

Abstract
Atherosclerosis is a chronic inflammatory disease promoted by hyperlipidemia. Several studies support FOXP3-positive regulatory T cells (Tregs) as inhibitors of atherosclerosis; however, the mechanism underlying this protection remains elusive. To define the role of FOXP3-expressing Tregs in atherosclerosis, we used the DEREG mouse, which expresses the diphtheria toxin (DT) receptor under control of the Treg-specific Foxp3 promoter, allowing for specific ablation of FOXP3+ Tregs. Lethally irradiated, atherosclerosis-prone, low-density lipoprotein receptor–deficient (Ldlr–/–) mice received DEREG bone marrow and were injected with DT to eliminate FOXP3+ Tregs. Depletion of Tregs caused a 2.1-fold increase in atherosclerosis without a concomitant increase in vascular inflammation. These mice also exhibited a 1.7-fold increase in plasma cholesterol and an atherogenic lipoprotein profile with increased levels of VLDL. Clearance of VLDL and chylomicron remnants was hampered, leading to accumulation of cholesterol-rich particles in the circulation. Functional and protein analyses complemented by gene expression array identified reduced protein expression of sortilin-1 in liver and increased plasma enzyme activity of lipoprotein lipase, hepatic lipase, and phospholipid transfer protein as mediators of the altered lipid phenotype. These results demonstrate that FOXP3+ Tregs inhibit atherosclerosis by modulating lipoprotein metabolism.
Introduction
Cardiovascular disease remains a major cause of death and morbidity despite currently available drug therapies (1). Atherosclerosis constitutes the underlying cause of the clinical manifestations of myocardial infarction, stroke, and gangrene, and is recognized as a chronic inflammatory disease involving a variety of immunological features (2). Subendothelial lipid retention and oxidative modification in the arterial wall elicit a chronic inflammatory process characterized by the formation of an immune cell infiltrate dominated by lipid-laden macrophage foam cells and T cells (2–4). The accumulation and activation of these immunological cells play a pivotal role in the development of atherosclerotic lesions.
Both innate and adaptive immunity are involved in the pathogenesis of atherosclerosis. Pattern recognition receptors of innate immunity account for cholesterol uptake and contribute to the activation of macrophages and endothelial cells. Antigen-specific T cells recognizing LDL in the intima constitute the adaptive immunity component in the pathogenesis of atherosclerosis and elicit proinflammatory stimuli that further exacerbate and propagate this disease (5). Inherent atheroprotective immune mechanisms include natural antibodies to LDL-derived phospholipids and antiinflammatory immune cells such as regulatory T cells (Tregs). Strategies to mobilize and stimulate immunosuppression may provide novel therapeutic approaches to reduce atherosclerotic cardiovascular disease (6, 7).
Tregs were initially characterized as CD4+CD25+ T cells that could suppress the activity of effector T cells (8). Further studies have identified several varieties of Tregs (9). The forkhead box transcription factor, FOXP3, was identified as the key lineage marker and master switch in the regulation of Treg development and function (10–12). FOXP3 is now accepted as the “gold standard” for defining thymic-derived Tregs (9). Accordingly, selective depletion of FOXP3+ Tregs reproduces the hyperinflamed, scurfy phenotype observed in Treg-deficient mice (13). In contrast, depletion of CD4+CD25+ T cells by administration of an antibody targeting the interleukin-2 (IL-2) receptor α-chain (CD25) on the surface of T cells failed to reproduce the fulminant disease phenotype of scurfy mice (14, 15). This indicates that the CD4+CD25+ T cell population only partly overlaps with the FOXP3+ Treg cell population.
Transfer experiments in immune-deficient, hyperlipidemic mice show that CD4+CD25+ cells with features of natural Tregs are atheroprotective (16). Accordingly, IL-10 and TGF-β, the 2 major cytokine products of Tregs, are powerful inhibitors of the atherosclerotic process in similar mouse models (17–20). Several questions remain, however, with respect to the mechanism of how Tregs impact atherosclerosis progression (21). To address these questions, we have adapted a model that permits conditional depletion of Tregs in a mouse model of atherosclerosis. The recently described DEREG (depletion of regulatory T cells) mouse (13) expresses a fusion protein of the human diphtheria toxin (DT) receptor (hDTR) and enhanced green fluorescent protein (eGFP) under control of the Foxp3 promoter introduced by BAC technology. The expression of eGFP under control of the Foxp3 promoter permits tracking of transgenic eGFP+ Tregs. FOXP3+ Tregs can be selectively depleted upon DT administration in DEREG mice, since wild-type mice do not express the hDTR receptor and are thus insensitive to DT.
