

Abstract
Concurrent activation of RAS/ERK and PI3K/AKT pathways is implicated in prostate cancer progression. The negative regulators of these pathways, including sprouty2 (SPRY2), protein phosphatase 2A (PP2A), and phosphatase and tensin homolog (PTEN), are commonly inactivated in prostate cancer. The molecular basis of cooperation between these genetic alterations is unknown. Here, we show that SPRY2 deficiency alone triggers activation of AKT and ERK, but this is insufficient to drive tumorigenesis. In addition to AKT and ERK activation, SPRY2 loss also activates a PP2A-dependent tumor suppressor checkpoint. Mechanistically, the PP2A-mediated growth arrest depends on GSK3β and is ultimately mediated by nuclear PTEN. In murine prostate cancer models, Pten haploinsufficiency synergized with Spry2 deficiency to drive tumorigenesis, including metastasis. Together, these results show that loss of Pten cooperates with Spry2 deficiency by bypassing a novel tumor suppressor checkpoint. Furthermore, loss of SPRY2 expression correlates strongly with loss of PTEN and/or PP2A subunits in human prostate cancer. This underlines the cooperation between SPRY2 deficiency and PTEN or PP2A inactivation in promoting tumorigenesis. Overall, we propose SPRY2, PTEN, and PP2A status as an important determinant of prostate cancer progression. Characterization of this trio may facilitate patient stratification for targeted therapies and chemopreventive interventions.
Introduction
Prostate cancer (PC) is the most common malignancy among adult men in the developed world. Despite early diagnosis and treatment, many patients relapse and develop aggressive castrate-resistant PC (CRPC). Recent studies have implicated the activation of both PI3K/AKT and RAS/ERK pathways by aberrant receptor tyrosine kinase (RTK) signaling in the development of aggressive CRPC (1). Failing to detect significant mutations or amplifications of RTKs, integrated genomic analyses have highlighted the involvement of PI3K/AKT and RAS/ERK pathways at a relatively high frequency (43%) in primary PC and almost universally in metastatic disease (2). Dual activation of PI3K and RAS pathways has been implicated in bypassing primary tumor-suppressive responses such as growth arrest and senescence (3, 4). Interestingly, expression of phosphatase and tensin homolog (PTEN) and sprouty2 (SPRY2), key negative feedback regulators of the PI3K and RAS cascades, is decreased at higher frequencies than mutational activation of oncogenes such as PI3KCA, KRAS, and BRAF (2, 5). This strongly suggests cooperative interactions among these pathway-specific lesions in PC progression, and insight into how these alterations confer selective advantage to tumorigenesis will facilitate biology guided patient stratification and therapeutic design.
RTK-mediated activation of PI3K increases phosphatidylinositol 3,4,5 phosphate (PIP3) levels, leading to AKT activation (6). The lipid phosphatase PTEN acts as the central negative regulator of the PI3K/AKT pathway by dephosphorylating PIP3. PTEN is a commonly mutated tumor suppressor in human cancer (7). Phosphorylated PTEN is unable to associate with the membrane due to conformational change and hence fails to inhibit PI3K signaling (8). In addition to its role at the cell membrane, PTEN can also act as a tumor suppressor in the nucleus by binding and stabilizing tumor protein p53 (TP53) to induce growth arrest and apoptosis (9, 10). Extensive in vivo studies show that Pten is a haploinsufficient tumor suppressor, and the extent of its inactivation can determine PC progression (11). Pten heterozygous mice develop prostate intraepithelial neoplasia, whereas complete loss of functional Pten leads to tumor formation (12). In agreement with this, while mutations in the PTEN gene are rare, primary prostate tumors often show loss or alteration in one of the PTEN alleles. Complete inactivation of PTEN appears to be a late event in cancer progression (13). Recent studies have suggested that deregulated expression of PTEN interacting proteins and altered PTEN regulation by posttranslational modifications may also contribute to prostate carcinogenesis (14). It could be postulated that loss of PTEN activity may play an important role in tumor initiation, while further reduction in its expression can aid carcinogenesis. Hence it becomes important to study regulation of PTEN and how disruption of this is involved in tumorigenesis.
Sprouty proteins (SPRY1–4) are a family of well-conserved negative regulators of RTKs. Sprouty was originally identified in Drosophila as an antagonist of FGF signaling (15). Overall, SPRY2 is a tumor suppressor, and its expression is repressed in breast, lung, and liver cancers (16). However, in certain cancers, such as colonic adenocarcinoma, SPRY2 can also function as an oncogene (17). Consistent with its tumor-suppressive role, SPRY2 overexpression suppressed proliferation, motility, and invasion in cultured tumor cells (18). SPRY2 is a well-established inhibitor of the RAS/ERK signaling cascade, and it can act as a tumor suppressor in lung cancer by inhibiting ERK activation (19). SPRY2 also binds to and interacts with other tumor suppressors such as protein phosphatase 2A (PP2A) (20). However, how SPRY2 influences the function of PP2A remains unclear. PP2A is a ubiquitously expressed multimeric serine/threonine phosphatase, accounting for a large fraction of the phosphatase activity in eukaryotic cells (21). Suppression of PP2A activity can transform multiple cell types by activating key pathways such as PI3K and RAS (22).
Genomic profiling of clinical PC showed SPRY2 inactivation in almost 18% of primary and over 70% of metastatic tumors, supporting a probable tumor-suppressive role for SPRY2 in PC (2). We have previously reported that epigenetic silencing of SPRY2 is a common event in prostate tumors (5). Edwin et al. showed that, in addition to the RAS/ERK pathway, SPRY2 can also regulate PI3K signaling by enhancing lipid phosphatase function of PTEN (23). Thus, SPRY2 can act as a tumor suppressor by inhibiting both RAS/ERK and PI3K/AKT signaling cascades. Understanding the molecular basis and functional consequence of tumor suppressors, especially when molecular and genomic analyses implicate 2 tumor suppressors, e.g., SPRY2 and PTEN within the same pathway, poses an important problem in cancer biology. Here, by profiling the tumor-suppressive roles of SPRY2 and PTEN in a coherent set of in vitro and in vivo systems, we showed that SPRY2 deficiency is sufficient to activate both PI3K/AKT and RAS/ERK cascades. However, despite such dual pathway activation, SPRY2-deficient cells exhibit growth arrest induced by nuclear PTEN, independent of its canonical lipid phosphatase activity. Mechanistically, our data revealed important interplay among established tumor suppressors PTEN, PP2A, GSK3B, and TP53 in restraining PC. Overall, we have identified the cooperative role of concomitantly inactivated components of PI3K and RAS pathways, namely SPRY2, PTEN, and PP2A, to drive PC progression.
Results
SPRY2 knockdown decreases cell proliferation despite activation of mitogenic signaling.
The effects of SPRY2 expression on activation of mitogenic signaling effectors such as AKT and ERK were investigated in DU145 PC cells with stable SPRY2 knockdown (KD). DU145 clones with stable SPRY2 KD (CL13 and CL61) showed increased PTEN phosphorylation and decreased membrane localization with an associated activation of PDK-1 and AKT when compared with nonsilencing (Nsi) vector control (VC) cells (Figure 1A and Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI63672DS1). Stable overexpression of SPRY2 in DU145 cells enhanced membrane localization of PTEN with a corresponding reduction in phosphorylation of PTEN, PDK-1, and AKT (Supplemental Figure 1, A and B). To confirm the role of PTEN phosphorylation in AKT activation, WT and phosphorylation-deficient PTEN mutants (PTENS380F and PTENT382/383A) were expressed in control and SPRY2-expressing (PTEN deficient) LNCaP cells. In the presence of WT PTEN, SPRY2-deficient LNCaP cells showed increased phosphorylated PTEN (p-PTEN) and p-AKT levels. However, p-AKT levels were significantly decreased in the presence of phosphorylation-deficient PTEN mutants in SPRY2-deficient LNCaP cells (Supplemental Figure 1C). In the presence of WT PTEN, SPRY2 expression in LNCaP cells reduced p-PTEN and p-AKT levels. Thus, SPRY2 deficiency decreased PTEN activity to inhibit PI3K/AKT signaling. Furthermore, SPRY2 KD elevated and SPRY2 overexpression decreased p-ERK1/2 levels in DU145 cells, underscoring the role of SPRY2 as a negative regulator of RAS/ERK signaling (Figure 1A and Supplemental Figure 1B). Similarly, Spry2 heterozygous (Spry2+/–) mouse embryonic fibroblasts (MEFs) showed enhanced Pten, Akt, and Erk phosphorylation when compared with WT MEFs (Figure 1B). The increased phosphorylation of PTEN, Akt, and Erk was also observed in prostatic tissue from PB-Cre4: Spry2+/– mice when compared with WT (Figure 1C).
Figure 1. SPRY2 KD decreases cell proliferation despite activation of mitogenic signaling.

