

Abstract
The spindle assembly checkpoint (SAC) ensures correct chromosome segregation during mitosis by preventing aneuploidy, an event that is detrimental to the fitness and survival of normal cells but oncogenic in tumor cells. Deletion of SAC genes is incompatible with early mouse development, and RNAi-mediated depletion of SAC components in cultured cells results in rapid death. Here we describe the use of a conditional KO of mouse Mad2, an essential component of the SAC signaling cascade, as a means to selectively induce chromosome instability and aneuploidy in the epidermis of the skin. We observe that SAC inactivation is tolerated by interfollicular epidermal cells but results in depletion of hair follicle bulge stem cells. Eventually, a histologically normal epidermis develops within ∼1 mo after birth, albeit without any hair. Mad2-deficient cells in this epidermis exhibited abnormal transcription of metabolic genes, consistent with aneuploid cell state. Hair follicle bulge stem cells were completely absent, despite the continued presence of rudimentary hair follicles. These data demonstrate that different cell lineages within a single tissue respond differently to chromosome instability: some proliferating cell lineages can survive, but stem cells are highly sensitive.
Keywords: mouse models, consequences of aneuploidy, whole chromosome instability, epidermal stem cell biology
At each cell division, newly replicated chromosomes are evenly distributed between daughter cells. If this process goes awry, daughters inherit an aneuploid chromosome content. Aneuploidy causes early developmental abnormalities in mice and is a leading cause of mental retardation and miscarriage in humans (1, 2). At the cellular level, imbalance in chromosome numbers disrupts metabolic homeostasis, decreasing the rate of proliferation (3). The mitotic spindle assembly checkpoint (SAC) is a key component of the machinery that guards against aneuploidy. The SAC functions by ensuring that all kinetochores are properly attached to spindle microtubules before the onset of anaphase (4). Mad2 is a key signal transducer in the SAC pathway, shuttling between unattached kinetochores and the anaphase promoting complex. Germ-line deletion of Mad2, or of any other SAC gene, results in massive aneuploidy and early embryonic lethality. In cell lines, depletion of SAC components causes apoptosis (5). However, the vast majority of human cancers exhibit extensive aneuploidy, suggesting that aneuploidy can be tolerated by tumor cells in vivo (5–9). In this paper, we investigate the in vivo consequences of chromosomal instability by conditionally abrogating the SAC, using the epidermis as a model.
The mammalian skin is a highly organized and mitotic tissue consisting of layers of keratinocytes [interfollicular epidermis (IFE)] interspersed with hair follicles. In the IFE, keratinocytes divide in the basal layer and subsequently differentiate into stratified cells while migrating toward the surface of the skin. Hair follicles harbor several stem cell populations essential for hair formation and capable of repopulating the IFE after wounding (10). In normal conditions, homeostasis within the skin is maintained by progenitor cells that reside in the basal layer of the IFE (11–13).
Results
To study the consequences of induced aneuploidy in the skin, we generated a conditional allele of Mad2 in which loxP sites flank exons 2 and 5, enabling Cre-mediated deletion of virtually the entire Mad2 coding region. The structure of the deleted locus is identical to that of the conventional Mad2 deletion we have previously shown to be embryonic lethal and eliminates detectable Mad2 mRNA and protein (14). Mice carrying the loxP-Mad2 allele (Mad2f) were intercrossed with Keratin14-Cre (Krt14-Cre) transgenic mice (15) to generate animals in which Mad2 was selectively deleted from multiple epidermal lineages. Mad2f/f, Krt14-Cre+ mice were born at normal Mendelian ratios, but unlike WT littermates, which acquired hair within 3–4 d after birth, mutant mice remained completely devoid of hair (Fig. 1A; Fig. S1A). Other than hair loss, the epidermis of mutant mice appeared largely normal despite the complete absence of Mad2 RNA or protein in the epidermis (Fig. 1 B and C). However, 80% of Mad2f/f, Krt14-Cre+neonates died within the first month of life because of malnutrition that apparently arose from a combination of poor mothering and competition with control littermates for food. The remaining 20% of mice survived into adulthood (Fig. 1D).
