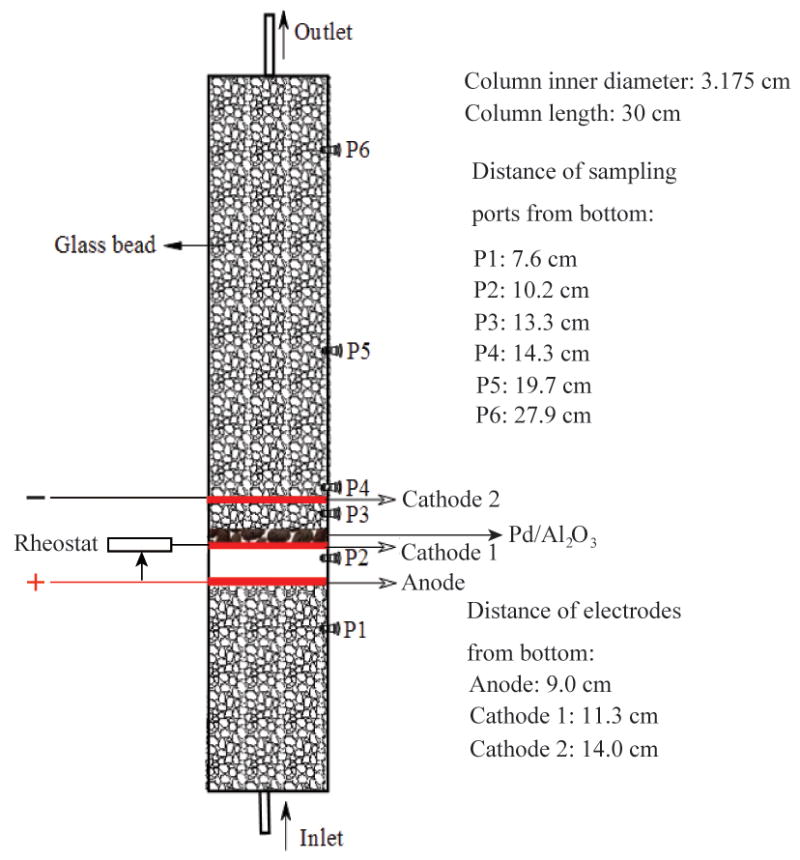
A schematic of three-electrode column
Four sets of column experiments were conducted with testing parameters as listed in Table 1. The first set (Columns C1 to C4) evaluates the automatic pH-regulation at different operation conditions and different bicarbonate concentrations. The second set (Columns C5 to C9) evaluates the performance of contaminant oxidation with automatic pH-regulation in the presence of different concentrations of Fe(II). The third set (Columns C10 to C11) investigates the influence of sulfite and sulfite on TCE degradation. And the last set (Column C12) assesses the long-term performance of this process on resisting fouling by sulfide.
Table 1.
Parameters and results associated with column experiments
| no.a | Column description | Steady-state pH | Fitted rate constants k (min−1) | Removal efficiency b | ||
|---|---|---|---|---|---|---|
|
| ||||||
| Pd vicinity | Effluent | At electrodes | Between anode and Cathode 2 | |||
| C1 | 3 mM Na2SO4 + 0.5 mM CaSO4 | 2.5–3.0 | 7.5–8.0 | – | – | – |
| C2c | 3 mM Na2SO4 + 0.5 mM CaSO4 | 3.5–4.0 | 6.5–7.5 | – | – | – |
| C3 | 3 mM Na2SO4 + 2 mM NaHCO3 | 3.5–4.5 | 7.5–8.5 | – | – | – |
| C4 | 3 mM Na2SO4 + 5 mM NaHCO3 | 3.5–5.0 | 8.0–8.5 | – | – | – |
| C5 | 5.3 mg/L TCE, no Pd/Al2O3 | – | – | 0.090 d 0.045 e |
– | 31% |
| C6 | 5.3 mg/L TCE, no Pd/Al2O3, 10 mg/L Fe(II) | – | – | 0.090 d 0.045 e |
0.007 | 36% |
| C7 | 5.3 mg/L TCE, 2 g Pd/Al2O3 | 3.0–3.5 | 6.5–7.0 | 0.090 d 0.045 e |
0.018 | 44% |
| C8 | 5.3 mg/L TCE, 2 g Pd/Al2O3, 5 mg/L Fe(II) | 2.5–4.0 | 6.5–7.5 | 0.090 d 0.045 e |
0.042 | 59% |
| C9 | 5.3 mg/L TCE, 2 g Pd/Al2O3, 10 mg/L Fe(II) | 2.7–4.0 | 6.5–7.5 | 0.090 d 0.045 e |
0.069 | 68% |
| C10 | 5.3 mg/L TCE, 2 g Pd/Al2O3, 5 mg/L Fe(II), 1 mM sulfite | 2.5–4.5 | 6.5–7.5 | 0.090 d 0.045 e |
0.072 | 71% |
| C11 | 5.3 mg/L TCE, 2 g Pd/Al2O3, 5 mg/L Fe(II), 31.3 μM sulfide | 2.5–4.5 | 6.5–7.5 | 0.090 d 0.045 e |
0.042 | 57% |
| C12 f | 5.3 mg/L TCE, 2 g Pd/Al2O3, 5 mg/L Fe(II), 31.3 μM sulfide | 2.5–4.5 | 6.5–7.5 | 0.090 d 0.045 e |
0.020–0.041 g | 46% |
For C5–C11, 3 mM Na2SO4 and 0.5 mM CaSO4 were dissolved in groundwater as background electrolytes.