(A) Whole cell lysates (WCL) of Nsi VC and SPRY2 KD DU145 clones were analyzed by Western blot (WB). (B and C) WCLs of (B) MEFs and (C) prostatic tissue from mice as indicated were analyzed by Western blot (n = 3). (D and E) BrdU incorporation analysis on (D) DU145 and (E) MEFs with indicated genetic alterations (*P < 0.001, **P < 0.05; n = 3; analyzed by Mann-Whitney test). Data are presented as mean ± SEM. (F) Representative IHC images and quantification for Ki67 staining in prostates of indicated mice. Scale bars: 100 μm. Arrows indicate Ki67-positive nuclei. (*P < 0.01, **P < 0.001; number of mice analyzed = 5 analyzed by Dunnett’s multiple comparison test). Box and whisker plots show median (lines within boxes), interquartile range (bounds of boxes), and upper and lower range (whiskers). All the Western blots were quantified using ImageJ, and the values represent relative immunoreactivity of each protein normalized to respective loading control.
We next investigated the role of SPRY2 expression on cell proliferation. When compared with the Nsi control, BrdU incorporation was significantly decreased in stable SPRY2 KD DU145 clones (CL13 and CL61) (Figure 1D). These effects were also validated by knocking down SPRY2 transiently in DU145 and normal prostatic RWPE-1 cells (Supplemental Figure 1D). As expected, SPRY2 KD–induced growth arrest in DU145 cells was rescued by expressing WT SPRY2 (Supplemental Figure 1E). The Spry2+/– MEFs also showed decreased cell proliferation with concomitant upregulation of p21 when compared with WT MEFs, supporting the notion that SPRY2 loss induced growth arrest despite increased Akt and Erk activation (Figure 1, B and E). The prostatic tissue of PB-Cre4: Spry2+/– mice also showed increased p21 (Waf1/Cip1) levels and decreased Ki67 staining indicative of probable growth arrest despite increased activation of mitogenic signaling (Figure 1, C and F). Interestingly, PB-Cre4: Ptenfl/+Spry2+/– (dual Pten and Spry2 heterozygous) prostates and MEFs showed elevated Ki67 and decreased p21 levels with associated increases in Akt and Erk activation when compared with PB-Cre4: Spry2+/– (Figure 1, B, C, E, and F). Overall, this suggests a potential growth-suppressive function of PTEN in SPRY2-deficient cells independent of its inhibitory effects on the PI3K/AKT pathway.
SPRY2 deficiency induces PTEN-mediated growth arrest.
To investigate the tumor-suppressive functions of PTEN in SPRY2-deficient cells, cell proliferation was assayed in PTEN-deficient PC-3 cells with stable SPRY2 KD. When compared with Nsi controls, BrdU incorporation was significantly increased in stable SPRY2 KD PC-3 clones (CL1 and CL10) (Figure 2A). This was also confirmed by knocking down SPRY2 transiently in PC-3 cells (Supplemental Figure 2A). Furthermore, stable WT SPRY2 expression significantly decreased BrdU incorporation in SPRY2- and PTEN-deficient LNCaP cells (Figure 2B). We next investigated the impact of SPRY2 and PTEN status on prostate tumorigenesis using an orthotopic prostate tumor model. SPRY2 KD DU145 (PTEN proficient) (CL61) cells and SPRY2-expressing LNCaP (PTEN deficient) cells along with the respective controls were injected into the dorsolateral prostate of nude mice. As expected, after 8 weeks, tumor incidence in mice with DU145 Nsi cells was 100%, with significant increase in tumor burden, whereas tumor incidence in mice with SPRY2 KD cells was under 30% with no significant increase in tumor burden (Figure 2C). In PTEN-deficient LNCaP cells, VC cells formed larger tumors when compared with corresponding SPRY2-expressing LNCaP cells (Figure 2D). The cell-cycle analyses showed that SPRY2 KD in DU145 cells resulted in slower G1 to S transit and accumulation of cells in G1 indicating pronounced G1 arrest (Figure 2E and Supplemental Figure 2B). To confirm the role of PTEN in observed G1 arrest, PTEN was transiently knocked down in SPRY2 KD DU145 cells. PTEN KD released SPRY2 KD DU145 cells from G1 arrest and increased their proliferation (Figure 2F and Supplemental Figure 2C). This was further confirmed in SPRY2- and PTEN-deficient LNCaP cells where SPRY2 and PTEN expression induced G1 arrest only in SPRY2-deficient cells (VC) (Supplemental Figure 2D). Also, Spry2+/– MEFs showed increased G1 arrest, which was significantly reduced in Spry2 and Pten heterozygous (Ptenfl/+Spry2+/–) MEFs (Figure 2G). Overall, PTEN inhibited tumorigenesis in SPRY2-deficient cells by inducing growth arrest.
Figure 2. SPRY2 loss induces PTEN-mediated G1 arrest.

(A and B) Western blot and BrdU incorporation analysis on (A) PC3 and (B) LNCaP cells (*P < 0.001; n = 3, analyzed by Mann-Whitney test). (C and D) Representative orthotopic tumor images and the weights of prostate following injection with (C) DU145 and (D) LNCaP cells as indicated. (*P < 0.01, number of mice = 5, analyzed by Dunnett’s multiple comparison test). Scale bars: 0.5 cm. Box and whisker plots show median (lines within boxes), interquartile range (bounds of boxes), and upper and lower range (whiskers). (E) SPRY2 KD DU145 were analyzed for cell-cycle profiles (top panel) after thymidine block release, and percentage of G1 cells are in bar chart (bottom panel; *P < 0.01; n = 3, analyzed by Mann-Whitney test). (F) SPRY2 KD DU145 transfected with PTEN siRNA were analyzed by Western blot and the G1 cells quantified. (*P < 0.01; n = 3, analyzed by Mann-Whitney test). (G) MEFs as indicated were quantified for cells in G1 (*P < 0.001; **P < 0.01; n = 3, analyzed by Mann-Whitney test). All Western blots were quantified using ImageJ, and the values represent relative immunoreactivity of each protein normalized to respective loading control. Data (A, B, F, and G) presented as mean ± SEM.
Nuclear PTEN mediates SPRY2 deficiency–induced growth arrest.
Since the SPRY2-deficient DU145 cells show decreased membrane levels of PTEN, cellular distribution of PTEN was assayed. The SPRY2 KD DU145 cells showed increased nuclear PTEN staining with associated increases in nuclear p21 staining when compared with Nsi cells (Figure 3A). This was further confirmed by cell fractionation in which nuclear fractions showed elevated PTEN levels in SPRY2 KD cells and decreased PTEN levels in SPRY2-overexpressing cells (Supplemental Figure 3A). SPRY2-mediated regulation of nuclear PTEN was also confirmed in other prostate cells (Supplemental Figure 3, B and C). Similarly, in SPRY2-deficient LNCaP cells, GFP-PTEN showed enhanced nuclear localization (Supplemental Figure 3D). Increased nuclear Pten staining was also observed in Spry2+/– MEFs and prostatic tissue when compared with WT (Figure 3, B and C, and Supplemental Figure 3E). The Spry2+/– MEFs also showed associated increase in nuclear transformation–related protein 53 (Trp53) and p21 staining relative to WT MEFs, underscoring the G1 arrest observed in these cells (Figure 3D, Supplemental Figure 3F, and Figure 2G). The smaller orthotopic tumors formed by SPRY2 KD DU145 cells showed lower BrdU labeling and enhanced nuclear PTEN staining when compared with Nsi tumors (Figure 3E). To confirm the relative impact of nuclear and cytoplasmic PTEN in inducing growth arrest, LNCaP cells were transfected with WT-PTEN tagged with either nuclear exclusion sequence (NES) or nuclear localizing sequence (NLS). Since SPRY2 influenced PTEN phosphatase function (23), phosphatase-dead PTEN-G129R mutant was also studied. As expected, WT-PTEN expression induced G1 arrest only in SPRY2-deficient cells (Figure 3F). In LNCaP cells, irrespective of SPRY2 expression, nuclear (NLS) PTEN (WT and G129R) induced G1 arrest, indicating that nuclear PTEN was sufficient to induce G1 arrest in a phosphatase-independent manner (Figure 3F). Irrespective of SPRY2 expression, cytosolic expression of WT PTEN enhanced sub-G1 population, indicating cell death, while the G1 population was relatively unaffected (Figure 3F). Thus, the observed growth arrest in SPRY2-deficient cells is mediated by nuclear PTEN.
Figure 3. SPRY2 loss–induced G1 arrest is mediated by nuclear accumulation of PTEN.

(A) Representative immunofluorescence (IF) images of indicated DU145 cells stained for PTEN (green), p21 (red), and DAPI (blue) (n = 3). Scale bars: 50 μm. (B and C) Representative immunofluorescence images of WT and Spry2+/– (B) MEFs and (C) prostate tissue stained for Pten (green). Red arrows indicate selected nuclei positive for Pten (n = 5). Scale bars: 100 μm. (D) Representative immunofluorescence images of indicated MEFs stained for Trp53 (green), p21 (red), and DAPI (blue). Scale bars: 100 μm. (E) Representative images and quantitation of DU145 orthotopic tumors stained for H&E, BrdU, and PTEN (*P < 0.01 **P < 0.001; n = 5, analyzed by Mann-Whitney test). Scale bars: 100 μm. Box and whisker plots show median (lines within boxes), interquartile range (bounds of boxes), and upper and lower range (whiskers). (F) SPRY2-LNCaP cells transfected with indicated plasmids were analyzed for cell-cycle profile after 24 hours (*P < 0.01; **P < 0.05 n = 3, analyzed by Mann-Whitney test). Data are presented as mean ± SEM.
