

Abstract
Members of the RAS superfamily of small guanosine triphosphatases (GTPases) transition between GDP-bound, inactive and GTP-bound, active states and thereby function as binary switches in the regulation of various cellular activities. Whereas HRAS, NRAS, and KRAS frequently acquire transforming missense mutations in human cancer, little is known of the oncogenic roles of other small GTPases, including Ras-related C3 botulinum toxin substrate (RAC) proteins. We show that the human sarcoma cell line HT1080 harbors both NRAS(Q61K) and RAC1(N92I) mutant proteins. Whereas both of these mutants were able to transform fibroblasts, knockdown experiments indicated that RAC1(N92I) may be the essential growth driver for this cell line. Screening for RAC1, RAC2, or RAC3 mutations in cell lines and public databases identified several missense mutations for RAC1 and RAC2, with some of the mutant proteins, including RAC1(P29S), RAC1(C157Y), RAC2(P29L), and RAC2(P29Q), being found to be activated and transforming. P29S, N92I, and C157Y mutants of RAC1 were shown to exist preferentially in the GTP-bound state as a result of a rapid transition from the GDP-bound state, rather than as a result of a reduced intrinsic GTPase activity. Activating mutations of RAC GTPases were thus found in a wide variety of human cancers at a low frequency; however, given their marked transforming ability, the mutant proteins are potential targets for the development of new therapeutic agents.
Keywords: oncogene, resequencing
The identification of transforming proteins and the development of agents that target them have markedly influenced the treatment and improved the prognosis of individuals with cancer. Chronic myeloid leukemia (CML), for example, has been shown to result from the growth-promoting activity of the fusion tyrosine kinase breakpoint cluster region-Abelson murine leukemia viral oncogene homolog 1 (BCR-ABL1), and treatment with a specific ABL1 inhibitor, imatinib mesylate, has increased the 5-y survival rate of individuals with CML to almost 90% (1). Similarly, the fusion of echinoderm microtubule associated protein like 4 gene (EML4) to anaplastic lymphoma receptor tyrosine kinase (ALK) is responsible for a subset of non–small-cell lung cancer cases (2), and therapy targeted to EML4-ALK kinase activity has greatly improved the progression-free survival of affected individuals compared with that achieved with conventional chemotherapies (3). Therapies that target essential growth drivers in human cancers are thus among the most effective treatments for these intractable disorders.
V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS), v-Ha-ras Harvey rat sarcoma viral oncogene homolog (HRAS), and neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS) are the founding members of the rat sarcoma (RAS) superfamily of small guanosine triphosphatases (GTPases) that is known to comprise >150 members in humans (4). Five subgroups of these small GTPases have been identified and designated as the RAS; ras homolog family member (RHO); RAB1A, member RAS oncogene family (RAB); RAN, member RAS oncogene family (RAN); and ADP-ribosylation factor (ARF) families. All small GTPases function as binary switches that transition between GDP-bound, inactive and GTP-bound, active forms and thereby contribute to intracellular signaling that underlies a wide array of cellular activities, including cell proliferation, differentiation, survival, motility, and transformation (5). Somatic point mutations that activate KRAS, HRAS, or NRAS have been identified in a variety of human tumors, with KRAS being the most frequently activated oncoprotein in humans. Somatic activating mutations of KRAS are thus present in >90% of pancreatic adenocarcinomas, for example (6). Surprisingly, however, mutational activation of small GTPases other than KRAS, HRAS, and NRAS has not been widely reported.
Ras-related C3 botulinum toxin substrate (RAC) 1, RAC2, and RAC3 belong to the RHO family of small GTPases (7). RAC proteins orchestrate actin polymerization, and their activation induces the formation of membrane ruffles and lamellipodia (8), which play essential roles in the maintenance of cell morphology and in cell migration. Accumulating evidence also indicates that RAC proteins function as key hubs of intracellular signaling that underlies cell transformation. RAC1, for example, serves as an essential downstream component of the signaling pathway by which oncogenic RAS induces cell transformation, and artificial introduction of an amino acid substitution (G12V) into RAC1 renders it oncogenic (9). Furthermore, suppression of RAC1 activity induces apoptosis in glioma cells (10), and loss of RAC1 or RAC2 results in a marked delay in the development of BCR-ABL1–driven myeloproliferative disorder (11). Despite such important roles of RAC proteins in cancer, somatic transforming mutations of these proteins have not been identified in cancer specimens.
