

Abstract
AIM: To examine the clinical features and analyze prognostic factors in a prospective study of primary biliary cirrhosis (PBC) patients.
METHODS: From 1995 to 2010, PBC patients without hepatic decompensation seen at the Peking Union Medical College Hospital were enrolled. Clinical signs and manifestations (pruritus, persistent fatigue, jaundice and pain in the right hypochondrium), laboratory parameters (auto-antibodies for autoimmune hepatic disease, biliary and hepatic enzymes, immunoglobulin, bilirubin, and albumin) and imaging findings were recorded at entry and at specific time points during follow-up. Cox regression and Kaplan-Meier analyses, respectively, assessed the risk factors for hepatic decompensation and survival.
RESULTS: Two hundred and sixty-two PBC patients were enrolled with a median follow-up of 75.2 mo (range, 21-201 mo). The 240 patients were aged 51.5 ± 10.2 years at diagnosis and 91.6% were female. Two hundred and forty-five (93.5%) were seropositive for anti-mitochondrial antibodies. At presentation, 170 patients (64.9%) were symptomatic, while 96 patients (36.6%) had extra-hepatic autoimmune disease. During the follow-up period, 62 (23.7%) patients developed hepatic decompensation of whom four underwent liver transplantation and 17 died. The cumulative survival rate and median survival time were 83.9% and 181.7 mo, respectively. Cox regression analysis revealed that an incomplete ursodeoxycholic acid (UDCA) response or inconsistent treatment [P < 0.001; hazard risk (HR) 95%CI = 2.423-7.541], anti-centromere antibodies (ACA) positivity (P < 0.001; HR 95%CI = 2.516-7.137), alanine aminotransferase ratio (AAR) elevations (P < 0.001; HR 95%CI = 1.357-2.678), and histological advanced liver disease (P = 0.006; HR 95%CI = 1.481-10.847) were predictors of hepatic decompensation. The clinical features and survival of PBC in China are consistent with those described in Western countries.
CONCLUSION: Incomplete UDCA response or inconsistent treatment, ACA positivity, AAR elevations, and advanced histological stage are predictors of decompensation.
Keywords: Primary biliary cirrhosis, Risk factor, Hepatic decompensation, Survival, Ursodeoxycholic acid response, Anti-centromere antibodies, Histological stage
INTRODUCTION
Primary biliary cirrhosis (PBC) is a chronic cholestatic autoimmune liver disease that causes lymphocytic portal hepatitis and bile duct destruction, leading to cirrhosis and liver failure[1]. The annual incidence rates range between 0.7 and 49 cases per million persons, while prevalence rates range between 6.7 and 402 cases per million[2,3]. Currently, treatment with ursodeoxycholic acid (UDCA) at a dose of 13-15 mg/kg/d is the recommended first-line therapy.
According to clinical observations, the progression and prognosis of PBC vary significantly among patients, including a relatively slow progression in some patients, while others have a more rapid progression to hepatic failure[4,5]. Therefore, knowledge of the factors affecting disease progression and prognosis would be of great value in clinical management.
Several prognostic models have been developed for PBC. The most important is the Mayo score model, which has been widely used to predict the survival of PBC patients, and assess the timing of liver transplantation[6,7]. However, some patients are asymptomatic at the time of diagnosis, which limits the application of the Mayo model; and this model was not originally developed to assess disease progression in the early stages of PBC[8]. In terms of treatment, UDCA has been reported to delay histological progression and improve long-term prognosis[9]. Previous studies have shown that anti-gp210 and anti-centromere antibodies (ACA) were associated with severe disease course and poor prognosis. Bilirubin, prothrombin time, hypoalbuminemia and serum immunoglobulin (Ig) G have also been reported as prognostic markers[10-12]. However, these studies were mainly retrospective and had small sample sizes.
We aimed to assess the clinical features and risk factors for hepatic decompensation in a large prospective cohort study in China.
MATERIALS AND METHODS
Study population and data collection
Patients with PBC seen at the Peking Union Medical College Hospital (PUMCH) from 1995 to 2010 were recruited. The ethics committee at PUMCH approved the study, and written informed consent was obtained from all participants prior to enrollment. The diagnosis of PBC was made if at least two of the following three criteria were fulfilled: (1) elevated serum alkaline phosphatase (ALP) (at least 1.5 times the upper limit of normal); (2) the presence of anti-mitochondrial antibodies (AMAs) in serum; and (3) representative histological manifestations of portal area inflammation and bile duct injury. Early PBC was defined by seropositivity for AMA, and the presence of histological manifestations, but normal liver function[4]. The liver histology was graded according to the Ludwig classification[13]. Patients with liver decompensation, autoimmune hepatitis (AIH), viral hepatitis and other causes of liver damage were excluded. Patients with PBC/AIH overlap syndrome were identified according to the criteria proposed by Chazouillères et al[14]. In the current study, serum antinuclear antibody (ANA), AMA, anti-smooth muscle antibody, anti-liver kidney microsomal antibody, and anti-parietal cell antibody were tested. In patients who were ANA positive, the level of alanine aminotransferase (ALT), aspartate aminotransferase (AST), IgG and autoantibodies for AIH were assessed to exclude PBC/AIH. The extra-hepatic autoimmune diseases were diagnosed according to the generally accepted criteria.
The following information was recorded: (1) demographic features, clinical signs and manifestations of liver disease including pruritus, persistent fatigue, jaundice and pain in the right hypochondrium; (2) laboratory parameters, including ANA, AMA, ACA, ALT, AST, ALP, γ-glutamyl-transferase (GGT), total bilirubin (TBil), direct bilirubin (DBil), albumin (ALB), IgG, IgA and IgM; and (3) imaging findings: liver ultrasound or CT demonstrating morphologic changes of liver and spleen, and portal vein blood flow. UDCA at the dose of 13-15 mg/kg per day was prescribed for all patients.