Nuclear PTEN induces p53-dependent growth arrest.
Since both SPRY2 KD cells and Spry2+/– MEFs showed increased p21 levels and nuclear PTEN staining, the effects of PTEN on TP53-mediated G1 arrest were investigated. Although DU145 cells express mutant TP53 alleles, the mutant TP53 retains its ability to induce p21-mediated growth arrest (24). Compared with Nsi, SPRY2 KD DU145 cells showed elevated levels of TP53 and p21, which were decreased by PTEN KD (Figure 4A). Similarly, Ptenfl/+Spry2+/– MEFs showed significantly lower nuclear Trp53 and p21 than Spry2+/– MEFs (Figure 3D). The endogenous PTEN only associated with TP53 in SPRY2-deficient DU145 and MEFs (Figure 4B and Supplemental Figure 4A). Also, only the nuclear expression of both WT and G129R PTEN increased TP53 and p21 levels in PTEN-deficient LNCaP cells (Figure 4C and Supplemental Figure 4B). SPRY2-deficient DU145 cells and MEFs with TP53 KD showed diminished G1 arrest with associated decrease in p21 and increase in proliferation (Figure 4D and Supplemental Figure 4, C and D). To confirm the role of TP53 in nuclear PTEN–mediated growth arrest, WT and G129R PTEN tagged with either NES or NLS were expressed in PTEN- and TP53-deficient PC-3 cells with SPRY2 KD and WT-TP53 expression. Importantly, the induction of G1 arrest by nuclear PTEN (WT and G129R) was observed only in WT TP53 expressing PC-3 cells, confirming that, in a TP53-proficient context, the phosphatase function of PTEN is dispensable for its antitumor effects (Supplemental Figure 4E). The WT PTEN-NES induced cell death independent of SPRY2 expression in TP53-null PC-3 cells (Supplemental Figure 4E).
Figure 4. SPRY2 deficiency induces TP53-dependent G1 arrest via nuclear PTEN.

(A) Western blot analysis of indicated DU145 cells transfected with PTEN siRNA. (B) Western blot analysis of PTEN IP from DU145. (C) Western blot analysis for LNCaP cells transfected with indicated plasmids. (D) DU145 cells transfected with TP53 siRNA were analyzed by Western blot and quantified for cells in G1. (*P < 0.01; n = 3, analyzed by Mann-Whitney test). Data are presented as mean ± SEM. (E) Representative IHC images for Srpy2, Trp53, and p21 in prostates of mice as indicated (n = 4). Scale bars: 50 μm. All Western blots were quantified using ImageJ, and the values represent relative immunoreactivity of each protein normalized to respective loading control.
The prostates from PB-Cre4:Spry2+/– showed increased nuclear TP53 and p21 staining when compared with PB-Cre4:Ptenfl/+Spry2+/– mice (Figure 4E and Supplemental Figure 4, F and G). Also, the prostates from PB-Cre4:Spry2+/– Trp53fl/+ showed increased Ki67 staining when compared with PB-Cre4:Trp53fl/+ mice (Supplemental Figure 4H). Overall, SPRY2 deficiency induced growth arrest via nuclear PTEN-mediated induction of TP53 and p21 independent of its phosphatase activity.
SPRY2 deficiency–induced ROS increases nuclear accumulation of PTEN.
Consistent with a previous report (25), nuclear PTEN in DU145 cells was upregulated following H2O2 treatment (Supplemental Figure 5A), suggesting a possible role of intracellular ROS in nuclear localization of PTEN. Hence, ROS levels were assayed in SPRY2-deficient DU145 and MEF cells. SPRY2 KD increased ROS levels in the presence of 10% serum supplement, an effect not observed in cells maintained in serum-free medium (Figure 5A). SPRY2 is a negative regulator of FGFR signaling, while PTEN’s phosphatase function is essential for negative feedback regulation of activated IGFR (15, 26). Consistent with this, IGF-1 and FGF-2 activated AKT and ERK more potently in SPRY2 KD DU145 cells, indicating increased RTK signaling (Supplemental Figure 5B). SPRY2-deficient cells showed increased ROS production upon FGF-2 and IGF-1 treatment, which was abolished by treatment with specific inhibitors, namely BIBF1120 and PPP (inhibitors for FGFR and IGF-1R respectively; Figure 5B and Supplemental Figure 5C). Thus, SPRY2 loss increased intracellular ROS by enhancing RTK activation. RTK-mediated induction of intracellular ROS is reported to involve activation of the NADPH oxidase enzyme complex (27). As expected, in SPRY2 KD cells, treatment with the NADPH oxidase inhibitor apocynin significantly reduced the ROS levels and PTEN nuclear localization (Supplemental Figure 5D). Treatment with a ROS quencher DTT potently decreased nuclear localization of PTEN, implicating a significant role of ROS in elevating nuclear PTEN levels in SPRY2 KD cells (Supplemental Figure 5E). Furthermore, treatment with the antioxidant N-acetyl cysteine (NAC) decreased nuclear Pten and p21 staining in Spry2+/– MEFs (Figure 5C). Similarly, NAC treatment in SPRY2 KD DU145 cells also abolished nuclear accumulation of PTEN, TP53, and p21 with associated decrease in G1 arrest (Figure 5D). In an orthotopic PC model, mice injected with DU145 (Nsi or SRPY2 KD CL61) cells were treated with NAC for 5 weeks to test its effects on tumor growth. Consistent with our in vitro findings, NAC treatment increased the tumor incidence of SPRY2 KD cells to 100% (7/7), compared with 42% (3/7) in vehicle control–treated mice, with a significant increase in tumor burden and proliferation (Figure 5, E and F, and Supplemental Figure 5F). The tumor incidence, burden, and proliferation were unaltered by NAC treatment in mice injected with Nsi cells (Figure 5, E and F, and Supplemental Figure 5F). NAC treatment also decreased nuclear PTEN, TP53, and p21 staining in SPRY2 KD orthotopic tumors (Figure 5F and Supplemental Figure 5F). To confirm that the growth-promoting effects of NAC are dependent on PTEN in SPRY2-deficient cells, mice injected with control and SPRY2-expressing LNCaP (PTEN and SPRY2 deficient) cells were treated with NAC for 5 weeks. As expected, LNCaP cells with stable SPRY2 expression showed significantly lower tumor burden and proliferation (P < 0.01, respectively). NAC treatment in these mice did not alter the tumor incidence, burden, or proliferation (Supplemental Figure 5, G and H). Also, NAC-induced increase in cell proliferation in SPRY2 KD DU145 cells was abrogated upon PTEN KD (Supplemental Figure 5I). Overall, we show that SPRY2 loss–induced ROS drives nuclear PTEN–mediated growth arrest.
Figure 5. SPRY2 deficiency–induced ROS increases nuclear accumulation of PTEN.

(A) DU145 and MEFs were analyzed for intracellular ROS using DCFDA dye in the presence and absence of 10% serum (*P < 0.01; n = 3, analyzed by Mann-Whitney test). (B) Intracellular ROS was detected using DCFDA dye and analyzed in 24-hour serum-starved DU145 cells following treatment with growth factors (FGF2 [10 ng/ml], IGF-1 [100 ng/ml]) along with respective inhibitors (FGFR inhibitor- BIBF1120 [0.4 μM], IGF-1R inhibitor, PPP [2 nM]) for 30 minutes. SS, serum starved. (*P < 0.01; n = 3, analyzed by Mann-Whitney test). (C) Representative immunofluorescence images of MEFs stained for Pten (green) and p21 (red) after 5 μM NAC treatment for 48 hours. Scale bars: 100 μm. (D) Nuclear extracts from DU145 cells treated with 5 μM NAC for 48 hours were analyzed by Western blot, and G1 cells were quantified (*P < 0.001; n = 3, analyzed by Mann-Whitney test). (E) Representative images and prostate weights of nude mice orthotopically injected with Nsi or SPRY2 KD (CL61) DU145 cells and treated with NAC. Scale bars: 0.5 cm. (*P < 0.01, number of mice = 7, analyzed by Dunnett’s multiple comparison test). Box and whisker plots show median (lines within boxes), interquartile range (bounds of boxes), and upper and lower range (whiskers). (F) Representative images of IHC for Ki67, PTEN, TP53, and p21 in indicated DU145 orthotopic tumors. Scale bars: 100 μm. All the Western blots were quantified using ImageJ, and the values represent relative immunoreactivity of each protein normalized to respective loading control. Data are presented as mean ± SEM (A, B, and D).
GSK3B activation drives phosphorylation and nuclear accumulation of PTEN.
