

Abstract
Nine cancer patients were treated with adoptive cell therapy using autologous anti-MAGE-A3 TCR engineered T cells. Five patients experienced clinical regression of their cancers including two on-going responders. Beginning 1–2 days post-infusion, three patients (#’s 5, 7, and 8) experienced mental status changes, and two patients (5 and 8) lapsed into comas and subsequently died. Magnetic resonance imagining analysis of patients 5 and 8 demonstrated periventricular leukomalacia, and examination of their brains at autopsy revealed necrotizing leukoencephalopathy with extensive white matter defects associated with infiltration of CD3+/CD8+ T cells. Patient 7, developed Parkinson-like symptoms, which resolved over 4 weeks and fully recovered. Immunohistochemical staining of patient and normal brain samples demonstrated rare positively staining neurons with an antibody that recognizes multiple MAGE-A family members. The TCR used in this study recognized epitopes in MAGE-A3/A9/A12. Molecular assays of human brain samples using Q-RT-PCR, Nano string quantitation, and deep-sequencing indicated that MAGE -A12 was expressed in human brain (and possibly MAGE-A1, MAGE-A8, and MAGE-A9). This previously unrecognized expression of MAGE-A12 in human brain was possibly the initiating event of a TCR-mediated inflammatory response that resulted in neuronal cell destruction and raises caution for clinical applications targeting MAGE-A family members with highly active immunotherapies.
Keywords: Cancer Testes Antigen, Immunotherapy, TCR, Gene Therapy
Introduction
Adoptive immunotherapy using naturally occurring tumor infiltrating lymphocytes or cytotoxic T lymphocytes can mediate regression of metastatic melanoma when combined with non-myeloablative chemotherapy and IL-21, 2. Motivated by the potential to treat common malignancies multiple investigators have explored the genetic modification of normal peripheral T cells with anti -tumor antigen receptors, based on T cell receptors (TCR) or chimeric antigen receptors (CAR)that target antigens expressed on tumor cells 3. While long-term tumor regressions have been observed in patients bearing a variety of malignancies that include neuroblastoma, melanoma, colorectal cancer, synovial cell sarcoma, and lymphoma4–11, these responses were, in some cases, associated with significant off-tumor/on-target toxicities (i.e., recognition of normal tissues expressing the tumor-associated antigen) including patient deaths12, 13.
In an attempt to limit normal tissue toxicities, investigators have targeted the cancer -testes class of tumor-associated antigens. The first member of this family to be identified, MAGE-A1 (melanoma associated antigenA1) was cloned by Boon and colleagues in 199114. Soon thereafter it was found that this gene was a member of a larger family of tumor-associated antigens called cancer-testes or cancer-germ line antigens (herein abbreviated CTA)15–19. The hallmark of this class of tumor-associated antigens is their restricted expression to germ-line tissues (such as the non-MHC expressing adult testes), trophoblasts, placenta, and a limited subset of normal adult tissues. Importantly, many of the CTAs were found to be expressed in a variety of common epithelial malignancies including cancers of the lung, breast, ovary, bladder, and melanoma20–24. The frequency of CTA expression in these common cancers is generally in the range of 30%–50%, but in some malignancies such as synovial cell carcinoma, it can be as high as 80%25. Based on their immunogenicity and frequency of expression, CTAs have been targeted in multiple cancer vaccine trials, as well as, adoptive T cell transfer trials using either CTL or TCR gene modified T cells7, 26–29.
Based on our clinical success in treating melanoma and synovial cell sarcoma using TCR gene-modified T cells targeting CTA NY -ESO-17, we sought to develop a T cell receptor targeting the widely expressed CTA, MAGE-A330. The MAGE-A gene family has been shown to be one of the most widely expressed of the CTA genes with greater than 30% expression in common epithelial malignancies including esophageal cancer, melanoma, head and neck cancer, breast, and lung cancer15–17. To generate a high avidity TCR against MAGE-A3 we immunized HLA-A*0201 transgenic mice with MAGE-A3 peptide:112–120 (KVAELVHFL, an epitope shared with MAGE-A9). We subsequently determined that T cells transduced with this TCR also recognized MAGE-A12 (epitope KMAELVHFL) and to a lesser extent, MAGE-A2 (KMVELVHFL) and MAGE-A6 (KVAKLVHFL). This TCR was further modified by site-directed mutagenesis in the TCR CDR3 region in order to further increase its avidity while maintaining MAGE-A gene specificity30.
Herein we report the clinical application of MAGE-A3 TCR gene-modified T cells in an adoptive cell therapy trial in nine cancer patients.
Methods
Clinical Protocol
The clinical trial under which these patients were treated is listed on-line Cancer Treatment With Anti-MAGE-A3/12 TCR-Gene Engineered Lymphocytes (www.clinicaltrials.gov) as protocol NCT01273181, title MAGE-A3/12 Metastatic. The protocol was reviewed and approved by the National Institutes of Health Institutional Biosafety Committee, the National Institutes of Health Recombinant DNA Advisory Committee, the National Cancer Institute Institutional Review Board, and the Food and Drug Administration (all Bethesda, MD). All patients gave informed consent for protocol enrolment. Patient inclusion criteria included metastatic cancer that expressed MAGE-A3/12 as assessed by one of the following methods: RT -PCR on tumor tissue defined as 30,000 copies of MAGE-A3/12 per 106 GAPDH copies, or by immunohistochemistry of resected tissue defined as 10% or greater of cells being 2–3+ reactive with anti-MAGE-A antibody (mAb 6C1, Santa Cruz Biotechnologies, Santa Cruz, CA). Patients were HLA-A*0201, 18 years of age or older, and Eastern Cooperative Oncology Group status 0 or 1. Patients must have previously received systemic standard care (or effective salvage chemotherapy regimens) for metastatic disease, and have been either nonresponders (progressive disease) or have recurred.
Before receiving treatment with transduced peripheral blood lymphocytes (PBLs), patients were transiently lymphoablated using a nonmyeloablative lymphodepleting regimen as previously described31, by intravenous administration of cyclophosphamide 60 mg/kg for 2 days followed by fludarabine 25 mg/m2 for 5 days. One day after completion of their lymphodepleting regimen, patients received transduced lymphocytes infused intravenously followed by high -dose (720,000 IU/kg) IL-2 (Aldesleukin; Prometheus, San Diego, CA) every 8 hours to tolerance. The protocol was designed as a cell dose escalation starting with an initial cohort of three patients treated with 5 × 109 to 3 × 1010 cells followed by a cohort of 3 × 1010 up to 1 × 10 11 total cells. Patients received baseline computed tomography (CT) and/or magnetic resonance imaging (MRI) and, positron emission tomography (PET)before treatment. Tumor size was evaluated monthly by CT, MRI, or documented with photography for cutaneous/subcutaneous lesions. Tumor measurements and patient responses were determined according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.0)32 .
Gene Transfer Procedure
The γ-retroviral vector targeting MAGE-A3/A12 has been previously described in detail30. In brief, MSGV-1 is derived from MSGV vector that utilizes the murine stem cell virus long terminal repeat and contains an extended gag region and Kozak sequence. The TCR transgene construct was arranged in the following order of configuration:TCR α chain, linker peptide furinSGSGP2A and TCR β chain. The TCR was derived from immunization of HLA-A*0201 transgenic mice using peptide MAGE-A3: 112–120 (KVAELVHFL) and was further modified by an Ala to Thr substitution in CDR3 region of the TCR α chain at position 118 30. A high-titer PG13 cell-based producer cell line was selected and using current good manufacturing practice grade retroviral vector supernatant produced by the NCI Surgery Branch Vector Production Facility (Bethesda, MD). The vector supernatant was tested and passed all currently required US Food and Drug Administration guidelines for the production of recombinant γ-retroviral vectors for clinical application.
The transduction procedure was initiated by stimulating peripheral blood mononuclear cells (PBMCs) with anti-CD3 mAb OrthocloneOKT3 (Centocor Ortho Biotech, Raritan, NJ) at a final concentration of 50 ng/ml with recombinant human IL -2 at a final concentration of 300 IU/ml in AIM-V medium (Invitrogen, Carlsbad, CA) containing 5% human serum (Valley Biomedical, Winchester, VA). Cells were harvested for retroviral transduction on day 2 and resuspended in the same medium without OKT3. Retroviral vector supernatant was thawed and diluted with two parts of medium before being loaded onto RetroNectin (CH-296; Takara Bio, Otsu, Japan) coated (using 10 μg/ml of CH-296) non-tissue culture treated six-well plates. Vector supernatant was “spin loaded” onto coated plates by centrifugation at 2,000 g for 2 hours at 32 °C. Retroviral vector was aspirated from the wells and 2 × 106 activated PBMC were added pre -well followed by centrifugation at 1,000 g for 10 minutes. Plates are incubated at 37°Covernight and the next day all wells are harvested, pooled, and the transduction procedure repeated. Following the second transduction, cells were collected and maintained in medium at 0.5–2.0 × 106 cells/ml for a total of 10 days after stimulation. At day 10 after stimulation, cells were subject to a rapid expansion procedure for an additional 14 days using 3,000 IU/ml IL-2 with 50 ng/ml anti-CD3 mAb OKT3 and 100-fold excess 4 Gy irradiated allogeneic PBMC feeder cells. Treatment cells were washed in saline before infusion and resuspended in 125 ml containing 300 IU/ml IL-2 then administered to the patient intravenously over 30 min. Before treatment, TCR-transduced PBLs from all patients were evaluated for expression of the appropriate TCR by tetramer staining and mouse beta TCR chain using flow cytometric analysis, and cell function was evaluated by overnight coculture with cognate antigen-bearing target cells (1 × 105:1 × 105) and enzyme-linked immunosorbent assay (ELISA) measurement (Thermo Scientific, Rockford, Il) of interferon-γ (IFN-γ) produced in the culture supernatant as previously described11.
Flow Cytometry Analysis
To assess the phenotype of the MAGE-A3 TCR transduced cells in the infusion sample, cells were stained with allophycocyanin (APC)-H7-conjugated anti-human CD3 antibody (clone Sk7; BD biosciences, San Jose, CA), phycoerythrin (PE)-TR-conjugated anti-human CD8 antibody (clone 3B5; San Diego, CA Invitrogen), PE conjugated anti-mouse TCR beta chain (clone H57-597; eBiosciences,), APC-conjugated anti-human CD62L antibody (clone DREG-56; BD biosciences) and PE-Cy7-conjugated anti-human CD45RO antibody. Differentiation phenotype (CD62L by CD45RO expression) was assessed after excluding aggregates, and dead cells using propidium iodide (PI) and gating on CD3+/CD8+/murine TCR beta chain + cells. Patient PBMCs obtained approximately1 month after adoptive transfer were analyzed for TCR expression, following over-night culture in IL- containing media. Anti-MAGE-A3 TCR expression Core Facility at Emory University (Atlanta, GA) was determined using a HLA-A*0201 specific tetramer produced, with PE as fluorophore along by the NIH Tetramer with a fluorescein isothiocyanate (FITC)-labeled anti-human CD8 (BD Pharmingen), or FITC-conjugated monoclonal antibody against the constant region of the murine TCR β chain (eBioscience) and P E-conjugated anti-CD8 antibody. Cells were analyzed using a FACScanto II flow cytometer with CellQuest software (BD Biosciences) or FlowJo software (Tree Star, Inc, Ashland, OR).