Fig. 1.
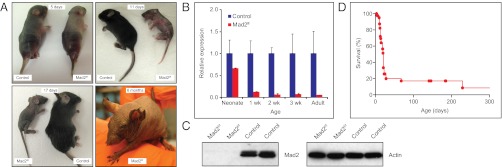
Mad2 deficiency provokes hair loss in mouse epidermis. (A) Macroscopic phenotype of Mad2 conditional KO (cKO) mice in neonates and adults. (B) Quantitative PCR comparing Mad2 RNA levels in Mad2f/f, Krt14-Cre+ (Mad2-deficient), and control epidermis in neonates and adults. Error bars indicate SE of the mean (SEM) of at least two biological replicates. (C) Western blot for Mad2 in Mad2-deficient and control epidermis. (D) Kaplan-Meier curve depicting survival of Mad2-deficient mice within the first year.
To determine whether Mad2 deficiency indeed resulted in aneuploidy, we harvested back skin epidermis from neonates (1 and 3 wk of age) and adults, generated single cell suspensions, and measured DNA content by flow cytometry. Whereas epidermal samples from control littermates exhibited a narrow G1 peak indicative of euploid DNA content, Mad2-deficient cells consistently exhibited a wider distribution with a significant increase in the number of cells with a 4n DNA content. This phenotype was most prominent at 3 wk of age and is suggestive of aneuploid cell division (Fig. 2A). To confirm this hypothesis we assayed chromosome number in the mutant epidermis using interphase FISH against chromosomes 4, 15, and 19 (Fig. 2 B and C; Fig. S1B; Table S1). We observed a two- to threefold increase in the number of cells trisomic for the assayed chromosomes compared with WT controls, confirming widespread aneuploidy in Mad2-deficient epidermis. We measured up to 10% of the WT IFE cells to be aneuploid, which is similar to aneuploidy rates observed in WT mouse embryonic fibroblasts (MEFs) but higher than aneuploidy rates in for instance hematopoietic cells (6–8, 16), suggesting that IFE cells tolerate aneuploidy better than some other cell types.
Fig. 2.

Krt14-Cre; Mad2 conditional mice display dramatic aneuploidy in the epidermis. (A) DNA content distribution in control and Mad2-deficient epidermal cells in neonates and adults. The CV is used as a measure of the variance of DNA content within the G1 peak. (B) Interphase FISH for chromosomes 15 (red) and 19 (green) on freshly isolated control and Mad2-deficient keratinocytes. (C) Percentage of cells showing more than two copies of chromosome 15 or 19 assessed by interphase FISH (aneuploidy index) in Mad2-deficient or control epidermis. Error bars indicate SE of the mean (SEM) values for at least three biological replicates. Individual values can be found in Table S1.
Histological examination of Mad2-null epidermis revealed hyperkeratosis during the first 3 wk of life, followed by the acquisition of a relatively normal morphology at later stages (Fig. 3A). Consistent with hyperkeratosis, we observed that some keratinocytes in the Mad2-null epidermis in neonates coexpressed the basal marker Krt14 along with the differentiation marker Krt10. Expression of these markers is normally mutually exclusive, with proliferating basal cells exhibiting Krt14 staining and cells in the stratified, differentiated layer Krt10 staining (Fig. 3B), but coexpression can occur in hyperproliferative epidermis (17). Hair follicles appeared abnormal at all ages, explaining the persistent absence of hair. In agreement with the increased cellularity of the epidermis, we observed an increase in proliferation in the basal layers of the IFE and in hair follicles (Fig. S1C). However, both the pattern of Krt10/Krt14 and levels of Ki67 staining returned to normal in the subset of Mad2f/f, Krt14-Cre+ animals that survived until adulthood.
Fig. 3.

Mad2 deficiency provokes a wave of hyperkeratosis in neonates, which largely normalizes in adults. (A) H&E staining of epidermal samples at various time points showing epidermal morphology with hyperkeratotic features in neonates. (B) Hyperkeratotic epidermis shows signs of increased differentiation (evidenced by thickened Krt10 positive epidermis).