PB-Cre4:Spry2+/– mice treated with NAC also showed decreased nuclear Pten and p21, with significant increase in Ki67 staining in the prostatic tissues (Figure 6, A and B, and Supplemental Figure 6, A and B). Interestingly, NAC treatment decreased PTEN phosphorylation in the prostates of PB-Cre4:Spry2+/– mice and SPRY2 KD DU145 cells (Figure 5, C and D). Similarly, induction of ROS by H2O2 treatment in DU145 cells increased and treatment of SPRY2 KD cells with DTT (ROS quencher) decreased PTEN phosphorylation (Supplemental Figure 6, C and D). To test the significance of PTEN phosphorylation on its nuclear localization, WT and phosphorylation-deficient PTEN-GFP (PTENS380F and PTEN T382/383A) mutants were expressed in SPRY2-null LNCaP cells. Both S380F and T382/383A PTEN mutants showed decreased nuclear accumulation when compared with WT PTEN (Supplemental Figure 6E), suggesting a role for PTEN phosphorylation at Ser380/Threo382/Threo383 residues in its nuclear localization. Since PTEN is phosphorylated by GSK3B (28), GSK3B expression was knocked down in SPRY2 KD DU145 cells. GSK3B KD potently decreased nuclear localization and phosphorylation of PTEN in SPRY2 KD cells with an associated increase in cell proliferation (Figure 6, E and F, and Supplemental Figure 6F). Treatment of SPRY2-deficient MEFs and DU145 cells with specific GSK3B inhibitor (SB216763) significantly decreased PTEN phosphorylation (Figure 6G and Supplemental Figure 6G). To further confirm the role of GSK3B activity in DU145 cells, we generated SPRY2 KD and Nsi DU145 cells with stable expression of either kinase-defective (K85A) or constitutively active (S9A) GSK3B. GSK3BK85A expression significantly decreased PTEN phosphorylation and nuclear localization in SPRY2 KD cells with associated decrease in G1 arrest (Figure 6H and Supplemental Figure 6, H and I). Conversely, GSK3BS9A expression significantly elevated PTEN phosphorylation and nuclear localization with associated increased in G1 arrest in Nsi control cells (Figure 6H and Supplemental Figure 6, H and I). Here, we show that GSK3B-mediated phosphorylation of PTEN at S380/T382/T383 governs its nuclear localization.
Figure 6. GSK3B activation drives phosphorylation and nuclear accumulation of PTEN.

(A and B) Representative images of (A) PTEN (red) immunofluorescence and of (B) Ki67 and p21 IHC on prostate tissue from WT and Spry2+/– mice treated with 10 mM NAC for 75 days in drinking water ad libitum. Scale bars: 50 μm. (C) Western blot analysis of WCL from prostates of indicated mice treated with 10 mM NAC for 75 days in drinking water ad libitum. (D) Western blot analysis of indicated DU145 cells treated with 10 μM NAC for 12 hours. (E) Western blot analysis of DU145 cells treated with GSK3B siRNA. (F) Representative PTEN immunofluorescence images of DU145 cells treated with GSK3B siRNA. Scale bars: 50 μm. (G) Western blot analysis of indicated MEFs treated with Gsk3b inhibitor SB216763 (5 μM for 12 hours). (H) Western blot analysis of DU145 SPRY2 KD clones with stable expression of kinase dead (GSK3BK85A) and constitutively active (GSK3BS9A) GSK3B. All the Western blots were quantified using ImageJ, and the values represent relative immunoreactivity of each protein normalized to respective loading control.
ROS-mediated PP2A activation increases phosphorylation and nuclear localization of PTEN via GSK3B.
GSK3B activity is tightly regulated by the phosphorylation status at serine 9, which is induced by inactivating kinases such as AKT and reduced by activating phosphatases such as PP2A (29). Despite increased AKT activation in SPRY2-deficient cells (Figure 1, A–C), GSK3B showed decreased inactivating phosphorylation at serine 9 in SPRY2-deficient cells, indicating enhanced phosphatase function to activate GSK3B (Figure 6, C and D, and Supplemental Figure 6D). Therefore, the potential involvement of PP2A was investigated. PP2A activity was significantly elevated in SPRY2 KD DU145 cells; this activity could be further enhanced by FTY720 (PP2A activator) or inhibited by okadaic acid (a selective PP2A inhibitor), respectively (Figure 7A and Supplemental Figure 7A). Also, Spry2+/– MEFs and murine prostatic tissues showed increased PP2A activity (Figure 7A and Supplemental Figure 7B). The role of PP2A in GSK3B-mediated PTEN phosphorylation was tested using okadaic acid and knocking down structural (PP2A-A) or catalytic (PP2A-C) subunits of PP2A. All the treatments that diminished PP2A function enhanced inactivating phosphorylation of GSK3B, decreased p-PTEN, and diminished nuclear localization of PTEN in SPRY2 KD cells (Figure 7, B–D, Supplemental Figure 7, C–F, and Supplemental Figure 8, H and I). Endogenous SPRY2 was able to bind to both A and C core subunits of PP2A in both DU145 and MEF cells (Supplemental Figure 8A). In SPRY2-deficient cells, GSK3B showed increased association with PP2A-A (Figure 7E). Moreover, in SPRY2 KD DU145 cells, GSK3B-associated PP2A activity was significantly increased; this increase could be further enhanced by FTY720 or inhibited by okadaic acid treatment (Figure 7F). Thus, in addition to regulating overall PP2A activity, SPRY2 loss may also alter PP2A substrate specificity.
Figure 7. ROS-mediated PP2A activation increases phosphorylation of PTEN.

(A) PP2A activity was measured in DU145 and MEFs (*P < 0.05, analyzed by Mann-Whitney test), and PP2A-C IP was analyzed by Western blot. (B–D) Western blot analysis of indicated DU145 cells treated with (B) 1 μM okadaic acid for 2 hours, (C) 5 nmol PP2A-A siRNA for 42 hours, and (D) 5 nmol PP2A-C siRNA for 42 hours. (E) PP2A-A IP from DU145 and MEFs was analyzed by Western blot. (F) PP2A activity and Western blot of GSK3B IP in DU145 cells treated with 1 μM okadaic acid and 10 μM FTY720 for 2 hours (*P < 0.05, n = 3, analyzed by Mann-Whitney test). All Western blots were quantified using ImageJ, and the values represent relative immunoreactivity of each protein normalized to respective loading control. Data are presented as mean ± SEM (A and F).
While SPRY2 deficiency is associated with increased PP2A activity in DU145 cells, MEFs, and prostatic tissues, we observed that treatment with the antioxidant NAC abolished such PP2A activation (Figure 7A and Supplemental Figure 7B). Induction of ROS by H2O2 treatment significantly increased PP2A activity in DU145 cells (Supplemental Figure 7H). Similarly, decreasing ROS levels by apocynin or DTT treatment abolished PP2A activation in SPRY2 KD cells (Supplemental Figure 7, I and J). Thus, ROS may regulate nuclear PTEN levels in DU145 SPRY2 KD cells by sequential activation of PP2A and GSK3B. In SPRY2 KD DU145 cells, both PP2A inhibitor (okadaic acid) and GSK3B inhibitor (SB216763) treatments decreased nuclear PTEN localization and nuclear p-PTEN levels (Figure 8A and Supplemental Figure 7K). In contrast, activation of PP2A by FTY720 treatment enhanced total and phosphorylated levels of nuclear PTEN in both Nsi and SPRY2 KD cells (Figure 8A and Supplemental Figure 7K). Similarly, in Spry2+/– MEFs, inhibition of PP2A and Gsk3b decreased nuclear Pten and p21 staining, while FTY720 treatment increased their staining in WT MEFs (Supplemental Figure 7M). Furthermore, FTY720-induced effects on PTEN required both PP2A and GSK3B, as FTY720-induced PTEN phosphorylation was eliminated by KD of either PP2A-A (Figure 8B and Supplemental Figure 7D) or GSK3B (Figure 8C and Supplemental Figure 7L). Overall, we show that SPRY2 loss–induced ROS drives nuclear accumulation of PTEN by PP2A-mediated activation of GSK3B.
Figure 8. PP2A activation drives PTEN phosphorylation and nuclear localization via GSK3B.

(A) Representative immunofluorescence images of okadaic acid–treated (0.5 μM for 1 hour), FTY720-treated (10 μM for 6 hours) and SB216763-treated (5 μM for 12 hours) DU145 cells stained for PTEN (green). Scale bars: 50 μm. (B) Western blot analysis of DU145 cells treated with 5 nmol PP2A-A siRNA for 24 hours followed by 5 μM FTY720 for 12 hours as indicated. (C) Western blot analysis of DU145 cells treated with GSK3B siRNA for 24 hours followed by 5 μM FTY720 for 12 hours. All Western blots were quantified using ImageJ, and the values represent relative immunoreactivity of each protein normalized to respective loading control.
PP2A activation suppresses tumorigenesis by increasing nuclear localization of PTEN.