We have now discovered a mutant form of RAC1 with the amino acid substitution N92I in a human sarcoma cell line, HT1080, and have found that this mutation renders RAC1 constitutively active and highly oncogenic. Even though HT1080 cells also harbor the NRAS(Q61K) oncoprotein, RAC1(N92I) is the essential growth driver in this cell line, given that RNA interference (RNAi)-mediated knockdown of RAC1(N92I) markedly suppressed cell growth. Further screening for RAC1, RAC2, and RAC3 mutations among cancer cell lines as well as public databases identified additional transforming mutations of RAC1 and RAC2. Our data thus reveal oncogenic amino acid substitutions for the RAC subfamily of small GTPases in human cancer.
Results
Discovery of the RAC1(N92I) Oncoprotein.
To identify transforming genes in the fibrosarcoma cell line HT1080 (12), we isolated cDNAs for cancer-related genes (n = 906) from HT1080 cells and subjected them to deep sequencing with the Genome Analyzer IIx (GAIIx) system. Quality filtering of the 92,025,739 reads obtained yielded 45,325,377 unique reads that mapped to 843 (93.0%) of the 906 target genes. The mean read coverage for the 843 genes was 495× per nucleotide, and ≥70% of the captured regions for 568 genes were read at ≥10× coverage.
Screening for nonsynonymous mutations in the data set with the use of our computational pipeline (13) revealed a total of five missense mutations with a threshold of ≥30× coverage and a ≥30% mutation ratio (Table S1). One of these mutations, a heterozygous missense mutation of NRAS that results in a Gln-to-Lys substitution at amino acid position 61 (Q61K), was described previously in this cell line (14) and is the most frequent transforming mutation of NRAS (5). We also discovered a missense mutation in another small GTPase, RAC1 (Fig. S1 and Table S1). An A-to-T transversion at position 516 of human RAC1 cDNA (GenBank accession no. NM_006908.4), resulting in an Asn-to-Ile substitution at position 92 of the encoded protein, was thus identified in 11,525 (47.5%) of the 24,238 total reads covering this position.
To examine the transforming potential of RAC1(N92I), we infected mouse 3T3 fibroblasts and MCF10A human mammary epithelial cells (15) with a retrovirus encoding wild-type or N92I mutant form of human RAC1 and then seeded the cells in soft agar for evaluation of anchorage-independent growth. Neither 3T3 nor MCF10A cells expressing wild-type RAC1 grew in soft agar (Fig. 1A), indicating the lack of transforming potential of RAC1. In contrast, the cells expressing RAC1(N92I) readily grew in soft agar (Fig. 1A), showing that this RAC1 mutant confers the property of anchorage-independent growth on both 3T3 and MCF10A cells. We also confirmed the transforming potential of an artificial mutant of RAC1, RAC1(G12V) (8), which harbors an amino acid substitution corresponding to that of the oncogenic G12V mutant form of RAS proteins.
Fig. 1.

