

Significance
The structure–function relationship of proteases is central to our understanding of biochemistry. Nature has evolved at least 23 independent solutions to this problem, using an acylation mechanism. We examined the structures of these proteases, using a new framework to characterize the geometric relationships within each active site. This analysis revealed the orientation of the base determines the stereochemistry of catalysis and elucidated why threonine does not substitute for serine in the catalytic triad. These observations explain how the absolute stereostructures of natural protease inhibitors prevent off-target inhibition and serve as boundary conditions to enzyme design.
Keywords: peptidase, antibiotic stereochemistry, N-terminal nucleophile
Abstract
The study of proteolysis lies at the heart of our understanding of biocatalysis, enzyme evolution, and drug development. To understand the degree of natural variation in protease active sites, we systematically evaluated simple active site features from all serine, cysteine and threonine proteases of independent lineage. This convergent evolutionary analysis revealed several interrelated and previously unrecognized relationships. The reactive rotamer of the nucleophile determines which neighboring amide can be used in the local oxyanion hole. Each rotamer–oxyanion hole combination limits the location of the moiety facilitating proton transfer and, combined together, fixes the stereochemistry of catalysis. All proteases that use an acyl-enzyme mechanism naturally divide into two classes according to which face of the peptide substrate is attacked during catalysis. We show that each class is subject to unique structural constraints that have governed the convergent evolution of enzyme structure. Using this framework, we show that the γ-methyl of Thr causes an intrinsic steric clash that precludes its use as the nucleophile in the traditional catalytic triad. This constraint is released upon autoproteolysis and we propose a molecular basis for the increased enzymatic efficiency introduced by the γ-methyl of Thr. Finally, we identify several classes of natural products whose mode of action is sensitive to the division according to the face of attack identified here. This analysis of protease structure and function unifies 50 y of biocatalysis research, providing a framework for the continued study of enzyme evolution and the development of inhibitors with increased selectivity.
The acceleration of chemical reactions is essential to all biochemistry. The generation of reactive species by enzymes is tightly controlled and limited by the chemical makeup of the 20 proteinogenic amino acids. In isolation, no protein residue harbors a strong nucleophile at physiological pH. Rather, enzymatic activity arises after protein folding introduces cooperative interactions that selectively amplify the reactivity of their otherwise weakly active functional groups. The preeminent system through which this phenomenon has been studied is the Ser-His-Asp catalytic triad of the Ser proteases (1). Several distinct molecular strategies have been identified that contribute to rate enhancement. Substrate binding increases the local concentration of interacting species, general acids and bases facilitate proton transfer, and a high-energy step can be split into multiple lower-energy steps (2). These distinct molecular strategies may be unified through the formalism of transition-state theory (3) and the idea that the active site is electrostatically preorganized for transition-state stabilization (4, 5). Although the biophysics of rate acceleration are intricate and sensitive to even minor structural perturbations, evolution has converged on a catalytic triad (or diad) with a reactive Ser, Cys, or Thr nucleophile more than 25 separate times to facilitate central biochemical reactions such as hydrolysis (6), transacylation (7), and phosphorylation (8).
Our understanding of biochemistry has progressed into an increasingly complex picture but many basic questions about enzyme evolution remain unanswered. Because evolution operates through random forces, rationalizing why a particular outcome occurs is a difficult challenge. For example, the hydroxyl nucleophile of a Ser protease was swapped for the thiol of Cys at least twice in evolutionary history (9). However, there is not a single example of Thr naturally substituting for Ser in the protease catalytic triad, despite its greater chemical similarity (9). Instead, the Thr proteases generate their N-terminal nucleophile through a posttranslational modification: cis-autoproteolysis (10, 11). These facts constitute clear evidence that there is a strong selective pressure against Thr in the catalytic triad that is somehow relieved by cis-autoproteolysis. Because the catalytic triad is the principal system through which enzymatic rate acceleration has been studied, the inability to rationalize the evolutionary selection of the catalytic nucleophile is a fundamental weakness in our understanding of biocatalysis. Motivated by this simple, yet unexplained structure–function relationship, we sought to answer the question: How do the chemical and structural constraints of proteolysis combine to shape the evolution of a protease active site?
Two strategies can be undertaken to probe the properties of diverse catalytic architectures. The first is the contemporary experimental approach whereby all parameters of interest in an active site are systematically varied and analyzed in silico for catalytic viability. This technique was used by Smith et al. to show that the catalytic triad of esterases adopts a consensus geometry that minimizes reorganization during the multistep mechanism (12). Computational explorations are unsurpassed in their detail and broad applicability, but hindered by their need to explicitly anticipate the structural and mechanistic diversity generated through natural selection. We elected to pursue an alternative strategy to elucidate the constraints that guide active site evolution that is not computational, but observational. Studying the large set of naturally occurring structures limits analyses to coarser features, but greatly increases the universality of the results. This technique requires only a diverse set of enzyme structures and a rudimentary understanding of organic and biochemistry. By recognizing the constraints imposed by bond geometries and steric clashes, it is easy to compare the potential catalytic architectures for a given reaction.
Results from the veritable explosion of gene sequencing and protein structure determination have identified at least 23 folds related through convergent evolution that support proteolysis through an acyl-enzyme intermediate (9). We cross-compared the active site architecture among these “analogous” enzymes (13), each of which has been optimized for catalysis through natural selection, to interrogate the entire known structural diversity that supports efficient proteolysis. We first identified key parameters that define active site architecture, such as the orientation of substrate binding, the φ, ψ angles of the nucleophile, and the stereochemistry of catalysis. Analysis of all Ser, Cys, and Thr proteases of independent lineage showed they were all highly interrelated. By rationalizing these correlations between analogous enzymes in terms of their underlying chemistry, we describe how the function of proteases has guided the evolution of their structure. These data show that proteases naturally divide into two mechanistically distinct classes that are inhibited by separate groups of naturally occurring antibiotics. By identifying prohibitive steric interactions of the Thr γ-methyl within each class, we explain why evolution selected Ser, and not Thr, as the nucleophile of the catalytic triad.
Results and Discussion
Chemical Constraints of Proteolysis.
To understand how the chemical constraints of proteolysis affect the evolution of a protease active site, it is necessary to invoke the chemical principles that underlie its mechanism. As the present work is not a review of the protease literature, we limit this discussion to only the most central facts (Fig. 1). The Ser hydroxyl group is not itself strongly nucleophilic and enzymes must supply a general base to facilitate its deprotonation. The traditional residue for this role is His, although other residues are also used (6, 14). Nucleophilic attack into a peptide bond generates an oxyanion intermediate that is stabilized by the “oxyanion hole” composed of two hydrogen bond donors, one of which typically comes from a backbone NH flanking the nucleophilic residue (14, 15). The nitrogen of the scissile bond becomes basic upon formation of the tetrahedral intermediate and proton transfer to the amine leaving group occurs concomitantly with collapse of the oxyanion, yielding the acyl-enzyme intermediate. Subsequent hydrolysis of this enzyme-bound species also proceeds through a tetrahedral intermediate. Although the energetics of hydrolysis are different from enzyme acylation, they share many structural constraints. As is shown in this investigation, it is sufficient to consider only the acylation step of the reaction, as displayed in Fig. 1, to rationalize much of the observable convergent evolution of proteases.