Our work so far showed that PP2A activation may act as a check point to inhibit tumorigenesis by activating tumor suppressors GSK3B and PTEN. To further confirm this, we tested the effects of overexpressing the structural A subunit and the catalytic C subunit of the PP2A enzyme complex in DU145 cells, which express relatively low levels of PP2A sub-units (Supplemental Figure 8A). As expected, restoring PP2A-A and PP2A-C expression to higher levels increased overall PP2A activity with an associated decrease in serine 9 phosphorylation of GSK3B and increase in p-PTEN levels (Supplemental Figure 8, B–E). DU145 cells with PP2A-A overexpression also showed elevated nuclear staining of PTEN and increased G1 arrest (Supplemental Figure 8, F and G). We next tested the role of GSK3B in PP2A-mediated effects on PTEN by knocking down GSK3B in PP2A-A–overexpressing DU145 cells. GSK3B KD abrogated PP2A–mediated increase in phosphorylation and nuclear localization of PTEN and associated growth arrest without affecting overall PP2A activity (Figure 9, A and B, and Supplemental Figure 8J). Also, Nsi and SPRY2 KD cells with stable expression of constitutively active GSK3B (S9A) showed elevated p-PTEN levels irrespective of PP2A status or activity (Figure 9C). Similarly, in Nsi and SPRY2 KD cells with stable expression of kinase-deficient GSK3B (K85A), FTY720-mediated increase in PP2A activity failed to increase PTEN phosphorylation (Supplemental Figure 8K). Furthermore, suppression of PP2A function by okadaic acid treatment significantly decreased growth arrest in SPRY2-deficient DU145 and MEFs (Figure 9D). In contrast, activation of PP2A by FTY720 enhanced G1 population in both control and SPRY2 KD cells (Figure 9D). Importantly, FTY720-treated mice with s.c. xenografts of DU145 cells showed significantly reduced tumor growth with increased PP2A activity when compared with vehicle-treated mice (Figure 9, E and F, and Supplemental Figure 8L). These xenografts also showed significantly less Ki67 and enhanced nuclear PTEN and p21 staining (Figure 9G). Overall, we have identified a PP2A-induced checkpoint in SPRY2-deficient cells. Mechanistically, PP2A-induced growth arrest depends on GSK3B, which is ultimately mediated by nuclear PTEN.
Figure 9. PP2A activation suppresses tumorigenesis by increasing nuclear localization of PTEN.

(A) DU145 cells infected with control and PP2A-A–expressing viruses were treated with GSK3B siRNA for 24 hours and analyzed by Western blot and WST-1 assay for measuring cell proliferation (*P < 0.01; n = 3). (B) Representative immunofluorescence images of PP2A-A–expressing DU145 cells treated with GSK3B siRNA for 24 hours and stained for PTEN. Scale bars: 50 μm. (C) Western blot analysis of DU145 SPRY2 KD clones with stable expression of constitutively active GSK3B (S9A) treated with 5 nmol PP2A-A siRNA for 42 hours. (D) DU145 and MEF cells treated with 0.2 μM okadaic acid or 10 μM FTY720 for 12 hours were quantified for percentage of G1 cells (*P < 0.01; n = 3). (E) Representative images and PP2A activity measurement in s.c. xenografts of DU145 cells in nude mice treated with PP2A activator FTY720 (10 mg/kg/d i.p. injection) for 5 days. (*P < 0.01; n = 5). (F) Representative images and tumor burden of s.c. injected DU145 cells in nude mice treated as indicated. Red line indicates schedule of FTY720 treatment (10 mg/kg/d i.p. injection). (G) Representative IHC images and quantification of Ki67 (white), PTEN (gray), and p21 (blue) in DU145 s.c. xenografts treated as above. Scale bars: 50 μm. Box and whisker plots (E and G) show median (lines within boxes), interquartile range (bounds of boxes), and upper and lower range (whiskers). The values of Western blot represent relative immunoreactivity of each protein normalized to respective loading control. Data are presented as mean ± SEM and analyzed by Mann-Whitney test (A and D–F).
In murine PC model, Pten loss bypasses Spry2 deficiency–induced growth arrest to synergistically drive tumorigenesis.
Our data suggest that in the context of SPRY2 loss, regulation of PTEN status represents a key factor in controlling prostate carcinogenesis. Confirming this, we find that the dual heterozygous PB-Cre4:Ptenfl/+Spry2+/– mice showed increased number of hyperplastic lesions and prostatic intraneoplasia (PIN) when compared with PB-Cre4:Ptenfl/+ mice at 6 months (Figure 10A and Supplemental Figure 9A). As previously reported, the prostates of PB-Cre4:Ptenfl/+ mice developed hyperplastic lesions with increased frequency of PIN at 1 year. In contrast, PB-Cre4:Ptenfl/+Spry2+/– mice developed invasive tumors at 1 year (Figure 10, A and B, and Supplemental Figure 9B). The prostate tissue of PB-Cre4:Spry2+/– mice showed increased p21 staining despite enhanced Akt and Erk1/2 activation at both 6 and 12 months (Figure 10C and Supplemental Figure 9, C–E). In contrast, the prostates of PB-Cre4:Ptenfl/+Spry2+/– mice showed decreased p21 staining (Figure 10C and Supplemental Figure 9, C–E) and the tumors from PB-Cre4:Ptenfl/+Spry2+/– mice showed increased Ki67 staining and Akt activation relative to prostatic tissue from PB-Cre4:Ptenfl/+ mice (Figure 10C and Supplemental Figure 9, E and F). The PIN lesions in PB-Cre4:Ptenfl/+Spry2+/– mice showed enhanced Erk activation, while PINs in PB-Cre4:Ptenfl/+ mice showed minimal Erk activation (Figure 10C and Supplemental Figure 9D). However, the majority of the prostate tumors from PB-Cre4:Ptenfl/+Spry2+/– mice at 12 months showed enhanced Akt activation with localized Erk activation (Figure 10C). Interestingly, PB-Cre4:Ptenfl/+Spry2+/– mice also showed enlarged lymph nodes, with evidence of metastasis as seen by pan-cytokeratin staining (Figure 10D). Consistent with probable epithelial-to-mesenchymal transition, prostate tumors from PB-Cre4:Ptenfl/+Spry2+/– mice showed delocalized (nonmembranous) E-cadherin and elevated vimentin staining (Supplemental Figure 9G). Similarly, in a mouse orthotopic prostate model using DU145 cells, NAC treatment, which enhanced overall tumor incidence of SPRY2 KD cells, also increased the incidence of metastasis and distant organ (liver) metastasis (Supplemental Figure 9H and Supplemental Table 1). Thus, Spry2 and Pten inactivation synergistically drive murine PC progression.
Figure 10. Spry2 and Pten inactivation synergistically drive murine prostate carcinogenesis.

(A) Representative H&E-stained images of alterations in prostates from mice as indicated (n = 6). Scale bars: 50 μm. (B) Representative prostate image and relative prostate weight of indicated mice at 12 months (*P < 0.01, n = 6, analyzed by Mann-Whitney Test). SV, seminal vesicles; AP, anterior prostate, DLP, dorsolateral prostate; B, bladder. Box and whisker plot shows median (lines within boxes), interquartile range (bounds of boxes), and upper and lower range (whiskers). (C) Representative IHC images of prostate tissue from 12-month-old mice as indicated (n = 6). Scale bars: 50 μm. (D) Representative IHC images for pancytokeratin staining in lymph nodes from indicated mice (n = 5). Scale bars: 50 μm.
Impact of SPRY2, PTEN, and PP2A status in clinical PC.