Removal efficiency refers to total fitted removal percentages of TCE in columns under steady state.
Two electrodes were used.
Reaction rate at Cathode 1.
Reaction rate at Cathode 2.
A much longer period of 10 d was lasted.
Time-dependent rate constants in the region between Anode and Cathode 2.
2.3. Degradation in batch mode
The same experimental setup as reported previously (Yuan et al., 2012) is used for degradation of TCE in batch setup at ambient temperature (25 ± 1 °C). Experimental details are described in Section S1 in the Supporting Information.
2.4. Chemical analysis
TCE, cis-DCE, phenol and toluene were measured by a 1200 Infinity Series HPLC (Agilent) equipped with an 1260 DAD detector and a Thermo ODS Hypersil C18 column (4.6 × 50 mm). The mobile phase was a mixture of acetonitrile and water (60:40, v/v) at 1 mL/min. The detection wavelength was 210 nm. Sulfite at concentration higher than 1 mM was also measured by HPLC using the same procedure as for TCE analysis. Sulfide was detected at 665 nm on a spectrometer (Spectronic 20D+, Caley & Whitmore Corp.) after coloration with dimethyl-p-phenylene (Environmental Protection Administration of China, 2002), and H2O2 was analyzed at 405 nm after coloration with TiSO4 (Eisenberg, 1943).
The •OH levels were determined by DMSO trapping and HPLC according to the literature (Section S2, Tai et al., 2004). In order to identify the generation of new radicals, 100-μL sample collected from the batch degradation system without contaminants was immediately mixed with 25 μL of 0.2 M DMPO to form DMPO-radical adduct, which was then measured by electron spin resonance (ESR) assay. The ESR spectra were obtained on a Bruker EMX ESR spectrum with microwave bridge (receiver gain, 5020; modulation amplitude, 2 Gauss; microwave power, 6.35 mW; modulation frequency, 100 kHz; center field: 348.5 mT).
2.5. Numerical simulation of contaminant reactive transport
The reactive transport of contaminants under the advection, dispersion and first-order transformation in one-dimensional column is governed by
| (5a) |
The following initial and boundary conditions are assumed for contaminants in the column tests in this study:
| (5b) |
| (5c) |
| (5d) |
where C(x, t) stands for the contaminant concentration at location x and time t; k denotes the first-order transformation rate; v is the pore water velocity; D is the hydrodynamic dispersion coefficient; C0 is the initial concentration in the reactive transport domain; ΩCD(x, t) is the specified concentration on the Dirichlet boundary ΓD; F(x, t) is the dispersive flux across the Neuman boundary ΓN, and n(x) is the outward unit vector normal to the boundary ΓN. The hydrodynamic dispersion tensor in one-dimensional flow field can be simplified as (Burnett and Frind, 1987)
| (6) |
where αL is the longitudinal dispersivity, and can be estimated by fitting the column tracer test; D* presents the molecular diffusion coefficient of the contaminant in water.
First-order transformation kinetics are assumed for TCE transformation because the generated reactive species in the column experiments are significantly less than those generated in the batch experiment (Yuan et al., 2012) and the degradation of TCE in the presence of RSCs follows pseudo-first-order kinetics. NUFT (Nonisothermal Unsaturated-saturated Flow and Transport), a code developed at Lawrence Livermore National Laboratory (Hao et al., 2011; Nitao, 1998), was used to perform numerical simulations of TCE reactive transport. NUFT has been demonstrated as a robust and accurate model in many applications for simulating mass transfer and reactive transport (Buscheck et al., 2012; Sun et al., 2000). The details of the model setup are presented in Section S3.
3. Results and discussion
3.1. Automatic pH-regulation in three-electrode columns
Using the three-electrode electrolytic column, groundwater pH within the Pd vicinity quickly dropped to about 2.5 in the absence of bicarbonate (Fig. 2a). As one third of the cathodic current is shared by Cathode 2, the H+ produced at Anode is theoretically one third more than the OH− produced at Cathode 1. Moreover, because no beads were filled in the space between Anode and Cathode 1, OH− produced at Cathode 1 was immediately neutralized by H+ produced at Anode under the assistance of electro-generated bubbling of O2 and H2. In contrast, the space between Cathodes 1 and 2 was fully packed with Pd/Al2O3 pellets and glass beads, OH− produced at Cathode 2 was difficult to migrate to the Pd vicinity in an opposite direction to groundwater flow. Consequently, the automatic development of low pH within the Pd vicinity was attained. As a comparison, the pH in the Pd vicinity dropped to only 3.5 in the conventional two-electrode column.