Following a wave of cell proliferation at postnatal week 2, we observed increasing levels of apoptosis in hair follicles and the IFE; this continued through postnatal week 3 (Fig. S1D). We also observed infiltration of immune cells into the underlying dermis, suggesting inflammation. Such inflammation can arise if the barrier function of the skin is compromised, thereby allowing microorganisms to penetrate into the dermis. However, when we tested epidermal barrier integrity in mutant mice, we observed that the skin barrier was intact (Fig. S2), implying that immune infiltration was a consequence of abnormal mitosis and cell death. We conclude that Mad2 deficiency provokes aneuploidy and apoptosis in IFE and hair follicle cells and that this promotes a temporary inflammatory reaction in the dermis, explaining the hyperkeratosis.
To analyze the responses of epidermal cells to Mad2 deletion, we isolated mRNA from samples of epidermis [including both hair follicle (HF) and IFE] of newborns at 3, 6, 13, and 21 d after birth and of adults and then compared the transcript profiles of Mad2-deficient skin with Cre− Mad2-positive littermates using Illumina microarrays (Fig. 4A). We next extracted all probes that were significantly deregulated (P < 0.05) at two or more time points. To assess the quality of the microarray data, we also validated a selection of outlier genes (Fig. S3A) by qPCR, in all cases confirming our microarray results (Fig. S3B). Data from Mad2-null 3-d-old neonates were excluded from the analysis because transcript profiles were very similar to aged-matched controls, presumably because Mad2 transcripts had not completely disappeared yet [Fig. 1B, postnatal day (P)3]. The analysis of days 6–21 and adult mice generated a set of 1,619 down-regulated and 1,347 up-regulated genes (Dataset S1), which we processed to recover enriched biological pathways based on Wiki pathways and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (using Bonferroni-corrected P < 0.05) and gene ontologies using Webgestalt (18) (Fig. 4B; Table S2; Datasets S2 and S3). This analysis revealed significant up-regulation of genes involved in apoptosis and inflammation (most clearly at days 6 and 13) and the appearance of an aneuploidy fingerprint involving enrichment for genes and pathways known to be altered by aneuploidy. This fingerprint was first described in yeast strains and MEFs carrying supernumerary chromosomes and involves a substantial deregulation of genes that play a role in basic metabolic processes (19, 20). We now show that this response also occurs in vivo, because our dataset showed enrichment for similar pathways in Mad2-deficient skin: up-regulation of metabolic pathways and endocytosis and down-regulation of cell cycle genes, mRNA processing genes, and genes encoding spliceosome components. The latter pathways were recently associated with the response to aneuploidy in other organisms as well (plants, yeast, and humans) (21).
Fig. 4.

Mad2 abrogation provokes a general aneuploidy response, keratinocyte differentiation, and depletion of HF stem cell markers. (A) Overview of microarray study with number of significantly deregulated probes and number of samples per time point. (B) Schematic overview of deregulated pathways grouped into four main responses: aneuploidy inflammation, aneuploidy fingerprint, keratinocyte differentiation, and stem cell and hair follicle markers.
We also observed significant up-regulation of genes involved in epidermal differentiation and epidermal growth factor receptor (EGFR) signaling, and down-regulation of genes involved in Sonic Hedgehog (Shh), Wnt signaling, and hair follicle development. This down-regulation suggests that in skin, aneuploidy leads to depletion of the stem cell compartment. To test this hypothesis, we quantified the levels of stem cell lineage markers Krt15, Lgr5, Lrig1, and Sox9 by qPCR. We observed substantial reductions in the levels of all four genes, showing that HF stem cells that normally reside in the bulge compartment of the hair follicle are depleted (Fig. 5A). We also labeled whole-mount tail epidermal samples with antibodies against Krt14 and α-6-integrin, which label the IFE, and Krt15 and CD34, which label bulge HF stem cells. This confirmed depletion of the bulge stem cell compartment as a result of Mad2 depletion, whereas the IFE cells survived abrogation of the SAC. We conclude that HF stem cells are specifically depleted in the skin of Mad2-null animals (Fig. 5B; Fig. S3C). The differential response between these two cell lineages is not the result of differential Mad2 expression, because FACS-sorted WT HF stem cells and IFE cells express similar levels of Mad2 as determined by qPCR (Fig. S4 A and B). We conclude that stem cells cannot survive Mad2 loss, whereas IFE cells can.