Evaluation of cell activity and persistence
Enzyme-linked immunosorbent spot (ELISPOT) assays were carried out by incubating PBMCs overnight in the absence of exogenous cytokine, followed by culturing 105 PBMCs with 105 target cells for 18 hours and evaluating the number of cells secreting IFN-γ as previously described 11. Cell activity was evaluated by coculturing patient PBLs with cognate antigen on T2 target cells, or HLA-matched and mismatched melanomas mel526, mel624 (HLA-A*0201), or mel888 and mel938 (non-HLA-A*0201) or H1299 and H1299-HLA-A*0201 lung cancer cell lines. ELISPOT reagents were purchased from Mabtech Inc (Cincinnati, OH), Millipore Corp (Billerica, MA), and Kirkegaard & Perry (Gaithersburg, MD). Intracellular cytokine staining was performed using a BD cytofix/cytoperm™ (BD Biosciences) according to the manufacturer’s instructions. Briefly, cells were first stained with cell surface markers CD3 and CD8 and mouse TCR beta chain antibody and then stained with PE-conjugated anti-IFN-γ (BD Bioscience) and peridinin chlorphyll protein cytochrome 5.5 (PerCP/Cy5.5) conjugated anti-TNF (Biolegend San Diego, CA.) antibodies for intracellular detection of the cytokines. Cells were analyzed by flow cytometry using a FACSCanto II flow cytometer (BD Biosciences), and data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Patient pathologic analysis and immunohistochemistry
Brain biopsy and autopsy specimens from two patients and control patients were evaluated. Sections from various regions were stained with Hematoxylin and Eosin (H&E). Selected sections were additionally stained with Luxol Fast Blue/Periodic Acid Schiff (LFB/PAS), and immunohistochemistry was performed on deparaffinized, formalin-fixed tissue sections using a panel of antibodies: MAGE A (6C1, 1:100; Santa Cruz Biotechnology, Santa Cruz, CA), CD20 (L26, 1:500; Dako Corporation, Carpinteria, CA), CD3 (F7.2.38, 1:800; Dako), CD4 (1F6, 1:40, Novocastra, Buffalo Grove, IL), CD8 (144B, 1:50, Dako), KP-1 (CD68, 1:400, Dako); NFTP (2F11, 1:800, Dako), GFAP (6F2, 1:200, Dako), CMV (CC42, 1:25, Dako), SV40 (Ab-2, 1:100, CalBiochem, Billerica, MA) and Toxoplasma (strain C56, 1:100, Biogenex, Freemont, CA). Following antigen retrieval, immunostaining was performed on an automated platform (Ventana Medical Systems, Tucson, AZ or Dako Corporation), using one of the manufacturers” recommended detection systems. Each staining batch was run with an external negative and positive control.
Evaluation of non-specific peptide reactivity
To analyze potential cross-reactivity with similar peptides, MAGE-A3/A9 peptide sequence KVAELVHFL was searched against the human genome (standard protein BLAST, blastp algorithm, non-redundant human protein sequence database, http://blast.ncbi.nlm.nih.gov/). Resultant protein matches were then screened against a database of genes expressed in normal human brain33. Additionally, only peptides with a calculated binding affinity >0.1nM (NetMHCpan 2.4 Server, www.cbs.dtu.dk/services/NetMHCpan/) were selected. Eight peptides were synthesized based on these criteria. Peptides were pulsed onto T2 cells at various concentrations for 2 hours at 37°C, washed and then incubated with MAGE -A3 TCR-gene engineered T cells (100,000 cells each) in 200μl media in 96-well plates and incubated overnight. Resultant IFN-γ was measured by ELISA. The gene coding for EPS8L2 that demonstrated reactivity at 100ng/ml was obtained and transfected into 293T cells along with positive and negative control gene (MAGE-A3, GFP). 48 hours post-transfection, cells were co-cultured with TCR engineered T cells (100,000 cells each) in 200μl media in 96-well plates and incubated overnight. Resultant IFN-γ was measured by ELISA.
Pre-treatment PBMC and ex vivo MAGE-A3 TCR-gene engineered T cells from patients 5, 8 and a patient treated with NY-ESO-1 TCR-gene engineered T cells were tested for reactivity against a HLA-A*0201 self-peptide library. This library consisted of 114 peptides eluted and characterized from various cell types, including normal human brain by Stefan Stevanovic and Hans-Georg Rammensee (University of Tubingen, Tubingen, Germany). Peptides were loaded onto T2 cells at 10 μg/ml co-cultured with T cells (5,000 cells each with about 15–36% IFN-γ positive cells after relevant peptide stimulation in intracellular antibody staining) in 200 μl media in 96-well plates and incubated for 24 hours. Titrations of the control peptides (positive and negative controls) ionomycin (750 ng/ml) (both Sigma-Aldrich, and the melanoma cell line 624.38 were included Saint each Louis, MO) were used for TCR- test. PMA (5 ng/ml) and independent stimulation. Resultant IFN-γ was measured by ELISA. Any cultures that demonstrated reactivity above background levels were retested.
Recognition of in vitro generated neural cells
All cell culture reagents were purchased from Invitrogen (Carlsbad, CA) if not otherwise specified. The human neural progenitor cells (NPC) were cultured as previously described34. Briefly, human fetal brain specimens of 7–8 weeks gestation were obtained from Birth Defects Research Laboratory of University of Washington with permission of the Office of Human Subjects Research at the National Institutes of Health (NIH). The tissues were disassociated after removing meninges and blood vessels. After centrifugation at 1000 rpm for 10 minutes, cells were resuspended in NPC maintaining media [DMEM/F12 media containing 8 mM glucose, 1x N2 supplement, 1% antibiotics, 0.1% albumin (Sigma), human fibroblast growth factor-beta (hFGFb; 20 ng/mL) and human epithelial growth factor (hEGF) (20 ng/mL)] and seeded on poly-D-lysine (Sigma) coated T 25 cm2 tissue culture flasks. When cell cultures reached 60% confluence, they were treated with 0.0125% trypsin (Sigma) and replated at a density of 2×104 cells in 100 μl NPC maintaining media on poly-D-lysine coated 96 well plates. Media was replaced every other day. For lentivirus infection, 10 μl of supernatants containing either Pak HLA-A2 or Pak GFP virus were added into the media in each well for 72 hours. The media were then replaced with neuronal differentiation media (DMEM/F12 containing 2% fetal bovine serum, Gemini, West Sacramento, CA) for neuronal differentiation. The NPCs were capable of differentiating into neurons (10–50%) and astrocytes (50–90%) after 4–7 days.
Autologous dendritic cells (DC) recognition of brain homogenates
Generation of DC was performed as described previously35. Briefly, CD14+ monocytes were isolated by positive selection (Miltenyi Biotech, Bergisch Gladbach, Germany) from PBMC and plated in IMDM containing 200 ng/ml rhGM-CSF, 100 ng/ml rhIL-4 (PeproTech, Rocky Hill, NJ) and 5% human serum. Medium was replenished every three days. Immature DC were washed and co-incubated with complex antigens (Ag) brain homogenate (5 μg/ml) and SK-N-SH cell lysate (1 μg/ml). Maturation cocktail (50 ng/ml TNF-α, 10 ng/ml IL-1β, 150 ng/ml IL-6 (all PeproTech) and 1 μg/ml 1 PGE2) was added 24 hours after complex antigens and cells were incubated for additional 48 hours. SSX241–49 peptide, used as negative control, and MAGE-A3112–120 peptide (both 1 μg/ml) were loaded onto mature DC (mDC) 1 hour before co-cultures were set up. DC phenotype was characterized by flow cytometric staining for MHC-II, CD11c, CD25, CD80, and CD83 (eBioscience and BD Biosciences). Peripheral T cells were isolated from CD14-depleted PBMC by negative selection (Pan T cell Isolation Kit II; Miltenyi Biotech) and activated with anti-CD3/CD28 beads (2:1 T cell to bead ratio) for 48 hours. Subsequently, beads were depleted and T cells cultured in IL-2 media (30 IU/ml) for 6 days till assembly of co-culture. At the day of co-culture cryopreserved expanded CSF T cells, transduced and non-transduced peripheral T cells were thawed and rested in T cell media. All T cells were cultured in round bottom 96-well plates with autologous Ag-loaded mDC in a 4:1 ratio in a total volume of 200 μl x-vivo. For degranulation assays, 10 μl of FITC-conjugated anti-CD107a antibody (BD Biosciences) was added directly to unstained T cell-mDC co-cultures for 24 hours. T cells were then analyzed for surface marker expression of CD3, CD4, and CD8 and binding of CD107a. For proliferation assays, peripheral and cerebrospinal fluid (CSF) derived T cells were labeled with carboxy fluorscein succinimidyl ester (CFSE) as described previously 36 and cultured with autologous Ag-loaded mDC for 9 days. Then, T cell proliferation was assessed by means of CFSE dilution as described previously36. Data were analyzed with BD FACS Diva 6.1 and results were standardized to negative control.
Molecular assays
RNA was isolated from flash-frozen tissue samples using RNeasy mini kit (Qiagen, Valencia, CA) according to the manufacture’s instruction. One μg of total RNA was reverse transcribed using the ThermoScript RT -PCR system (Invitrogen) and 100 ng of each cDNA was used for the real-time quantitative-PCR (Q-RT-PCR) assay (TaqMan; Applied Biosystems, Foster City, CA). All PCR were performed using an ABI 7500 Fast Real-time PCR System instrument (Applied Biosystems/Life Technologies, Carlsbad, CA). The TaqMan gene-specific assay was designed by ABI Assays-by-Designs software (Applied 6-carboxyfluorescein Biosystems). Primers and probe used for detection were: MAGE-A3/A6-Probe ((FAM)) 5′-CCAGGTCAGCCTGTCCCC-3′,MAGE-A3/A6-F:5′-GAAGCCGGCCCAGGCTCG-3′; MAGE-A3/A6 R: 5′-TCCTCCGGGGGCCTCT-3′; MAGE-A9-Probe(FAM) 5′-CCACAGGCAGGTCTTC-3′,MAGE-A9-F 5′-CCCCAGAGCAGCACTGA-3′, MAGE-A9-R 5′-GGAGCTGGGCGATGGA-3′; MAGE-A12 Probe-(FAM) 5′-AGTGTGGGCAGGAGCTAGTGCTGCTCCG-3′, MAGE12-F 5′-TCCGTGAGGAGGCAAGGTTC-3′, MAGE A12-R 5′-CTCTGGTCAGGGCAGCAGGTA-3′. The reference standard curves were established using plasmid DNA. TaqMan β-actin control reagents kit (Applied Biosystems) was used to verify the quality of input RNA. Human brain reference RNA, cerebellum, occipital cortex, brain stem, liver and heart RNAs were purchased from commercial sources and were derived from pools of multiple donors (Invitrogen, Agilent Technologies, Santa Clara, CA, and Clontech, Mountain View, CA). 2.0μg of each RNA was analyzed using the nCounter Analysis System37, 38 (NanoString Technologies, Seattle, WA) with a custom designed reporter code set and capture probes with nCounter master kit hybridization solutions for 12 hours at 65°C. The post-hybridization processing was undertaken using the nCounter Prep Station and analyzed with an nCounter Digital Analyzer. nSolver Analysis software version 1.0 was used to carry out normalization compared to internal controls. For RNA deep-sequencing, total RNA was extracted from the dorsolateral prefrontal cortex (DLPFC) of post-mortem brain tissue from 6 psychiatrically normal controls. Three of these were obtained from the Stanley Medical Research Institute (Chevy Chase, MD) and 3 from the Clinical Brain Disorders Branch (CBPB), National Institute of Mental Health (Bethesda, MD). Details on the pathology and dissections of the CBPB post -mortem brains were as described39. Deep-sequencing of mRNA was performed at the NIH Intramural Sequencing Center (Bethesda, MD). Paired-end sequencing San Diego, CA). (read length = 100bp) was carried out on an Illumina HiSeq instrument (Illumina, Quality control, mapping and data -analysis were as described (Akula et al, manuscript in preparation).