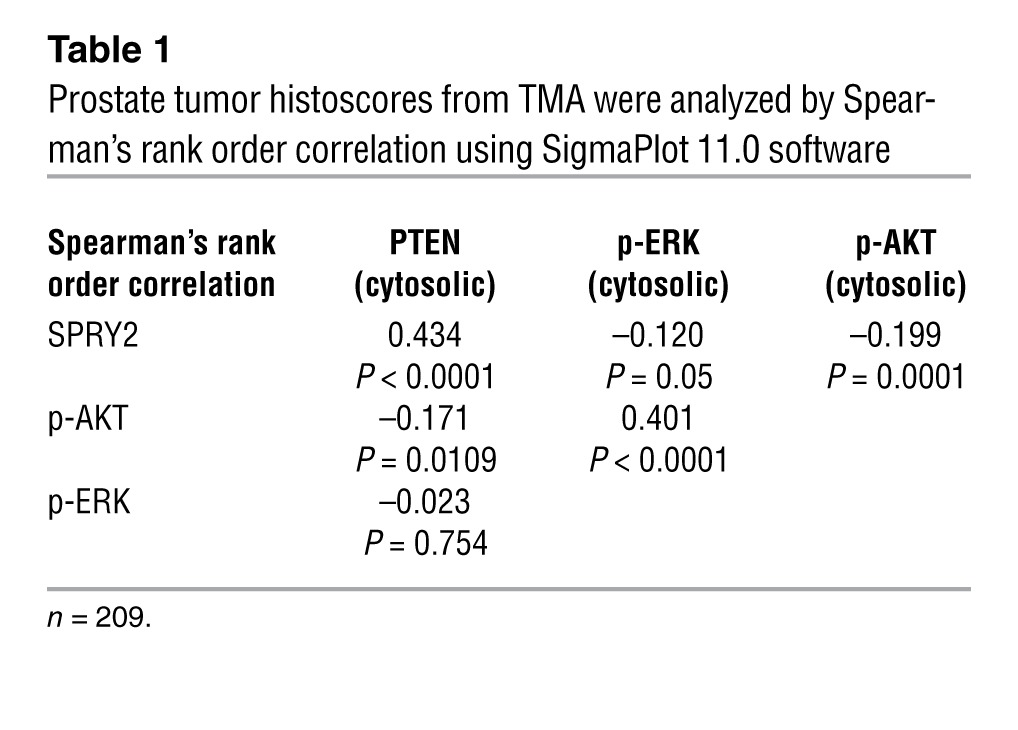
We next tested the clinical relevance of SPRY2 loss and nuclear PTEN in PC. In a cohort of clinical PC (n = 244), SPRY2 staining was significantly reduced in the malignant epithelium when compared with benign prostatic hyperplasia (BPH) (Figure 11A). Consistent with its role as a tumor suppressor, nuclear and cytosolic levels of PTEN in tumors were significantly lower in PC (Figure 11A). Confirming our in vitro and in vivo data, SPRY2 showed a significant inverse correlation with nuclear PTEN and p21 staining in BPH (Figure 11B and Supplemental Table 2). Interestingly, in BPH, nuclear PTEN showed a significant correlation with p-AKT levels, suggesting that nuclear localization of PTEN may increase PI3K-AKT activation (Figure 11B and Supplemental Table 2). We also confirmed a strong correlation between SPRY2 and PTEN in our tumor cohort (Table 1). As previously shown, PTEN showed significant inverse correlation to p-AKT, whereas there was no evidence of association with p-ERK (Table 1). As seen in our in vivo models, SPRY2 expression showed a significant inverse correlation with both p-AKT and p-ERK, which were strongly associated mutually, underscoring the nature of dual pathway activation (Table 1). Among patients bearing tumors with low SPRY2 levels, cases with nuclear PTEN-positive tumors have a significantly better survival outcome than those with tumors negative for nuclear PTEN, supporting our model that, by enhancing nuclear PTEN, SPRY2 loss in isolation has a tumor-suppressing, rather than tumor-promoting effect (Figure 11C). Overall, the expression status of PTEN and SPRY2 did not significantly influence patient survival as individual factors (Supplemental Figure 10A). Expression analysis of SPRY2 in a clinical PC data set from cBio Cancer Genomics Portal (2, 30) showed overall decrease in expression in tumors (Supplemental Figure 10B). In the same data set of primary prostate tumors SPRY2 expression strongly correlated with both PTEN (ρ = 0.367; P < 0.01) and PPP2CB (PP2A-C) (ρ = 0.423; P < 0.01). In this data set, 20% of the cases showed significant (more than 2-fold) decrease in SPRY2, and, among these tumors, approximately 90% showed associated loss of either PTEN or PP2A subunits (Supplemental Figure 10C). Furthermore, 38% of metastatic prostate tumors showed loss of SPRY2 expression and the majority of these tumors showed concomitant decrease in either PTEN or PPP2CB (PP2A-C) expression (Figure 11D). These data suggest that loss of either PTEN or PP2A is necessary and sufficient to synergistically cooperate with SPRY2 deficiency to drive PC (Figure 11E).
Figure 11. Impact of SPRY2, PTEN, and PP2A status in clinical PC.

(A) TMA of clinical PC and BPH cohorts was analyzed for expression of SPRY2 and nuclear and cytoplasmic PTEN. Box and whisker plots show median (lines within boxes), interquartile range (bounds of boxes), and upper and lower range (whiskers). Data were analyzed by ANOVA using Dunnett’s multiple comparison test. (B) Representative IHC images in clinical BPH samples. Scale bars: 100 μm. (C) Kaplan-Meier survival plot for PC patients with reduced SPRY2 expression (below median histoscore); analysis was according to the levels of nuclear PTEN. (D) Heat map of alterations in SPRY2, PTEN, and PPP2CB (PP2A catalytic subunit) generated from metastatic tumors (27 cases) using MSKCC Prostate Oncogenome Project data set from cBio genomic portal. (E) Schematic of PC progression. SPRY2 loss leads to tumor suppression by inducing growth arrest via PP2A-mediated nuclear accumulation of PTEN. Subsequent inactivation of PTEN or PP2A as observed in clinical PC may drive tumor progression.
Table 1.
Prostate tumor histoscores from TMA were analyzed by Spearman’s rank order correlation using SigmaPlot 11.0 software
Discussion
The recent report by Ding et al. has highlighted the complex nature of disruptive events during PC progression and suggested the need to understand the genesis of PC in the context of events such as PTEN inactivation (31). Our work has identified SPRY2 loss and PP2A inactivation as additional lesions that cooperate with PTEN in PC progression (Figure 11E). This provides insights into how interactions among major tumor suppressors influencing key signaling pathways may facilitate the development of therapeutic strategy and biomarkers. Concurrent mutations in components of the PTEN/PI3K/AKT and SPRY2/RAS/ERK pathways leading to simultaneous activation of these oncogenic cascades have been described in a number of cancer types, including colon, endometrium. and pancreas (32). Molecular analyses have shown that concomitant activation of both PI3K/AKT and RAS/ERK pathways synergize to bypass the primary tumor-suppressive responses such as senescence (3). In this study, we present evidence to suggest that, surprisingly, mere activation of oncogenic signaling cascades is insufficient to drive tumorigenesis. We have identified a network of important tumor suppressors involving PTEN, PP2A, GSK3B, and TP53 that functions to provide a primary fail-safe response of growth arrest to PI3K/AKT and RAS/ERK activation by oncogenic stimulus of SPRY2 loss.
Using a range of in vitro models as well as in vivo murine and clinical prostate tissues, we show that loss of SPRY2 regulates PTEN localization to induce growth arrest despite paradoxical and simultaneous activation of AKT and ERK. Since phosphorylated PTEN is unable to associate with plasma membrane and inhibit PI3K signaling, phosphorylation of PTEN can act as an important oncogenic stimulus (8). Here we show that the activation of PI3K signaling upon SPRY2 loss is at least in part mediated by PTEN phosphorylation. SPRY2 has been previously shown to inhibit cell proliferation by enhancing PTEN activity (23). In contrast with this report, despite increased AKT activity, SPRY2 KD PTEN-proficient PC cells proliferate more slowly and undergo G1 arrest. Beyond its cytoplasmic role in inhibition of the PI3K pathway, it is now clear that when localized to the nucleus, PTEN can suppress tumor progression by inducing growth arrest (10, 33). For the first time, loss of SPRY2 has been shown to induce G1 arrest via nuclear accumulation of PTEN. In agreement with previous reports, nuclear PTEN, independent of its lipid phosphatase activity, physically interacts and stabilizes TP53 to induce G1 arrest (34). This suggests that in addition to the previously reported role of SPRY2 in inhibition of cell proliferation by PTEN activation and subsequent inhibition of the PI3K pathway, SPRY2 inactivation can also trigger tumor-suppressive effects of PTEN by enhancing its nuclear accumulation. Supporting this, Chang et al. have shown that, independent of its phosphatase activity, nuclear expression of PTEN in PC cells such as LNCaP can inhibit tumorigenesis (25). Also, the phosphatase mutant PtenC124R/+ heterozygous mice showed fewer proliferative lesions in the prostate when compared with Pten+/– mice, underscoring the phosphatase-independent functions of Pten (35).
Deregulated RTK signaling can enhance intracellular ROS as an intrinsic part of their signaling cascade (36). Consistent with this and considering the role of SPRY2 as a negative regulator of RTK signaling, we show that the observed increase in ROS upon SPRY2 loss is mediated by enhanced RTK signaling. We have also identified SPRY2 loss–induced oxidative stress as a key regulator of nuclear localization of PTEN and tumor suppression. Of note, antioxidants have been proposed as chemopreventive agents in prostate carcinogenesis (37). The role of antioxidants as chemopreventive agents in clinical trial has been disappointing, with data from the discontinued SELECT PC prevention trial suggesting an unexpected trend (P = 0.06) toward increased risk, rather than protection from, clinical PC in men taking the antioxidant vitamin E (38). Based on our findings, the use of antioxidants can potentially promote tumor progression in the context of reduced SPRY2 expression. Hence, we propose that future trials should incorporate expression status of SPRY2 and PTEN for patient stratification in their design. Alternatively, the fallback tumor-suppressive pathways such as PP2A activation as identified here can be exploited for cancer prevention.