Follow-up
Patients were followed up in the outpatient clinic, and data were collected at 3-mo intervals during the first year, 6-mo intervals or yearly thereafter. During each visit, the clinical signs, symptoms and laboratory parameters (AST, ALT, ALP, GGT, ALB, TBil, DBil, IgG, IgA, IgM, white blood cell, hemoglobin and platelet count) were assessed. Liver ultrasound was performed annually. Gastroscopy was carried out if necessary. If the patients had symptoms of hepatic decompensation (ascites, esophageal varices, variceal bleeding, hypersplenism, coagulant function abnormality, hypoproteinemia, and hepatic encephalopathy), they were permitted to visit doctors at any time or to go to emergency departments. After treatment for one year, patients whose ALP levels decreased by more than 40% of baseline values or returned to normal were defined as UDCA responders[15].
Outcome evaluation
Two clinical outcomes were studied: hepatic decompensation, and liver-related death. Hepatic decompensation was defined as the occurrence of severe functional damage of liver and one or more complications of liver cirrhosis[16]. We investigated the following features of decompensation: hypersplenism, ascites, esophageal varices (variceal bleeding), encephalopathy, hypoproteinemia, coagulant function abnormality, and spontaneous bacterial peritonitis. Figure 1 shows a diagram of the study design.
Figure 1.
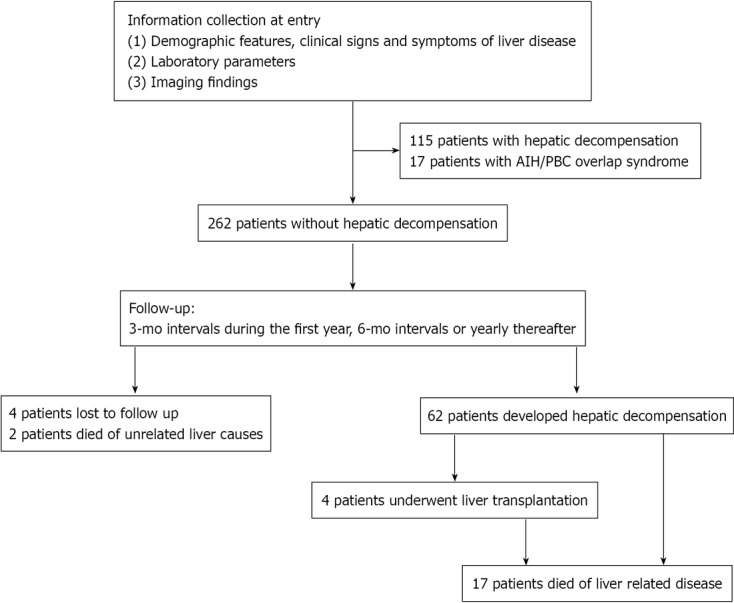
Diagram showing the study enrollment.
Statistical analysis
Data were expressed as median (range) or mean ± SD. Analysis was performed using SPSS 17.0 statistical software (Chicago, IL, United States). A two-sided P-value < 0.05 was considered statistically significant. Categorical variables were compared by the χ2 test while continuous variables were compared by the Student’s t-test or Mann-Whitney U test. Variables that were significant in univariate analysis were submitted to Cox regression analysis to predict the independent risk factors. Hazard risk (HR) and 95%CI were presented. Kaplan-Meier analysis was used to assess survival. Patients who were lost to follow-up or died of causes unrelated to liver failure were censored.
RESULTS
Clinical profiles of the study group at the entry
In this study, 262 patients with a mean age of 51.5 ± 10.2 years, including 240 (91.6%) women, were enrolled. The median duration of follow-up was 75.2 mo (range, 21-201 mo). The demographic and clinical features are shown in Tables 1 and 2.
Table 1.
Demographic and clinical laboratory characteristics of primary biliary cirrhosis patients at the time of entry (n = 262) n (%)
| Age (yr), mean ± SD | 51.5 ± 10.2 |
| Gender (female/male), n | 240/22 |
| The symptomatic | 170 (64.9) |
| Jaundice | 78 (29.8) |
| Pruritus | 69 (26.3) |
| Persistent fatigue | 63 (24.0) |
| Pain in the right hypochondrium | 40 (15.3) |
| UDCA responders | 190 (72.5) |
| Early PBC | 19 (7.3) |
| Extra-hepatic autoimmune diseases | 96 (36.6) |
| Sjögren's syndrome | 54 (20.6) |
| Autoimmune thyroid diseases | 25 (9.5) |
| CREST/systemic sclerosis | 8 (3.1)/3 (1.1) |
| Polymyositis/dermatomyositis | 8 (3.1)/1 (0.4) |
| Systemic lupus erythematosus | 4 (1.5) |
| Psoriasis | 4 (1.5) |
| Rheumatoid arthritis | 3 (1.1) |
| Takayasu’s arteritis | 2 (0.8) |
| Histological stages | 110 (42.0) |
| I-II | 97 (88.2) |
| III-IV | 13 (11.8) |
UDCA: Ursodeoxycholic acid; PBC: Primary biliary cirrhosis; PBC: Primary biliary cirrhosis; CREST: Comprehensive overview covers symptoms, causes, treatment of this autoimmune disorder.
Table 2.