Fig. 1.
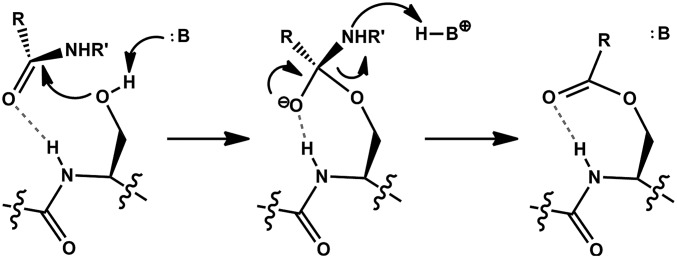
The acylation mechanism of a Ser protease. After substrate binding, the Ser side chain attacks the scissile peptide bond to generate a tetrahedral oxyanion intermediate. Protonation of the amine leaving group allows collapse of the intermediate and formation of an acyl-enzyme species. Dashed bonds represent the hydrogen bond of the oxyanion hole. See text for details.
Evolution has selected for many different residues to activate the nucleophile, but the cooperative interactions that engender reactivity are effectively “tuned” differently for each individual enzyme. This principle was first tested by the chemical swapping of the hydroxyl nucleophile of subtilisin for a thiol and observing loss of proteolytic activity (16, 17). More exhaustive studies using site-directed mutagenesis of both subtilisin and trypsin showed that alteration of any component of the catalytic triad results in a large decrease in enzyme efficiency (18, 19). Interestingly, the Ser→Thr variant of trypsin created a worse enzyme than simultaneously mutating all three residues of the catalytic triad. Conversely, when the N-terminal Thr nucleophile of the proteasome β-subunit was mutated to Ser, there was a reduction in its catalytic power (20). The juxtaposition of these results and the inability to rationalize the opposite effects of the Thr γ-methyl further stimulated our desire to understand how evolution follows the chemical constraints of proteolysis.
Reactive Rotamer Determines the Identity of the Local Oxyanion Hole.
To our knowledge, the extent of active site variation among different proteases has not been systematically explored. Relying on the clan/family nomenclature developed in the MEROPS database (9) and several excellent reviews (6, 14, 21, 22), we used a simple strategy to analyze the existing collection of protease structures in the Protein Data Bank (PDB). Because our investigation is based upon structural analysis of actual proteases, the residues facilitating this chemistry are evolutionarily optimized for catalysis. Cross-comparison of these structures enables us to probe the amount of structural diversity present when the biophysical constraints for rate acceleration are met and ask how this diversity relates to the underlying chemistry.
There are three staggered rotamers of Ser and they are denoted according to whether the γOH is gauche or trans to its peptide NH (Fig. 2A). Table 1 shows that all three staggered rotamers have been exploited for enzyme acylation and we denote the state from which chemistry arises as the “reactive rotamer.” Table 1 also shows that all but one clan of Ser proteases (23) use an amide NH flanking the nucleophile to stabilize the oxyanion hole. It has been established that utilization of a local oxyanion hole moiety forces a suboptimal geometry of oxyanion stabilization (24, 25). Although the geometry of the oxyanion holes is well understood, it was proposed that the identity of the oxyanion hole “… is a direct consequence of the handedness of the catalytic triad” (ref. 26, p. 165). “Handedness” refers to the direction from which the base approaches the nucleophile, but is a limited descriptor because it cannot be extended to the Thr proteases. We introduce a term that both encompasses the handedness of the active site and is generalizable: the dihedral angle between the side chain of the nucleophile and the basic atom, designated χcat (Fig. 2B). Contrary to the earlier hypothesis, the identity of the oxyanion hole is not determined by the handedness of the catalytic triad, but is correlated with the reactive rotamer. When either of the gauche (g+ or g−) rotamers is used, it is the amide NH of the nucleophilic residue that stabilizes the oxyanion intermediate (Fig. 2 D–H). When the trans (t) rotamer is used, it is the amide NH of the residue C-terminal to the nucleophile (denoted the N + 1 residue) that is accessible (Fig. 2 F and I). The molecular basis for this relationship is simple: Only the backbone NH that is adjacent to the nucleophile can be used to facilitate catalysis.
Fig. 2.

Active site architectures of Ser Proteases. (A) Newman projection down the Cβ-Cα bond of a side chain on a peptide. The rotamers of the γ-atom are named g+, g−, or t, as shown. (B) Newman projection down the Cβ-Oγ bond of a Ser nucleophile. The dihedral angle χcat is measured between Cα, Cβ, Oγ, and the base (B). αcat is defined as the angle between the base, Oγ, and carbonyl carbon. (C) The dihedral angle between Cβ, Oγ of the nucleophile, and the carbonyl of the scissile bond, χoxy, is used to measure the relative orientation of the substrate. (D-F) Schematic of the three possible active site arrangements when a local oxyanion hole is used. (G–I) Representative examples of a Ser protease with each reactive rotamer. Dashes represent hydrogen bonds between the nucleophile and the base (green) or the protein backbone and substrate (orange). PDB IDs: G, 3VGC, γ-chymotrypsin with a boronic acid inhibitor; H, 2EEP, signal peptidase I with a β-lactam inhibitor; I, 1B12, prolyl aminopeptidase with a boronic acid inhibitor.
Table 1.
Active site parameters of convergently related Ser proteases
| Clan | Enzyme | PDB | Complex | Reactive rotamer | ϕ, ψ angles | Catalytic motif | Oxyanion hole | Face of attack | χcat | αcat | χoxy | Reference |
| PA | Elastase | 3HGN | Hemiketal | g− | (−43, 140) | S-H-D | N | re | −131 | 125 | −32 | (68) |
| SB | Sedolisin | 1GA4 | Ester | g− | (−62, −24) | S-E-D | N | re | −147 | 108 | −18 | (69) |
| SE | D-Ala-D-Ala peptidase | 1MPL | Phophonate | g− | (−72, 0) | S-Y-K | N | re | −156 | 97 | −48 | (70) |
| SC | Prolyl aminopeptidase | 1QFM | Hemiketal | t | (66, −115) | S-H-D | N + 1 | si | 117 | 106 | 32 | (26) |
| SS | L,D-carboxypeptidase | 1ZRS | t | (64, −130) | S-H-E | N + 1 | si | 109 | (49) | |||
| PC | Aspartyl dipeptidase | 1FYE | t | (57, −122) | S-H-E | N + 1 | si | 84 | (71) | |||
| SH | CMV protease | 1NKM | α-Ketoamide | t | (−177, 132) | S-H-H | Extrinsic | re | −125 | 111 | −39 | (23) |
| SK | ClpP | 1FZS | HMK | t | (59, −122) | S-H-D | N + 1 | si | 130 | 74 | 50 | (29) |
| SF | Signal peptidase | 1B12 | Ester | g+ | (−68, −11) | S-K | N | si | 148 | 109 | 1 | (60) |
| SJ | VP4 protease | 3R0B | Ester | g+ | (−65, −13) | S-K | N | si | 173 | 114 | −9 | (72) |
| ST | Rhomboid-1 | 2IC8 | g+ | (−63, −29) | S-H | N | si | 113 | (73) | |||
Halomethyl ketone (HMK) inactivators give rise to unusually small αcat values. See text for details.