Fig. 2.

(a) Temporal variations of pH within the Pd vicinity and (b) steady-state variations of pH along column. Pd vicinity refers to samples collected form Port P2. Operation conditions are: a flow rate of 2 mL/min and total current of 60 mA with 40 and 20 mA are distributed through Cathodes 1 and 2, respectively. Note for C2 (2 electrodes only) no current passed through Cathode 2.
The automatic pH-regulation is further evaluated using bicarbonate buffering groundwater. Fig. 2a shows that pH within the Pd vicinity quickly declined to 3.5 in the presence of 2 mM bicarbonate, and the decrease occurred slowly when 5 mM bicarbonate was present. Theoretical calculation of bicarbonate concentrations that can be acidified by the three-electrode column indicate that 20 mA of total 60 mA shared by Cathode 2 is capable of decreasing the pH of a 6.3 mM bicarbonate solution to a value of 4.0 (Table S1). Fig. 2b shows that neutral effluent can be maintained in this three-electrode column. The local acidity developed around Pd catalyst provides the possibility of contaminant oxidation in the presence of Fe(II) (Yuan et al., 2012). This is the first study to report automatic pH-regulation of groundwater using a three-electrode column system.
3.2. TCE degradation in columns
TCE degradation reached steadystate after 120 min of operation (1.5 PVs) in a typical column (Fig. S3). As shown in Fig. 3a, the steady-state TCE concentrations along columns demonstrate a quick decrease when passing through Pd/Al2O3 fillings. Note that the decrease in TCE concentration before Pd is due to the diffusion process because no fillings were packed between Anode and Cathode 1. Without Pd/Al2O3 packing, TCE removals attained 31 and 36% in the absence (C5) and presence (C6) of 10 mg/L Fe(II), respectively. This suggests that the electro-Fenton process based on Ti/MMO electrode could slightly contribute to TCE oxidation. With Pd/Al2O3 packing, the removals of TCE increased from 44% in the absence of Fe(II) (C7) to 59% in the presence of 5 mg/L Fe(II) (C8), and further to 68% in the presence of 10 mg/L Fe(II) (C9) (Table 1). This trend is in accordance with those reported in batch experiment (Yuan et al., 2012), suggesting a dominant oxidation pathway for TCE degradation in the presence of Fe(II). Under the acidic conditions that automatically develop, more H2O2 was produced on Pd surface (Yuan et al., 2011, 2012), and more Fe(II) was in the form of free Fe2+. Thus, more oxidizing •OH radicals were generated by the classical Fenton reaction resulting in efficient oxidation of TCE. Negligible degradation was observed after groundwater passed through Cathode 2 because negligible •OH can be generated from Fe(II) and H2O2 under neutral and alkaline conditions (Yuan et al., 2012).
Fig. 3.

Steady-state variation of TCE along the columns (a) at different column conditions and (b) in the presence of RSCs. Curves refer to the simulated results corresponding to the data point in the same color. Initial concentrations of TCE was 5.3 mg/L. Operation conditions are: a flow rate of 2 mL/min and total current of 60 mA with 40 and 20 mA distributed through Cathodes 1 and 2, respectively.
The reactive transport of contaminants in the column was simulated by incorporating reaction term into convection dispersion equation (smooth curves in Fig. 2). The reactions responsible for contaminant degradation in different regions are discussed in Section S3. As summarized in Table 1, with the increase in Fe(II) concentration from 0 to 5 and further to 10 mg/L, the degradation rate constants in Pd vicinity increased from 0.018 to 0.042 and further to 0.069 min−1, respectively. Note that the apparent high rate constants at the electrodes are due to the small region for electrode reaction. As a result, it can be concluded that the hybrid electrolysis and Pd-catalytic process with automatic pH-regulation is capable of oxidizing TCE in groundwater with Fe(II) at concentration levels on the order of mg/L.