Laboratory characteristics of primary biliary cirrhosis patients at the time of entry (n = 262)
| ANA | 181(69.1) |
| AMA | 245 (93.5) |
| ACA | 73 (27.9) |
| ALT (IU/L) | 91.0 (9-598) |
| AST (IU/L) | 86.2 (15-428) |
| AST/ALT | 1.10 (0.22-4.85) |
| ALP (IU/L) | 348.5 (21-1486) |
| GGT (IU/L) | 428.2 (9-2614) |
| ALB (g/L) | 40.7 (25-69) |
| TBil (μmol/L) | 28.05 (4.90-334.40) |
| DBil (μmol/L) | 14.39 (0.34-237.40) |
| IgG (g/L) | 16.81 (5.12-56.60) |
| IgA (g/L) | 3.15 (0.19-8.84) |
| IgM (g/L) | 4.66 (0.41-22.30) |
Continuous data were expressed as n (%) or median (range). ANA: Anti-nuclear antibody; AMA: Anti-mitochondrial antibody; ACA: Anti-centromere antibody; ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; ALP: Alkaline phosphatase; GGT: γ-glutamyl-transferase; ALB: Albumin; TBil: Total bilirubin; DBil: Direct bilirubin; Ig: Immunoglobulin.
On entry into the study, 170 (64.9%) patients presented with manifestations of liver disease, and 96 (36.6%) patients had extra-hepatic autoimmune disease. In addition, we enrolled 19 (7.3%) patients with early PBC. Two hundred and forty-five (93.5%) patients were seropositive for AMA. ANA and ACA were detected in 181 (69.1%) and 73 (27.9%) patients, respectively. Among 110 patients who underwent needle liver biopsies, 97 (88.2%) were at stages I-II, while 13 (11.8%) were at stages III-IV. One hundred and ninety (72.5%) patients received UDCA regularly and responded, while 46 patients (17.6%) took UDCA consistently, but failed to respond. Of the latter, eight patients terminated treatment on their own. Others (9.9%) did not take UDCA consistently, as prescribed.
Incidence of outcomes and survival
During the study period, 62 (23.7%) patients developed adverse events, and the median duration from diagnosis to hepatic decompensation was 57.4 mo (range, 7-151 mo). Among these patients, four (1.5%) patients underwent liver transplantation at the end of follow-up or before death, and 17 (6.5%) patients died of liver failure. The median duration from diagnosis to death was 63.6 mo (range, 29-112 mo). According to Kaplan-Meier analysis, the cumulative survival rate and estimated median survival time were 83.9% and 181.7 mo, respectively (Figure 2 and Table 3).
Figure 2.
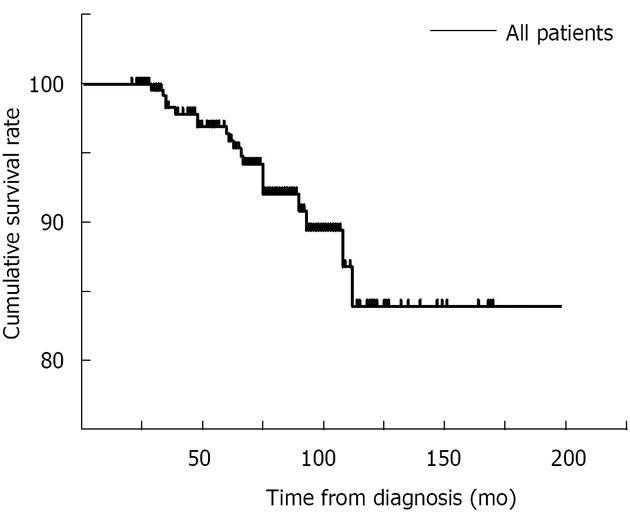
Survival curve of all patients in this cohort.
Table 3.
Complications of chronic liver disease in the primary biliary cirrhosis cohort (n = 262) n (%)
| Hepatic decompensation | 62 (23.7) |
| Portal hypertension | 62 (23.7) |
| Hypersplenism | 44 (16.8) |
| Esophageal varices | 47 (17.9) |
| Variceal bleeding | 35 (13.4) |
| Ascites | 43 (16.4) |
| Encephalopathy | 19 (7.3) |
| Coagulant function abnormality | 12 (4.6) |
| Hypoproteinemia | 35 (13.4) |
| Spontaneous bacterial peritonitis | 3 (1.1) |
| Liver transplantation | 4 (1.5) |
| Death-liver related | 17 (6.5)1 |
| Censor | 6 (2.3) |
| Lost to follow-up | 4 (1.5) |
| Death from causes unrelated to liver failure | 2 (0.8) |
One patient who underwent liver transplantation died.
Risk factors for hepatic decompensation
Possible risk factors were first identified by univariate analysis. Subsequently, all variables, except histological stage, that showed statistical differences in univariate analysis were introduced into the Cox regression model. Incomplete UDCA response or inconsistent UDCA treatment (P < 0.001; HR 95%CI = 2.423-7.541), ACA positivity (P < 0.001; HR 95%CI = 2.516-7.137), alanine aminotransferase ratio (AAR) elevations (P < 0.001; HR 95%CI = 1.357-2.678), TBil (P = 0.004; HR 95%CI = 1.002-1.009), and ALP (P = 0.008; HR 95%CI = 1.000-1.002) showed statistical significance in the model (Figure 3 and Tables 4 and 5). Although histological stage was not introduced into the model because of limited numbers, Cox regression analysis indicated that it was also a prognostic factor (P = 0.006; HR 95%CI = 1.481-10.847; Figure 3).
Figure 3.