Results
Patient characteristics and infused cell properties
This phase I/II clinical trial was designed to determine if the administration of anti-MAGE-A3 TCR gene-modified peripheral blood lymphocytes and IL-2 to patients following non-myeloablative but lymphoid depleting preparative regimen would result in clinical tumor regression in patients with metastatic cancer that express the MAGE-A3 (or A12) antigen. The first three patients in the trial were limited to a total cell dose of up to 3 × 1010 cells with subsequent patients being permitted to receive up to 1 × 10 11 total cells. Table 1 summarizes the patient characteristics, cell treatment, and clinical findings. Nine patients were treated (four male, five female), seven with metastatic melanoma, one with synovial sarcoma, and one with esophageal cancer. They ranged in age from 21 to 71 and had been extensively pretreated, including enrolment in previous T cell gene-modified T cell adoptive cell transfer (ACT) protocols (patients 1, 2, 3, 4, and 9).
Table 1.
Patient characteristics, therapy, and response.
| Patient | Age/Sex | Body weight (kg) | Diagnosis | Sites of disease | MAGE-A IHC | Prior therapy | Cells (× 109) | IL-2 doses | Response | Neurological toxicity |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 59/M | 102.6 | Melanoma | lung | 3+, >80% | Surgery, IL-2, NY ESO-1 TCR, Chemo, XRT | 28 | 6 | CR (15+) | none |
| 2 | 38/F | 110 | Melanoma | R axilla, L periadrenal, R breast, retrocaval, brain | 2+, >70% | Surgery, IFN, IL-2, MART-1 TCR, Zanolimumab, TIL, Chemo, XRT | 30 | 5 | NR | none |
| 3 | 56/F | 59.5 | Melanoma | Subcu, intraabd, R axilla, omentum | 3+, >90% | Surgery, IL-2, TIL-IL12, Chemo | 30 | 7 | PR (4) | none |
| 4 | 21/F | 74.2 | Synovial sarcoma | lung | 3+, >90% | Surgery, XRT, Chemo, NY ESO-1 TCR, ALVAC, IL2 | 41 | 1 | PR (5) | none |
| 5 | 54/M | 94.8 | Melanoma | lung, subcu, mesenteric nodes, brain, R axilla, spleen, paratracheal | 2–3+, >70% | Surgery, IFN, XRT, IL2, Zanolimumab | 79 | 5 | PR(4) | Seizures, coma, TRM |
| 6 | 44/M | 64.6 | Melanoma | Right thigh, R iliac nodes | 3+, >95% | Surgery, Chemo, GM-CSF, TIL, IL-2 | 53 | 4 | NR | none |
| 7 | 62/M | 82.7 | Melanoma | lymph node, rib | 3+, >90% | Surgery, INF, IL2 | 62 | 6 | PR (12+) | Seizure, MS |
| 8 | 71/F | 43.5 | Esophageal cancer | Mediastinal; bilat lung | 3+, >90% | Surgery, XRT, Chemo | 61 | 1 | NR | Coma, TRM |
| 9 | 62/F | 99.2 | Melanoma | Liver, subcu and L axilla, intra-abd, neck/cervical, vertebral | 3+, >50% | Surgery, IFN, IL-2, NY ESO-1 TCR, Chemo, ALVAC | 30 | 0 | NR | TIA |
Abbreviations: IL-2, interleukin-2; IFN, interferon; Chemo, chemotherapy; TCR, T cell receptor; ALVAC, modified canary pox virus vaccine; GM-CSF, granulocyte-macrophage colony-stimulating factor; M, male; ln, lymph node; XRT, radiation; F, female; subc, subcutaneous; L, left; R, right; bilat, bilateral; abd, abdominal; MS, mental status; TRM, treatment related mortality; IHC, immunohistochemistry; IL12, interleukin-12; TIA, transient ischemic attack; CR, complete response; PR, partial response; NR, no response.
Table 2 lists the properties of the infused cell products. Robust gene transfer efficiency was achieved, with an average of 85% CD3+T cells expressing the murine TCR beta chain and an average of 70% CD8+T cells positive for the MAGE -A3 tetramer (Table 2). Ex vivo cell expansion resulted in slightly more CD8+ than CD4 + T cells on average (43% CD4+ and 50% CD8+). All transduced T cell products demonstrated specific IFN-γ produced following co-culture with antigen and HLA matched tumor cell lines. Table 2 also lists the total CD4+ and CD8 + T cells in fused, the number of T cells/kg body weight and the total number of CD3+/CD8+/TCR+ administered. The infused T cell product displayed a phenotype consistent with our previous observations11 that the majority of ex vivo expanded T cells have a T effector cell (Teff) phenotype (CD45RO+/CD62L−), with a smaller number of cells with a T central memory phenotype (Tcm, CD45RO+/CD62L+) and a T naïve phenotype (Tn, CD45R0−/CD62L+) (Fig. 1).
Table 2. Properties of infused cell product.
Percent transduction determined as CD3+/murine TCR beta chain+ cells.
| Patient | Tumor Cell Targets (pg/ml, IFN-γ) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % of CD3 | Percent Transduction | % CD3+/CD8+ | MAGE-A3 Positive | MAGE-A3 Negative | Total cells (×109) Per Kg BW | Total (×109) | Total (×109)CD3+/CD8+ | |||||
| CD4 | CD8 | Tet | mTCR | CD4 | CD8 | Tet | mTCR | |||||
| 1 | 36 | 64 | 92 | 78 | 92 | 23,543 | <30 | 0.273 | 10.08 | 17.92 | 13.98 | 16.49 |
| 2 | 50 | 18 | 90 | 75 | 90 | 9,668 | 41 | 0.273 | 15 | 5.4 | 4.05 | 4.89 |
| 3 | 59 | 41 | 92 | 85 | 95 | 19,883 | <30 | 0.504 | 17.7 | 12.3 | 10.46 | 11.69 |
| 4 | 42 | 58 | 61 | 58 | 90 | >109,600 | 53 | 0.553 | 17.22 | 23.78 | 13.79 | 21.28 |
| 5 | 39 | 56 | 81 | 53 | 88 | 23,850 | 47 | 0.833 | 30.81 | 44.27 | 23.46 | 38.96 |
| 6 | 55 | 43 | 77 | 50 | 89 | 8,223 | <30 | 0.82 | 29.15 | 22.79 | 11.40 | 20.28 |
| 7 | 29 | 58 | 95 | 85 | 96 | 21,767 | <30 | 0.75 | 17.98 | 40.92 | 34.78 | 39.28 |
| 8 | 26 | 66 | 87 | 70 | 91 | 23,867 | 69 | 1.402 | 15.86 | 40.26 | 28.18 | 36.64 |
| 9 | 54 | 45 | 90 | 73 | 93 | 7,035 | <30 | 0.302 | 16.2 | 13.5 | 9.86 | 12.56 |
Tet, HLA-*0201-specific MAGE-A3 tetramer. mTCR, anti-murine TCR beta chain antibody. IFN-γ, gamma-interferon. BW, body weight.
Figure 1.
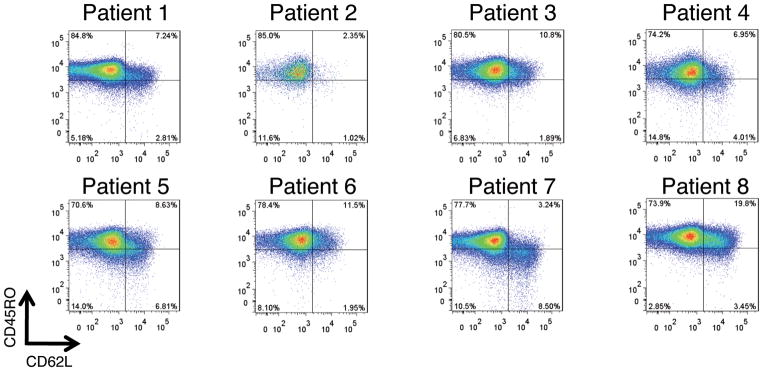
Phenotype of infused cell products. Shown are FACS dot plots for patients 1–8 for cells surface marker proteins CD45RO and CD62L with the percentages of positive cells indicated in each quadrant. T cells subsets are defined as follows, T effector cells (Teff, CD45RO+/CD62L−), T central memory cells (Tcm, CD45RO+/CD62L+), and T naïve cells (Tn, CD45RO−/CD62L+).
In vivo properties of MAGE-A3 TCR engineered T cells and clinical response
At one-month post-treatment peripheral blood samples were obtained to determine the presence and biological activity of the TCR gene-modified T cells (it should be noted that samples from patients 7 and 8 were obtained after the administration of high-dose steroids, see below). Using an antibody specific for the murine TCR beta chain permitted the accurate determination of these T cells by FACS analysis (Fig. 2). All patients demonstrated TCR+ cells in the circulation in the range of 11%-84% (mean 41%). The biological activity of the persistent T cells was determined by intracellular cytokine staining and Elispot assays. Cells from patients 1–8 were co-cultured with the MAGE-A3+ positive lung cancer line H1299 (or the same line engineered to express HLA -A*0201) for four hours and then stained for TNF and IFN-γ production (Fig. 3A). FACS analysis following gating on CD3+/CD8+ T cells showed that TCR -engineered cells from all patients stained positive for IFN-γ. Patients 1,2,4,5, and 6 demonstrated TNF positive cells, and patients 4, 5, and 6 had cell populations co-expressing both IFN-γ and TNF. We next performed Elispot assays using both peptide pulsed T2 cells and MAGE-A3+ tumor cell lines (Fig. 3B). Every patient analyzed was reactive with both peptide pulsed cells and the tumor cell line targets (patients7 and 8 had low levels of antigen reactive cells in the circulation, e.g., for patient 7; 127 IFN-γ spots/1 × 10 5 PBMC with peptide MAGE-A3 peptide pulsed cells, compared to control peptide <10).