Natural Limits on the Location of the Proton Transfer Residue.
We observed that the identity of the reactive rotamer is tightly correlated with the location of the base, as indicated by χcat (Table 1). The only exception to this correlation is clan SH, which does not use the local oxyanion hole. Therefore, we hypothesized that constraints introduced by the reactive rotamer and oxyanion hole combine to severely limit the catalytically viable location of the base. For proteases that share a common reactive rotamer, there is ∼50° variability in χcat. Some of this variation may be due to the fact that conformational heterogeneity in the ground state can hide features that are used to stabilize the transition state(s) (12).
We identified a common limit, |χcat| > 90°, which is shared among the vast majority of protease structures. To explore the basis of this limit and whether divergent evolution may have altered active site architecture, we extended our analysis to every structurally characterized Ser protease family (Table S1). We observed that all outliers to the trend |χcat| > 90° also contained features that indicated an inactive state had been crystallized (SI Text). Hence, |χcat| > 90° is a naturally occurring constraint in protease architecture. A partial explanation for this limit may be attributed to the protein backbone flanking the nucleophile, which enforces an insurmountable barrier to an approach from more acute angles. Although additional work will be needed to ascertain the exact origin of this constraint on χcat, the fact that it occurs within all Ser proteases marks it as an intrinsic consequence of their activity.
A second constraint on the location of the base is the angle defined by the basic atom, nucleophile, and electrophile, designated αcat (Fig. 2B). With respect to deprotonation, αcat ∼ 116° because the γO of the nucleophile is sp3 hybridized during the concerted deprotonation and nucleophilic attack that initiates catalysis (27). Because this is inherent to proteolysis with Ser, all such proteases must converge on this geometry. Inspection of Table 1 shows this parameter is faithfully reported by most structures with a mechanism-based analog. An interesting exception is the family of halomethyl ketone (HMK) inactivators, which covalently link to both the nucleophile and the base and show αcat ∼ 75°, distorting the system that has moved away from its catalytic orientation (28–30).
These simple geometric constraints combine to define the catalytic architecture for all proteases that use the local oxyanion hole. For each reactive rotamer, only one candidate backbone amide can contribute to the oxyanion hole. Because the substrate and base must both be adjacent to the nucleophile, this limitation on the position of the substrate translates into a restriction on the location of the base. Consequently, χcat must be positive for the g+ and t rotamers and negative for the g−rotamer, as was experimentally observed (Fig. 2 D–F). The location of the base is further defined by the requirements that |χcat| > 90° and αcat ∼116°. Although additional constraints are imposed by the nuances of transition state stabilization to define the reaction trajectory, these fundamental constraints are sufficient to explain the convergent evolution of all proteases that use a local oxyanion hole.
Ser Proteases Divide into Two Mechanistic Classes.
The most conspicuous feature of Table 1 is the interrelationship between χcat and the orientation of substrate binding. In principle, a peptide substrate can bind to present either the re or the si face of the scissile bond. For re-face attacking enzymes, our measurements show that χcat is always negative. Moreover, measurement of the dihedral angle between Cβ and Oγ of the nucleophile and the carbonyl electrophile (χoxy, Fig. 2C) shows these enzymes bind their substrate in a similar, staggered, position. When the local backbone is used in oxyanion hole formation, only the g− rotamer is observed to support re-face attack. Conversely, χcat is always positive for the si-face–attacking enzymes and only the g+ and t rotamers are used.
This complex convergence was wholly unexpected and we sought to rationalize this phenomenon in terms of the underlying structural and mechanistic constraints. For a protease to be catalytically productive, the leaving group must be positioned within hydrogen-bonding distance of the proton-transfer moiety. For χcat < −90° this constraint is satisfied only when a re-face attack occurs and, by symmetry, the opposite holds for χcat > 90° (Fig. 3). Because χcat is predetermined for each reactive rotamer/oxyanion hole pair, the convergence on a common stereochemical mechanism for each protease is the inevitable evolutionary consequence of using the local backbone to facilitate proteolysis.
Fig. 3.

Stereospecificity of proteolysis. (A) The substrate carbonyl points toward the local backbone for stabilization with a local oxyanion hole. When χcat > 90°, the substrate must expose the si face of the peptide for attack, because a re-face attack is nonproductive for proton transfer. (B) When χcat < 90°, the opposite holds. Only a re-face attack is viable for enzyme acylation and a si-face attack leads to an inactive complex.
As noted earlier, the proteases of clan SH do not use the local backbone in oxyanion hole formation but still attack into the re face of the scissile bond and have χcat and χoxy values similar to those of other re-face attackers (23). This prompted us to consider whether some additional mechanistic factor might preclude alternative geometries. For example, the oxyanion intermediate that is generated during enzyme acylation is highly basic and must be shielded from the acid moiety used in catalysis. In the framework described here, this chemical imperative translates into a requirement that χoxy and χcat must have the same sign (Fig. S1). We suggest additional constraints might be identified by in silico interrogation of nonobserved, hypothetical active site configurations. In sum, despite the great diversity in Ser protease structure and substrate specificity, they may be partitioned into precisely two distinct mechanistic classes according to which face of the peptide bond is attacked during catalysis.
Extension of Convergent Analysis to the Cys Proteases.
The mechanistic constraints discussed above are not unique to the Ser proteases. The availability of the local backbone to contribute to oxyanion hole formation, the relevance of the reactive rotamer, and the importance of χcat are all applicable to the Cys proteases. To validate these principles, we extended our analysis to each of the 11 structurally characterized clans of Cys proteases (Table 2). Like the Ser proteases, the Cys proteases evolved to use all three staggered rotamers of the nucleophile and we observed the same correlation between the reactive rotamer and the identity of the local oxyanion hole residue. We found that χcat did not correlate well with the face of attack because most Cys proteases use a thiolate/acid pair that forms before substrate binding and the larger radius of sulfur means the thiol is deprotonated from farther away (31). This important distinction from the Ser proteases allows the acid to be preorganized for protonation of the leaving group and not activation of the nucleophile. More generally, the thiol of Cys is intrinsically more reactive than the hydroxyl of Ser and the structural constraints for efficient catalysis are correspondingly less stringent. These positional differences are made clearest by analysis of clans PA and PC, which contain both Ser and Cys proteases that are related through divergent evolution (9). In each case, the Cys proteases show geometries that would be ill-suited for deprotonation of Ser, but the acidic moiety is well positioned for protonation of the amine leaving group. Although thiols and hydroxyls require different geometries of proton transfer, the orientation of substrate binding is conserved between these enzymes. Table 2 shows that all Cys proteases use the local oxyanion hole and |χoxy| < 90°. Moreover, the relationship between the reactive rotamer and the face of attack is identical to that of the Ser proteases (with the exception of clan SH). Although divergent evolution may change the substrate specificity or the identity of the catalytic triad of a protease, we report here that the stereochemistry of catalysis is absolutely conserved.
Table 2.