3.3. Effect of RSCs on TCE degradation
RSCs are recognized as Pd fouling agents in groundwater remediation (Davie et al., 2008; Lowry and Reinhard, 2000, 2001; Schueth et al., 2004). However, in the present study 1 mM sulfite greatly enhanced TCE degradation; that is, the transformation rate constants increased from 0.042 to 0.072 min−1, while 31.3 μM sulfide (1 mg/L) did not result in any significant fouling effect (Fig. 3b). The mechanism for this unexpected influence of sulfite is discussed in the following section. For the influence of sulfide on TCE degradation, the results from batch experiment suggest increasing inhibition with increasing sulfide concentration (Fig. S4). However, this inhibition is much less pronounced when compared with Pd-catalytic hydrodechlorination under anaerobic condition (Lowry and Reinhard, 2000; Schueth et al., 2004). Both precipitation and oxidation may lead to the removal or transformation of sulfide. According to the solubility product constant of FeS (Ksp = 3.7 × 10−19) and dissociation constants of H2S (pKa1 = 7.04, pKa2 = 11.96), the precipitation of sulfide was thermodynamically favorable but was calculated to be negligible at groundwater conditions of 5 mg/L Fe(II) and pH 6. Oxidation of sulfide on the MMO electrodes was addressed recently (Pikaar et al., 2011), and was also confirmed herein (Fig. S5). Furthermore, the in situ generated reactive oxidizing species, i.e., O2, H2O2 and •OH (Chen and Morris, 1972; Hoffmann, 1977), may also oxidize sulfide, thus preventing Pd deactivation.
3.4. Mechanism for sulfite-enhanced degradation
A series of batch experiments were conducted to elucidate the unexpected enhancement caused by sulfite. TCE degradation increased remarkably with the increase in sulfite concentration from 0 to 1 mM (Fig. 4a), while the production of •OH was approximately within the same concentration range (Fig. 4b). In the presence of 3 mM sulfite, TCE degradation is slightly inhibited within initial 30 min compared with the absence of sulfite, and afterwards the degradation promptly increased (Fig. 4a). At the inflexion time of 30 min, sulfite concentration decreased to less than 1 mM (Fig. S6). The production of •OH was negligible within 40 min at 3 mM sulfite (Fig. 4b), indicating the minor contribution of •OH oxidation to TCE degradation at this stage. It is noted that the degradation kinetics deviated from zero order in the absence of sulfite to first order in the presence of sulfite (≤1 mM), whereas the generation of •OH still followed zero order. This further suggests a new mechanism for TCE degradation in addition to •OH oxidation in the presence of sulfite.
Fig. 4.

Effect of sulfite on (a) TCE degradation and (b) •OH radical accumulation. The degradation conditions for (a) are based on 198 μM initial TCE concentration, pH 4, 13.7 mg/L Fe2+, 1 g/L Pd/Al2O3 and 10 mM Na2SO4 background electrolyte. The reaction conditions for (b) are the same as for (a) except that no TCE was included. Lines and curves in (a) refer to pseudo-zero-order and pseudo-first-order kintic fittings, respectively. Pseudo-zero-order reaction kinetics is given by Ct = C0−k0t, where t is the reaction time (min), k0 is the zero-order rate constant (μM/min), and c0 and ct are the concentrations (μM) at times of t = 0 and t = t, respectively. Pseudo-first-order reaction kinetics is expressed as ln(Ct/C0) = k1t + b, where b is a constant and k1 is the first-order rate constant (min−1).
Since inhibition by sulfite is recognized for Pd-catalytic hydrodechlorination under anaerobic conditions (Davie et al., 2008; Lowry and Reinhard, 2000; Schueth et al., 2004), the enhancement herein must be related to the generation of oxidizing species. It is reported that SO32− can be induced by oxidizing species to produce SO3•−, which combines with O2 generating a strong oxidizing radical of SO4•− (2.5–3.1 V/SHE, Guan et al., 2011) through a series of free radical chain reactions (Neta and Huie, 1985; Razskazovskii and Sevilla, 1996). To identify the possible contribution of SO4•−, the degradation was observed by the addition of methanol and TBA, respectively, as methanol scavenges both •OH and SO4•− while TBA scavenges only •OH (Guan et al., 2011; Liang and Su, 2009). The different inhibitory effect (Fig. 5a) implies the generation of SO4•−. Note that TCE degradation with TBA addition again conformed to zero order kinetics implying a constant generation rate of SO4•− (Yuan et al., 2012). ESR assay was further employed to identify the generation of new radicals (Fig. 5b). Characteristic spectrum of •OH with hyperfine coupling constants of aN = 14.9 G and aH = 14.9 G was evident by the addition of Fe(II) (Yuan et al., 2011). When 1 mM sulfite was introduced at the beginning of the experiment, several new signals appeared, which can be presumably attributed to the generation of SO4•− and unknown S-centered radicals (Guan et al., 2011; Shi et al., 1994). When sulfite concentration was increased to 3 mM, the characteristic signals for SO3•− with hyperfine coupling constants of aN = 14.7 G and aH = 16.0 G were measured along with •OH (Ranguelova et al., 2012; Shi et al., 1994). Because of the low concentration and weak response of SO4•−, the ESR signals were not observed. However, the transformation of SO3•− to SO4•− in the presence of O2 was proved by ESR simulation (Ranguelova et al., 2012). Note that the signals of OH were enhanced compared with Fe(II) addition initially, which may also contribute to TCE degradation.