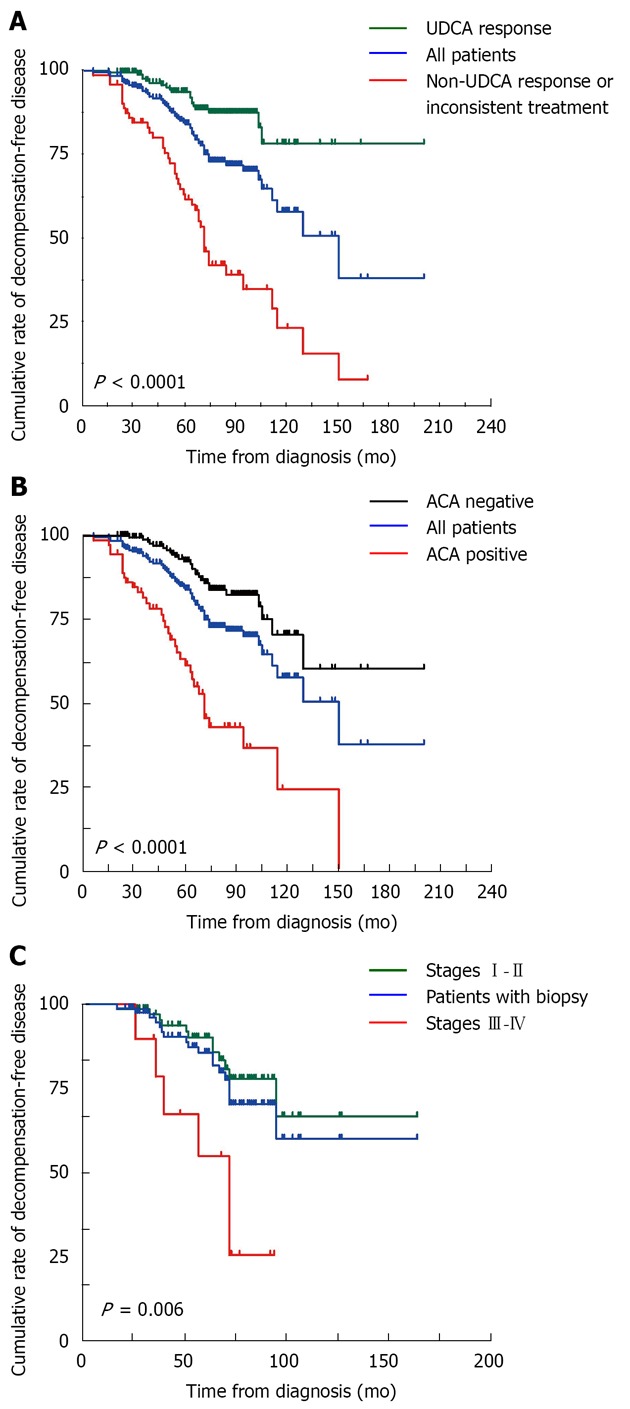
Cumulative rates of decompensation-free disease in primary biliary cirrhosis patients stratified by ursodeoxycholic acid treatment, anti-centromere antibody, and histological stage, respectively. A: Ursodeoxycholic acid (UDCA); B: Anti-centromere antibody (ACA); C: Histological stage.
Table 4.
Univariate analysis of possible risk factors for hepatic decompensation in primary biliary cirrhosis
| Decompensation free (n = 196) | Decompensation (n = 62) | P value | |
| Age at diagnosis (yr) | 50.2 | 55.6 | < 0.001 |
| Gender (female/male) | 178/18 | 59/3 | 0.275 |
| Symptoms | 60.20% | 80.65% | 0.003 |
| UDCA treatment | 85.20% | 30.65% | < 0.001 |
| ANA | 64.80% | 80.65% | 0.019 |
| AMA | 92.86% | 95.16% | 0.731 |
| ACA | 19.39% | 56.45% | < 0.001 |
| ALT (IU/L) | 89.6 | 96.6 | 0.109 |
| AST (IU/L) | 80.6 | 103.2 | < 0.001 |
| AST/ALT | 1.05 | 1.27 | 0.013 |
| ALP (IU/L) | 311.6 | 454.1 | < 0.001 |
| GGT (IU/L) | 395.4 | 524.3 | 0.001 |
| ALB (g/L) | 41.5 | 38.4 | < 0.001 |
| TBil (μmol/L) | 20.92 | 48.84 | < 0.001 |
| DBil (μmol/L) | 9.27 | 29.06 | < 0.001 |
| IgG (g/L) | 16.61 | 17.43 | 0.386 |
| IgA (g/L) | 2.99 | 3.62 | 0.004 |
| IgM (g/L) | 4.61 | 4.83 | 0.285 |
| Histological stage1 (I-II) | 92.22% | 66.67% | 0.008 |
| Early PBC | 9.70% | 0.00% | 0.023 |
| Extra-hepatic autoimmune disease | 37.24% | 37.10% | 0.983 |
n = 108. ANA: Anti-nuclear antibody; AMA: Anti-mitochondrial antibody; ACA: Anti-centromere antibody; ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; ALP: Alkaline phosphatase; GGT: γ-glutamyl-transferase; ALB: Albumin; TBil: Total bilirubin; DBil: Direct bilirubin; UDCA: Ursodeoxycholic acid; Ig: Immunoglobulin.
Table 5.
Cox regression analysis of risk factors for hepatic decompensation in primary biliary cirrhosis (n = 262)
| HR | HR 95%CI | P value | |
| Poor UDCA response or inconsistent treatment | 4.275 | 2.423-7.541 | < 0.001 |
| ACA positivity | 4.237 | 2.516-7.137 | < 0.001 |
| AAR | 1.906 | 1.357-2.678 | < 0.001 |
| TBil | 1.005 | 1.002-1.009 | 0.004 |
| ALP | 1.001 | 1.000-1.002 | 0.008 |
ACA: Anti-centromere antibody; ALP: Alkaline phosphatase; TBil: Total bilirubin; UDCA: Ursodeoxycholic acid; AAR: Alanine aminotransferase ratio; HR: Hazard risk.
DISCUSSION
PBC is a chronic autoimmune liver disease that has the potential to progress to cirrhosis and, eventually, hepatic failure. Although it is less common in East Asia[17], there is a rising frequency attributable to a more widespread awareness of this disease among physicians[18]. In this study, with a follow-up for 17 years, we identified the clinical profiles and risk factors for hepatic decompensation of PBC patients. To the best of our knowledge, this is the first prospective study with such a large number of patients in China.