Active site parameters of convergently related Cys proteases
| Clan | Enzyme | PDB | Complex | Reactive rotamer | ϕ, ψ angles | Catalytic motif | Oxyanion hole | Face of attack | χcat | αcat | χoxy | Reference |
| PA | SARS protease | 3SND | Thioacetal | g− | (−56, 142) | C-H | N | re | −168 | 82 | −41 | (74) |
| CD | Caspase 8 | 1QTN | Thioacetal | g− | (−73, 159) | C-H | N | re | NA | NA | −2 | (75) |
| CF | Pyrrolidone carboxylate peptidase | 1IU8 | g− | (−64, -52) | C-H-E | N | re | −169 | (76) | |||
| CL | L,D-transpeptidase | 1ZAT | g− | (−65, 164) | C-H | N | re | 179 | (77) | |||
| CM | HCV NS2 protease | 2HD0 | g− | (−66, 141) | C-H-E | N | re | 153 | (78) | |||
| PC | PH1704 | 1G2I | t | (60, −115) | C-E | N + 1 | si | 180 | (79) | |||
| CA | Papain | 1POP | Thioacetal | g+ | (−56, −32) | C-H | N | si | −177 | 76 | 30 | (80) |
| CE | Ulp1 | 1EUV | Thioacetal | g+ | (−52, −46) | C-H-D | N | si | −169 | 82 | 36 | (81) |
| CN | Equine encephalitis alphavirus nsP2 | 2HWK | g+ | (−54, −26) | C-H | N | si | ND | (82) | |||
| CO | Diamino endopeptidase | 2HBQ | g+ | (−64, −47) | C-H-H | N | si | −160 | (83) | |||
| CP | deSUMOylase DeSI-1 | 2WP7 | Unmodeled | g+ | (−59, −32) | C-H | N | si | 179 | (84) |
Members of clan CD do not use the same general base for activation of the nucleophile and protonation of the leaving group. The structure 2HWK crystallized with the base in an inactive state and 2WP7 has a ligand covalently bound to the nucleophile that was not modeled.
Mechanistic Constraints Imposed by the Thr γ-Methyl.
With this comprehensive framework for dissecting proteolysis in place, we can address the question that first motivated this investigation: Why do the Ser proteases not use Thr? For every system we examined, the Thr γ-methyl was modeled onto the Ser nucleophile and its interactions with the environment were recorded. Surprisingly, only two types of interference were necessary to account for the inability of Thr to function in the catalytic triad. For Ser proteases with χcat > 90°, which attack into the si face of the scissile bond, the γ-methyl of Thr would form a preclusive steric clash with the oxyanion generated during catalysis (Fig. 4 C and D). Alternatively, when χcat < −90°, the γ-methyl clashes with the residue facilitating proton transfer (Fig. 4 A and B). Because this residue is the active site component subject to the most variation, we expanded our test of the γ-methyl to every structurally characterized Ser protease family, which accounts for divergent evolutionary pressures. For the few structures in which no clash was observed there was strong evidence that a catalytically incompetent conformation had been crystallized (Fig. S2 and SI Text). Hence, a clash with a hypothetical γ-methyl was observed for all Ser proteases irrespective of the identity of the catalytic triad, the reactive rotamer, or the components of the oxyanion hole. Because this clash is intrinsic to protease structure, evolution always selects against a Thr nucleophile in all clans of Ser proteases.
Fig. 4.

Steric clashes of the Thr γ-methyl. (A) γ-Chymotrypsin with a boronic acid inhibitor (PDB ID: 3VGC). The γ-methyl of Thr was modeled (black) and its steric clash with the base is indicated by overlapping spheres drawn with the van der Waals radii. (B) CMV protease with an α-ketoamide inhibitor (PDB ID: 1NKM) shows a steric clash still occurs when the local oxyanion hole is not used. (C) Prolyl aminopeptidase with a boronic acid inhibitor (PDB ID: 2EEP). The “nucleophilic elbow” motif is incompatible with the Thr γ-methyl, which clashes with the oxyanion substrate (red sphere). (D) A similar clash is observed for signal peptidase I with a β-lactam inhibitor (PDB ID: 1B12).
In light of this conclusion, the report that trypsin may be converted to a weak, but functional, Thr protease with as few as three mutations is particularly striking (32). This variant was never tested against a true protein substrate and we hypothesize that additional experiments will show it avoids enzyme acylation or does not feature direct proton transfer to the leaving group. There do exist natural Thr nucleophiles in biochemistry but without posttranslational modification, none of them catalyze proteolysis. The most similar reaction is that of the bacterial asparaginase enzymes, which hydrolyze the terminal amide of asparagine to yield aspartate and ammonia. Evidence clearly supports the use of a Thr nucleophile in a Thr-Tyr-Glu catalytic triad. However, removal of the phenolic OH of the Tyr base has a relatively small 100-fold reduction on kcat, compared with the ∼1,000,000-fold reduction upon removal of the His residue of the Ser protease catalytic triad (18, 33). This and other circumstantial evidence suggest more than one residue or a bridging water may be facilitating proton transfer during acylation (SI Text).
When the chemistry in question is fundamentally altered from hydrolysis to phosphoryl group transfer, so are the mechanistic imperatives. Protonation of the leaving group is no longer necessary, and the structure of phosphothreonine lyase shows the longer P-O bonds may accommodate the Thr γ-methyl without a steric clash (8). Although some reactions have sufficiently weak chemical constraints to allow a Thr nucleophile, the vast majority of enzymes that function with an acyl-enzyme intermediate do not use Thr. Covalent catalysis is a common theme in biochemistry and the structural constraints identified here for the proteases also apply to many other enzymatic reactions including amidohydrolases (34), esterases (35), β-lactamases (36), thioesterases (37), and nitrilases (38), as well as more sophisticated enzymes such as glyceraldehye-3-phosphate dehydrogenase (39).
Positive Selection for an N-Terminal Thr Nucleophile.
Of course, the structural barrier to the Thr nucleophile is not insurmountable. Seminal research into the mechanism of the proteasome β-subunit (PβS) established it as the first member of the “Thr proteases” (11, 40, 41). In a departure from the canonical Ser proteases, PβS and related enzymes require an intramolecular posttranslational modification, cis-autoproteolysis, to generate the active site (10, 42, 43). The resultant N-terminal nucleophile (Ntn) harbors an α-amine that acts as the proton transfer catalyst, analogous to the role played by His in the canonical catalytic triad. These enzymes are found to use all three possible nucleophiles (Cys, Ser, Thr). As indicated by the results discussed above, catalysis with an Ntn must be geometrically distinct from that of the Ser and Cys proteases to accommodate a Thr nucleophile. Furthermore, a thorough explanation of Ntn function should also account for the fact that mutational removal of the γ-methyl decreases the activity of PβS (20) and other Thr-using enzymes (44). Therefore, we extended our analysis to the Ntn enzymes in an effort to resolve the unusual evolutionary preference for a Thr nucleophile.
There are at least two distinct protein folds that support autoproteolysis to generate an Ntn enzyme active site: the original Ntn-fold (10) and the DOM-fold (45). As described above, autoproteolysis generates an α-amine base with χcat = 30°, which tightly constrains the potential catalytic geometries. Consequently, all Ntn-using enzymes bind their substrates with a common orientation and attack into the si face of their substrates through the g− rotamer. More importantly, because the base approaches from a previously disallowed trajectory, it no longer has a steric clash with the γ-methyl, thereby allowing Thr to function as a catalytic nucleophile.