Fig. 5.

(a) Effect of radical scavengers on TCE degradation, (b) radical generation measured by ESR. The degradation conditions for (a) are based on 198 μM initial TCE concentration, pH 4, 13.7 mg/L Fe2+, 1 g/L Pd/Al2O3 and 10 mM Na2SO4 background electrolyte. Concentrations of methanol and TBA were 60 and 50 mM, respectively. The degradation conditions for (b) are based on pH 4, 13.7 mg/L Fe2+, 1 g/L Pd/Al2O3 and 10 mM Na2SO4 background electrolyte unless otherwise specified. A standard ESR spectrum for •OH was obtained for reference by Fe(II) addition after 10 min electrolysis.
Then, what species are responsible for the generation of SO3•− and SO4•−? Degradation of TCE in the anodic compartment with 1 g/L Pd/Al2O3 and 1 mM sulfite addition was negligible (Fig. S7a), and the influence of sulfite on conventional Fenton degradation is minimal (Fig. S7b). As a result, anodic oxidation, free •OH and H2O2 in solution can not transform SO32− to SO4•− in this study. Based on these validation results, the combination of oxidizing species with Pd catalyst, probably the as-generated H2O2 and •OH chemisorbed on Pd surface, were assumed to be responsible for the transformation of SO32−. As a consequence, the following mechanism is proposed for sulfite-enhanced degradation. SO32− donates one electron to the as-generated H2O2 or •OH chemisorbed on Pd surface producing SO3•− (7), which further combines with O2 with the production of SO5•− (8). The one-electron reduction of SO5•− yields an oxidizing species of HSO5− (peroxymonosulfate, 1.82 V/SHE) (9), which may be transformed to •OH (10) and SO4•− (11) in the presence of Fe2+. Both •OH and SO4•− contribute to TCE oxidation. Because the generation of SO4•− is dependent on instantaneous SO32− concentration, the total concentrations of •OH and SO4•− varied during degradation process, resulting in the deviation of degradation kinetics from zero to first order.
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
3.5. Long-term performance evaluation
The long-term performance of the hybrid electrolysis and Pd-catalytic oxidation process in resisting RSC was further evaluated. Fig. 6 shows that TCE degradation in the presence of 31.3 μM sulfide can be sustained at a stable level of about 50% for 10 d. The rate constants fitted for TCE degradation in Pd vicinity decreased from 0.041 min−1 in initial stage to a constant value of 0.020 min−1 after 4 d. The field application of Pd-catalytic hydrodechlorination of TCE in groundwater suggests that the catalytic activity of Pd can be maintained for several years (Davie et al., 2008). Therefore, a long longevity can be proposed for this hybrid electrolysis and Pd-catalytic oxidation process even in the presence of RSCs.
Fig. 6.

Long-term performance of the column in TCE degradation in the presence of 31.3 μM sulfide. Curvers refere to the simulated results corresponding to the data point in the same color. Operation conditions are: initial concentration of TCE was 5.3 mg/L, flow rate of 2 mL/min and total current of 60 mA with 40 and 20 mA shared in Cathodes 1 and 2, respectively.
3.6. Implications for groundwater remediation
Using phenol and toluene as representatives of other groundwater contaminants, significant degradations are also achieved (Fig. S8). Therefore, it is technologically feasible to apply this three-electrode system for the degradation of a wide range of organic contaminants in groundwater. However, the application is dependent on groundwater conditions, i.e., buffering capacity, Fe(II) and Cl− concentrations, etc. A low buffering capacity (<5 mM bicarbonate) is preferred for the automatic development of low pH in the Pd zone. The concentration of Fe(II) in groundwater at the ppm levels is required to catalyze the production of •OH, while the concentration of Cl- can not be too high (>20 mM) to scavenge the •OH radicals (Yuan et al., 2012). In the case of very low concentrations of Fe(II), iron-containing materials such as Fe3O4, zerovalent iron or iron anode with small fraction of or periodical anodic current can be mixed with Pd catalysts to supply Fe(II) (Joo et al., 2005; Mao et al., 2011; Navalon et al., 2010).
As Pd catalyst is supported on the first cathode in this application, the corrosion and release of Pd (which can be toxic) will be insignificant due to the cathodic protection. The leaching of Pd is measured to be minimal in our recent study using Pd and cathodic H2 for contaminant hydrodechlorination (Zheng et al., 2012). The local low pH developed in the reaction zone may also avoid or alleviate the precipitation of groundwater cations, i.e., Ca2+ and Mg2+, which is found to be significant and detrimental under electrochemically developed alkaline conditions (Lohner et al., 2011). The local acidity and in situ generated oxidizing species, H2O2 and •OH have the potential to inhibit the growth of microorganism on the catalyst surface.