In the current cohort, the demographic features and autoantibody profiles were consistent with those of previous studies[19]. Although PBC is an organ-specific disease, association with extra-hepatic autoimmune diseases is an important characteristic. The prevalence of such co-existing diseases was high (36.6%), the most common of which were Sjögren’s syndrome (SS) (20.6%) and autoimmune thyroid diseases (9.5%). These findings were similar to the results obtained by Marasini et al[20] in Italy and Silveira et al[21] in the United States.
Clinically, PBC patients can progress from an asymptomatic stage to a symptomatic stage because of liver damage[22]. In the current cohort, 64.9% patients had hepatic-related symptoms at diagnosis, which is higher than that reported by Prince et al[23], but lower than the results by Su et al[11]. In the current study, the most common manifestation was jaundice, which was different from other reports in which the most common manifestation was fatigue[22,24,25]. The median survival time for asymptomatic patients has been reported to be longer than that for symptomatic patients[26]. In addition, other studies indicated that asymptomatic patients had shorter life spans than people in the general population[26,27]. However, several studies have indicated that asymptomatic patients did not have a better prognosis[23,28].
In the current study, at the end of follow-up, sixty-tow (23.7%) patients developed hepatic decompensation. Among them, all had portal hypertension, and 17 died of liver failure. Only four patients underwent transplantation because of the enormous financial cost for patients, and limited medical resources. However, the survival rate of our patients was similar to those presented in other recent studies[29-31].
Although there has been controversy about efficacy, UDCA is currently accepted as the first-line drug for PBC. Several studies have shown that UDCA not only ameliorates laboratory indices, but also delays histological progression, and improves survival without transplantation[9,15,32-35], especially for patients at early histological stages[33]. In the current study, 72.5% patients took UDCA as prescribed and responded according to the Barcelona criteria; their prognoses were significantly better than those of non-responders, which is consistent with results of a previous study[9]. Therefore, the data suggest that all PBC patients should take UDCA regularly, but novel therapeutic options are needed for patients who do not respond to UDCA.
ACA had been observed in patients with various rheumatic disorders, including CREST syndrome (comprehensive overview covers symptoms, causes, treatment of this autoimmune disorder)[36], SS[37], Raynaud’s phenomenon[38], interstitial pneumonia in systemic lupus erythematosus[39], SS overlap syndrome[40] and 15%-30% PBC patients[41-44]. In the current study, ACA was detected in 27.9% patients and 56.5% of decompensated patients. It was found to be an independent prognostic factor for patients. Previous cross-sectional studies have also indicated that ACA was associated with the development of liver failure[45] and progression to portal hypertension in PBC[10]. However, Rigamonti et al[46] found that PBC patients with or without ACA had the same rate of liver-related death, while Parveen et al[47] reported that ACA was not correlated with either laboratory or histological manifestations.
Elevation of AAR may be explained by a reduction in AST clearance, and by mitochondrial injury in severe liver disease[48]. A previous study showed that AAR exhibited modest correlations to Mayo and Child scores for evaluating the severity of PBC, and it was higher in histological stages III-IV than I-II disease. Patients with an AAR of 1 or less had better prognoses than their counterparts[49]. These results are consistent with the current data.
The beneficial value of liver biopsy in patients with PBC is controversial because the pattern of cirrhosis in PBC is irregular, and sampling error may lead to misinterpretation[50]. However, several studies have indicated a relationship between histology and clinical profile. AST and bilirubin were positively related to portal fibrosis for the AMA-positive patients[51]. Prognosis was found to be correlated with the histological stages of hepatic fibrosis, cholestasis and periportal cell necrosis[52]. Cirrhosis was found to have developed within four years in 31% and 50% of patients who were initially at stage I and stage II, respectively[53]. Cox analysis demonstrated that histological stage was a predictor of liver decompensation.
Although TBil and ALP were included in our model, the HR approached one. Therefore, they had a relatively low correlation with survival compared with other factors, and their value for this purpose still needs to be confirmed by other, large long-term studies.
There are some limitations of the current study: (1) only patients without decompensation were enrolled, which resulted in the expected better survival rates compared to studies that included patients with decompensation; and (2) this was a single center study with a relatively short follow-up. Consequently, these risk factors still need to be confirmed further.
In summary, the clinical features and survival of patients with PBC in China are consistent with those described in Western countries. Incomplete UDCA response or inconsistent treatment, ACA positivity, higher AAR, and advanced histological stage are risk factors for hepatic decompensation.
ACKNOWLEDGMENTS
We thank Xu-Hua Shi and Li-Xia Gao for the collection of data and patient information.
COMMENTS
Background
Primary biliary cirrhosis (PBC) is a chronic cholestatic autoimmune liver disease with a rising frequency, attributable to a widespread awareness from physicians. The progression and prognosis of PBC vary significantly among patients. However, the clinical features and risk factors for hepatic decompensation have not yet been well-documented in a large prospective cohort study in China.
Research frontiers
In the current study, the authors identified the clinical profiles and risk factors for hepatic decompensation of PBC patients. In the authors’ view, this is the first prospective study with such a large number of patients in China.
Innovations and breakthroughs
Recent reports have highlighted the importance of ursodeoxycholic acid (UDCA) treatment for improving prognosis in patients with PBC. This is the first prospective cohort study showing that incomplete UDCA response or inconsistent treatment, anti-centromere antibodies positivity, higher alanine aminotransferase ratio, and advanced histological stage are risk factors for hepatic decompensation.
Applications
Knowledge of the factors for hepatic decompensation in PBC patients would be of great value in clinical management. Novel treatments are needed for patients with poor prognostic factors.
Terminology
PBC is an autoimmune liver disease characterized by the presence of anti-mitochondrial antibodies, and destruction of intrahepatic bile ducts, which can ultimately lead to cirrhosis and hepatic failure. Early UDCA treatment, excellent biochemical response to UDCA, and histological stages are predictors of survival.