Transforming potential of RAC1 and RAC2 mutants. (A) 3T3 or MCF10A cells were infected with recombinant retroviruses encoding enhanced green fluorescent protein (EGFP) as well as wild-type or mutant forms of RAC1 or RAC2 and were then assayed for anchorage-independent growth in vitro under the presence of 10% (vol/vol) FBS. After 14 d (3T3) or 20 d (MCF10A) of culture, the cells were stained with crystal violet and examined by conventional microscopy (Left: left image of each pair), and they were monitored for EGFP expression by fluorescence microscopy (Left: right image of each pair). (Scale bars, 0.5 mm.) The numbers of cell colonies were also determined as means ± SD from three independent experiments (Right). (B) 3T3 cells expressing wild-type or mutant forms of RAC1 or RAC2 were injected s.c. into the shoulder of nude mice, and the size of the resulting tumors [(length × width)/2] was determined at the indicated times thereafter. Tumor size for 3T3 expressing NRAS(Q61K) was similarly monitored. Data are means ± SD for tumors at four injection sites. (C) HEK293T cells were transfected with expression vectors for wild-type or mutant forms of RAC1 or RAC2 together with the SRE.L reporter plasmid and pGL-TK. The activity of firefly luciferase in cell lysates was then measured and normalized by that of Renilla luciferase. Data are means ± SD from three independent experiments. (D) Lysates of 3T3 cells expressing wild-type or mutant forms of RAC1 or RAC2 were subjected to a pull-down assay with PAK1-PBD. The precipitated proteins as well as the total cell lysates were then subjected to immunoblot analysis with antibodies to RAC1 or to RAC2. The relative amounts of pulled-down RAC proteins compared with their corresponding expression levels in total cell lysate were normalized to that of wild-type RAC1 (for the RAC1 mutants) or RAC2 (for the RAC2 mutants) and are shown at the bottom.
Other Transforming Mutations of RAC1 and RAC2.
We next searched for other transforming mutations of RAC proteins. Human RAC1, RAC2 (GenBank accession no. NM_002872.3), and RAC3 (GenBank accession no. NM_005052.2) cDNAs were isolated from 40 cancer cell lines (Table S2), and their nucleotide sequences were determined by Sanger sequencing, resulting in the discovery of RAC1(P29S), RAC2(P29Q), and RAC2(P29L) in the breast cancer cell line MDA-MB-157, the CML cell line KCL-22, and the breast cancer cell line HCC1143, respectively (Fig. S1 and Table S3). Further searching for RAC1, RAC2, and RAC3 mutations in the COSMIC database of cancer genome mutations (Release V59; http://cancer.sanger.ac.uk/cancergenome/projects/cosmic) revealed various amino acid substitutions detected in human tumors, namely RAC1(P29S), RAC1(C157Y), RAC1(P179L), RAC2(I21M), RAC2(P29L), RAC2(D47Y), and RAC2(P106H) (Table S3). Importantly, all of these RAC1 and RAC2 mutations identified in clinical specimens were confirmed to be somatic, given that the corresponding mutations were absent in the genome of paired normal cells.
To examine the transforming potential of these various RAC1 and RAC2 mutants, we expressed each protein in 3T3 and MCF10A cells and evaluated anchorage-independent growth. Whereas the wild-type form of RAC2 did not transform 3T3 or MCF10A cells, growth in soft agar was apparent for 3T3 cells expressing RAC1(P29S), RAC1(C157Y), RAC2(P29L), or RAC2(P29Q), but not for those expressing RAC1(P179L), RAC2(I21M), RAC2(D47Y), or RAC2(P106H) (Fig. 1A). Of interest, colony number in the assay varied substantially in a manner dependent on the type of amino acid substitution as well as on cell type. RAC1(C157Y), for example, yielded fewer colonies in soft agar compared with the other transforming mutants. Furthermore, RAC1(P29S), which was identified in a breast cancer cell line, generated a larger number of colonies with MCF10A cells than with 3T3 cells. Conversely, RAC1(N92I), which was identified in a fibrosarcoma cell line, yielded a larger number of colonies with 3T3 cells than with MCF10A cells. The oncogenic activity of RAC1(P29S), RAC1(N92I), RAC1(C157Y), RAC2(P29L), and RAC2(P29Q) mutants was further confirmed with a tumorigenicity assay in nude mice (Fig. 1B), with the activity of RAC1(N92I) being the most pronounced with regard to the transformation of 3T3 cells in this assay.
The colony number in soft agar for 3T3 cells expressing NRAS(Q61K) was fewer than that for the cells expressing oncogenic RAC1 or RAC2 mutants (Fig. 1A), whereas expression of these small GTPases was readily confirmed in 3T3 (Fig. S2). Interestingly, s.c. tumors from the same 3T3 cells expressing NRAS(Q61K) grew more rapidly than tumors expressing the RAC1/RAC2 mutants (Fig. 1B), indicating that the measured intensity of the transforming potential of GTPases may vary in a dependent manner on assay systems.