We recently reported that the Thr γ-methyl may accelerate the autoactivation of Ntn enzymes through the destabilization of inactive ground state conformations (46) through a phenomenon known as the reactive rotamer effect (RRE). Although the RRE is defined for intramolecular cyclizations (47), in principle it will apply to any reaction for which equilibration between active and inactive conformational states is fast relative to catalysis. The apparent rate of the reaction (kcat) can be increased if the addition of steric bulk shifts the ground state ensemble to favor the reactive state. This is precisely the situation presented by Thr→Ser mutation of PβS and related enzymes. As described by Fig. 5, the γ-methyl accelerates catalysis by intrinsically favoring the reactive g− rotamer. These rotamer distributions may be further shifted by interactions with residues proximal to the active site (48). This rationale explains the reduction in kcat observed upon Thr→Ser mutation of PβS (20) and glycosylasparaginase (44). Hence, the close association of multiple functional groups forces a steric clash with the Thr γ-methyl that precludes its use as a nucleophile for most enzymes. These constraints are relieved upon posttranslational autoproteolysis, whereupon the γ-methyl is not only tolerated but may intrinsically accelerate catalysis, explaining why it is the evolutionarily favored nucleophile in PβS and other Ntn enzymes.
Fig. 5.

Gauche interactions of an N-terminal nucleophile (Ntn). The g− rotamer of an Ntn Thr has the fewest gauche interactions. When the γ-methyl is removed by mutation to Ser, the t rotamer becomes favored. This effect is exacerbated by repulsive interactions with the neighboring carbonyl. Consequently, the Thr γ-methyl shifts the rotamer distribution toward a reactive state, which increases kcat. These effects are intrinsic to Ntn structure and may contribute to the evolutionary selection of a Thr nucleophile.
Structural Constraints for Catalysis with a Local Oxyanion Hole.
In addition to affecting the selection of the catalytic residue, the mechanistic constraints of enzyme acylation with a local oxyanion hole limit the structural variation surrounding the nucleophile. For example, a Ramachandran plot showing the φ, ψ angles of the nucleophile is clustered by the reactive rotamer (Fig. 6). For the two gauche rotamers, φ ∼ −55° is necessary to use the local backbone in the formation of an oxyanion hole. This in turn limits the possible ψ angles. When ψ ∼ 150°, the carbonyl of the nucleophilic residue has a repulsive interaction with the g+ rotamer and only the g− rotamer is catalytically viable. Hence, all enzymes that use a local oxyanion hole and acylate the g+ rotamer of the nucleophile have φ, ψ ∼ −55°, −20°, where the g− rotamer is also allowed. Far more limiting, though, is use of the backbone of the N + 1 residue in the oxyanion hole, which requires ψ ∼ −120°. This angle is allowed only in a small region of the φ, ψ space. When φ, ψ ∼ 60°, −120°, a g+ rotamer has a strong steric repulsion with the N − 1 carbonyl, explaining why this rotamer is never associated with an enzyme that uses an N + 1 oxyanion hole.
Fig. 6.

Plot of the φ, ψ angles of the nucleophilic residue for Ser and Cys overlaying the Ramachandran plot. Proteases cluster according to the reactive rotamer in catalysis. The outlier, marked X, does not use the local backbone for oxyanion stabilization. Ramachandran plot was generated in MolProbity: light blue, strongly favored; dark blue, allowed (85).
These restrictions on the conformation of the local backbone have larger structural consequences. When the nucleophile has φ, ψ ∼ −55°, 150°, there is no correlation in the local secondary structure between analogous enzymes (Fig. 7A). For φ, ψ ∼ −55°, −20° there is a clear trend for the nucleophile to be positioned at the N terminus of a helix (Fig. 7B). When this circumstance occurs, the oxyanion is further stabilized by the helix dipole. Catalysis through the t rotamer with the N + 1 oxyanion hole shows evidence of the strongest structural constraints because ψ ∼ −120° is allowed when φ ∼ 60°. For these enzymes, the nucleophile is always preceded by a β-strand that leads into a helix (Fig. 7C). This motif has been recognized previously as the “nucleophilic elbow” and has strong sequence requirements for its formation (37, 49). This is the only viable conformation when a single residue separates a strand–helix transition in protein structure (50), which we suggest could be the first feature formed in a t rotamer proto-enzyme active site. Previously, there was no explanation for the convergent evolution of this elaborate motif for the proteases and more sophisticated nitrilases (37, 49, 51). When this question is posed in the framework described here, the nucleophilic elbow is simply a consequence of using the t rotamer and the N + 1 oxyanion hole for catalysis.
Fig. 7.
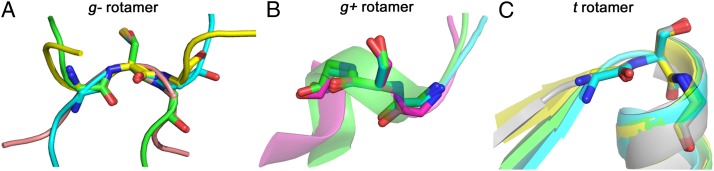
Structural overlay of proteases according to their reactive rotamer. (A) Enzymes that use the g− rotamer show no structural similarity apart from their φ, ψ angles. (B) A reactive g+ rotamer has a loop at its N-terminal direction and a helix at it C-terminal end. (C) Use of the t rotamer and the N + 1 oxyanion hole imparts stringent structural constraints that are satisfied by a β-strand turning into an α-helix, the “nucleophilic elbow” motif. Structures shown are listed in Tables 1 and 2.
Recently, there has been great interest in developing artificial enzymes (52) and enzymes with novel function (53, 54). Landmark progress have been made with the design of Kemp Eliminases (55) and Diels-Alderases (56). For comparable success with enzymes that use an acylation mechanism, it would be beneficial to understand the interactions between the minimal active site components: nucleophile, oxyanion hole, and proton transfer catalyst. These were recently explored by the rational design of novel enzymes with esterase activity (57); but these enzymes had significantly less catalytic power than natural esterases. We believe that active site designs featuring the angles, distances, and structural motifs defined here should lead to catalysts with improved activity.
Stereospecific Mechanism-Based Inhibitors.
Enzymes that use an acylation mechanism have a long and successful history as therapeutic targets for drug development. Since the discovery of penicillin, there has been an unceasing effort in the scientific community to both understand these enzymes and develop new molecules that inhibit them (58). Traditionally, these enzymes were classified according to the residue or metal ion used to generate the nucleophile (6). Unfortunately, this organization affords no perspective for the design of inhibitors that possess a mechanism of action that is sensitive to the stereochemistry of catalysis. Therefore, we sought to understand how nature exploits the distinction between re- and si-face–attacking enzymes by analyzing naturally occurring, stereospecific inhibitors in the context of our mechanism-based classification.
The β-lactam antibiotics inhibit penicillin-binding proteins responsible for the final cross-linking step in bacterial cell wall biosynthesis (59), a reaction that occurs through a re-face attack. Conspicuously, all classes of the naturally occurring bicyclic β-lactam antibiotics feature stereochemistry that occludes the si face of the β-lactam moiety (Fig. 8A). The bacterial signal peptidase (SPase) is also present in the periplasm, but functions through a si-face attack. Consistent with our analysis, it has been shown that incubation of traditional β-lactam antibiotics fails to inhibit these enzymes (60). However, a β-lactam–based SPase inhibitor was successfully generated when the bridgehead stereochemistry of a penicillin-like molecule was inverted, to expose the si face of the synthetic β-lactam.