Peer review
The authors have documented the clinical profiles and analyzed risk factors for hepatic decompensation in patients with PBC. It is a relatively large prospective study and gives important information on the prognosis of PBC patients.
Footnotes
Supported by National Science Technology Pillar Program in the 11th Five-Year Plan, No. 2008BAI59B03; and Research Special Fund for the Public Welfare Industry of Health, No. 201202004
P- Reviewers Caboclo JLF, Beltran MA, Shimizu Y, Weekitt K S- Editor Gou SX L- Editor Stewart G E- Editor Xiong L
References
- 1.Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–1273. doi: 10.1056/NEJMra043898. [DOI] [PubMed] [Google Scholar]
- 2.Prince MI, James OF. The epidemiology of primary biliary cirrhosis. Clin Liver Dis. 2003;7:795–819. doi: 10.1016/s1089-3261(03)00102-8. [DOI] [PubMed] [Google Scholar]
- 3.Lazaridis KN, Talwalkar JA. Clinical epidemiology of primary biliary cirrhosis: incidence, prevalence, and impact of therapy. J Clin Gastroenterol. 2007;41:494–500. doi: 10.1097/01.mcg.0000225653.07932.8f. [DOI] [PubMed] [Google Scholar]
- 4.Ishibashi H, Komori A, Shimoda S, Ambrosini YM, Gershwin ME, Nakamura M. Risk factors and prediction of long-term outcome in primary biliary cirrhosis. Intern Med. 2011;50:1–10. doi: 10.2169/internalmedicine.50.4462. [DOI] [PubMed] [Google Scholar]
- 5.Poupon R. Primary biliary cirrhosis: a 2010 update. J Hepatol. 2010;52:745–758. doi: 10.1016/j.jhep.2009.11.027. [DOI] [PubMed] [Google Scholar]
- 6.Dickson ER, Grambsch PM, Fleming TR, Fisher LD, Langworthy A. Prognosis in primary biliary cirrhosis: model for decision making. Hepatology. 1989;10:1–7. doi: 10.1002/hep.1840100102. [DOI] [PubMed] [Google Scholar]
- 7.Grambsch PM, Dickson ER, Wiesner RH, Langworthy A. Application of the Mayo primary biliary cirrhosis survival model to Mayo liver transplant patients. Mayo Clin Proc. 1989;64:699–704. doi: 10.1016/s0025-6196(12)65350-6. [DOI] [PubMed] [Google Scholar]
- 8.Mayo MJ, Parkes J, Adams-Huet B, Combes B, Mills AS, Markin RS, Rubin R, Wheeler D, Contos M, West AB, et al. Prediction of clinical outcomes in primary biliary cirrhosis by serum enhanced liver fibrosis assay. Hepatology. 2008;48:1549–1557. doi: 10.1002/hep.22517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuiper EM, Hansen BE, de Vries RA, den Ouden-Muller JW, van Ditzhuijsen TJ, Haagsma EB, Houben MH, Witteman BJ, van Erpecum KJ, van Buuren HR. Improved prognosis of patients with primary biliary cirrhosis that have a biochemical response to ursodeoxycholic acid. Gastroenterology. 2009;136:1281–1287. doi: 10.1053/j.gastro.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Gao L, Tian X, Liu B, Zhang F. The value of antinuclear antibodies in primary biliary cirrhosis. Clin Exp Med. 2008;8:9–15. doi: 10.1007/s10238-008-0150-6. [DOI] [PubMed] [Google Scholar]
- 11.Su CW, Hung HH, Huo TI, Huang YH, Li CP, Lin HC, Lee PC, Lee SD, Wu JC. Natural history and prognostic factors of primary biliary cirrhosis in Taiwan: a follow-up study up to 18 years. Liver Int. 2008;28:1305–1313. doi: 10.1111/j.1478-3231.2008.01715.x. [DOI] [PubMed] [Google Scholar]
- 12.Zhao DT, Liao HY, Liu YM, Zhao Y, Feng X, Yan HP. Prognostic factors and survival analysis of antimitochondrial antibody-positive primary biliary cirrhosis in Chinese patients. Dig Dis Sci. 2011;56:2750–2757. doi: 10.1007/s10620-011-1661-7. [DOI] [PubMed] [Google Scholar]
- 13.Ludwig J, Dickson ER, McDonald GS. Staging of chronic nonsuppurative destructive cholangitis (syndrome of primary biliary cirrhosis) Virchows Arch A Pathol Anat Histol. 1978;379:103–112. doi: 10.1007/BF00432479. [DOI] [PubMed] [Google Scholar]
- 14.Chazouillères O, Wendum D, Serfaty L, Montembault S, Rosmorduc O, Poupon R. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome: clinical features and response to therapy. Hepatology. 1998;28:296–301. doi: 10.1002/hep.510280203. [DOI] [PubMed] [Google Scholar]
- 15.Parés A, Caballería L, Rodés J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic Acid. Gastroenterology. 2006;130:715–720. doi: 10.1053/j.gastro.2005.12.029. [DOI] [PubMed] [Google Scholar]
- 16.Murray-Lyon IM, Pugh RN, Nunnerley HB, Laws JW, Dawson JL, Williams R. Treatment of bleeding oesophageal varices by infusion of vasopressin into the superior mesenteric artery. Gut. 1973;14:59–63. doi: 10.1136/gut.14.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farrell GC. Primary biliary cirrhosis in Asians: less common than in Europeans, but just as depressing. J Gastroenterol Hepatol. 2008;23:508–511. doi: 10.1111/j.1440-1746.2008.05379.x. [DOI] [PubMed] [Google Scholar]