Mechanistically, we show that SPRY2 loss–mediated increase in ROS induced nuclear accumulation of PTEN via PP2A activation. Posttranslational modifications (PMTs) of PTEN such as ubiquitylation and phosphorylation have been previously implicated in cellular distribution of PTEN (14, 25). Here, we show that phosphorylation of PTEN at Ser380/Threo382/Threo383 can promote nuclear accumulation of PTEN. Consistent with a previous report, we show GSK3B as an important mediator of PTEN phosphorylation (27). The counterintuitive activation of GSK3B despite AKT activation was mediated by PP2A activation. SPRY2 KD induced intracellular ROS-mediated sequential activation of PP2A and GSK3B to increase PTEN phosphorylation and nuclear accumulation. Interestingly, we also observed increased binding of the core PP2A subunit PP2A-A to GSK3B in SPRY2-deficient cells, suggesting an altered PP2A substrate specificity in the absence of SPRY2. Since our data and previous reports indicate association of SPRY2 with core PP2A subunits, we hypothesize that loss of SPRY2 may change substrate specificity of PP2A to deregulate various signaling cascades (20). The core structural (PP2A-A) and catalytic (PP2A-C) subunits of PP2A are essential for its tumor-suppressive activity. The MSKCC clinical PC data set shows decreased expression of PP2A-A in almost 19% of primary prostate tumors (30). Consistent with the haploinsufficient tumor-suppressive functions of PP2A-A, we observed lower levels of PP2A-A in DU145 and PC-3 PC cells relative to RWPE-1 normal prostate cells. Hence, by restoring the PP2A-A levels in DU145 cells, we observed enhanced PP2A activity as well as tumor-suppressive effects. Most importantly, for what we believe is the first time, our data indicate that PP2A may play an important tumor-suppressive role by enhancing nuclear accumulation of PTEN. Hence, activation of PP2A can be considered a novel strategy to prevent prostate carcinogenesis.
Nuclear PTEN has been implicated in maintaining stem cell quiescence (39, 40). Interestingly, prostatic epithelial cells in Spry2+/– mice showed an increased tendency for nuclear Pten localization and decreased proliferation. This is consistent with our clinical survival data indicating a significantly better outcome for patients with tumors showing reduced SPRY2 expression in the presence of nuclear PTEN (P = 0.001). In contrast, the prostatic epithelium of PB-Cre4:Ptenfl/+Spry2+/– mice showed enhanced proliferation, suggesting that simultaneous loss of Spry2 and Pten may synergize to promote PC. Supporting this hypothesis, we find that Spry2 heterozygosity in PB-Cre4:Ptenfl/+Spry2+/– mice promotes PC from precancerous lesions as observed in PB-Cre4:Ptenfl/+ mice to invasive tumors. Spry2 deficiency also increased metastasis in murine PC models, which is supported by the global genomic analysis by Taylor et al., showing reduced SPRY2 expression in 74% of metastatic tumors, significantly more prevalent than in primary tumors (18%) (2). Our in vivo data indicate that haploinsufficiency of Spry2 and Pten leads to invasive prostate tumors with associated Akt and localized ERK activation.
The clinical PC data sets show a decrease in overall expression of all 3 tumor suppressors, SPRY2, PTEN, and PP2A (PPP2CB). Supporting our in vitro and in vivo data, the expression analysis in clinical PC data sets showed significant correlation of SPRY2 with PTEN and PP2A (P < 0.001). Furthermore, altered SPRY2, PTEN, and PPP2CB expression was associated with aggressive PC and decreased disease-free survival. We also observed an inverse correlation between SPRY2 and nuclear PTEN levels in BPH samples. In primary PC, almost all cases with SPRY2 loss show either loss of expression PTEN or PP2A subunits. Thus, it is plausible that loss of SPRY2 may represent an early event in prostate carcinogenesis whereby the presence of nuclear PTEN inhibits further progression. Such a state of “growth arrest” may be overcome by subsequent inactivation of PTEN, TP53, or PP2A.
In summary, our work has identified an intricate tumor-suppressive network of SPRY2, PTEN, and PP2A in prostate carcinogenesis. We show what we believe to be a novel role of SPRY2 in regulating cell fate by modulating PTEN function and its cellular distribution besides its documented role in suppressing the PI3K and RAS pathways. PC with depleted SPRY2 levels may progress due to subsequent loss of PTEN or PP2A. We propose that the status of the SPRY2, PTEN, and PP2A trio may serve as a predictor of PC progression.
Methods
Cell lines and MEFs.
Human PC cell lines DU145, PC-3, LNCaP, and CWR-22 were authenticated by LGC standards. For MEFs, the stable recombinations for PTEN were achieved by infecting MEFs with pBABE puro and pBABE puro-CRE (gift from Kevin Ryan, The Beatson Institute for Cancer Research) expressing adenoviruses and selecting the stable clones with 3 μg/ml puromycin (Sigma-Aldrich). The appropriate genotypes selected for this study were checked by genotyping (Transnetyx Inc.) and Western blotting. Cell proliferation assays were carried out using WST-1 reagent (Roche).
Animal studies.
For genetically modified mice, ARR2Probasin-Cre (PB-Cre4) mice with the Cre recombinase under the control of a modified rat probasin promoter that is expressed in prostatic epithelium were crossed with Ptenfl/+ mice (41). These were further crossed with Spry2+/– mice obtained from Gail Martin (UCLA, Los Angeles, California, USA). For controls, WT littermates were used. For the xenograft model, 107 DU145 cells were injected in 100 μl of serum-free RPMI s.c. in CD-1 nude mice. For the orthotopic model, dorsolateral prostates of male CD-1 nude mice were injected with 3 × 106 cells/25 μl serum free RPMI as previously described (42). The mice were gavaged daily for 5 weeks with NAC (10 mM) as indicated. WT and Spry2+/– mice were treated with 10 mM NAC (pH 7.5) in drinking water ad libitum for 75 days, and the NAC water was changed twice weekly. For the xenograft model, 107 DU145 cells were injected in 100 μl of serum-free medium s.c. in CD-1 nude mice. During the second week after injections, mice were randomized and treated with vehicle or FTY720 until the end of fifth week. The tumor volume was measured weekly using venire calliper. For the orthotopic tumor model, NAC and vehicle treatments were initiated 3 weeks after orthotopic injections. After a period of 8 weeks, mice were scarified after BrdU injection. S.c. and prostate tumors were excised, weighted, and fixed in formalin for 24 hours. Mice were sacrificed at time points of 3 months for proliferation studies, with a further cohort aged to 6 and 12 months for tumorigenic studies. The excised prostates were placed in formalin for overnight fixation before paraffin embedding.
Plasmids and siRNA.
A 19-mer SPRY2 target sequence (5′-AACACCAATGAGTACACAGAG-3′) from the 5′ end of the SPRY2 cDNA previously identified employing RNAi on-line tools (QIAGEN) and validated in successful siRNA KD experiments was inserted as a hairpin sequence into pTER+ plasmid (ShSPRY2). Alternatively, a 19-mer Nsi control sequence from QIAGEN was also inserted into the pTER+ as a Nsi shRNA control (shNsi). The shRNA vector pTER+ was made by inserting a modified H1 promoter cassette between the BglII and HindII sites of pcDNA4/TO (Invitrogen) as previously described (43). Cells stably expressing shSPRY2 and shNsi were selected using zeocin (300 μg/ml; Invitrogen). SPRY2 expression construct used for stable and transient expression was a gift from Graeme Guy (Institute of Molecular and Cell Biology, Singapore). Cells stably expressing SPRY2 were selected using geneticin (500 μg/ml; Gibco, Invitrogen). The PTEN siRNA (6251) and p53 siRNA (6562) were obtained from Cell Signaling. The PTEN expression construct (EGFP-PTEN) was generated by cloning PTEN cDNA (OriGene) into EcoRI/SalI-cut EGFPC2 (Clontech) vector. The PTEN expression constructs pSG5L HA NLS PTEN (Addgene 864), pSG5L HA NES PTEN (Addgene 863), pSG5L HA NES PTENG129R (Addgene 867) and pSG5L HA NLS PTENG129R (Addgene 868) were obtained from Addgene (William Sellers, Dana Farber Cancer Institute, Boston, Massachusetts, USA). Phosphorylation mutant (S380F and T382/383A) PTEN plasmids were generated from EGFP-PTEN plasmid using a site-directed mutagenesis kit (Alligent) and primers 5′-CCTGATCATTATAGATATTTTGACACCACTGACTCTGATCC-3′ (for S380F), 5′-CCTGATCATTATAGATATTCTGACGCCACTGACTCTGATCC -3′ (for T382A), and 5′-CCTGATCATTATAGATATTCTGACGCCGCTGACTCTGATCC-3′ (for T382/383A using PTENT382A plasmid as the target). GSK3B expression constructs HA GSK3 β S9A pcDNA3 (14754) and HA GSK3 β K85A pcDNA3 (14755) were obtained from Addgene (Jim Woodgett, Samuel Lunenfeld Research Institute, Toronto, Ontario, Canada). Retroviral PP2A-A expression construct pMIG-Aα WT (10884), empty backbone pMIG (9044), and PP2A-C expression construct pBABE PPP2CA WT (10689) were obtained from Addgene (William Hahn, Dana Farber Cancer Institute).
Flow cytometry analysis.
A total of 105 cells were acquired using a flow cytometer (FACScan, BD) and analyzed using Cell Quest Pro software.
PP2A activity assay.
PP2A activity was measured using PP2A immunoprecipitation phosphatase assay kit by Millipore (#17-313) according to the manufacturer’s protocol. PP2A activity in cells with either PP2A-C KD or overexpression was measured using assay components from the kit and PP2A-A antibody by Millipore (# 07-250).