4. Conclusions
This study shows that a three-electrode system is capable of developing local low pH conditions for Pd-catalytic oxidation of contaminant along with neutral effluent, even in the presence of bicarbonate. In theory, any desired pH values can be developed by adjusting total current and current partition. This configuration provides an alternative approach to developing appropriate pH conditions in the subsurface wherein chemical addition is difficult. The column performance in the degradation of TCE, phenol and toluene proves the feasibility of this hybrid electrolysis and Pd-catalytic oxidation process for groundwater remediation. As oxidizing rather than reducing conditions are developed, RSCs can be oxidized by the reactive oxidizing species, i.e., O2, H2O2 and •OH. As a consequence, this process has certain potential to resist the fouling caused by RSCs. SO4•−, a strong oxidizing radical, and more •OH can be produced in the presence of low concentration of sulfite in the process, which greatly contributes to contaminant oxidation. This mechanism is quite different from that under reducing conditions.
Supplementary Material
Acknowledgments
This work was supported by the US National Institute of Environmental Health Sciences (NIEHS, Grant No. P42ES017198), the Natural Science Foundation of China (NSFC, No. 41172220), and the Fundamental Research Funds for the Central Universities, China University of Geosciences (Wuhan) (No. CUGL110608). We appreciate the assistance in ESR assay by Prof. David Budil and Mr. Xianzhe Wang in Department of Chemistry & Chemical Biology, Northeastern University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIEHS, the National Institutes of Health or Lawrence Livermore National Security, LLC.
Appendix. Supplementary Information
The Supporting information associated with this article can be found in the on-line version.
References
- Anglada A, Urtiaga A, Ortiz I. Pilot scale performance of the electro-oxidation of landfill leachate at boron-doped diamond andoes. Environ Sci Technol. 2009;43:2035–2040. doi: 10.1021/es802748c. [DOI] [PubMed] [Google Scholar]
- Angeles-Wedler D, Mackenzie K, Kopinke FD. Permanganate oxidation of sulfur compounds to prevent poisoning of Pd catalysts in water treatment processes. Environ Sci Technol. 2008;42:5734–5739. doi: 10.1021/es800330s. [DOI] [PubMed] [Google Scholar]
- Brillas E, Sirés I, Oturan A. Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry. Chem Rev. 2009;109:6570–6631. doi: 10.1021/cr900136g. [DOI] [PubMed] [Google Scholar]
- Burnett RD, Frind EO. An alternating direction Galerkin technique for simulation of groundwater contaminant transport in three dimensions: 2 Dimensionality effects. Water Res Res. 1987;23:695–705. [Google Scholar]
- Buscheck TA, Sun Y, Chen M, Hao Y, Wolery TJ, Bourcier WL, Court B, Celia MA, Friedmann JS, Aines RD. Active CO2 reservoir management for carbon storage: Analysis of operational strategies to relieve pressure buildup and improve injectivity. Int J Greenh Gas Con. 2012;6:230–245. [Google Scholar]
- Carter KE, Farrell J. Electrochemical oxidation of trichloroethylene using boron-doped diamond film electrodes. Environ Sci Technol. 2009;43:8350–8354. doi: 10.1021/es9017738. [DOI] [PubMed] [Google Scholar]
- Chaplin BP, Shapley JR, Werth CJ. Regeneration of sulfur-fouled bimetallic Pd-based catalysts. Environ Sci Technol. 2007;41:5491–5497. doi: 10.1021/es0704333. [DOI] [PubMed] [Google Scholar]
- Chaplin BP, Reinhard M, Schneider WF, Schueth C, Shapley JR, Strathmann T, Werth CJ. A critical review of Pd-based catalytic treatment of priority contaminants in water. Environ Sci Technol. 2012;46:3655–3670. doi: 10.1021/es204087q. [DOI] [PubMed] [Google Scholar]
- Chen GH. Electrochemical technologies in wastewater treatment. Sep Purif Technol. 2004;38:11–41. [Google Scholar]
- Chen G, Betterton EA, Arnold RG, Ela WP. Electrolytic reduction of trichloroethylene and chloroform at a Pt- or Pd-coated ceramic cathode. J Appl Electrochem. 2003;33:161–169. [Google Scholar]
- Chen KY, Morris JC. Kinetics of oxidation of aqueous sulfide by O2. Environ Sci Technol. 1972;6:529–537. [Google Scholar]