To examine whether such oncogenic potential is linked directly to the activation of RAC1 or RAC2, we investigated the activity of the mutant proteins with the use of a luciferase reporter plasmid that selectively responds to intracellular signaling evoked by RHO family GTPases (16). In concordance with the data from the soft agar and tumorigenicity assays, only the transforming mutants of RAC1 and RAC2 yielded a substantial level of luciferase activity in transfected HEK293T cells (Fig. 1C).
Activated RAC1 or RAC2 would be expected to be loaded with GTP. We therefore examined the GTP-binding status of the RAC1 and RAC2 oncoproteins with the use of a pull-down assay based on the p21-binding domain (PBD) of PAK1. All of the transforming RAC1 and RAC2 mutants were found to exist preferentially in the GTP-bound state (Fig. 1D), indicative of their constitutive activation. Furthermore, these RAC1 and RAC2 mutants induced marked reorganization of the actin cytoskeleton in 3T3 cells, resulting in the accumulation of polymerized actin in ruffles at the plasma membrane (Fig. 2).
Fig. 2.

Actin reorganization induced by the RAC1/RAC2 mutants. 3T3 cells infected with retroviruses encoding enhanced green fluorescent protein (EGFP) as well as wild-type or mutant forms of RAC1 or RAC2 were stained with Alexa Fluor 594-labeled phalloidin to visualize actin organization (Left image of each pair). The same cells were also examined for EGFP fluorescence (Right image of each pair). (Scale bars, 20 µm.)
RAC1 and RAC2 as Therapeutic Targets.
Given that NRAS(Q61K) is also known to transform 3T3 cells (17) (Fig. 1A), our data show that HT1080 cells harbor two independent oncogenic GTPases. We therefore examined whether RAC1(N92I) or NRAS(Q61K) is the principal growth driver in this sarcoma cell line. Among several small interfering RNAs (siRNAs) designed to attenuate the expression of RAC1 or NRAS, we selected two independent siRNAs that specifically target each mRNA (Fig. 3A). Whereas transfection of HT1080 cells with either NRAS siRNA resulted in a moderate inhibition of cell proliferation under the presence of 10% (vol/vol) FBS, that with either RAC1 siRNA almost blocked cell growth (Fig. 3B). Transfection with an NRAS siRNA in addition to either RAC1 siRNA did not result in an additional effect on cell proliferation (Fig. 3B). Similar data were observed in a culture with 1% (vol/vol) FBS (Fig. S3A) or under FBS-free conditions (Fig. S3B). To further examine the effects of silencing RAC1/NRAS, we quantitated cell cycle distribution of HT1080 transfected with siRNAs against either RAC1 or NRAS. As shown in Fig. S4A, DNA synthesis was equally suppressed by the knockdown of RAC1 or NRAS. Interestingly, however, CASP3/CASP7 activity (a surrogate marker for apoptosis) was markedly induced only by RAC1 depletion (Fig. S4B). Therefore, RAC proteins are likely to provide RAS-independent cell survival signals, which is supported by the fact that, even under FBS-free conditions, RAC1 depletion has more antiproliferative effects in HT1080 than NRAS depletion (Fig. S3B). These data show that active RAC1 may be the essential growth driver in HT1080 cells and is therefore a potential therapeutic target. Furthermore, our data suggest that oncogenic RAS proteins may require additional transforming hits to give rise to full-blown cancer.
Fig. 3.