Human tissue microarray.
Formalin-fixed, paraffin-embedded (FFPE) sections from 244 patients were studied. Tissue microarray (TMA) consisting of 209 primary PCs and 35 BPH samples was generated and used for IHC.
Statistics.
Data are presented as mean ± SEM. Statistical significance was assessed by Student’s t test (2-tailed) with Mann-Whitney test and ANOVA with Dunnett’s multiple comparison test using GraphPad Prism 5. Correlation and Kaplan-Meier survival analyses on human data were performed using SigmaPlot 11.0. P ≤ 0.05 was considered significant.
Study approval.
All the animal experiments conducted for this study were carried out in compliance with Animal (Scientific Procedures) Act 1986 and approved by United Kingdom Home Office (London, United Kingdom). All the human samples for TMA were taken from PC patients at the time of transurethral resection of the prostate, and their use in this study was approved by the Multicentre Research Ethics Committee for Scotland (MREC/01/0/36) (Edinburgh, United Kingdom). These archival clinical samples were “surplus to diagnostic need,” and specific consent from individual patients was not deemed required by the Ethics Committee.
Detailed information is supplied in Supplemental Methods. The uncut gels for the respective figures are also included.
Supplementary Material
Acknowledgments
This work was funded by Cancer Research UK. We thank Beatson Institute for Cancer Research core services and the Think Pink charity for provision of the Aperio slide scanner and the Slidepath software. We are particularly grateful to Peter Adams for helpful discussions. I. Ahmad was a Medical Research Council fellow.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(3):1157–1175. doi:10.1172/JCI63672.
References
- 1.Yap TA, Zivi A, Omlin A, de Bono JS. The changing therapeutic landscape of castration-resistant prostate cancer. Nat Rev Clin Oncol. 2011;8(10):597–610. doi: 10.1038/nrclinonc.2011.117. [DOI] [PubMed] [Google Scholar]
- 2.Taylor BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kennedy AL, et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumourigenesis. Mol Cell. 2011;42(1):36–49. doi: 10.1016/j.molcel.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmad I, et al. HER2 overcomes PTEN (loss)-induced senescence to cause aggressive prostate cancer. Proc Natl Acad Sci U S A. 2011;108(39):16392–16397. doi: 10.1073/pnas.1101263108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKie AB, et al. Epigenetic inactivation of the human sprouty2 (hSPRY2) homologue in prostate cancer. Oncogene. 2005;24(13):2166–2174. doi: 10.1038/sj.onc.1208371. [DOI] [PubMed] [Google Scholar]
- 6.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 7.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133(3):403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 8.Ross AH, Gericke A. Phosphorylation keeps PTEN phosphatase closed for business. Proc Natl Acad Sci U S A. 2009;106(5):1297–1298. doi: 10.1073/pnas.0812473106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker SJ. PTEN enters the nuclear age. Cell. 2007;128(1):25–28. doi: 10.1016/j.cell.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 10.Song MS, et al. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell. 2011;144(2):187–199. doi: 10.1016/j.cell.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alimonti A, et al. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010;42(5):454–458. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trotman LC, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1(3):E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whang YE, et al. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci U S A. 1998;95(9):5246–5250. doi: 10.1073/pnas.95.9.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trotman LC, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128(1):141–156. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow MA. Sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell. 1998;92(2):253–263. doi: 10.1016/S0092-8674(00)80919-8. [DOI] [PubMed] [Google Scholar]
- 16.Lo TL, et al. 2006. Sprouty and cancer: the first terms report. Cancer Lett. 2006;242(2):141–150. doi: 10.1016/j.canlet.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 17.Holgren C, et al. Sprouty-2 controls c-Met expression and metastatic potential of colon cancer cells: sprouty/c-Met upregulation in human colonic adenocarcinomas. Oncogene. 2010;29(38):5241–5253. doi: 10.1038/onc.2010.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee CC, et al. Overexpression of sprouty2 inhibits HGF/SF-mediated cell growth, invasion, migration, and cytokinesis. Oncogene. 2004;23(30):5193–5202. doi: 10.1038/sj.onc.1207646. [DOI] [PubMed] [Google Scholar]
- 19.Guy GR, Jackson RA, Yusoff P, Chow SY. Sprouty proteins: modified modulators, matchmakers or missing links? J Endocrinol. 2009;203(2):191–202. doi: 10.1677/JOE-09-0110. [DOI] [PubMed] [Google Scholar]
- 20.Lao DH, et al. Direct binding of PP2A to Sprouty2 and phosphorylation changes are a prerequisite for ERK inhibition downstream of fibroblast growth factor receptor stimulation. J Biol Chem. 2007;282(12):9117–9126. doi: 10.1074/jbc.M607563200. [DOI] [PubMed] [Google Scholar]
- 21.Mumby M. PP2A: unveiling a reluctant tumor suppressor. Cell. 2007;130(1):21–24. doi: 10.1016/j.cell.2007.06.034. [DOI] [PubMed] [Google Scholar]
- 22.Junttila MR, et al. CIP2A inhibits PP2A in human malignancies. Cell. 2007;130(1):51–62. doi: 10.1016/j.cell.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 23.Edwin F, Singh R, Endersby R, Baker SJ, Patel TB. The tumor suppressor PTEN is necessary for human Sprouty2-mediated inhibition of cell proliferation. J Biol Chem. 2006;281(8):4816–4822. doi: 10.1074/jbc.M508300200. [DOI] [PubMed] [Google Scholar]
- 24.Gurova KV, et al. Cooperation of two mutant p53 alleles contributes to Fas resistance of prostate carcinoma cells. Cancer Res. 2003;63(11):2905–2912. [PubMed] [Google Scholar]
- 25.Chang CJ, Mulholland DJ, Valamehr B, Mosessian S, Sellers WR, Wu H. PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Mol Cell Biol. 2008;28(10):3281–3289. doi: 10.1128/MCB.00310-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao H, Dupont J, Yakar S, Karas M, LeRoith D. PTEN inhibits cell proliferation and induces apoptosis by downregulating cell surface IGF-IR expression in prostate cancer cells. Oncogene. 2004;23(3):786–794. doi: 10.1038/sj.onc.1207162. [DOI] [PubMed] [Google Scholar]
- 27.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194(1):7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem. 2005;280(42):35195–35202. doi: 10.1074/jbc.M503045200. [DOI] [PubMed] [Google Scholar]
- 29.Seeling JM, Miller JR, Gil R, Moon RT, White R, Virshup DM. Regulation of beta-catenin signaling by the B56 subunit of protein phosphatase 2A. Science. 1999;283(5410):2089–2091. doi: 10.1126/science.283.5410.2089. [DOI] [PubMed] [Google Scholar]
- 30.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding Z, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 2011;470(7333):269–273. doi: 10.1038/nature09677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeang CH, McCormick F, Levine A. Combinatorial patterns of somatic gene mutations in cancer. FASEB J. 2008;22(8):2605–2622. doi: 10.1096/fj.08-108985. [DOI] [PubMed] [Google Scholar]
- 33.Planchon SM, Waite KA, Eng C. The nuclear affairs of PTEN. J Cell Sci. 2008;121(pt 3):249–253. doi: 10.1242/jcs.022459. [DOI] [PubMed] [Google Scholar]
- 34.Freeman DJ, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3(2):117–130. doi: 10.1016/S1535-6108(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, et al. Allele-specific tumor spectrum in pten knockin mice. Proc Natl Acad Sci U S A. 2010;107(11):5142–5147. doi: 10.1073/pnas.0912524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rhee SG, Bae YS, Lee SR, Kwon J. Hydrogen peroxide: a key messenger that modulates protein phosphorylation through cysteine oxidation. Sci STKE. 2000;2000(53):pe1. doi: 10.1126/stke.2000.53.pe1. [DOI] [PubMed] [Google Scholar]
- 37.Trottier G, Boström PJ, Lawrentschuk N, Fleshner NE. Nutraceuticals and prostate cancer prevention: a current review. Nat Rev Urol. 2010;7(1):21–30. doi: 10.1038/nrurol.2009.234. [DOI] [PubMed] [Google Scholar]
- 38.Lippman SM, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2009;301(1):39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He XC, et al. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat Genet. 2007;39(2):189–198. doi: 10.1038/ng1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang S, Garcia AJ, Wu M, Lawson DA, Witte ON, Wu H. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc Natl Acad Sci U S A. 2006;103(5):1480–1485. doi: 10.1073/pnas.0510652103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lesche R, et al. 2002. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32(2):148–149. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- 42.Somers KD, et al. Orthotopic treatment model of prostate cancer and metastasis in the immunocompetent mouse: efficacy of flt3 ligand immunotherapy. Int J Cancer. 2003;107(5):773–780. doi: 10.1002/ijc.11464. [DOI] [PubMed] [Google Scholar]
- 43.van de Wetering M, et al. Specific inhibition of gene expression using a stably integrated, inducible small-interfering-RNA vector. EMBO Rep. 2003;4(6):609–615. doi: 10.1038/sj.embor.embor865. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.