- Davie MG, Cheng H, Hopkins GD, Lebron CA, Reinhard M. Implementing heterogeneous catalytic dechlorination technology for remediating TCE-contaminated groundwater. Environ Sci Technol. 2008;42:8908–8915. doi: 10.1021/es8014919. [DOI] [PubMed] [Google Scholar]
- Eisenberg G. Colorimetric determination of hydrogen peroxide. Ind Eng Chem Anal Ed. 1943;15:327–328. [Google Scholar]
- Environmental Protection Administration of China. Water and Wastewater Monitoring and Analysis Methods. 4. China Environmental Science Press; 2002. [Google Scholar]
- Franz JA, Williams RJ, Flora JRV, Meadows ME, Irwin WG. Electrolytic oxygen generation for subsurface delivery: Effect of precipitation at the cathode and an assessment of side reactions. Water Res. 2002:2243–2254. doi: 10.1016/s0043-1354(01)00443-2. [DOI] [PubMed] [Google Scholar]
- Gent DB, Wani AH, Davis JL, Alshawabkeh A. Electrolytic redox and electrochemical generated alkanline hydrolysis of hexahydro-1,3,5-trinitro-1,3,5 triazine (RDX) in sand columns. Environ Sci Technol. 2009;43:6301–6307. doi: 10.1021/es803567s. [DOI] [PubMed] [Google Scholar]
- Guan YH, Ma J, Li XC, Fang JY, Chen LW. Influence of pH on the formation of sulfate and hydroxyl radicals in the UV/peroxymonosulfate system. Environ Sci Technol. 2011;45:9308–9314. doi: 10.1021/es2017363. [DOI] [PubMed] [Google Scholar]
- Hao Y, Sun Y, Nitao JJ. Overiew of NUFT: Aversatile numerical model for simulating flow and reactive transport in porous media. Lawrence Livermore National Laboratory, Groundwater Reactive Transport Models; 2011. pp. 213–240. [Google Scholar]
- Hildebrand H, Mackenzie K, Kopinke FD. Highly active Pd-on-magnetite nanocatalysts for aqueous phase hydrodechlorination reactions. Environ Sci Technol. 2009;43:3254–3259. doi: 10.1021/es802726v. [DOI] [PubMed] [Google Scholar]
- Hoffmann MR. Kinetics and mechanism of oxidation of hydrogen sulfide by hydrogen peroxide in acidic solution. Environ Sci Technol. 1977;11:61–66. [Google Scholar]
- Joo SH, Feitz AJ, Sedlak DL, Waite TD. Quantification of the oxidizing capacity of nanoparticulate zero-valent iron. Environ Sci Technol. 2005;39:1263–1268. doi: 10.1021/es048983d. [DOI] [PubMed] [Google Scholar]
- Liang CJ, Su HS. Identification of sulfate and hydroxyl radicals in thermally activated persulfate. Ind Eng Chem Res. 2009;48:5558–5562. [Google Scholar]
- Lohner ST, Tiehm A. Application of electrolysis to stimulate microbial reductive PCE dechlorination and oxidative VC biodegradation. Environ Sci Technol. 2009;43:7098–7104. doi: 10.1021/es900835d. [DOI] [PubMed] [Google Scholar]
- Lohner ST, Becker D, Mangold KM, Tiehm A. Sequential reductive and oxidative biodegradation of chloroethenes stimulated in a coupled bio-electro-process. Environ Sci Technol. 2011;45:6491–6497. doi: 10.1021/es200801r. [DOI] [PubMed] [Google Scholar]
- Lowry GV, Reinhard M. Pd-catalyzed TCE dechlorination in groundwater: Solute effects, biological control, and oxidative catalyst regeneration. Environ Sci Technol. 2000;34:3217–3223. [Google Scholar]
- Lowry GV, Reinhard M. Pd-catalyzed TCE dechlorination in water: Effects of [H2](aq) and H2-utilizing competitive solutes on the TCE dechlorination rate and product distribution. Environ Sci Technol. 2001;35:696–702. doi: 10.1021/es001623f. [DOI] [PubMed] [Google Scholar]
- Mao XH, Ciblak A, Amiri M, Alshawabkeh AN. Redox control for electrochemical dechlorination of trichloroethylene in bicarbonate aqueous media. Environ Sci Technol. 2011;45:6517–6523. doi: 10.1021/es200943z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcnab JRWW, Ruiz R. Palladium-catalyzed reductive dehalogenation of dissolved chlorinated aliphatics using electrolytically-generated hydrogen. Chemosphere. 1998;37:925–936. [Google Scholar]
- Mishra D, Liao Z, Farrell J. Understanding reductive dechlorination of trichloroethene on boron-doped diamond film electrodes. Environ Sci Technol. 2008;42:9344–9349. doi: 10.1021/es801815z. [DOI] [PubMed] [Google Scholar]
- Navalon S, Alvaro M, Garcia H. Heterogeneous Fenton catalysts based on clays, silicas and zeolites. Appl Catal B Environ. 2010;99:1–26. [Google Scholar]