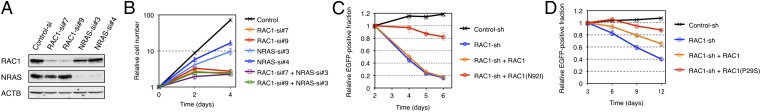
Oncogenic RAC proteins as therapeutic targets. (A) HT1080 cells were transfected with control, RAC1, or NRAS siRNAs; lysed; and subjected to immunoblot analysis with antibodies to RAC1, NRAS, or ACTB (loading control). (B) HT1080 cells were transfected with control, RAC1, or NRAS siRNAs, as indicated, and cultured under the presence of 10% (vol/vol) FBS. Cell number was counted at the indicated times after the onset of transfection. Data are means ± SD from three independent experiments. (C) HT1080 cells were infected with a retrovirus encoding green fluorescent protein (EGFP) as well as a control or RAC1 shRNA. They were also infected with a retrovirus encoding shRNA-resistant wild-type RAC1 or RAC1(N92I), as indicated. The number of EGFP-positive cells was determined by flow cytometry after culture of the cells for the indicated times, and the size of the EGFP-positive fraction relative to that at 2 d was calculated. Data are means ± SD from three independent experiments. (D) MDA-MB-157 cells were infected with a retrovirus encoding EGFP as well as a control or RAC1 shRNA. They were also infected with a retrovirus encoding shRNA-resistant wild-type RAC1 or RAC1(P29S), as indicated. The number of EGFP-positive cells was determined by flow cytometry after culture of the cells for the indicated times, and the size of the EGFP-positive fraction relative to that at 3 d was calculated. Data are means ± SD from three independent experiments.
We next infected HT1080 cells with a retrovirus expressing a short hairpin RNA (shRNA) targeted to RAC1 mRNA. Expression of the RAC1 shRNA markedly suppressed cell growth, whereas restoration of shRNA-resistant RAC1(N92I) expression reversed this effect (Fig. 3C and Fig. S5), showing that the effect of the RAC1 shRNA was not an off-target artifact. Forced expression of shRNA-resistant wild-type RAC1 failed to reverse the inhibitory effect of the RAC1 shRNA on cell growth, indicating that growth suppression by the shRNA was due to depletion of the N92I mutant, not to that of the wild-type protein. We performed similar experiments with the breast cancer cell line MDA-MB-157, which harbors RAC1(P29S). Again, the RAC1 shRNA inhibited cell growth, and this effect was reversed to a larger extent by restoration of the expression of shRNA-resistant RAC1(P29S) than by forced expression of the wild-type protein (Fig. 3D and Fig. S5).
RAC1(P29S), RAC1(N92I), and RAC1(C157Y) Are Rapid-Cycling Mutants.
Oncogenic mutations at G12, G13, or Q61 of RAS proteins found in human tumors reduce the intrinsic GTPase activity of these proteins and thereby maintain them in the GTP-bound state (18, 19). On the other hand, an artificial F28L substitution in HRAS or the RHO family protein Cdc42Hs was shown to confer constitutive activity by accelerating the transition from the GDP-bound to the GTP-bound state without the involvement of an exogenous guanine nucleotide exchange factor (GEF) (20, 21).
To determine how transforming mutations of RAC1 results in constitutive activation of these proteins, we examined their affinity for GTP and GDP. Compared with wild-type RAC1, all of RAC1(P29S), RAC1(N92I), and RAC1(C157Y) was found to bind GTPγS (nonhydrolyzable GTP analog) rapidly in vitro, even without the addition of a GEF protein (Fig. 4A). Likewise, the dissociation of GDP from the mutant forms of RAC1 was greatly accelerated (Fig. 4B). On the other hand, the intrinsic GTPase activity of these mutants was similar to (for P29S and N92I) or slightly higher (for C157Y) than that of the wild-type protein (Fig. 4C). These data thus indicated that, in contrast to transforming RAS mutants associated with human cancer, RAC1(P29S), RAC1(N92I), and RAC1(C157Y) are fast-cycling mutants, for which the probability of being in the GTP-bound state is increased as the result of an increased rate of GDP dissociation, rather than as the result of a loss of GTPase activity.
Fig. 4.