- 18.Liu H, Liu Y, Wang L, Xu D, Lin B, Zhong R, Gong S, Podda M, Invernizzi P. Prevalence of primary biliary cirrhosis in adults referring hospital for annual health check-up in Southern China. BMC Gastroenterol. 2010;10:100. doi: 10.1186/1471-230X-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hohenester S, Oude-Elferink RP, Beuers U. Primary biliary cirrhosis. Semin Immunopathol. 2009;31:283–307. doi: 10.1007/s00281-009-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marasini B, Gagetta M, Rossi V, Ferrari P. Rheumatic disorders and primary biliary cirrhosis: an appraisal of 170 Italian patients. Ann Rheum Dis. 2001;60:1046–1049. doi: 10.1136/ard.60.11.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Silveira MG, Mendes FD, Diehl NN, Enders FT, Lindor KD. Thyroid dysfunction in primary biliary cirrhosis, primary sclerosing cholangitis and non-alcoholic fatty liver disease. Liver Int. 2009;29:1094–1100. doi: 10.1111/j.1478-3231.2009.02003.x. [DOI] [PubMed] [Google Scholar]
- 22.Prince M, Chetwynd A, Newman W, Metcalf JV, James OF. Survival and symptom progression in a geographically based cohort of patients with primary biliary cirrhosis: follow-up for up to 28 years. Gastroenterology. 2002;123:1044–1051. doi: 10.1053/gast.2002.36027. [DOI] [PubMed] [Google Scholar]
- 23.Prince MI, Chetwynd A, Craig WL, Metcalf JV, James OF. Asymptomatic primary biliary cirrhosis: clinical features, prognosis, and symptom progression in a large population based cohort. Gut. 2004;53:865–870. doi: 10.1136/gut.2003.023937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Selmi C, Bowlus CL, Gershwin ME, Coppel RL. Primary biliary cirrhosis. Lancet. 2011;377:1600–1609. doi: 10.1016/S0140-6736(10)61965-4. [DOI] [PubMed] [Google Scholar]
- 25.Milkiewicz P, Heathcote EJ. Fatigue in chronic cholestasis. Gut. 2004;53:475–477. doi: 10.1136/gut.2003.025155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahl TC, Shockcor W, Boyer JL. Primary biliary cirrhosis: survival of a large cohort of symptomatic and asymptomatic patients followed for 24 years. J Hepatol. 1994;20:707–713. doi: 10.1016/s0168-8278(05)80139-4. [DOI] [PubMed] [Google Scholar]
- 27.Springer J, Cauch-Dudek K, O’Rourke K, Wanless IR, Heathcote EJ. Asymptomatic primary biliary cirrhosis: a study of its natural history and prognosis. Am J Gastroenterol. 1999;94:47–53. doi: 10.1111/j.1572-0241.1999.00770.x. [DOI] [PubMed] [Google Scholar]
- 28.Floreani A, Caroli D, Variola A, Rizzotto ER, Antoniazzi S, Chiaramonte M, Cazzagon N, Brombin C, Salmaso L, Baldo V. A 35-year follow-up of a large cohort of patients with primary biliary cirrhosis seen at a single centre. Liver Int. 2011;31:361–368. doi: 10.1111/j.1478-3231.2010.02366.x. [DOI] [PubMed] [Google Scholar]
- 29.Baldursdottir TR, Bergmann OM, Jonasson JG, Ludviksson BR, Axelsson TA, Björnsson ES. The epidemiology and natural history of primary biliary cirrhosis: a nationwide population-based study. Eur J Gastroenterol Hepatol. 2012;24:824–830. doi: 10.1097/MEG.0b013e328353753d. [DOI] [PubMed] [Google Scholar]
- 30.Ngu JH, Gearry RB, Frampton CM, Stedman CA. Mortality and the risk of malignancy in autoimmune liver diseases: a population-based study in Canterbury, New Zealand. Hepatology. 2012;55:522–529. doi: 10.1002/hep.24743. [DOI] [PubMed] [Google Scholar]
- 31.Myers RP, Shaheen AA, Fong A, Burak KW, Wan A, Swain MG, Hilsden RJ, Sutherland L, Quan H. Epidemiology and natural history of primary biliary cirrhosis in a Canadian health region: a population-based study. Hepatology. 2009;50:1884–1892. doi: 10.1002/hep.23210. [DOI] [PubMed] [Google Scholar]
- 32.Angulo P, Batts KP, Therneau TM, Jorgensen RA, Dickson ER, Lindor KD. Long-term ursodeoxycholic acid delays histological progression in primary biliary cirrhosis. Hepatology. 1999;29:644–647. doi: 10.1002/hep.510290301. [DOI] [PubMed] [Google Scholar]
- 33.Corpechot C, Carrat F, Bahr A, Chrétien Y, Poupon RE, Poupon R. The effect of ursodeoxycholic acid therapy on the natural course of primary biliary cirrhosis. Gastroenterology. 2005;128:297–303. doi: 10.1053/j.gastro.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 34.Poupon RE, Lindor KD, Parés A, Chazouillères O, Poupon R, Heathcote EJ. Combined analysis of the effect of treatment with ursodeoxycholic acid on histologic progression in primary biliary cirrhosis. J Hepatol. 2003;39:12–16. doi: 10.1016/s0168-8278(03)00192-2. [DOI] [PubMed] [Google Scholar]
- 35.Corpechot C, Abenavoli L, Rabahi N, Chrétien Y, Andréani T, Johanet C, Chazouillères O, Poupon R. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology. 2008;48:871–877. doi: 10.1002/hep.22428. [DOI] [PubMed] [Google Scholar]
- 36.Miyawaki S, Asanuma H, Nishiyama S, Yoshinaga Y. Clinical and serological heterogeneity in patients with anticentromere antibodies. J Rheumatol. 2005;32:1488–1494. [PubMed] [Google Scholar]