We transplanted bone marrow from DEREG mice into lethally irradiated, atherosclerosis-prone, LDL receptor-deficient (Ldlr–/–) mice, identified eGFP+ Tregs in atherosclerotic lesions, administered DT to ablate FOXP3+ Tregs, and analyzed the effects on immunity, metabolism, and atherosclerosis. Depletion of Tregs led to a dramatic increase in atherosclerotic lesions, but did not increase vascular inflammation. However, it associated with impaired clearance of large lipoproteins, leading to a 70% increase in plasma cholesterol, with a robust increase in VLDL levels. These data identify a previously unknown function of Tregs as modulators of lipid metabolism.
Results
Long-term DT treatment is tolerated in DEREG mice.
We achieved effective depletion of 99% of transgenic eGFP+ Tregs in peripheral blood for more than 3 days after i.p. administration of a single dose (0.125 μg/mouse) of DT to DEREG mice (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI63891DS1). Based on these findings, a twice-weekly DT administration regimen was chosen for all subsequent experiments. To evaluate the long-term safety in DEREG mice, DT was administered i.p. twice weekly for 8 weeks to DEREG and wild-type mice at 0.125 or 0.25 μg/mouse (20 g body weight). The higher DT dose was found to be associated with signs of toxicity (fatigue, changes in fur appearance, loss of appetite), while no obvious adverse effects were observed at the lower dose in DEREG or wild-type mice. Accordingly, the low DT dose was chosen for long-term administration in the atherosclerosis study.
DT administration eliminates functional Tregs in chimeric DEREG/Ldlr–/– mice.
DEREG bone marrow was transplanted into lethally irradiated Ldlr–/– mice that received an atherogenic diet to promote hypercholesterolemia and atherosclerosis. One group of the chimeric DEREG/Ldlr–/– mice received DT twice weekly for 8 weeks to eliminate FOXP3+ Tregs. The FOXP3-driven coordinate expression of eGFP and hDTR permitted an identification of FOXP3+ cells in bone marrow chimeras not receiving DT and an evaluation of the efficiency of Treg depletion in the DT-treated mice.
Effective depletion of transgenic (eGFP+) Tregs in inguinal lymph nodes (Figure 1A) and lymphoid organs (not shown) was achieved by DT administration to chimeric DEREG/Ldlr–/– mice. The suppressor function of total Tregs was markedly impaired, as documented by robustly increased proliferation of lymph node cells from mice treated with DT versus PBS (Figure 1B).
Figure 1. Effective depletion of transgenic Treg cells is achieved in chimeric DEREG/Ldlr–/– mice.

(A) Proportions of cells expressing eGFP within the CD3+CD4+ population in inguinal lymph nodes from chimeric DEREG/Ldlr–/– mice treated with DT or PBS harvested at 4 and 8 weeks, respectively (n = 6–7 mice per group). (B) Proliferation of lymph node cells from chimeric DEREG/Ldlr–/– mice treated for 8 weeks with DT or PBS and stimulated in vitro with anti-CD3. Data show stimulation index based on 3H-thymidine uptake (n = 4 mice per group). *P < 0.05.
In a separate experiment, chimeric DEREG/Ldlr–/– mice were treated with DT versus PBS and bled at different intervals after initiation of treatment. Flow cytometric data on the kinetics of Tregs revealed that over time, depletion of eGFP+ Tregs became less effective. Ten days after initiating DT administration, complete depletion of transgenic Tregs was achieved, followed by a gradual recovery of these GFP+FOXP3+ cells. The total number of FOXP3+ cells was not substantially reduced due to a compensatory increase in GFP–FOXP3+ cells (Supplemental Figure 1, B–D). Plasma levels of the inflammatory marker, serum amyloid A (SAA), did not differ between groups (Table 1). DT treatment thus led to persistently perturbed immunoregulation with increased T cell activity, though without signs of overt systemic inflammation after 8 weeks of treatment.
Table 1.