Figure 2.

Persistence of MAGE-A3 TCR post-infusion. Shown are the FACS density dot plots from patients PBMC isolated approximately one-month post-infusion. Scatter plots depict CD3+ and murine MAGE -A3 TCR beta chain expression on lymphoid cells, with the percentages of positive cells indicated in each quadrant. muTCR, murine beta chain TCR.
Figure 3.

Biological activity of MAGE-A3 TCR engineered T cells in patient PBMC. A. PBMC samples from patients 1–8 were obtained approximately one month post-infusion and co-cultured with control tumor line H1299 (MAGE-A3+/HLA-A*0201−) or reactive tumor line H1299-A2 (MAGE-A3+/HLA-A*0201+). Shown are the resultant FACS density plots gated on CD3+/mouse-TCR beta+ for effector cytokines IFN-γ and TNF. The percentages of positive cells were as indicated in each quadrant. B. PBMC samples from patients 1–9 were obtained approximately one month post-infusion and subject to Elispot analysis. Number of Elispot reactive cells are plotted following PBMC co-culture with MAGE-A3-peptide pulsed cells (left side) or MAGE-A3+/HLA-A*0201+ tumor cell line (right side). Results from control peptide pulsed cells and MAGE-A3+/HLA-A*0201− tumor cell lines were <10 spots/1 × 105 PBMC (not shown).
Five of 9 patients demonstrated cancer regression using RECIST criteria following infusion of anti-MAGE-A3 TCR gene-engineered T cells (Table 1, Fig. 4). Patient 1 had metastatic melanoma in the lung that decreased at one month and is an on-going complete responder 15+ months following treatment. Patient 3 had metastatic melanoma in the subcutaneous and intra-abdominal sites and had a partial regression lasting 4 months. Patient 4 with metastatic disease in the lung from synovial cell sarcoma demonstrated partial regression for 5 months. Patient 5 with metastatic melanoma to the lung, subcutaneous, and mesenteric lymph nodes had a partial regression for 4 months. Patient 7, with metastatic melanoma to lymph nodes and rib has demonstrated an on-going partial regression (89% decrease in target lesions) 12+ months post-treatment.
Figure 4.

Clinical anti-tumor response. Shown are CT scans for patients 1 and 5 and CT/PET scans for patient 7. The timing of the images were as listed.
Neurologic toxicity following the infusion of MAGE-A3 TCR engineered T cells
Although our observations of cancer regressions were encouraging, three patients developed severe neurologic toxicity. Patient 5 received 79 ×109 anti -MAGE-A3 TCR gene engineered cells followed by 5 doses of IL-2, which was stopped on day three for mental status changes, tachycardia, and hypotension. In the prior five months before receiving MAGE-A3 TCR transduced T cells, patient 5 had undergone a craniotomy to remove three brain metastases followed by whole brain irradiation. The patient’s clinical condition continued to deteriorate with loss of consciousness and he was placed on ventilator support. Sedation was discontinued on day seven but the patient remained unresponsive with a brain CT on day eight showing no intracranial abnormalities. The patient experienced grand mal seizures on days 11, 13 and 41, and an EEG on day 11 demonstrated diffuse background slowing, with no epileptiform activity. A brain MRI on day 25 showed bi-occipital edema, with repeat MRIs on day 41 showing prominent bilateral white matter abnormalities involving the cerebral hemispheres and on day 57 continued progression of leukomalacia (Fig. 5). A brain biopsy on day 68 revealed vaccuolation of white matter, with sparing of gray matter. Inflammatory infiltrates (T cells) and reactive changes were also identified. The patient could open his eyes to voice command and tracked movements but could not follow commands. During this period the patient was treated with high dose steroids, levetiracetam, and carbidopa-levodopa with no change in clinical course. The initial regression of melanoma lesions was followed by progression, however, and the family decided to withdraw life support, and the patient died on day 167.
Figure 5.
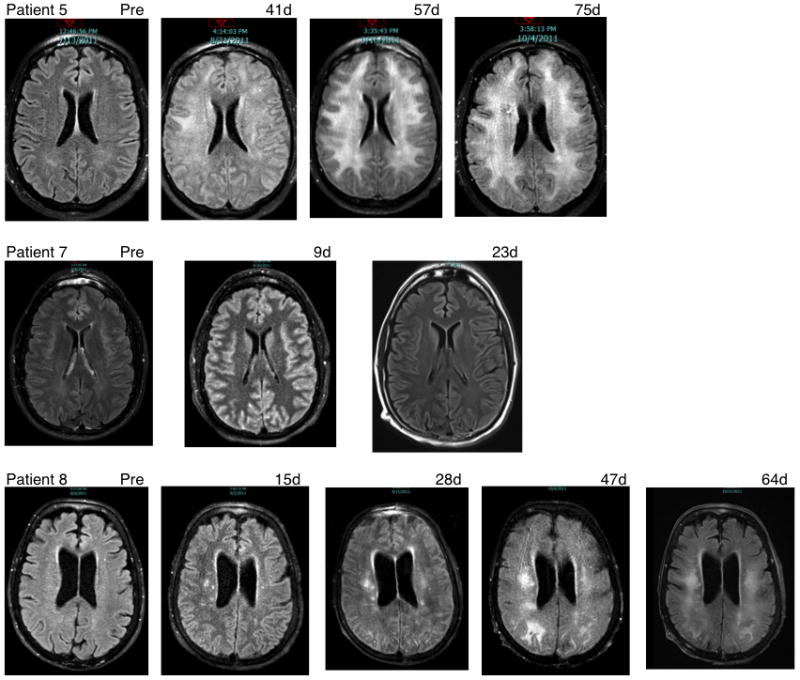
Neurological imaging studies. Shown are MRI scans for patients 5, 7 and 8, with the timing of the images as listed.
Patient 6 was subsequently treated with a similar T cell product administered to patient 5, and did not manifest any signs of neurologic complications. It was thus unclear whether the neurological complications experienced by patient 5 were related to the cell infusion or were the results of the interaction of IL-2 with the existing brain disease and the recent surgery and radiation therapy. Patients 7 and 8 were treated the same week. Patient 7 received 62 × 109 gene modified T cells, followed by 6 dose of IL-2 stopping for shortness of breath and pulmonary edema. On day three the patient was noted to have mental status changes and had a generalized tonic-clonic seizure and was intubated for airway support. A head CT scan at this time was normal. Between days 3–8 the patient continued to display seizure activity with EEG documented activity in the left temporal lobe, and brain MRI scans carried out on day nine and day 23 were normal (Fig. 5). The patient was administered phenytoin, levetivacetam, and midazolam in an attempt to break seizure activity, which resolved and he was awake and following commands by day 11 and was extubated on day 12. The patient’s mental status was decreased from baseline with difficulty in word finding and demonstrated significant loss of fine-motor coordination. The patient was treated with dexamethasone and carbidopa-levodopa beginning on day 21; these were gradually discontinued over the next few weeks. The patient made rapid improvement in mental and physical condition and was discharged on day 37. The patient made a full neurological recovery by day 64 and has demonstrated a near complete regression of his melanoma, which is on-going at 12 months following treatment.
Patient 8 received 61 × 109 cells followed by 1 dose of IL -2 stopping for mental status changes and hypotension. She was placed on ventilator and vasopressor support. Vasopressors were ultimately weaned on day 7 and sedation was discontinued at that time as well. During this time, the patient had rising creatinine and worsening oliguria requiring continuous veno-venous hemofiltration therapy from day 12 through day 20. The patient failed to demonstrate improvement in mental status following correction of metabolic abnormalities and a brain MRI on day 15 demonstrated ischemic lesions and areas of infarct (Fig. 5). A stroke work-up failed to reveal an embolic source. Steroids were initiated with minimal improvement in mental status. Follow-up MRI studies (days 28, 47, and 64) demonstrated progressive white matter changes consistent with leukomalacia. A brain biopsy was performed on day 34 and demonstrated significant vaccuolation of white matter, a small infarct area, with sparing of gray matter. Inflammatory infiltrates (lymphocytes) and reactive changes were also identified. Although the patient had an initial regression of her esophageal cancer at 1 month, there was clear disease progression at 2 months. Given the poor neurological prognosis and overall disease progression, the patient was withdrawn from life support and died on day 94.
Patient 9 developed a transient aphasia six days after receiving cells. She received a short course of steroids and all symptoms resolved over 48 hours. Forty-six days later she experienced another episode of transient aphasia. Carotid ultrasound and magnetic resonance angiography revealed complete occlusion of the left internal carotid artery and she received anti-coagulation therapy. We have thus attributed these symptoms to transient ischemic attacks, although a contribution of the MAGE -A3 TCR transduced cells cannot be ruled out.
Consent was given for autopsy on patients 5 and 8. Microscopic examination of patient 5’s brain revealed widespread white matter damage, diffusely involving the whole brain (cerebrum, cerebellum, and brainstem). A spectrum of myelin/axonal damage was present with morphological changes including white matter spongy vaccuolation, myelin pallor, and axonal injury characterized by thickening and axonal spheroids; frankly necrotic areas with myelin and axonal loss and mineralization were also present (Fig. 6). Extensive gliosis and microglia activation was seen. There were high numbers of CD3+/CD8+ lymphocytes infiltrated into the parenchyma and perivascular spaces with frequent KP1/CD68+ monocytic/macrophage lineage cells pre sent (microglia, histiocytes) (Fig. 6). CD4+ T cells and CD20+ B cells were rare (Fig. 6). The gray matter was more preserved and had significantly fewer infiltrating lymphocytes. There was a small focus of melanophages observed in the left frontal cortex, but no viable tumor was present. Examination of patient 8’s brain revealed strikingly similar morphological changes at autopsy. Again damage was mainly confined to white matter and there were numerous CD3+CD8+ T cells present. The patient’s brain also displayed areas of frank necrosis with myelin and axonal loss and mineralization. There was no microscopic evidence of tumor in the brain. These histological features and the extent of CNS lesions in both patients are most consistent with multifocal necrotizing leukoencephalopathy.
Figure 6.

Neurologic toxicity. Autopsy samples from patients 5 and 8 had microscopic sections that displayed similar changes consistent with necrotizing leukoencephalopathy, multifocal. There was diffuse white matter damage with sparing of gray matter (panel A, hematoxylin and eosin (HE) stain, patient 8, 10X). Marked white matter vaccuolation (patient 8, panel B, HE-20X) to frank areas of infarct/necrosis with mineralization (patient 5, panel C, HE -20X) were identified. Inflammatory infiltrates were present around small vessels and parenchyma and consisted of CD3+/CD8+ lymphocytes and KP1+ histiocytes (patient 5, panel D: CD3 immunostain at 2x, panel E: CD8 immunostain at 2x, panel F : KP-1 immunostain at 2× and patient 8; panel G: CD3 immunostain at 2x, panel H: CD8 immunostain at 2x)
Cerebral spinal fluid (CSF) samples were obtained from patients 5, 7, and 8 and showed elevated protein levels. Small numbers of CD3+ T cells were recovered from CSF and observed to have a preponderance of CD4+ T cells. Following ex vivo cell expansion of the CSF cells, the introduced TCR genes were found expressed in a higher percentage of CD4+ than CD8+ T cells (Fig. 7A). A sample from the peripheral blood of patient 7 obtained at the same time as the CSF sample (and subjected to the same cell expansion) did not show this preponderance of MAGE-A3 TCR+CD4+ T cells. Expanded CSF cells from patient 5 produced IFN-γ in response to MAGE -A3+ tumor target cell lines demonstrating that these cells were biologically active (Fig. 7B). The TCR beta gene variable regions were directly amplified from the CSF sample obtained from patient 8, cloned and subject to DNA sequence determination. Twenty-four individual TCR beta chain gene clones were analyzed with no significant TCR repertoire skewing observed in this limited analysis (Fig. 7C).