Fig. 5.
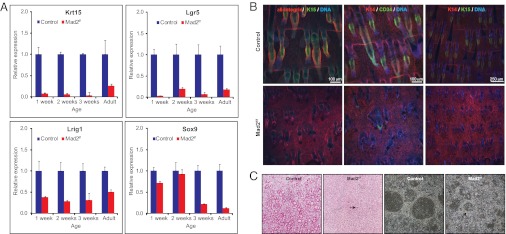
Aneuploidy provokes bulge HF stem cell depletion leaving IFE cells unaffected. (A) Quantitative PCR reveals a dramatic decrease of established HF stem cell markers Krt15, Lgr5, Lrig1, and Sox9. (B) Whole mount tail epidermis labeled for α-6 integrin or Krt14 (labeling IFE in red) and Krt15 or CD43 (both labeling bulge HF stem cells in green) showing HF stem cell depletion in vivo at P25. (C) Mad2-deficient primary keratinocytes do not form colonies in vitro. (Left) 1% Rhodamine-B–stained cultures, showing bright red colonies in control cultures. (Right) Phase contrast images, showing epidermal colonies in control cultures, but not in Mad2-deficient cultures. Note that cells were cultured on a MEF feeder layer. Arrows indicate one microcolony that emerged in a Mad2-deficient keratinocyte culture.
To test whether we could propagate Mad2 deficient keratinocytes in vitro, we transferred primary keratinocytes (not enriched for any epidermal cell lineage) from WT and Mad2-deficient back skin into tissue culture (22) and propagated these cells for 3 wk. Whereas control cultures contained many colonies that grew to confluence, no viable colonies were present in mutant cultures (Fig. 5C) despite having plated similar numbers of viable cells from mutant and control animals (viability was assessed by trypan blue staining; Fig. S4 C and D). These findings are in line with earlier findings that Mad2 deficiency provokes rapid cell death in vitro (5) and, even more important, emphasize that the in vivo response to chromosomal instability is fundamentally different from the in vitro response.
Discussion
Chromosomal instability (CIN) and the resulting aneuploidy are hallmarks of cancer cells, yet they are detrimental to untransformed cells in vitro, where they result in decreased proliferation capacity, disrupted cell physiology, and ultimately when CIN is very severe, cell death (5, 19, 20, 23). In this study, we report the consequences of SAC abrogation and the resulting aneuploidy in vivo. To circumvent the embryonic lethality associated with SAC inactivation through the germ line (6–8), we created a conditional KO allele for Mad2, a key player in the SAC and used this allele to abrogate the SAC in the epidermis. Remarkably, we found that Mad2 deletion is surprisingly well tolerated by IFE cells, resulting in mice with a functional Mad2-deficient epidermis, albeit without hair. The ability of IFE cells to tolerate Mad2 loss is not a reflection of their limited proliferative potential and reduced requirement for mitosis, because basal cells that contribute to the IFE are at least equally proliferative in Mad2 KO animals compared with controls, and they can generate all of the structures in the adult skin with the exception of functional hair follicles. Moreover, the majority of IFE cells divide asymmetrically, producing one proliferating and one differentiated cell (24), further arguing that a subpopulation of IFE cells is highly proliferative. Last, we show that Mad2-deficient IFE cells are highly aneuploid, arguing that they must have undergone several rounds of cell division in the absence of a functional SAC.
However, cell survival does not simply imply that aneuploidy is inconsequential for IFE cells: we observe significant changes in gene transcription including up-regulation of metabolic pathways and down-regulation of mRNA processing, changes that have previously been proposed to be part of an aneuploidy signature in yeast and cultured murine cells. Presumably, this aneuploidy fingerprint is caused by a stress response induced by the burden of extra transcripts and proteins from the supernumerary chromosomes (19, 20), and our data provide evidence that this response also occurs in vivo.