Biochemical properties of RAC1 mutants. (A) Bacterially expressed and purified proteins of the wild-type, P29S, N92I, or C157Y mutant of RAC1 (5 pmol each) were incubated with [35S]GTPγS in the presence of 0.8 mM Mg2+, and the amounts of [35S]GTPγS-bound proteins were determined at the indicated times. (B) [3H]GDP dissociation from [3H]GDP-bound RAC1 proteins was initiated by the addition of unlabeled GTPγS in the presence of 0.8 mM Mg2+, and the amounts of [3H]GDP-bound proteins were determined at the indicated times. (C) RAC1 proteins were preloaded with [γ32P] GTP, and then GTP hydrolysis reactions were initiated by the addition of unlabeled GTP in the presence of 0.8 mM Mg2+. Pi released from the proteins was isolated and measured at the indicated times. (D) [35S]GTPγS dissociation from [35S]GTPγS-bound RAC1 proteins was initiated by the addition of unlabeled GTPγS in the presence of 0.8 mM Mg2+, and the amounts of [35S]GTPγS-bound proteins were determined at the indicated times. (E) Schematic representation of the structure of the GTP-binding pocket of human RAC1 (ID 1mh1 in the Protein Data Bank; www.pdb.org) with α-helices and β-sheets shown in magenta and orange, respectively. The GTP analog guanosine 5′-(β,γ-imido)-triphosphate (GppNp) and Mg2+ are depicted in red and green, respectively. D11, P29, N92, and C157 amino acid residues are in orange, blue, yellow and purple, respectively. The positions of switch I and switch II regions and the P-loop are also indicated.
Interestingly, dissociation of GTPγS was also accelerated only for RAC1(C157Y), but not for the wild-type, P29S, or N92I form of RAC1 (Fig. 4D). Thus, RAC1(C157Y) is a unique mutant in that both association and dissociation for GTP are accelerated, which may provide the molecular basis for its modest transforming potential compared with that of RAC1(P29S) or RAC1(N92I) (Fig. 1).
In the 3D structure of RAC1 (Fig. 4E), P29 is located in the switch I region, whereas C157 is positioned adjacent to the guanine ring of bound GTP. Substitution of these residues would thus likely affect the affinity of the protein for GDP or GTP (Fig. S6), a phenomenon that has been demonstrated recently for RAC1(P29S) (22). In contrast, N92 is located distant from the binding pocket for GDP/GTP, and so the structural mechanism by which the N92I substitution renders RAC1 constitutively active remains elusive (Fig. 4E and Fig. S6). Residue N92 is located close to D11 in the P-loop of RAC1, however (Fig. 4E and Fig. S7), and substitution with isoleucine at this position would abolish the interaction between the amino group of N92 and the carboxyl group of D11. It is thus possible that the N92I mutation affects the binding of GDP/GTP through an effect on the P-loop.
Discussion
We have here demonstrated the transforming potential of mutated RAC proteins. Our analysis of cell lines resulted in the identification of transforming mutants of RAC1 and RAC2, namely RAC1(N92I) and RAC2(P29Q), and we also revealed the transforming potential of the RAC1(P29S), RAC1(C157Y), and RAC2(P29L) mutants deposited the COSMIC database of cancer genome mutations (Release V59; http://cancer.sanger.ac.uk/cancergenome/projects/cosmic) (Table S3). In contrast, the soft agar assay did not reveal a transforming potential of the RAC1(P179L), RAC2(I21M), RAC2(D47Y), or RAC2(P106H) mutants found in the database, suggesting a possibility that they are “passenger mutations.” It may also be possible, however, that these mutants may still contribute to cancer development by modifying tumor properties (such as metastasis ability), given that they were somatically acquired and clonally selected in cancer.
An important finding of our study was that the oncogenic effects of RAC1(N92I) may be more pronounced than those of NRAS(Q61K), at least with regard to survival signals in HT1080 cells (Fig. 3B). It should be noted, however, that HT1080 expresses RAC1 almost exclusively among the RAC family proteins, whereas HRAS and KRAS are weakly expressed in addition to NRAS (Fig. S8). It is thus possible that the effects of NRAS knockdown in Fig. 3B may be partly complemented by the residual HRAS/KRAS proteins.