Figure 7.

MAGE-A3 TCR engineered T cells in CSF. A. Shown are FACS dot plots for patients 5, 7, and 8 for T cells obtained from CSF and expanded ex vivo (and PBL for patient 7). Samples were stained and plotted for murine TCR beta (muTCR) and CD8. B. Ex vivo expanded cells from patient 5’s CSF sample were co-cultured with control tumor lines 888 mel and 938 mel (MAGE-A3+/HLA-A*0201−) or reactive tumor cell lines 526 mel, 624 mel, and H1299-A2 (MAGE-A3+/HLA-A*0201+). Shown is the resultant IFN-γ production following overnight culture. C. Samples taken directly from the CSF of patient 8 were subject to rapid amplification of cDNA ends (5′-RACE) PCR to amplify the variable region of human TCR beta chain genes. Shown are the results of DNA sequence analysis as a pie chart showing the specific TCR vbeta gene (color) as well as its frequency (size of pie slice).
Multiple immunological assays failed to demonstrate off-target reactivity
Analysis of the IFN-γ serum levels from patients receiving the MAGE -A3 TCR gene engineered T cells did not demonstrate unusually high levels of serum IFN-γ (Fig. 8). Cytokine levels were similar in patients with and without neurologic toxicity and were comparable, or less, than those reported for patients receiving anti-gp100 and anti-MART-1 TCR gene engineered T cells11. To further investigate the potential cause for the observed neurologic toxicity, we performed a series of experiments designed to test for off-target recognition by this TCR.
Figure 8.

Serum cytokine production. Shown are the gamma-interferon (IFN-γ) levels (pg/ml) found in the serum of patients 1–8 during the times indicated (time = 0 is the day of infusion).
The peptide recognized by the MAGE-A3 reactive TCR (KVAELVHFL) is shared with MAGE-A9, with the MAGE-A12 gene having a similar peptide (KMAELVHFL) that is more avidly recognized due to a higher binding affinity with HLA-A*0201 (calculated binding affinity 12.47nM and 2.09nM respectively, NetMHCpan server 2.4). Based on a bioinformatics search of the human genome, we synthesized eight additional non-MAGE-A gene family peptides based on similar amino acid sequences to the MAGE -A3 peptide epitope plus appropriate HLA-A*0201 binding affinities (predicted 0.25–7.0 nM) and reported gene expression in human brain. These peptides were pulsed onto T2 cells and co-cultured with MAGE-A3 TCR gene modified T cells (Fig. 9). One of the non-MAGE-A peptides from gene EPS8L2, induced TCR gene-engineered T cells to produce IFN-γ when pulsed at high concentration (>100 ng/ml). Transfection of this gene into HLA-A2*0201 positive cells did not cause release of IFN-γ in co -culture assays (not shown), suggesting lack of protein processing and/or HLA presentation.
Figure 9.

Recognition of MAGE-A3-like peptides. The human genome was searched for peptide similar to MAGE-A3 that were predicted to have physiologically relevant HLA-A*0201 binding affinities and were expressed in human brain. These eight peptides were synthesized and pulsed onto T2 cells along with MAGE-A3 and MAGE-A12 peptides at the indicated concentrations. Shown is the resultant gamma-interferon (IFN-γ) production (pg/ml) following overnight co-culture.
We next tested for reactivity associated with the top 114 most prevalent peptides that had been eluted directly from HLA-A*0201 positive normal human tissues, including human brain. This human peptidome screen was performed with MAGE-A3 TCR gene modified cells from patient 5 and 8 and as a control, T cells from a patient that had been previously treated with NY-ESO-1 TCR gene-modified T cells. None of the 114 human peptides that were tested, however, stimulated cytokine release from TCR-transduced T cells (Fig. 10).
Figure 10.

Analysis of human peptidome. The 114 most highly prevalent peptides found on the surface of HLA-A*0201+ normal tissues including brain were used to test for reactivity of MAGE-A3 TCR transduced T cells. Shown are the resultant IFN-γ levels produced in overnight co-cultures with peptide pulsed cells, open bars-untransduced T cells, solid bars-MAGE-A3 TCR transduced T cells. Shown is data for patient 5, similar results were obtained with patient 8 and a patient treated with NY-ESO-1 TCR transduced T cells. Co-cultures with melanoma cells mel 624.38 (+) and stimulation with PMA/ionomycin (P) served as positive controls per 96 -well plate to exclude differences between plates. Medium alone, PBL alone, T2 cells alone and PBL co-cultured with unpulsed T2 cells (−) were used as negative controls.
To test for potential recognition of naturally expressed and presented peptides from normal human brain tissue we took fetal neural progenitor cells, transduced them with the HLA-A*0201 gene and then differentiated them into mature neural cells (Fig. 11A). These normal human neural cells were then co-cultured with MAGE-A3 TCR gene-transduced T cells along with peptide-pulsed positive and negative controls. Again, the MAGE-A3 TCR-transduced T cells failed to recognize normal human neural cells. As a final test for off-target reactivity, autologous dendritic cells from patient 5 were produced and pulsed with normal human brain tissue homogenate and then co-cultured with MAGE-A3 TCR gene-transduced cells and the MAGE-A3 reactive T cells (mainly CD4+) that had been isolated and expanded from this patients’ CSF sample. There was no evidence for the recognition of normal human brain tissue homogenate by these assays, as demonstrated by either their ability to mobilize degranulation marker CD107a (Fig. 11B) or to proliferate in response to tissue homogenates (Fig. 11C).
Figure 11.

Test for recognition of neuronal cells. A. Fetal-derived neural progenitor cells were transduced with a lentiviral vector expressing GFP or HLA-A*0201 and induced to differentiate into neural cells. Image of resultant GFP-transduced neural cells was as shown on the left. These cells were co-cultured with MAGE-A3 TCR transduced or untransduced (UnTd) T cells. As control for recognition, neural cell cultures were pulsed with MAGE-A3 peptide or as a negative control, SSX-2 peptide. B and C. Autologous dendritic cells were prepared from patient 5 and loaded with cell lysates from neuroblastoma cell line SK-N-SH, whole brain homogenate (BH), or pulsed with MAGE-A3 peptide (MAGE-A3). These were then co-cultured with T cells derived from patient 5; pre-treatment PBMC (periph), T cells derived from the ex vivo expanded CSF (CSF), the MAGE-A3 TCR transduced infusion cell product (TCR), or ex vivo cultured, untransduced T cells (UnTd). Reactivity was measured by CD107 amobilization (B) or 8-day cell proliferation measured by CFSE dilution (C). Reactivity in both CD8+ (left) and CD4+ (right) T cells was as shown.
Detection of MAGE-A gene expression in human brain
Brain samples obtained from patients 5 and 8 during biopsy and subsequent autopsy were subjected to immunohistochemical staining with an anti-MAGE-A3 antibody. The antibody used (6C1) was reported to detect multiple MAGE-A family gene members and we verified by western blot analysis that it recognizes MAGE-A1, A2, A3, A4, A6, A9, A10, and A12 (not shown). We are not aware of a report of an antibody specific for MAGE-A3. Results form this analysis demonstrated rare MAGE-A positive staining of neurons and axons were observed in brain samples from both patients 5 and 8 and were also observed in the brains of control patients (Fig. 12).
Figure 12.

MAGE-A expression in human brain. MAGE-A family reactive antibody 6C1 was used for immunohistochemical staining of brain sections. Positive staining was observed in neuronal cell bodies and processes in three control patients (A at 10x, B at 20x, and C at 10x), in patient 5 (D at 20x, E at 40x, and F at 40x), and patient 8 (G at 10x, H at 20x, I at 20x).
RNA obtained from autopsy samples from patients 5 and 8 was subject to Q-RT-PCR using a set of primers and probes that were specific for the MAGE -A3 and MAGE-A6 gene products, as well as a primer and probe set that specifically detected the MAGE-A12 gene transcript (Table 3). Tumor samples from both patients expressed MAGE-A3/A6 transcripts at similar levels, whereas expression of MAGE-A12 transcripts appeared to be high in the tumor sample from patient 5 but relatively low in the tumor sample from patient 8. Five normal organ/tissue samples were negative or had < 6 copies of the MAGE-A genes. In comparison, multiple samples of neurological origin were found to have > 20 copies of MAGE-A12 but not MAGE-A3/A6. Because of the potential for contamination of normal brain tissue from these patients’ tumors, we obtained multiple commercially available RNA samples of normal human organs/tissues. Q-RT-PCR for the MAGE-A3/A6, MAGE-A9, and MAGE-A12 gene transcripts was performed on these samples with and without reverse transcriptase and the plasmid DNA standards were not amplified in these assays in an attempt to lessen the potential for cross-over contamination of samples. Comparison of cycle threshold values reported in Table 4 suggested that MAGE-A9 and MAGE-A12 gene transcripts were expressed in samples of human total brain and cerebellum (these RNAs were derived from pools of multiple donors, 23 and 11 respectively). To verify that the products amplified in these PCRs represented MAGE-A genes, the PCR reaction products were cloned and DNA sequence analysis was carried out. These results demonstrated that the MAGE -A9 and MAGE-A12 transcripts were amplified from these normal human brain samples.
Table 3. MAGE-A gene expression in patient autopsy samples.
Q-RT-PCR determination of MAGE-A3/A6 and MAGE-A12 copy numbers in clinical samples (100ng cDNA per sample) determined using plasmid DNA standard curves.
| Tissue | PATIENT 5 | PATIENT 8 | ||
|---|---|---|---|---|
| MAGE-A3/A6 | MAGE-A12 | MAGE-A3/A6 | MAGE-A12 | |
| Tumor | 6476 | 27686 | 5507 | 614 |
| Lung | nd | 5 | 1 | nd |
| Spleen | nd | 4 | 1 | nd |
| Kidney | nd | nd | nd | nd |
| Pancreas | nd | nd | nd | nd |
| Lymph node | 2 | nd | 1 | nd |
| Brain Stem | nd | 25 | 2 | 2 |
| Basal Ganglia | nd | 48 | nd | 38 |
| Frontal lobe-GM | nd | 53 | 2 | 26 |
| Frontal lobe-WM | nd | 7 | 3 | 14 |
| Cerebellum-GM | nd | 11 | 2 | 1 |
| Occipital lobe-GM | nd | 13 | 1 | 26 |
| Occipital lobe-WM | nd | nd | 1 | 2 |
| Parietal lobe-GM | nd | 15 | nd | 70 |
| Parietal lobe-WM | nd | 5 | 3 | 35 |
| Temporal lobe-GM | nd | 30 | 1 | 19 |
| Temporal lobe-WM | nd | 4 | 2 | 14 |
nd, Ct value of 40 or greater (not detected). GM, gray matter; WM white matter.