However, although SAC deficiency is well tolerated in IFE cells, we found that it is incompatible with survival of HF stem cells, resulting in the complete absence of bulge stem cells in Mad2-deficient hair follicles. This observation suggests that different cell lineages exhibit different responses to aneuploidy in vivo. Indeed, there is increasing evidence that some somatic cell lineages can tolerate high levels of aneuploidy. For instance, in the healthy brain, 1 in 10 neurons is reported to be aneuploid, with increasing rates upon aging and in pathologies such as Alzheimer’s disease (25–27). Furthermore, we observe up to 10% of aneuploid cells in normal mouse epidermis. On the other hand, other cell lineages (e.g., lymphocytes) show very little aneuploidy in vivo (6–8, 28).
What can explain this differential response toward Mad2 depletion? First, different cell lineages might exhibit different sensitivities toward apoptosis, with stem cells being more sensitive to apoptosis than more differentiated dividing cells. Alternatively, the signaling pathways that transduce the aneuploidy-induced stress response are more active in stem cells, thus triggering a stronger stress response and thereby driving them into apoptosis. A third possible explanation is that IFE cells spend more time in prometaphase than HF stem cells, providing them with more time to biorient their chromosomes properly and making them less dependent on a functional SAC. In this scenario, SAC inactivation would provoke more dramatic aneuploidy in HF stem cells than in IFE cells, specifically driving HF stem cells to cell death.
We speculate that aneuploidy specifically induces apoptosis in stem cells through a specific unknown mechanism that is absent, or less active, in other proliferating cells in the epidermis. This stem cell apoptosis triggers the marked inflammatory response observed in the skin of Mad2 KO mice, possibly as a consequence of the accumulation of cell debris. The inflammatory response then causes a wave of proliferation and apoptosis in neonates, explaining the hyperkeratosis and aberrant K14 expression (17). The inflammatory response dampens over time, ultimately allowing the appearance of a relatively normal adult epidermis. Although we were able to carefully map the response to aneuploidy in IFE cells, our current model precluded us from performing a similar analysis for HF stem cells, as all bulge stem cells had disappeared before we could isolate sufficient numbers. Therefore, we are now developing a unique mouse model in which Mad2 inactivation can be triggered in adults, which will allow us to isolate bulge stem cell and IFE cell populations directly following Mad2 inactivation, enabling us to identify the mechanism underlying the differential responses to Mad2 deletion–induced aneuploidy.
Altogether, we show that full SAC abrogation can be tolerated in vivo, despite a robust change in cell metabolism and despite the resulting aneuploidy. Up to two-thirds of all human cancers are aneuploid (29). It is likely that aneuploid cancer cells use similar pathways as IFE cells to cope with this aneuploidy. Our findings are a first step toward identifying such pathways by identifying the skin as a tissue that can cope with remarkable levels of aneuploidy and by carefully mapping the responses to aneuploidy in this tissue. Importantly, we found that the surviving IFE cells exhibit an aneuploidy-induced stress response in vivo, which might have a large impact on the future treatment of aneuploid cancers, because this stress response could be exploited to specifically target aneuploid tumor cells (30).
Materials and Methods
Animals and Keratinocyte Culture.
Animal protocols were approved by the Home Office (UK) and specified in the Home Office Project Licence. Mad2 conditional mice were generated by introducing LoxP sites surrounding exons 1 and 5, which when combined with Cre recombinase creates the same full KO as described previously (14). Krt14-Cre mice are described elsewhere (15). For genotyping, mice were ear clipped, and DNA was isolated using ear lysis buffer (Viagen). PCR primers are provided in Table S3, and primers were optimized for annealing temperatures at 60 °C. At selected time points, mice were killed, and back skin samples and tail samples were harvested for paraffin embedding or further processing. For further processing, subcutaneous fat was removed from the dermal side, and skin samples were incubated floating with the dermal side on Trypsin overnight at 4 °C. Epidermis was separated from the dermis using a scalpel, and epidermal samples were then prepared for interphase FISH, mRNA isolation, flow cytometry, or keratinocyte culture. In case of tail samples, skin was stripped from the bone and incubated in 5 mM EDTA for 1 h at 37 °C. The epidermis was then peeled from the dermis and fixed in 2% (vol/vol) paraformaldehyde for 1 h at room temperature. Keratinocytes for in vitro culture were harvested under sterile conditions and processed as described (22). Cell numbers were measured using an automated cell counter (Invitrogen) and viability by determining Trypan blue penetration.