Body weight, lipid profile, and hematological parameters in bone marrow chimeras: effect of Treg depletion (DT) versus control (PBS)
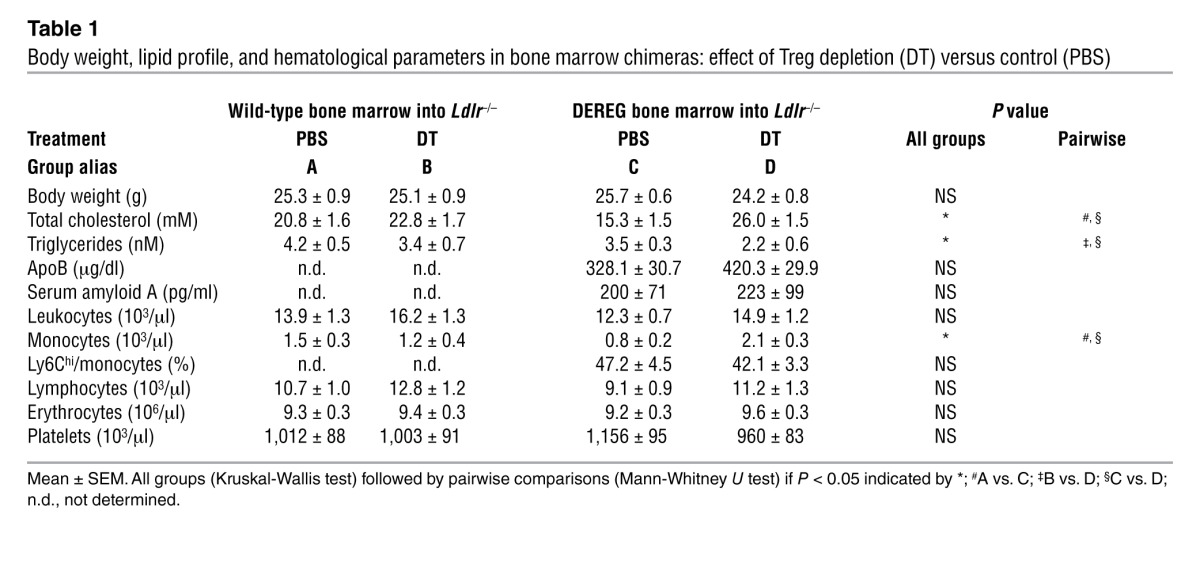
To define the source of eGFP– (nontransgenic) FOXP3+ Tregs, we transplanted DEREG bone marrow (on a C57Bl/6 background, thus expressing the CD45.2 isoform) into Ldlr–/– mice carrying the CD45.1 surface marker. FACS analysis of spleen cells showed that eGFP– (nontransgenic) FOXP3+ Tregs were predominantly derived from the CD45.1+ recipient Ldlr–/– mice (Supplemental Figure 2, A and B). Furthermore, nontransgenic CD45.1+ eGFP– cells predominated among FOXP3+ cells in the DT-treated mice, whereas CD45.2+ eGFP+ cells constituted the majority of FOXP3+ cells in PBS controls. This suggests a replenishment of the total FOXP3+ Treg pool from this niche that could only partially compensate for the loss of functional FOXP3+ Tregs upon DT treatment (Supplemental Figure 1, B–D). This interpretation is in line with the recent finding of functionally defective cell subsets carrying certain Treg markers in DEREG mice (22).
Treg depletion aggravates atherosclerosis in chimeric DEREG/Ldlr–/– mice.
DT treatment of chimeric DEREG/Ldlr–/– mice caused a pronounced 2.1-fold increase in the size of atherosclerotic lesions in the aortic sinus (3.23 × 105 ± 0.39 × 105 μm2 vs. 1.54 × 105 ± 0.21 × 105 μm2; P = 0.0026) and a 1.9-fold increase in relative lesion area (lesion cross-sectional area/total vessel area; 30.0 ± 3.1% vs. 16.0 ± 2.0%; P = 0.003) compared with PBS-treated controls (Figure 2). DT treatment did not affect lesion size in Ldlr–/– mice reconstituted with wild-type bone marrow (Figure 2C), indicating that the augmented lesion development upon DT treatment of DEREG/Ldlr–/– mice was due to the loss of Treg activity.
Figure 2. Depletion of transgenic Tregs aggravates atherosclerosis.