- Neta P, Huie RE. Free-radical chemistry of sulfite. Environ Health Persp. 1985;64:209–217. doi: 10.1289/ehp.8564209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitao JJ. User’s manual for the USNT module of the NUFT code, version 2 (NP-phase, NC-component, thermal) Lawrence Livermore National Laboratory; 1998. UCRL-MA-130653. [Google Scholar]
- Nutt MO, Hughes JB, Wong MS. Designing Pd-on-Au bimetallic nanoparticle catalysts for trichloroethene hydrodechlorination. Environ Sci Technol. 2005;39:1346–1353. doi: 10.1021/es048560b. [DOI] [PubMed] [Google Scholar]
- Pikaar I, Rozendal RA, Yuan Z, Keller J, Rabaey K. Electrochemical sulfide oxidation from domestic wastewater using mixed metal-coated titanium electrodes. Water Res. 2011;45:5381–5388. doi: 10.1016/j.watres.2011.07.033. [DOI] [PubMed] [Google Scholar]
- Ranguelova K, Rice AB, Khajo A, Triquigneaux M, Garantziotis S, Magliozzo RS, Mason RP. Formation of reactive sulfite-derized free radicals by the activation of human neutrophils: An ESR study. Free Radical Biol Med. 2012;52:1264–1271. doi: 10.1016/j.freeradbiomed.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razskazovskii Y, Sevilla MD. One-electron oxidation and reduction of sulfites and sulfinic acid in oxygenated media: The formation of sulfonyl and sulfuranyl peroxyl radicals. J Phys Chem. 1996;100:4090–4096. [Google Scholar]
- Reddy KR, Cameselle C. Electrochemical remediation technologies for polluted soils, sediments and groundwater. John Willey & Sons, Inc; 2009. [Google Scholar]
- Schueth C, Kummer NA, Weidenthaler C, Schad H. Field application of a tailored catalyst for hydrodechlorinating chlorinated hydrocarbon contaminants in groundwater. Appl Catal B. 2004;52:197–203. [Google Scholar]
- Shi XL, Dalal N, Kasprzak KS. Enhanced generation of hydroxyl radical and sulfur trioxide anion radical from oxidation of sodium sulfite, nickel(II) sulfite, and nickel subsulfide in the presence of nickel(II) complexes. Environ Health Persp. 1994;102:91–96. doi: 10.1289/ehp.94102s391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Demir Z, Delorenzo T, Nitao JJ. Application of the NUFT code for subsurface remediation by bioventing. Lawrence Livermore National Laboratory; 2000. UCRL-ID-137967. [Google Scholar]
- Tai C, Peng JF, Liu JF, Jiang GB, Zou H. Determination of hydroxyl radicals in advanced oxidation processes with dimethyl sulfoxide trapping and liquid chromatography. Anal Chim Acta. 2004;527:73–80. [Google Scholar]
- Vlyssides A, Barampouti EM, Mai S, Arapoglou D, Kotronarou A. Degradation of methylparathion in aqueous solution by electrochemical oxidation. Environ Sci Technol. 2004;38:6125–6131. doi: 10.1021/es049726b. [DOI] [PubMed] [Google Scholar]
- Weathers LJ, Parkin GF, Alvarez PJJ. Utilization of cathodic hydrogen as an electron donor for chloroform metabolism by a mixed methanogenic culture. Environ Sci Technol. 1997;31:880–885. [Google Scholar]
- Yang B, Yu G, Huang J. Electrocatalytic hydrodechlorination of 2,4,5-trichlorobiphenyl on a palladium-modified nickel foam cathode. Environ Sci Technol. 2007;41:7503–7508. doi: 10.1021/es071168o. [DOI] [PubMed] [Google Scholar]
- Yuan SH, Fan Y, Zhang YC, Tong M, Liao P. Pd-catalytic in situ generation of H2O2 from H2 and O2 produced by water electrolysis for the efficient electro-Fenton degradation of rhodamine B. Environ. Sci Technol. 2011;45:8514–8520. doi: 10.1021/es2022939. [DOI] [PubMed] [Google Scholar]
- Yuan SH, Mao X, Alshawabkeh AN. Efficient degradation of TCE in groundwater using Pd and electro-generated H2 and O2: A shift in pathway from hydrodechlorination to oxidation in the presence of ferrous ions. Environ Sci Technol. 2012;46:3398–3405. doi: 10.1021/es204546u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao GH, Zhang YG, Lei YZ, Lv BY, Gao JX, Zhang YA, Li DM. Fabrication and electrochemical treatment application of a novel lead dioxide anode with superhydrophobic surfaces, high oxygen evolution potential, and oxidation capability. Environ Sci Technol. 2010;44:1754–1759. doi: 10.1021/es902336d. [DOI] [PubMed] [Google Scholar]
- Zheng MM, Bao JG, Liao P, Wang K, Yuan SH, Tong M, Long HY. Electrogeneration of H2 for Pd-catalytic hydrodechlorination of 2,4-dichlorophenol in groundwater. Chemosphere. 2012;87:1097–1104. doi: 10.1016/j.chemosphere.2012.01.058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.