Paterson et al. previously isolated NRAS-attenuated subclones of HT1080 after treatment with an alkylating reagent (N-methyl-N′-nitro-N-nitrosoguanidine) and a subsequent culture with 5-fluorodeoxyuridine and 1-β-D-arabinofuranosylcytosine (23). Such subclones had a flat cell shape and a reduced ability for anchorage-independent growth. Likewise, we noted that transfection with NRAS siRNAs renders HT1080 a flatter shape (Fig. S9). As demonstrated in Fig. 3B and by Paterson et al. (23), however, such NRAS-depleted HT1080 was still viable and kept proliferation in vitro, suggesting the presence of other oncogene(s) in addition to NRAS(Q61K). Therefore, NRAS(Q61K) and RAC1(N92I) are likely to cooperate to fully transform this fibrosarcoma.
Regarding the coexistence of mutations within RAC family proteins and RAS-RAF-MAPK proteins, two studies independently reported recurrent P29S mutation of RAC1 in melanoma during the preparation of this article (22, 24). Of note, BRAF(V600E) was also detected in four of six and in two of seven of the RAC mutation-positive melanomas, respectively. These observations, together with our findings with HT1080 cells, thus indicate that activating mutations of RAC1 and those of the RAS-RAF–signaling pathway are not mutually exclusive.
Members of the RAC subfamily of GTPases show a high level of sequence identity in humans. The amino acid sequence of RAC1 is thus 92% identical to that of RAC2 or RAC3. Furthermore, all of the amino acid residues of RAC1 or RAC2 found to be mutated in cancer (Table S3) are completely conserved among RAC1, RAC2, and RAC3. Thus, transforming RAC3 mutants with similar nonsynonymous mutations may also exist in human cancer, although such mutations were not detected in the current screening. Of interest, none of the frequent mutation sites in RAS family proteins (G12, G13, and Q61) were found to be affected in RAC1 or RAC2, although an artificial G12V mutant of RAC1 did manifest constitutive GTP loading and transforming potential. Given that RAC proteins perform intracellular functions (such as orchestration of the actin cytoskeleton) that are distinct from those of RAS family members, RAC-driven activation of specific intracellular pathways may be advantageous for cancer development in vivo.
Given that we detected activation mutations of RAC1 or RAC2 in cell lines from sarcoma (HT1080), triple-negative breast cancer (MDA-MB-157 and HCC1143), and the blast crisis stage of CML (KCL-22), we performed deep sequencing of RAC1, RAC2, and RAC3 cDNAs with GAIIx for specimens of triple-negative breast cancer (n = 66), of RAC1 and RAC2 cDNAs for specimens of CML in blast crisis (n = 43), and of BCR-ABL1–positive acute lymphoblastic leukemia (n = 31), as well as of RAC1 cDNAs for specimens of sarcoma (n = 53). We failed, however, to detect any nonsynonymous mutations among these RAC cDNAs.
Our results have shown that RAC proteins have the potential to become oncogenic through amino acid substitution in a wide array of cancers. Although such RAC mutations may occur at a low frequency, the recent studies of Krauthammer et al. (22) and Hodis et al. (24) suggest that they may be enriched in melanoma (∼5%). Importantly, given that HT1080 cells are highly addicted to the increased activity of RAC1(N92I), the targeting of oncogenic RAC proteins or their downstream effectors with small compounds or RNAi may prove to be an effective approach to the treatment of cancer harboring such oncoproteins.
Materials and Methods
The human fibrosarcoma cell line HT1080 was obtained from American Type Culture Collection, and subjected to deep sequencing with GAIIx. Recombinant retrovirus expressing the wild-type or mutant forms of RAC1 or RAC2 was used to infect mouse 3T3 fibroblasts to examine its transforming potential. Detailed information for cDNA resequencing, transformation assays, biochemical analysis of RAC proteins, and RNAi are detailed in SI Materials and Methods.
Supplementary Material
Acknowledgments
This study was supported in part by a grant for Research on Human Genome Tailor-Made from the Ministry of Health, Labor, and Welfare of Japan; Grants-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science; and grants from The Yasuda Medical Foundation, The Sagawa Foundation for Promotion of Cancer Research, and The Mitsubishi Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. S.N. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1216141110/-/DCSupplemental.