Table 4. Cycle threshold values for MAGE-A gene transcripts from commercial RNA sources.
Q-RT-PCR data, average of duplicate determinations using 100ng cDNA per sample.
| Sample | MAGE-A3/A6 | MAGE-A9 | MAGE-A12 | Actin | ||||
|---|---|---|---|---|---|---|---|---|
| RT− | RT+ | RT− | RT+ | RT− | RT+ | RT− | RT+ | |
| Total Brain | 37.1 | 36.3 | nd | 33.6A | nd | 31.1C | 37.3 | 18.3 |
| Cerebellum | 36.7 | 36.4 | 37.6 | 31.5A | nd | 31.4C | nd | 18.2 |
| Thyroid | 36.4 | 36.8 | nd | 34.7B | nd | 38.9D | nd | 19.6 |
| Testes | 36.6 | 21.3 | nd | 21.9A | nd | 26.2C | nd | 18.6 |
RT+, RT−, reactions performed with and without reverse transcriptase addition.
sequence verified to be MAGE-A9.
unrelated sequence.
sequence verified to be MAGE-A12.
unrelated sequence.
nd, cycle threshold value of 40 or greater (not detected).
We next subjected normal human RNA samples to direct mRNA quantitation using solution-based multiplexed gene expression analysis using the NanoString nCounter methodology (Table 5). Multiple CTA genes were included in the NanoString probe set along with genes known to be expressed in neurological tissues (e.g., GD2) as well as a negative control probe designed to detect a glioblastoma-specific EGFR gene rearrangement (EGFRvIII) that is not know to be expressed in any normal tissue. In this assay the mean of the values obtained with 96 synthetic negative control probe samples was 8.7 (±8.6). Based on an arbitrary cut-off value of 20, these results suggest that MAGE-A1 and MAGE-A12 RNA may be present in commercial samples derived from normal human neurological tissues (total brain, cerebellum, or occipital cortex). Finally, we queried a human brain deep-sequencing database of RNA obtained from the DLPFC of control donors. A total of 6 individuals with >200 million mapped sequence reads per sample were analyzed (Table 6). Data from this large RNAseq database indicated that the MAGE-A12 and MAGE-A8 genes were expressed in samples from human frontal cortex, albeit at a low level.
Table 5. Nanostring quantitation of CT antigen expression in commercial brain RNA samples.
Shown are the averages of duplicate determinations using 2μg RNA/sample. Probes that defect multiple genes were as indicated in parenthesis. CT, cancer testes.
| Cancer Testes Gene | Gene Common Name | Brain reference | Cerebellum | Occipital cortex | Brain Stem | Liver | Heart |
|---|---|---|---|---|---|---|---|
| BAGE | CT antigen | 25 | 4 | 6 | 4 | 7 | 7 |
| CSAG2 | CT antigen (also detects CSAG3) | 153 | 58 | 111 | 54 | 5 | 12 |
| CTAB1B | NY-ESO-1 (also detects CTAG1A) | 6 | 2 | 2 | 3 | 4 | 5 |
| CTAG2 | LAGE1, CT antigen | 17 | 3 | 7 | 4 | 39 | 185 |
| CXorf48 | CT antigen | 192 | 34 | 402 | 73 | 41 | 38 |
| GAGE1 | CT antigen | 17 | 5 | 9 | 5 | 9 | 10 |
| GAGE4 | CT antigen | 106 | 180 | 58 | 56 | 5 | 57 |
| MAGEA1 | CT antigen | 31 | 25 | 28 | 18 | 54 | 31 |
| MAGEA10 | CT antigen | 8 | 8 | 12 | 6 | 19 | 14 |
| MAGEA11 | CT antigen | 6 | 4 | 7 | 4 | 2 | 1 |
| MAGEA12 | CT antigen | 33 | 15 | 25 | 19 | 9 | 3 |
| MAGEA2 | CT antigen | 2 | 2 | 1 | 2 | 2 | 1 |
| MAGEA3 | CT antigen (also detects MAGE-A6) | 6 | 2 | 5 | 3 | 1 | 27 |
| MAGEA4 | CT antigen | 7 | 2 | 3 | 3 | 4 | 6 |
| MAGEA9 | CT antigen | 6 | 2 | 4 | 12 | 3 | 2 |
| MAGEB1 | CT antigen | 7 | 6 | 8 | 5 | 5 | 4 |
| MAGEB2 | CT antigen | 6 | 1 | 2 | 2 | 5 | 2 |
| MAGEB3 | CT antigen | 21 | 21 | 21 | 34 | 33 | 26 |
| MAGEB4 | CT antigen | 2 | 2 | 3 | 5 | 4 | 2 |
| MAGEB5 | CT antigen | 2 | 3 | 4 | 3 | 3 | 2 |
| MAGEB6 | CT antigen | 104 | 55 | 119 | 82 | 67 | 124 |
| MAGEC1 | CT antigen | 5 | 3 | 5 | 1 | 2 | 1 |
| MAGEC2 | CT antigen | 8 | 4 | 3 | 4 | 1 | 3 |
| POTE-F | CT antigen | 8 | 3 | 3 | 3 | 3 | 2 |
| SAGE1 | CT antigen | 21 | 17 | 14 | 12 | 25 | 18 |
| SPANX-N3 | CT antigen | 44 | 25 | 54 | 34 | 37 | 63 |
| SPANXA1 | CT antigen (also detects A2,D,E, and C) | 22 | 12 | 18 | 15 | 25 | 17 |
| SSX1 | CT antigen (also detects SSX7,8) | 12 | 6 | 7 | 6 | 6 | 6 |
| SSX2 | CT antigen | 11 | 5 | 3 | 5 | 3 | 2 |
| SSX3 | CT antigen (also detects SSX2,3,4) | 7 | 5 | 8 | 4 | 3 | 2 |
| TSPY1 | CT antigen (also detects TPSY2,3, and 4) | 49 | 5 | 57 | 3 | 3 | 3 |
|
Other Tumor Genes
| |||||||
| B4GLNT1 | GD2 | 6051 | 2771 | 6585 | 3987 | 38 | 242 |
| MLANA | MART-1 | 10 | 5 | 1 | 8 | 5 | 1 |
| CEACAMS | CEA | 3 | 3 | 19 | 2 | 2 | 139 |
| EGFRvIII | EGFR variant III | 10 | 7 | 10 | 7 | 2 | 6 |
|
Control Genes
| |||||||
| GAPDH | Glyceraldehyde-3-phorsphate dehydrogenase | 291957 | 148676 | 344647 | 238596 | 207219 | 356034 |
| B2M | Beta2 microglobulin | 159331 | 90291 | 89879 | 111829 | 563882 | 295430 |
| HPRT1 | Hypoxanthine phosphoribosyl transferase | 35672 | 21778 | 45226 | 12548 | 3713 | 6111 |
| SYS1 | Golgi-localized integral membrane protein | 2975 | 3020 | 2277 | 1921 | 2243 | 1860 |
Table 6. RNA-Seq analysis of MAGE-A gene transcripts.
Shown are the numbers of mapped reads for the indicated MAGE-A gene transcripts determined by deep-sequencing of RNA isolated the dorsolateral prefrontal cortex of control (ctl) donors. Shown below each column is the total number of sequence reads mapped to the human reference genome (in millions).
| Transcript | ctl6 | ctl7 | ctl8 | ctl3063 | ctl4054 | ctl5841 |
|---|---|---|---|---|---|---|
| MAGEA1 | 0 | 0 | 0 | 0 | 0 | 0 |
| MAGEA10 | 0 | 1 | 6 | 0 | 3 | 1 |
| MAGEA11 | 0 | 0 | 0 | 0 | 0 | 0 |
| MAGEA12 | 11 | 8 | 22 | 15 | 10 | 4 |
| MAGEA2 | 0 | 3 | 0 | 0 | 0 | 1 |
| MAGEA2B | 0 | 0 | 2 | 0 | 1 | 0 |
| MAGEA3 | 0 | 0 | 0 | 0 | 0 | 0 |
| MAGEA4 | 0 | 0 | 1 | 0 | 0 | 2 |
| MAGEA5 | 3 | 2 | 0 | 2 | 0 | 0 |
| MAGEA6 | 0 | 0 | 0 | 0 | 0 | 0 |
| MAGEA8 | 7 | 23 | 18 | 8 | 15 | 7 |
| MAGEA9 | 0 | 0 | 0 | 0 | 0 | 0 |
| MAGEA9B | 0 | 0 | 0 | 0 | 0 | 0 |
|
| ||||||
| Reads (×106) | 282 | 288 | 277 | 274 | 232 | 228 |
Discussion
CT antigens have been extensively studied since their discovery more than 20 years ago, yet their biological function remains largely unknown. It has been suggested that the MAGE-A family of 12 genes (MAGE-A1 to A12, A7 is a pseudo gene) may function as adaptor proteins and may be involved in transcriptional regulation, protein ubiquitination, and regulation of the p53 pathway40–42. This interruption of normal p53 function, may be one of the reasons that MAGE-A expression has been associated with development of the malignant phenotype and why MAGE-A expressing tumors are associated with worse clinical outcomes43–46. The function of MAGE-A genes in normal cells outside of the germ cell lineage is unknown, with the exception of the primate-specific MAGE-A11 gene that may be involved in androgen receptor signaling47. The evolutionary ancestor of the MAGE-A genes, is thought to be one or more members of the MAGE-D family of somatically expressed genes48 and it is interesting to note that MAGE -D genes (e.g., MAGE-D1) are known to be involved in neuronal development and function49.
There continues to be intense interest in immune-based therapies targeting cancer testes antigens. In our first ACT-based trial targeting the CT antigen NY-ESO-1 we observed objective clinical responses in 5 of 11 melanoma patients and 4 of 6 patients with synovial cell sarcoma with no TCR transduced cell-associated toxicities7. In the trial reported herein, two of the five melanoma patients treated with MAGE-A3 TCR transduced T cells are on-going responders to this therapy, with patient 1 being a complete responder (15+ months) and patient 7 having a residual PET+ lymph node (12+ months). Of note, patient 7 was treated with high-dose steroids on day 21 post-infusion yet continued to demonstrate tumor regression, suggesting that either the anti-tumor antigen response was not sensitive to steroid administration or the mechanism(s) involved in tumor regression were initiated before steroids were given. At one month-post infusion, all patients were observed to have significant numbers of circulating MAGE-A3 TCR positive cells in their blood (Fig. 2) and these demonstrated anti-MAGE-A3 reactivity (Fig. 3). In agreement with our observations using NY-ESO-1 TCR gene modified T cells, we did not observe a correlation between clinical response and the presence or activity of MAGE-A3 TCR transduced cells in blood at one month.