Interphase FISH.
For interphase FISH, epidermal samples were minced and filtered through a 40-µm filter (Falcon) to obtain single cells. Cells were fixed in methanol/glacial acid after swelling in 0.075 M KCl for 30 min at 37 °C. Cells were dropped on slides, dehydrated, and treated with 0.1 mg/mL RNaseA (Sigma) in 2× SSC for 30 min at 37 °C, followed by 0.01% Pepsin (Sigma) treatment in 0.01 M HCl for 3 min at room temperature. Slides were then baked at 65 °C for 1 h and denatured at 63 °C for 80 s in 70% (vol/vol) formamide in 0.6× SSC. For probe preparation, the following bacterial artificial chromosomes (BACs) were used: RP23-341B14, RP23-32C12, RP23-463N9, RP23-24E11, RP23-67O11, and RP23-69G24 (recognizing chromosome 4); RP24-84H16, RP24-160M22, and RP24-160H6 (recognizing chromosome 15); and RP24-144K17, RP23-183J2, and RP23-292H23 (recognizing chromosome 19). Probes were labeled with green dUTP (Abott) or Texas red chromotide (Invitrogen) and hybridized at 37 °C overnight in hybridization buffer [62.5% formamide (Sigma), 2× SSC, 10% (wt/vol) dextran sulfate (Sigma), 0.5 M phosphate buffer, pH 7.4] containing 1 µL mouse Cot 1 DNA (Invitrogen) and 1.5 µL of each probe. Slides were mounted in the presence of DAPI-containing mounting media (Vectashield) following washing steps in 50% (vol/vol) formamide and 1× SSC and analyzed on a IX81 Olympus widefield microscope. Chromosome numbers (events per cell) were counted on an IX81 Olympus microscope using Excellence Software and scored aneuploid in the case of three or more events per cell by hand. Statistical analysis was performed in Microsoft Excel to calculate the SD of the mean values, and GraphPad Prism was used to calculate P values (t test).
Barrier Assay.
For barrier assays, neonates were killed and fixed in increasing series of ice-cold methanol in water. Next, carcasses were incubated in 0.1% toluidine blue (Sigma), and excess dye was washed away by three PBS washes.
Immunohistochemistry, Flow Cytometry, and Antibodies.
For DNA content analysis, freshly isolated epidermal cells were fixed in 70% (vol/vol) ethanol overnight and stained in 10 µg/mL propidium iodide (Invitrogen) in PBS in the presence of 0.1 mg/mL RNaseA (Sigma). For IFE and HF stem cells sorts, fresh keratinocytes were isolated from back skin and labeled with antibodies following sorting. Western blots were performed using standard protocols. For immunostaining of paraffin sections, slides were dewaxed, and epitopes were retrieved by boiling samples for 10 min in 10 mM sodium citrate. Samples were blocked using mouse serum (Vector Laboratories) and labeled overnight with primary antibodies in the presence of 1% BSA (Sigma), 10% (vol/vol) FCS (Invitrogen), and 0.01% TritonX100 (Sigma). Samples were then washed and incubated with secondary antibodies and mounted in the presence of DAPI-containing mounting media (Vectashield; Vector Laboratories) following confocal microscopy analysis (Zeiss). For whole mounts, epidermal samples were blocked [0.5% skimmed milk powder, 0.25% fish skin gelatin (Sigma), and 0.5% TritonX100 (Sigma) in 20 mM Hepes and 0.9% NaCl] for 1 h following overnight incubation at 4 °C and incubation with secondary antibodies. Samples were mounted in the presence of DAPI and analyzed by confocal microscopy (Leica). The primary antibodies used were Mad2 (BD), Actin (Cell Signaling), Krt14 (clone LL002; Abcam), Krt10 (Covance), Ki67 (Dako), CD34 (Ebioscience), α-6-integrin (Abcam), and Krt15 (clone LHK15). Secondary antibodies were Alexa 555– and 488–labeled anti-mouse and anti-rabbit antibodies (Invitrogen) and HRP-labeled goat anti-mouse (Cell Signaling). TUNEL assays on paraffin sections were performed using a Promega kit following the manufacturer’s protocol.
qPCRs and Microarrays.