- 37.Salliot C, Gottenberg JE, Bengoufa D, Desmoulins F, Miceli-Richard C, Mariette X. Anticentromere antibodies identify patients with Sjögren’s syndrome and autoimmune overlap syndrome. J Rheumatol. 2007;34:2253–2258. [PubMed] [Google Scholar]
- 38.Hossny E, Hady HA, Mabrouk R. Anti-centromere antibodies as a marker of Raynaud’s phenomenon in pediatric rheumatologic diseases. Pediatr Allergy Immunol. 2000;11:250–255. doi: 10.1034/j.1399-3038.2000.00066.x. [DOI] [PubMed] [Google Scholar]
- 39.Takada K, Suzuki K, Matsumoto M, Okada M, Nakanishi T, Horikoshi H, Higuchi T, Ohsuzu F. Clinical characteristics of patients with both anti-U1RNP and anti-centromere antibodies. Scand J Rheumatol. 2008;37:360–364. doi: 10.1080/03009740802116190. [DOI] [PubMed] [Google Scholar]
- 40.Pakozdi A, Nihtyanova S, Moinzadeh P, Ong VH, Black CM, Denton CP. Clinical and serological hallmarks of systemic sclerosis overlap syndromes. J Rheumatol. 2011;38:2406–2409. doi: 10.3899/jrheum.101248. [DOI] [PubMed] [Google Scholar]
- 41.Agmon-Levin N, Shapira Y, Selmi C, Barzilai O, Ram M, Szyper-Kravitz M, Sella S, Katz BS, Youinou P, Renaudineau Y, et al. A comprehensive evaluation of serum autoantibodies in primary biliary cirrhosis. J Autoimmun. 2010;34:55–58. doi: 10.1016/j.jaut.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 42.Muratori P, Muratori L, Ferrari R, Cassani F, Bianchi G, Lenzi M, Rodrigo L, Linares A, Fuentes D, Bianchi FB. Characterization and clinical impact of antinuclear antibodies in primary biliary cirrhosis. Am J Gastroenterol. 2003;98:431–437. doi: 10.1111/j.1572-0241.2003.07257.x. [DOI] [PubMed] [Google Scholar]
- 43.Granito A, Muratori P, Muratori L, Pappas G, Cassani F, Worthington J, Ferri S, Quarneti C, Cipriano V, de Molo C, et al. Antibodies to SS-A/Ro-52kD and centromere in autoimmune liver disease: a clue to diagnosis and prognosis of primary biliary cirrhosis. Aliment Pharmacol Ther. 2007;26:831–838. doi: 10.1111/j.1365-2036.2007.03433.x. [DOI] [PubMed] [Google Scholar]
- 44.Nakamura M, Kondo H, Mori T, Komori A, Matsuyama M, Ito M, Takii Y, Koyabu M, Yokoyama T, Migita K, et al. Anti-gp210 and anti-centromere antibodies are different risk factors for the progression of primary biliary cirrhosis. Hepatology. 2007;45:118–127. doi: 10.1002/hep.21472. [DOI] [PubMed] [Google Scholar]
- 45.Yang WH, Yu JH, Nakajima A, Neuberg D, Lindor K, Bloch DB. Do antinuclear antibodies in primary biliary cirrhosis patients identify increased risk for liver failure? Clin Gastroenterol Hepatol. 2004;2:1116–1122. doi: 10.1016/s1542-3565(04)00465-3. [DOI] [PubMed] [Google Scholar]
- 46.Rigamonti C, Shand LM, Feudjo M, Bunn CC, Black CM, Denton CP, Burroughs AK. Clinical features and prognosis of primary biliary cirrhosis associated with systemic sclerosis. Gut. 2006;55:388–394. doi: 10.1136/gut.2005.075002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parveen S, Morshed SA, Nishioka M. High prevalence of antibodies to recombinant CENP-B in primary biliary cirrhosis: nuclear immunofluorescence patterns and ELISA reactivities. J Gastroenterol Hepatol. 1995;10:438–445. doi: 10.1111/j.1440-1746.1995.tb01597.x. [DOI] [PubMed] [Google Scholar]
- 48.Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, Weinman SA. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–375. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- 49.Su CW, Chan CC, Hung HH, Huo TI, Huang YH, Li CP, Lin HC, Tsay SH, Lee PC, Lee SD, et al. Predictive value of aspartate aminotransferase to alanine aminotransferase ratio for hepatic fibrosis and clinical adverse outcomes in patients with primary biliary cirrhosis. J Clin Gastroenterol. 2009;43:876–883. doi: 10.1097/MCG.0b013e31818980ac. [DOI] [PubMed] [Google Scholar]
- 50.Garrido MC, Hubscher SG. Accuracy of staging in primary biliary cirrhosis. J Clin Pathol. 1996;49:556–559. doi: 10.1136/jcp.49.7.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drebber U, Mueller JJ, Klein E, Kasper HU, Schulze F, Schardt K, Quasdorff M, Schulte S, Odenthal M, Dienes HP. Liver biopsy in primary biliary cirrhosis: clinicopathological data and stage. Pathol Int. 2009;59:546–554. doi: 10.1111/j.1440-1827.2009.02405.x. [DOI] [PubMed] [Google Scholar]
- 52.Roll J, Boyer JL, Barry D, Klatskin G. The prognostic importance of clinical and histologic features in asymptomatic and symptomatic primary biliary cirrhosis. N Engl J Med. 1983;308:1–7. doi: 10.1056/NEJM198301063080101. [DOI] [PubMed] [Google Scholar]
- 53.Locke GR, Therneau TM, Ludwig J, Dickson ER, Lindor KD. Time course of histological progression in primary biliary cirrhosis. Hepatology. 1996;23:52–56. doi: 10.1002/hep.510230108. [DOI] [PubMed] [Google Scholar]