Fig. 8.

Stereospecific antibiotics. (A) The β-lactam molecule carbapenem contains stereochemistry that blocks the si face of the elecrophilic carbonyl and this molecule may be used only to inhibit re-face attacking enzymes. (B) Conversely, omuralide features a β-lactone–γ-lactam core that occludes the re face of the eletrophilic carbonyl. Consequently, this inhibitor scaffold is effective only against si-face–attacking enzymes.
Omuralide is a naturally occurring β-lactone–γ-lactam (Fig. 8B) antibiotic that functions through inhibition of PβS, a si-face–attacking Ntn enzyme (61). Despite its high binding affinity for PβS, it has been shown that incubation of omuralide with the Chlamydia protease CPAF, which uses the nucleophilic elbow motif, successfully inactivates the enzyme (62). This is consistent with the hypotheses that the γ-lactam blocks the re face of the molecule, thereby restricting the inhibition profile to si-face–attacking enzymes. Similar properties may be attributed to the salinosporamides (63), vibralactone (64), and avibactam (65). In total, we identified seven independent cases where nature selected for the biosynthesis of an inhibitor that is sensitive to the stereochemistry of the active site of the target enzyme (SI Text).
Currently, efforts to develop new therapeutics are often focused on optimizing interactions within the binding pocket that are distinct from the catalytic center itself. This is an extremely challenging pursuit because there are numerous enzymes with overlapping substrate specificities. We suggest that incorporation of moieties that preclude attack into the nontarget face of an inhibitor may be a useful negative-design parameter in the development of new drugs with improved specificity.
Conclusions
In the present investigation, we defined geometric enzyme–substrate parameters that enabled systematic examination of enzyme function and applied them to every structurally characterized family of the Ser, Cys, and Thr proteases. This study revealed surprising correlations between seemingly independent active site components. We explained that utilization of the local backbone in oxyanion stabilization is related to the reactive rotamer for catalysis and that the location of the base determines the orientation of substrate binding. We advocate division of the proteases into two classes according to the stereochemistry of their nucleophilic attack, as is common in the study of NAD(P)H-dependent enzymes (66). By understanding the active site constraints imparted by proteolysis through an acyl-enzyme mechanism, we accounted for the selection against a Thr nucleophile, established why the nucleophilic elbow forms, and provided a basis for understanding the evolution of stereospecific inhibitors. These relationships constitute incontrovertible links between structure and function and limit the fitness landscape upon which the evolution of biocatalysis occurs. This knowledge is distinct from, but no less valid than, that accessible through detailed quantum mechanical calculations, time, and computational expertise. Our method of convergent evolutionary analysis is broadly applicable and should aid in the continued study of enzymes and their elegant structure–function relationship.
Methods
Proteases of unrelated evolutionary lines were identified according to their classification in the MEROPS database (9). From this list, structures were selected according to their resolution and the presence of a mechanistically relevant ligand and uploaded into Coot for visual inspection of model quality within each active site (67). All of the measured values in Tables 1 and 2 contain error that is intrinsic to crystallography. These deviations are resolution dependent and, when possible, we have chosen the highest-resolution structure available. The face of attack for each enzyme was established by analysis of an inhibitor complex or inference from the location of the P1 site on the protein. Superimpositions and visualization of group van der Waals radii were performed in PyMol (Schrödinger, LLC).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Research Grant R01 AI014937.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1221050110/-/DCSupplemental.
References
- 1.Warshel A, Naray-Szabo G, Sussman F, Hwang JK. How do serine proteases really work? Biochemistry. 1989;28(9):3629–3637. doi: 10.1021/bi00435a001. [DOI] [PubMed] [Google Scholar]
- 2.Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: Freeman; 1999. [Google Scholar]
- 3.Kraut J. How do enzymes work? Science. 1988;242(4878):533–540. doi: 10.1126/science.3051385. [DOI] [PubMed] [Google Scholar]
- 4.Warshel A. Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J Biol Chem. 1998;273(42):27035–27038. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- 5.Warshel A. Energetics of enzyme catalysis. Proc Natl Acad Sci USA. 1978;75(11):5250–5254. doi: 10.1073/pnas.75.11.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ekici OD, Paetzel M, Dalbey RE. Unconventional serine proteases: Variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008;17(12):2023–2037. doi: 10.1110/ps.035436.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milkowski C, Strack D. Serine carboxypeptidase-like acyltransferases. Phytochemistry. 2004;65(5):517–524. doi: 10.1016/j.phytochem.2003.12.018. [DOI] [PubMed] [Google Scholar]
- 8.Chen L, et al. Structural basis for the catalytic mechanism of phosphothreonine lyase. Nat Struct Mol Biol. 2008;15(1):101–102. doi: 10.1038/nsmb1329. [DOI] [PubMed] [Google Scholar]
- 9.Rawlings ND, Barrett AJ, Bateman A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012;40(Database issue, D1):D343–D350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brannigan JA, et al. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature. 1995;378(6555):416–419. doi: 10.1038/378416a0. [DOI] [PubMed] [Google Scholar]
- 11.Seemüller E, et al. Proteasome from Thermoplasma acidophilum: A threonine protease. Science. 1995;268(5210):579–582. doi: 10.1126/science.7725107. [DOI] [PubMed] [Google Scholar]
- 12.Smith AJT, et al. Structural reorganization and preorganization in enzyme active sites: Comparisons of experimental and theoretically ideal active site geometries in the multistep serine esterase reaction cycle. J Am Chem Soc. 2008;130(46):15361–15373. doi: 10.1021/ja803213p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galperin MY, Walker DR, Koonin EV. Analogous enzymes: Independent inventions in enzyme evolution. Genome Res. 1998;8(8):779–790. doi: 10.1101/gr.8.8.779. [DOI] [PubMed] [Google Scholar]
- 14.Polgár L. The catalytic triad of serine peptidases. Cell Mol Life Sci. 2005;62(19–20):2161–2172. doi: 10.1007/s00018-005-5160-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertus JD, Kraut J, Alden RA, Birktoft JJ. Subtilisin; a stereochemical mechanism involving transition-state stabilization. Biochemistry. 1972;11(23):4293–4303. doi: 10.1021/bi00773a016. [DOI] [PubMed] [Google Scholar]
- 16.Neet KE, Koshland DE., Jr The conversion of serine at the active site of subtilisin to cysteine: A “chemical mutation”. Proc Natl Acad Sci USA. 1966;56(5):1606–1611. doi: 10.1073/pnas.56.5.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polgar L, Bender ML. A new enzyme containing a synthetically formed active site. Thiol-subtillisin. J Am Chem Soc. 1966;88(13):3153–3154. [Google Scholar]
- 18.Carter P, Wells JA. Dissecting the catalytic triad of a serine protease. Nature. 1988;332(6164):564–568. doi: 10.1038/332564a0. [DOI] [PubMed] [Google Scholar]