RNA was isolated using the RNeasy kit (Qiagen). Biotin-labeled cRNA was synthesized using the Illumina totalprep RNA amplification kit (Ambion). Biotinylated cRNA was hybridized to llumina Sentrix Bead Chips and labeled with streptavidin-conjugated Cy3 (Amersham) according to the manufacturer’s protocol. Data were quantile normalized (31) following scanning and analyzed using Bioconductor lumi (32) and limma packages (33). Data were P value adjusted to yield a sorted list of differentially expressed genes (34) and sorted in chromosomal position or frequency of deregulation in Microsoft Excel. Enrichment analyses were performed using Webgestalt software (18) using Bonferroni correction for multiple testing. For diagrams in Fig. 4B, genes in deregulated pathways were downloaded using Webgestalt, and matching expression Log2 rations were extracted from Dataset S1 using Excel. Extracted values (values per category shown in Dataset S3) were then imported into Genesis software (35) to generate expression diagrams. All raw and normalized Illumina microarray data can be found at the NCBI Gene Expression Omnibus (GEO) under accession no. GSE42698. For qPCR reactions, cDNA was generated from 1 µg of total RNA (Superscript II; Invitrogen) and used as a template for qPCR (ABI PRISM 7700 Sequence Detector) in the presence of SYBR green (Invitrogen) to label the products. Actin was used for normalization. The average expression values and SD of the mean were calculated as indicated in the figure legends and compared with the expression values in control mice (normalized to a value of 1). Primer sequences are provided in Table S3.
Supplementary Material
Acknowledgments
We thank N. Park, R. Bautista, and R. Andrews for help with microarray processing and bioinformatical analysis; E. Langley, R. Banerjee, W. Bensz, Y. Zhu, C. Rocchi, P. Bakker, and F. Yang for help with interphase FISH; R. Bronson for the initial pathology; the animal technicians for help with animal husbandry; and S. Bruggeman for critically reading the manuscript. This work was supported by the Dutch Cancer Society, the European Molecular Biology Organization (fellowships to F.F.), Australian Research Council and National Health and Medical Research Council fellowships (to I.S.), National Institutes of Health Grants CA084179 and CA139980 (to P.K.S.), and The Wellcome Trust (A.B.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE42698).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1217388110/-/DCSupplemental.
References
- 1.Hassold T, et al. Human aneuploidy: Incidence, origin, and etiology. Environ Mol Mutagen. 1996;28(3):167–175. doi: 10.1002/(SICI)1098-2280(1996)28:3<167::AID-EM2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 2.Brown S. Miscarriage and its associations. Semin Reprod Med. 2008;26(5):391–400. doi: 10.1055/s-0028-1087105. [DOI] [PubMed] [Google Scholar]
- 3.Sheltzer JM, Amon A. The aneuploidy paradox: Costs and benefits of an incorrect karyotype. Trends Genet. 2011;27(11):446–453. doi: 10.1016/j.tig.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8(5):379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 5.Kops GJ, Foltz DR, Cleveland DW. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci USA. 2004;101(23):8699–8704. doi: 10.1073/pnas.0401142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foijer F, Draviam VM, Sorger PK. Studying chromosome instability in the mouse. Biochim Biophys Acta. 2008;1786(1):73–82. doi: 10.1016/j.bbcan.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schvartzman JM, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: Mouse modelling of the human disease. Nat Rev Cancer. 2010;10(2):102–115. doi: 10.1038/nrc2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holland AJ, Cleveland DW. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10(7):478–487. doi: 10.1038/nrm2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker DJ, Chen J, van Deursen JM. The mitotic checkpoint in cancer and aging: What have mice taught us? Curr Opin Cell Biol. 2005;17(6):583–589. doi: 10.1016/j.ceb.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 10.Watt FM, Jensen KB. Epidermal stem cell diversity and quiescence. EMBO Molec Med. 2009;1(5):260–267. doi: 10.1002/emmm.200900033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pincelli C, Marconi A. Keratinocyte stem cells: Friends and foes. J Cell Physiol. 2010;225(2):310–315. doi: 10.1002/jcp.22275. [DOI] [PubMed] [Google Scholar]