References
- 1.Druker BJ, et al. IRIS Investigators Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 2.Soda M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 3.Shaw AT, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: A retrospective analysis. Lancet Oncol. 2011;12(11):1004–1012. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colicelli J. Human RAS superfamily proteins and related GTPases. Sci STKE. 2004;2004(250):RE13. doi: 10.1126/stke.2502004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010;1(1):2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaffee EM, Hruban RH, Canto M, Kern SE. Focus on pancreas cancer. Cancer Cell. 2002;2(1):25–28. doi: 10.1016/s1535-6108(02)00093-4. [DOI] [PubMed] [Google Scholar]
- 7.Wertheimer E, et al. Rac signaling in breast cancer: A tale of GEFs and GAPs. Cell Signal. 2012;24(2):353–362. doi: 10.1016/j.cellsig.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70(3):401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 9.Qiu RG, Chen J, Kirn D, McCormick F, Symons M. An essential role for Rac in Ras transformation. Nature. 1995;374(6521):457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]
- 10.Senger DL, et al. Suppression of Rac activity induces apoptosis of human glioma cells but not normal human astrocytes. Cancer Res. 2002;62(7):2131–2140. [PubMed] [Google Scholar]
- 11.Thomas EK, et al. Rac guanosine triphosphatases represent integrating molecular therapeutic targets for BCR-ABL-induced myeloproliferative disease. Cancer Cell. 2007;12(5):467–478. doi: 10.1016/j.ccr.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 12.Rasheed S, Nelson-Rees WA, Toth EM, Arnstein P, Gardner MB. Characterization of a newly derived human sarcoma cell line (HT-1080) Cancer. 1974;33(4):1027–1033. doi: 10.1002/1097-0142(197404)33:4<1027::aid-cncr2820330419>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 13.Ueno T, et al. High-throughput resequencing of target-captured cDNA in cancer cells. Cancer Sci. 2012;103(1):131–135. doi: 10.1111/j.1349-7006.2011.02105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall A, Marshall CJ, Spurr NK, Weiss RA. Identification of transforming gene in two human sarcoma cell lines as a new member of the ras gene family located on chromosome 1. Nature. 1983;303(5916):396–400. doi: 10.1038/303396a0. [DOI] [PubMed] [Google Scholar]
- 15.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30(3):256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 16.Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 1995;81(7):1159–1170. doi: 10.1016/s0092-8674(05)80020-0. [DOI] [PubMed] [Google Scholar]
- 17.Marshall CJ, Hall A, Weiss RA. A transforming gene present in human sarcoma cell lines. Nature. 1982;299(5879):171–173. doi: 10.1038/299171a0. [DOI] [PubMed] [Google Scholar]
- 18.Adari H, Lowy DR, Willumsen BM, Der CJ, McCormick F. Guanosine triphosphatase activating protein (GAP) interacts with the p21 ras effector binding domain. Science. 1988;240(4851):518–521. doi: 10.1126/science.2833817. [DOI] [PubMed] [Google Scholar]
- 19.Calés C, Hancock JF, Marshall CJ, Hall A. The cytoplasmic protein GAP is implicated as the target for regulation by the ras gene product. Nature. 1988;332(6164):548–551. doi: 10.1038/332548a0. [DOI] [PubMed] [Google Scholar]
- 20.Reinstein J, Schlichting I, Frech M, Goody RS, Wittinghofer A. p21 with a phenylalanine 28----leucine mutation reacts normally with the GTPase activating protein GAP but nevertheless has transforming properties. J Biol Chem. 1991;266(26):17700–17706. [PubMed] [Google Scholar]
- 21.Lin R, Bagrodia S, Cerione R, Manor D. A novel Cdc42Hs mutant induces cellular transformation. Curr Biol. 1997;7(10):794–797. doi: 10.1016/s0960-9822(06)00338-1. [DOI] [PubMed] [Google Scholar]
- 22.Krauthammer M, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44(9):1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paterson H, et al. Activated N-ras controls the transformed phenotype of HT1080 human fibrosarcoma cells. Cell. 1987;51(5):803–812. doi: 10.1016/0092-8674(87)90103-6. [DOI] [PubMed] [Google Scholar]
- 24.Hodis E, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.