- 19.Corey DR, Craik CS. An investigation into the minimum requirements for peptide hydrolysis by mutation of the catalytic triad of trypsin. J Am Chem Soc. 1992;114(5):1784–1790. [Google Scholar]
- 20.Kisselev AF, Songyang Z, Goldberg AL. Why does threonine, and not serine, function as the active site nucleophile in proteasomes? J Biol Chem. 2000;275(20):14831–14837. doi: 10.1074/jbc.275.20.14831. [DOI] [PubMed] [Google Scholar]
- 21.Rawlings ND, Barrett AJ. Families of serine peptidases. Methods Enzymol. 1994;244:19–61. doi: 10.1016/0076-6879(94)44004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paetzel M, Dalbey RE. Catalytic hydroxyl/amine dyads within serine proteases. Trends Biochem Sci. 1997;22(1):28–31. doi: 10.1016/s0968-0004(96)10065-7. [DOI] [PubMed] [Google Scholar]
- 23.Khayat R, et al. Structural and biochemical studies of inhibitor binding to human cytomegalovirus protease. Biochemistry. 2003;42(4):885–891. doi: 10.1021/bi027045s. [DOI] [PubMed] [Google Scholar]
- 24.Simón L, Goodman JM. Enzyme catalysis by hydrogen bonds: The balance between transition state binding and substrate binding in oxyanion holes. J Org Chem. 2010;75(6):1831–1840. doi: 10.1021/jo901503d. [DOI] [PubMed] [Google Scholar]
- 25.Simón L, Goodman JM. Hydrogen-bond stabilization in oxyanion holes: Grand jeté to three dimensions. Org Biomol Chem. 2012;10(9):1905–1913. doi: 10.1039/c2ob06717j. [DOI] [PubMed] [Google Scholar]
- 26.Fülöp V, Böcskei Z, Polgár L. Prolyl oligopeptidase: An unusual beta-propeller domain regulates proteolysis. Cell. 1998;94(2):161–170. doi: 10.1016/s0092-8674(00)81416-6. [DOI] [PubMed] [Google Scholar]
- 27.Wahlgren WY, et al. The catalytic aspartate is protonated in the Michaelis complex formed between trypsin and an in vitro evolved substrate-like inhibitor: A refined mechanism of serine protease action. J Biol Chem. 2011;286(5):3587–3596. doi: 10.1074/jbc.M110.161604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JS, et al. Navigation inside a protease: Substrate selection and product exit in the tricorn protease from Thermoplasma acidophilum. J Mol Biol. 2002;324(5):1041–1050. doi: 10.1016/s0022-2836(02)01153-1. [DOI] [PubMed] [Google Scholar]
- 29.Szyk A, Maurizi MR. Crystal structure at 1.9A of E. coli ClpP with a peptide covalently bound at the active site. J Struct Biol. 2006;156(1):165–174. doi: 10.1016/j.jsb.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Mac Sweeney A, et al. Crystal structure of delta-chymotrypsin bound to a peptidyl chloromethyl ketone inhibitor. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 3):280–286. doi: 10.1107/s0907444999016583. [DOI] [PubMed] [Google Scholar]
- 31.Storer AC, Ménard R. Catalytic mechanism in papain family of cysteine peptidases. Methods Enzymol. 1994;244:486–500. doi: 10.1016/0076-6879(94)44035-2. [DOI] [PubMed] [Google Scholar]
- 32.Baird TT, Jr, Wright WD, Craik CS. Conversion of trypsin to a functional threonine protease. Protein Sci. 2006;15(6):1229–1238. doi: 10.1110/ps.062179006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Derst C, Wehner A, Specht V, Röhm KH. States and functions of tyrosine residues in Escherichia coli asparaginase II. Eur J Biochem. 1994;224(2):533–540. doi: 10.1111/j.1432-1033.1994.00533.x. [DOI] [PubMed] [Google Scholar]
- 34.Mileni M, et al. Binding and inactivation mechanism of a humanized fatty acid amide hydrolase by alpha-ketoheterocycle inhibitors revealed from cocrystal structures. J Am Chem Soc. 2009;131(30):10497–10506. doi: 10.1021/ja902694n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cygler M, et al. Relationship between sequence conservation and three-dimensional structure in a large family of esterases, lipases, and related proteins. Protein Sci. 1993;2(3):366–382. doi: 10.1002/pro.5560020309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strynadka NCJ, et al. Molecular structure of the acyl-enzyme intermediate in beta-lactam hydrolysis at 1.7 A resolution. Nature. 1992;359(6397):700–705. doi: 10.1038/359700a0. [DOI] [PubMed] [Google Scholar]
- 37.Ollis DL, et al. The alpha/beta hydrolase fold. Protein Eng. 1992;5(3):197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 38.Brenner C. Catalysis in the nitrilase superfamily. Curr Opin Struct Biol. 2002;12(6):775–782. doi: 10.1016/s0959-440x(02)00387-1. [DOI] [PubMed] [Google Scholar]
- 39.Garavito RM, Rossmann MG, Argos P, Eventoff W. Convergence of active center geometries. Biochemistry. 1977;16(23):5065–5071. doi: 10.1021/bi00642a019. [DOI] [PubMed] [Google Scholar]
- 40.Löwe J, et al. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science. 1995;268(5210):533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 41.Groll M, et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386(6624):463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 42.Seemuller E, Lupas A, Baumeister W. Autocatalytic processing of the 20S proteasome. Nature. 1996;382(6590):468–471. doi: 10.1038/382468a0. [DOI] [PubMed] [Google Scholar]
- 43.Perler FB, Xu MQ, Paulus H. Protein splicing and autoproteolysis mechanisms. Curr Opin Chem Biol. 1997;1(3):292–299. doi: 10.1016/s1367-5931(97)80065-8. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y, Guan C, Aronson NN., Jr Site-directed mutagenesis of essential residues involved in the mechanism of bacterial glycosylasparaginase. J Biol Chem. 1998;273(16):9688–9694. doi: 10.1074/jbc.273.16.9688. [DOI] [PubMed] [Google Scholar]
- 45.Cheng H, Grishin NV. DOM-fold: A structure with crossing loops found in DmpA, ornithine acetyltransferase, and molybdenum cofactor-binding domain. Protein Sci. 2005;14(7):1902–1910. doi: 10.1110/ps.051364905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buller AR, Freeman MF, Wright NT, Schildbach JF, Townsend CA. Insights into cis-autoproteolysis reveal a reactive state formed through conformational rearrangement. Proc Natl Acad Sci USA. 2012;109(7):2308–2313. doi: 10.1073/pnas.1113633109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jung ME, Piizzi G. gem-disubstituent effect: Theoretical basis and synthetic applications. Chem Rev. 2005;105(5):1735–1766. doi: 10.1021/cr940337h. [DOI] [PubMed] [Google Scholar]
- 48.Buller AR, et al. Autoproteolytic activation of ThnT results in structural reorganization necessary for substrate binding and catalysis. J Mol Biol. 2012;422(4):508–518. doi: 10.1016/j.jmb.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Korza HJ, Bochtler M. Pseudomonas aeruginosa LD-carboxypeptidase, a serine peptidase with a Ser-His-Glu triad and a nucleophilic elbow. J Biol Chem. 2005;280(49):40802–40812. doi: 10.1074/jbc.M506328200. [DOI] [PubMed] [Google Scholar]
- 50.Street TO, Fitzkee NC, Perskie LL, Rose GD. Physical-chemical determinants of turn conformations in globular proteins. Protein Sci. 2007;16(8):1720–1727. doi: 10.1110/ps.072898507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pace HC, et al. Crystal structure of the worm NitFhit Rosetta Stone protein reveals a Nit tetramer binding two Fhit dimers. Curr Biol. 2000;10(15):907–917. doi: 10.1016/s0960-9822(00)00621-7. [DOI] [PubMed] [Google Scholar]
- 52.Motherwell WB, Bingham MJ, Six Y. Recent progress in the design and synthesis of artificial enzymes. Tetrahedron. 2001;57(22):4663–4686. [Google Scholar]
- 53.Kipnis Y, Baker D. 2012. Comparison of designed and randomly generated catalysts for simple chemical reactions. Protein Sci 21(9):1388–1395.