The lack of cell-associated on-target/off-tumor toxicity observed in our TCR gene therapy trial targeting NY-ESO-1 was in contrast to our previous experience when targeting tumor associated antigens MART-1, gp100, or CEA with TCR gene transduced T cells 6, 11. In the trials targeting the melanocyte differentiation antigens MART-1 and gp100, we reported that TCR gene-modified T cell infusions were associated with skin rash, uveitis, and auditory/vestibular dysfunction while the targeting of CEA resulted in the transient induction of colitis. On-target/off-tumor toxicity using receptor-modified T cells was first reported in 2006 by Lamers et al., 50 who observed liver toxicity associated with chimeric antigen receptor (CAR) targeting of carbonic anhydrase IX that was expressed in bile duct epithelial cells. CAR vector-transduced T cell trials targeting CD19 and ERBB2 were subsequently shown to be associated with elevated serum inflammatory cytokine levels and patient deaths in two case reports 12, 13. In the present study, serum IFN-γ cytokine levels were only mildly elevated in our patients (Fig. 8) and we did not detect IFN-γ in CSF samples (not shown).
Given that the anti-MAGE-A3 TCR used in this trial was of murine origin and had been further modified to increase peptide reactivity, we performed extensive studies to test for the recognition of non-MAGE-A family gene products. We found no evidence (Figs. 9–11) for off-target reactivity of the MAGE-A3 TCR used in this study, but cannot definitively exclude this possibility. We have used three murine-derived TCR in previous trials (targeting gp100, p53, and CEA) and did not observe off-target toxicities6, 11, 51. The specific modifications of the CDR3 region of this TCR were designed to increase MAGE-A3 reactivity and CD8-independence and similar modifications were made in the NY-ESO-1 TCR used in our first CTA targeted TCR gene therapy trial without issue. CD8-independent TCR reactivity permits this HLA class I restricted TCR to function in CD4+ T cells. CD4 + T cells are the mediators of autoimmune encephalomyelitis (EAE) in mice and while CD8+ T cells have been demonstrated in at least one mouse model of multiple sclerosis (MS), there have been several reports of the association of infiltrating CD8+ T cells with MS in humans 52–56. We observed an enrichment of CD4+ MAGE -A3 TCR+ T cells in CSF samples derived from patients 5, 7, and 8 and cells from the CSF of patient 5 recognized MAGE-A3/A12 expressing tumors (Fig. 7). CD8+ T cells were widely observed in the brains of patients 5 and 8 at autopsy, with CD4+ cells found much less frequently (Fig. 6). We do not have sufficient information to speculate which cell type (or both) were responsible for the neurologic toxicity, particularly because we have no data concerning T cell infiltrates during the period of acute toxicity.
Neurologic toxicity was not reported in any previous gene modified T cell ACT trial including the trials targeting antigens known to be expressed in the CNS such as GD24. In our patient population, six of nine patients receiving the MAGE-A3 TCR gene modified T cells did not manifest cell-related neurologic toxicity, suggesting either patient or treatment specific influences. A detailed analysis of the cell products infused into patients 5, 7, and 8 revealed differences potentially associated with toxicity (Tables 1 and 2, Fig. 1). Patients who developed neurologic toxicity received a higher total number of cells (mean 67.3 × 109 versus 36.4 × 109, p=0.036), more CD3+/CD8+/Tetramer+ cells (mean 28.81 × 109 versus 10.72 × 109, p=0.036), and more T cells with a naïve phenotype (mean 4.27 × 109 versus 0.88 × 109, p=0.036). For this statistical analysis (two-tailed p-values using an exact Wilcoxon rank sum test) we excluded patient 9, who suffered a transient ischemic attack (TIA) and could not be unambiguously categorized. Thus, although the number of patients in the trial was small, these data suggest a potential dose-related toxicity was associated with the administration of MAGE-A3 TCR gene engineered T cells.
Paraneoplastic syndrome can commonly manifest neurologic toxicity57 and chronic inflammatory demyelinating polyneuropathy (CIDP) has been observed in melanoma patients58–60. Furthermore, in an early melanoma immunotherapy trial using the administration of an anti-GD2 mAb, neurological toxicity including sensory and motor neuropathy and mental status changes were reported61, 62. Interestingly, there is one case report where a melanoma patient with minor oculomotor paresis developed severe proximal axonal motor neuropathy following administration of a MAGE-A3 vaccine63. Several other cancer vaccine studies targeting MAGE -A3 have been reported but to our knowledge, this is the only report of neurologic toxicity. There have been suggestive data that vaccination with MAGE-A3 peptides or protein may be associated with clinical responses in a small number of patients27, 64–67. Based on these results, a large Phase III vaccine trial of MAGE-A3 protein plus an immunological adjuvant is currently in progress (MAGRIT, NCT00480025, www.clinicaltrials.gov) as an adjuvant therapy in patients with resectable MAGE-A3 positive non-small cell lung cancer26. As T cell responses induced in humans to tumor-associated antigens by current vaccine technology have generally been of low avidity, it is unlikely that they will generate an anti-MAGE-A3 TCR similar to the high-avidity TCR used in this clinical trial.
We propose that the best explanation for the neurologic toxicity observed in three of the nine patients treated in this trial was the result of recognition of the MAGE-A12 protein expressed in a subset of neurons in the human brain, which initiated a destructive immune response resulting in severe damage to the white matter. MAGE -A12 expression was detected at the RNA level using three independent methods and MAGE-A protein expression was documented in rare neurons using a MAGE-A family antibody in our patients and multiple other individuals, including normal donors. To our knowledge, this is the first report of MAGE-A12 expression in human brain. The data from the RNA deep-sequencing study of the DLPFC of normal donors is highly suggestive that MAGE-A12 was the MAGE-A gene recognized by our T cells as the deep-sequencing results did not find MAGE-A3 or MAGE-A9 to be expressed. In fact, we have no evidence that MAGE-A3 is expressed in human brain, suggesting that if highly MAGE-A3 specific T cell epitopes could be found, this might avert the neurological toxicity observed here. Our results thus raise a caution in attempts to produce vaccine -induced anti-MAGE-A3 responses because of the homology of HLA-A2 restricted epitopes between MAGE-A3 and MAGE-A12, the later demonstrated here to be expressed in the brain.
The specific TCR used in these studies is a high-avidity anti-MAGE-A3 TCR that was known to recognize MAGE-A12 and because the MAGE-A12 peptide has higher affinity for HLA-A*0201, MAGE-A3 TCR transduced T cells could detect 10-fold lower MAGE-A12 peptide concentrations. The avidity of a specific TCR is known to be directly associated with tumor regression in animal models68. In our previous clinical experience using TCR gene modified T cells, we reported two separate trials using anti-MART-1 TCRs isolated from the same melanoma patient. In our first clinical trial5 using an anti-MART-1 TCR termed DMF4 we observed modest clinical responses (16%), and thus we undertook studies aimed at isolating a more avid TCR69. In the DMF4 TCR gene therapy trial, no on-target/off-tumor toxicities were noted in 31 treated patients. In a subsequent report using the high-avidity DMF5 TCR11, which recognized the same peptide epitope, 14 of 20 patients experienced skin rashes and 11 of 20 patients experienced eye or ear toxicity. These on-target/off-tumor toxicities were also associated with an increased response rate (30%). These results suggest that while higher avidity TCRs may be associated with better tumor responses, when the target antigen is a self -protein, the potential for on-target/off-tumor toxicities may also be increased. Modifying the affinity of an anti-myelin-specific class II-restricted TCR, has been reported to alter pathogenicity in the murine EAE model70.
We propose that caution needs to be exercised when targeting CT antigens using high avidity TCRs since the widely reported tumor specificity of this class of antigens may not be correct. New technologies such as deep-sequencing or large-scale immunohistochemical studies with gene-specific antibodies (or perhaps, in-situ hybridization) may better elucidate the expression pattern of CT genes in normal tissues. While this limited study suggested that the toxicity was associated with cell dose, it is difficult to directly relate this to a pharmacological dose as the infused product can expand in the patient and this is likely to be variable in different individuals. Whether or not a gene-based cell suicide switch71 would be of clinical utility in situations similar to those reported here is speculative, but worthy of consideration in future trials. TCRs that target MAGE-A3 gene-specific epitopes, not shared with other proteins, may yet be a productive and potentially safe method of adoptive cell therapy for common cancers, such as, lung cancer, for which treatment options are limited.
Acknowledgments
Support: This work was supported by the intramural program of the Center for Cancer Research, National Cancer Institute, Bethesda MD.
We thank Arnold Mixon and Shawn Farid for technical support for FACS analysis. We thank Takara Bio Inc, Otsu, Japan for providing RetroNectin. Tetramers were produced by NIH Tetramer Core Facility at Emory University, Atlanta, GA. We thank Drs. Stefan Stevanovic and Hans-Georg Rammensee, Department of Immunology, University of Tubingen, Tubingen, Germany, who provided unpublished data on the normal human peptidome. We thank the nursing staff on the 3NW ward and the Intensive Care Unit in the Clinical Center, National Institutes of Health who provided these patients with outstanding care. This manuscript is dedicated to memory of Nachimuthu “Samy” Chinnasamy.
Footnotes
Financial Disclosure: All authors have no financial disclosures in regards to this work.