- 12.Jones PH, Simons BD, Watt FM. Sic transit gloria: Farewell to the epidermal transit amplifying cell? Cell Stem Cell. 2007;1(4):371–381. doi: 10.1016/j.stem.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 13.Blanpain C, Fuchs E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10(3):207–217. doi: 10.1038/nrm2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobles M, Liberal V, Scott ML, Benezra R, Sorger PK. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 2000;101(6):635–645. doi: 10.1016/s0092-8674(00)80875-2. [DOI] [PubMed] [Google Scholar]
- 15.Dassule HR, Lewis P, Bei M, Maas R, McMahon AP. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development. 2000;127(22):4775–4785. doi: 10.1242/dev.127.22.4775. [DOI] [PubMed] [Google Scholar]
- 16.Weaver BA, Cleveland DW. Does aneuploidy cause cancer? Curr Opin Cell Biol. 2006;18(6):658–667. doi: 10.1016/j.ceb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Gareus R, et al. Normal epidermal differentiation but impaired skin-barrier formation upon keratinocyte-restricted IKK1 ablation. Nat Cell Biol. 2007;9(4):461–469. doi: 10.1038/ncb1560. [DOI] [PubMed] [Google Scholar]
- 18.Zhang B, Kirov S, Snoddy J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33(Web Server issue):W741–W748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams BR, et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322(5902):703–709. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Torres EM, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317(5840):916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- 21.Sheltzer JM, Torres EM, Dunham MJ, Amon A. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci USA. 2012;109(31):12644–12649. doi: 10.1073/pnas.1209227109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jensen KB, Driskell RR, Watt FM. Assaying proliferation and differentiation capacity of stem cells using disaggregated adult mouse epidermis. Nat Protoc. 2010;5(5):898–911. doi: 10.1038/nprot.2010.39. [DOI] [PubMed] [Google Scholar]
- 23.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 24.Clayton E, et al. A single type of progenitor cell maintains normal epidermis. Nature. 2007;446(7132):185–189. doi: 10.1038/nature05574. [DOI] [PubMed] [Google Scholar]
- 25.Yurov YB, et al. Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS ONE. 2007;2(6):e558. doi: 10.1371/journal.pone.0000558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iourov IY, Vorsanova SG, Liehr T, Yurov YB. Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: Differential expression and pathological meaning. Neurobiol Dis. 2009;34(2):212–220. doi: 10.1016/j.nbd.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 27.Thomas P, Fenech M. Chromosome 17 and 21 aneuploidy in buccal cells is increased with ageing and in Alzheimer’s disease. Mutagenesis. 2008;23(1):57–65. doi: 10.1093/mutage/gem044. [DOI] [PubMed] [Google Scholar]
- 28.Peterson SE, et al. Normal human pluripotent stem cell lines exhibit pervasive mosaic aneuploidy. PLoS ONE. 2011;6(8):e23018. doi: 10.1371/journal.pone.0023018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duijf PH, Schultz N, Benezra R. Cancer cells preferentially lose small chromosomes. Int J Cancer. 2012 doi: 10.1002/ijc.27924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell. 2011;144(4):499–512. doi: 10.1016/j.cell.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang YH, et al. Normalization for cDNA microarray data: A robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30(4):e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du P, Kibbe WA, Lin SM. lumi: A pipeline for processing Illumina microarray. Bioinformatics. 2008;24(13):1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 33.Smyth GK. Limma: Linear Models for Microarray Data. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 34.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57(1):289–300. [Google Scholar]
- 35.Sturn A, Quackenbush J, Trajanoski Z. Genesis: Cluster analysis of microarray data. Bioinformatics. 2002;18(1):207–208. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.