- 54.Gerlt JA, Babbitt PC. Enzyme (re)design: Lessons from natural evolution and computation. Curr Opin Chem Biol. 2009;13(1):10–18. doi: 10.1016/j.cbpa.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Röthlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453(7192):190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 56.Siegel JB, et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science. 2010;329(5989):309–313. doi: 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Richter F, et al. Computational design of catalytic dyads and oxyanion holes for ester hydrolysis. J Am Chem Soc. 2012;134(39):16197–16206. doi: 10.1021/ja3037367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Page MGP, Heim J. New molecules from old classes: Revisiting the development of beta-lactams. IDrugs. 2009;12(9):561–565. [PubMed] [Google Scholar]
- 59.Fisher JF, Meroueh SO, Mobashery S. Bacterial resistance to beta-lactam antibiotics: Compelling opportunism, compelling opportunity. Chem Rev. 2005;105(2):395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 60.Paetzel M, Dalbey RE, Strynadka NCJ. Crystal structure of a bacterial signal peptidase in complex with a beta-lactam inhibitor. Nature. 1998;396(6707):186–190. doi: 10.1038/24196. [DOI] [PubMed] [Google Scholar]
- 61.Fenteany G, et al. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268(5211):726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 62.Huang ZW, et al. Structural basis for activation and inhibition of the secreted chlamydia protease CPAF. Cell Host Microbe. 2008;4(6):529–542. doi: 10.1016/j.chom.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 63.Groll M, Huber R, Potts BCM. Crystal structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of beta-lactone ring opening and a mechanism for irreversible binding. J Am Chem Soc. 2006;128(15):5136–5141. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]
- 64.Zeiler E, et al. Vibralactone as a tool to study the activity and structure of the ClpP1P2 complex from Listeria monocytogenes. Angew Chem Int Ed Engl. 2011;50(46):11001–11004. doi: 10.1002/anie.201104391. [DOI] [PubMed] [Google Scholar]
- 65.Ehmann DE, et al. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc Natl Acad Sci USA. 2012;109(29):11663–11668. doi: 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fisher HF, Conn EE, Vennesland B, Westheimer FH. The enzymatic transfer of hydrogen. I. The reaction catalyzed by alcohol dehydrogenase. J Biol Chem. 1953;202(2):687–697. [PubMed] [Google Scholar]
- 67.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 68.Tamada T, et al. Combined high-resolution neutron and X-ray analysis of inhibited elastase confirms the active-site oxyanion hole but rules against a low-barrier hydrogen bond. J Am Chem Soc. 2009;131(31):11033–11040. doi: 10.1021/ja9028846. [DOI] [PubMed] [Google Scholar]
- 69.Wlodawer A, et al. Carboxyl proteinase from Pseudomonas defines a novel family of subtilisin-like enzymes. Nat Struct Biol. 2001;8(5):442–446. doi: 10.1038/87610. [DOI] [PubMed] [Google Scholar]
- 70.Silvaggi NR, Anderson JW, Brinsmade SR, Pratt RF, Kelly JA. The crystal structure of phosphonate-inhibited D-Ala-D-Ala peptidase reveals an analogue of a tetrahedral transition state. Biochemistry. 2003;42(5):1199–1208. doi: 10.1021/bi0268955. [DOI] [PubMed] [Google Scholar]
- 71.Håkansson K, Wang AHJ, Miller CG. The structure of aspartyl dipeptidase reveals a unique fold with a Ser-His-Glu catalytic triad. Proc Natl Acad Sci USA. 2000;97(26):14097–14102. doi: 10.1073/pnas.260376797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chung IYW, Paetzel M. Crystal structure of a viral protease intramolecular acyl-enzyme complex: Insights into cis-cleavage at the VP4/VP3 junction of Tellina birnavirus. J Biol Chem. 2011;286(14):12475–12482. doi: 10.1074/jbc.M110.198812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444(7116):179–180. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- 74.Zhu L, et al. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antiviral Res. 2011;92(2):204–212. doi: 10.1016/j.antiviral.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watt W, et al. The atomic-resolution structure of human caspase-8, a key activator of apoptosis. Structure. 1999;7(9):1135–1143. doi: 10.1016/s0969-2126(99)80180-4. [DOI] [PubMed] [Google Scholar]
- 76.Sokabe M, et al. The X-ray crystal structure of pyrrolidone-carboxylate peptidase from hyperthermophilic archaea Pyrococcus horikoshii. J Struct Funct Genomics. 2002;2(3):145–154. doi: 10.1023/a:1021257701676. [DOI] [PubMed] [Google Scholar]
- 77.Biarrotte-Sorin S, et al. Crystal structure of a novel beta-lactam-insensitive peptidoglycan transpeptidase. J Mol Biol. 2006;359(3):533–538. doi: 10.1016/j.jmb.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 78.Lorenz IC, Marcotrigiano J, Dentzer TG, Rice CM. Structure of the catalytic domain of the hepatitis C virus NS2-3 protease. Nature. 2006;442(7104):831–835. doi: 10.1038/nature04975. [DOI] [PubMed] [Google Scholar]
- 79.Du XL, et al. Crystal structure of an intracellular protease from Pyrococcus horikoshii at 2-A resolution. Proc Natl Acad Sci USA. 2000;97(26):14079–14084. doi: 10.1073/pnas.260503597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schröder E, Phillips C, Garman E, Harlos K, Crawford C. X-ray crystallographic structure of a papain-leupeptin complex. FEBS Lett. 1993;315(1):38–42. doi: 10.1016/0014-5793(93)81128-m. [DOI] [PubMed] [Google Scholar]
- 81.Mossessova E, Lima CD. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell. 2000;5(5):865–876. doi: 10.1016/s1097-2765(00)80326-3. [DOI] [PubMed] [Google Scholar]
- 82.Russo AT, White MA, Watowich SJ. The crystal structure of the Venezuelan equine encephalitis alphavirus nsP2 protease. Structure. 2006;14(9):1449–1458. doi: 10.1016/j.str.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 83.Xu Q, et al. Structural basis of murein peptide specificity of a gamma-D-glutamyl-l-diamino acid endopeptidase. Structure. 2009;17(2):303–313. doi: 10.1016/j.str.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Suh H-Y, et al. Crystal structure of DeSI-1, a novel deSUMOylase belonging to a putative isopeptidase superfamily. Proteins. 2012;80(8):2099–2104. doi: 10.1002/prot.24093. [DOI] [PubMed] [Google Scholar]
- 85.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.