References
- 1.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chapuis AG, Thompson JA, Margolin KA, et al. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proc Natl Acad Sci U S A. 2012;109:4592–4597. doi: 10.1073/pnas.1113748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park TS, Rosenberg SA, Morgan RA. Treating cancer with genetically engineered T cells. Trends Biotechnol. 2011;29:550–557. doi: 10.1016/j.tibtech.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pule MA, Savoldo B, Myers GD, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kochenderfer JN, Wilson WH, Janik JE, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter DL, Levine BL, Kalos M, et al. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brentjens R, Yeh R, Bernal Y, et al. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 2010;18:666–668. doi: 10.1038/mt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 15.Sang M, Lian Y, Zhou X, et al. MAGE-A family: attractive targets for cancer immunotherapy. Vaccine. 2011;29:8496–8500. doi: 10.1016/j.vaccine.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 16.Sang M, Wang L, Ding C, et al. Melanoma-associated antigen genes - an update. Cancer Lett. 2011;302:85–90. doi: 10.1016/j.canlet.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 17.Meek DW, Marcar L. MAGE-A antigens as targets in tumour therapy. Cancer Lett. 2012;324:126–132. doi: 10.1016/j.canlet.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 18.Hofmann O, Caballero OL, Stevenson BJ, et al. Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci U S A. 2008;105:20422–20427. doi: 10.1073/pnas.0810777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009;100:2014–2021. doi: 10.1111/j.1349-7006.2009.01303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roeder C, Schuler-Thurner B, Berchtold S, et al. MAGE-A3 is a frequent tumor antigen of metastasized melanoma. Arch Dermatol Res. 2005;296:314–319. doi: 10.1007/s00403-004-0527-7. [DOI] [PubMed] [Google Scholar]
- 21.Groeper C, Gambazzi F, Zajac P, et al. Cancer/testis antigen expression and specific cytotoxic T lymphocyte responses in non small cell lung cancer. Int J Cancer. 2007;120:337–343. doi: 10.1002/ijc.22309. [DOI] [PubMed] [Google Scholar]
- 22.Curigliano G, Viale G, Ghioni M, et al. Cancer-testis antigen expression in triple-negative breast cancer. Ann Oncol. 2011;22:98–103. doi: 10.1093/annonc/mdq325. [DOI] [PubMed] [Google Scholar]
- 23.Bergeron A, Picard V, LaRue H, et al. High frequency of MAGE-A4 and MAGE-A9 expression in high-risk bladder cancer. Int J Cancer. 2009;125:1365–1371. doi: 10.1002/ijc.24503. [DOI] [PubMed] [Google Scholar]
- 24.Mirandola L, JCM, Cobos E, et al. Cancer testis antigens: novel biomarkers and targetable proteins for ovarian cancer. Int Rev Immunol. 2011;30:127–137. doi: 10.3109/08830185.2011.572504. [DOI] [PubMed] [Google Scholar]
- 25.Jungbluth AA, Antonescu CR, Busam KJ, et al. Monophasic and biphasic synovial sarcomas abundantly express cancer/testis antigen NY-ESO-1 but not MAGE-A1 or CT7. Int J Cancer. 2001;94:252–256. doi: 10.1002/ijc.1451. [DOI] [PubMed] [Google Scholar]
- 26.Brichard VG, Lejeune D. GSK’s antigen-specific cancer immunotherapy programme: pilot results leading to Phase III clinical development. Vaccine. 2007;25(Suppl 2):B61–71. doi: 10.1016/j.vaccine.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 27.Corbiere V, Chapiro J, Stroobant V, et al. Antigen spreading contributes to MAGE vaccination-induced regression of melanoma metastases. Cancer Res. 2011;71:1253–1262. doi: 10.1158/0008-5472.CAN-10-2693. [DOI] [PubMed] [Google Scholar]
- 28.Brichard VG, Lejeune D. Cancer immunotherapy targeting tumour-specific antigens: towards a new therapy for minimal residual disease. Expert Opin Biol Ther. 2008;8:951–968. doi: 10.1517/14712598.8.7.951. [DOI] [PubMed] [Google Scholar]
- 29.Hunder NN, Wallen H, Cao J, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chinnasamy N, Wargo JA, Yu Z, et al. A TCR targeting the HLA-A*0201-restricted epitope of MAGE-A3 recognizes multiple epitopes of the MAGE-A antigen superfamily in several types of cancer. J Immunol. 2011;186:685–696. doi: 10.4049/jimmunol.1001775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dudley ME, Yang JC, Sherry R, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 33.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang T, Lee MH, Johnson T, et al. Activated T-cells inhibit neurogenesis by releasing granzyme B: rescue by Kv1.3 blockers. J Neurosci. 2010;30:5020–5027. doi: 10.1523/JNEUROSCI.0311-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wuest SC, Edwan JH, Martin JF, et al. A role for interleukin-2 trans-presentation in dendritic cell-mediated T cell activation in humans, as revealed by daclizumab therapy. Nat Med. 2011;17:604–609. doi: 10.1038/nm.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bielekova B, Catalfamo M, Reichert-Scrivner S, et al. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc. Natl Acad Sci U S A. 2006;103:5941–5946. doi: 10.1073/pnas.0601335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kulkarni MM. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr Protoc Mol Biol. 2011;Chapter 25(Unit25B):10. doi: 10.1002/0471142727.mb25b10s94. [DOI] [PubMed] [Google Scholar]
- 38.Geiss GK, Bumgarner RE, Birditt B, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 39.Lipska BK, Deep-Soboslay A, Weickert CS, et al. Critical factors in gene expression in postmortem human brain: Focus on studies in schizophrenia. Biol Psychiatry. 2006;60:650–658. doi: 10.1016/j.biopsych.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 40.Feng Y, Gao J, Yang M. When MAGE meets RING: insights into biological functions of MAGE proteins. Protein Cell. 2011;2:7–12. doi: 10.1007/s13238-011-1002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laduron S, Deplus R, Zhou S, et al. MAGE-A1 interacts with adaptor SKIP and the deacetylase HDAC1 to repress transcription. Nucleic Acids Res. 2004;32:4340–4350. doi: 10.1093/nar/gkh735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Monte M, Simonatto M, Peche LY, et al. MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc Natl Acad Sci U S A. 2006;103:11160–11165. doi: 10.1073/pnas.0510834103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim J, Reber HA, Hines OJ, et al. The clinical significance of MAGEA3 expression in pancreatic cancer. Int J Cancer. 2006;118:2269–2275. doi: 10.1002/ijc.21656. [DOI] [PubMed] [Google Scholar]
- 44.Karn T, Pusztai L, Ruckhaberle E, et al. Melanoma antigen family A identified by the bimodality index defines a subset of triple negative breast cancers as candidates for immune response augmentation. Eur J Cancer. 2012;48:12–23. doi: 10.1016/j.ejca.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 45.Suyama T, Shiraishi T, Zeng Y, et al. Expression of cancer/testis antigens in prostate cancer is associated with disease progression. Prostate. 2010;70:1778–1787. doi: 10.1002/pros.21214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shiraishi T, Terada N, Zeng Y, et al. Cancer/Testis Antigens as potential predictors of biochemical recurrence of prostate cancer following radical prostatectomy. J Transl Med. 2011;9:153. doi: 10.1186/1479-5876-9-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilson EM. Androgen receptor molecular biology and potential targets in prostate cancer. Ther Adv Urol. 2010;2:105–117. doi: 10.1177/1756287210372380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Artamonova, Gelfand MS. Evolution of the exon-intron structure and alternative splicing of the MAGE-A family of cancer/testis antigens. J Mol Evol. 2004;59:620–631. doi: 10.1007/s00239-004-2654-3. [DOI] [PubMed] [Google Scholar]
- 49.Wang X, Gao X, Xu Y. MAGED1: molecular insights and clinical implications. Ann Med. 2011;43:347–355. doi: 10.3109/07853890.2011.573806. [DOI] [PubMed] [Google Scholar]
- 50.Lamers CH, Sleijfer S, Vulto AG, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 51.Davis JL, Theoret MR, Zheng Z, et al. Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials. Clin Cancer Res. 2010;16:5852–5861. doi: 10.1158/1078-0432.CCR-10-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jadidi-Niaragh F, Mirshafiey A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand J Immunol. 2011;74:1–13. doi: 10.1111/j.1365-3083.2011.02536.x. [DOI] [PubMed] [Google Scholar]
- 53.Kebir H, Ifergan I, Alvarez JI, et al. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol. 2009;66:390–402. doi: 10.1002/ana.21748. [DOI] [PubMed] [Google Scholar]
- 54.Rothhammer V, Heink S, Petermann F, et al. Th17 lymphocytes traffic to the central nervous system independently of alpha4 integrin expression during EAE. J Exp Med. 2011;208:2465–2476. doi: 10.1084/jem.20110434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brucklacher-Waldert V, Stuerner K, Kolster M, et al. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain. 2009;132:3329–3341. doi: 10.1093/brain/awp289. [DOI] [PubMed] [Google Scholar]
- 56.Na SY, Hermann A, Sanchez-Ruiz M, et al. Oligodendrocytes enforce immune tolerance of the uninfected brain by purging the peripheral repertoire of autoreactive CD8(+) T cells. Immunity. 2012;37:134–146. doi: 10.1016/j.immuni.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 57.Giglio P, Gilbert MR. Neurologic complications of cancer and its treatment. Curr Oncol Rep. 2010;12:50–59. doi: 10.1007/s11912-009-0071-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dalakas MC. Advances in the diagnosis, pathogenesis and treatment of CIDP. Nat Rev Neurol. 2011;7:507–517. doi: 10.1038/nrneurol.2011.121. [DOI] [PubMed] [Google Scholar]
- 59.Bird SJ, Brown MJ, Shy ME, et al. Chronic inflammatory demyelinating polyneuropathy associated with malignant melanoma. Neurology. 1996;46:822–824. doi: 10.1212/wnl.46.3.822. [DOI] [PubMed] [Google Scholar]
- 60.Weiss MD, Luciano CA, Semino-Mora C, et al. Molecular mimicry in chronic inflammatory demyelinating polyneuropathy and melanoma. Neurology. 1998;51:1738–1741. doi: 10.1212/wnl.51.6.1738. [DOI] [PubMed] [Google Scholar]
- 61.Saleh MN, Khazaeli MB, Wheeler RH, et al. Phase I trial of the murine monoclonal anti-GD2 antibody 14G2a in metastatic melanoma. Cancer Res. 1992;52:4342–4347. [PubMed] [Google Scholar]
- 62.Saleh MN, Khazaeli MB, Wheeler RH, et al. Phase I trial of the chimeric anti-GD2 monoclonal antibody ch14.18 in patients with malignant melanoma. Hum Antibodies Hybridomas. 1992;3:19–24. [PubMed] [Google Scholar]
- 63.Kloos L, Sillevis Smitt P, Ang CW, et al. Paraneoplastic ophthalmoplegia and subacute motor axonal neuropathy associated with anti-GQ1b antibodies in a patient with malignant melanoma. J Neurol Neurosurg Psychiatry. 2003;74:507–509. doi: 10.1136/jnnp.74.4.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Atanackovic D, Altorki NK, Cao Y, et al. Booster vaccination of cancer patients with MAGE-A3 protein reveals long-term immunological memory or tolerance depending on priming. Proc Natl Acad Sci U S A. 2008;105:1650–1655. doi: 10.1073/pnas.0707140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coulie PG, Karanikas V, Lurquin C, et al. Cytolytic T-cell responses of cancer patients vaccinated with a MAGE antigen. Immunol Rev. 2002;188:33–42. doi: 10.1034/j.1600-065x.2002.18804.x. [DOI] [PubMed] [Google Scholar]
- 66.Kruit WH, van Ojik HH, Brichard VG, et al. Phase 1/2 study of subcutaneous and intradermal immunization with a recombinant MAGE-3 protein in patients with detectable metastatic melanoma. Int J Cancer. 2005;117:596–604. doi: 10.1002/ijc.21264. [DOI] [PubMed] [Google Scholar]
- 67.Connerotte T, Van Pel A, Godelaine D, et al. Functions of Anti-MAGE T-cells induced in melanoma patients under different vaccination modalities. Cancer Res. 2008;68:3931–3940. doi: 10.1158/0008-5472.CAN-07-5898. [DOI] [PubMed] [Google Scholar]
- 68.Zeh HJ, 3rd, Perry-Lalley D, Dudley ME, et al. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol. 1999;162:989–994. [PubMed] [Google Scholar]
- 69.Johnson LA, Heemskerk B, Powell DJ, Jr, et al. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol. 2006;177:6548–6559. doi: 10.4049/jimmunol.177.9.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alli R, Nguyen P, Geiger TL. Altered differentiation, diminished pathogenicity, and regulatory activity of myelin-specific T cells expressing an enhanced affinity TCR. J Immunol. 2011;187:5521–5531. doi: 10.4049/jimmunol.1102202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Di Stasi A, Tey SK, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]