(A) Representative photomicrographs showing Oil Red O- and H&E-stained sections from the proximal aorta of chimeric DEREG/Ldlr–/– mice treated for 8 weeks with PBS or DT. (B) Quantification of lesion size in cross-sections of the proximal aorta at different levels from the valves; n = 11–12 (PBS) mice and n = 5–8 (DT) mice, respectively. (C) Relative lesion area (lesion area/area inside external elastic lamina × 100) calculated from 4 sections per mouse (300–600 μm) for wild-type/Ldlr–/– and DEREG/Ldlr–/– mice treated for 8 weeks with PBS or DT, respectively. *P < 0.05; **P < 0.01. Scale bars: 100 μm.
DT-mediated Treg depletion led not only to increased atherosclerotic lesion size, but was also associated with reduced immune cell infiltration into atherosclerotic lesions. Immunohistochemical analysis identified eGFP+ Treg–expressing FOXP3 in atherosclerotic lesions of PBS-treated DEREG/Ldlr–/– mice (Figure 3A). DT treatment resulted in complete disappearance of eGFP+ cells, while residual FOXP3+ cells and total CD3+ T cells remained unaltered in the lesions and vessel wall (Figure 3, A and B, and Supplemental Figure 3). There was a significant reduction in CD68+-stained area (macrophages) per lesion surface area (Figure 3C) and similarly reduced areas staining positive for cells expressing the murine MHC class II molecule, IAb, respectively (Figure 3B). A decrease in total cellularity of lesions with fewer macrophages in DT-treated mice when calculated as the total number of CD68+ cells per lesion surface area was paralleled by an increase in the necrotic core size (Figure 3, D and E, and data not shown).
Figure 3. Depletion of Tregs affects cellular composition of atheroma.

(A) Representative fluorescence micrographs depicting eGFP+FOXP3+ cells (arrowheads) and eGFP–FOXP3+ (arrows) cells in aortic lesions of DEREG/Ldlr–/– mice treated for 8 weeks with PBS or DT. Anti-GFP was labeled with AlexaFluor 488 (green), anti-FOXP3 with AlexaFluor 555 (red), and nuclei with DAPI (blue). (B) Quantitation of immunohistochemical staining for T cells (CD3+), Tregs (FOXP3+), and I-Ab–expressing (MHCII-expressing) cells (all expressed as stained cells per lesion area) in the proximal aorta of chimeric DEREG/Ldlr–/– mice treated for 8 weeks with DT or PBS; n = 7–8 mice per group. (C) Quantitation of immunohistochemical staining for macrophages (CD68+), dendritic cells (CD11c+), and expression of the adhesion molecule VCAM-1 (all expressed as stained area per lesion area) in the aortic sinus of chimeric DEREG/Ldlr–/– mice treated for 8 weeks with DT or PBS; n = 7–8 mice per group. (D) Cellularity of atherosclerotic lesions (DAPI+ nuclei per lesion area) in the proximal aorta of chimeric DEREG/Ldlr–/– mice treated for 8 weeks with PBS or DT; n = 7–8 mice per group. (E) Necrotic core area relative to total atherosclerotic lesion area in the proximal aorta of chimeric DEREG/Ldlr–/– mice treated for 8 weeks with PBS or DT; n = 7–8 mice per group. *P < 0.05; **P < 0.01. Scale bars: 50 μm.
Treg depletion aggravates hypercholesterolemia and monocytosis in chimeric DEREG/Ldlr–/– mice.
Effects on plasma lipids were dramatic, with a 70% increase in plasma cholesterol concentration paralleled by a significant decrease in plasma triglycerides (Table 1 and Figure 4A). The increase in cholesterol was detectable already during the first month of treatment, suggesting that it was causally related to the increase in lesion size observed after 8 weeks (Figure 4A).
Figure 4. DT-induced depletion of transgenic Tregs promotes hypercholesterolemia.

(A) Changes in plasma cholesterol levels during treatment of DEREG/Ldlr–/– bone marrow chimeras with DT or PBS; n = 5 per group. *P < 0.05; **P < 0.01. (B) Fast protein liquid chromatographic (FPLC) analysis of plasma lipoprotein profiles from DEREG/Ldlr–/– bone marrow chimeras treated for 8 weeks with PBS (gray line) or DT (black line). The cholesterol concentration in each fraction (y axis) is plotted against retention time (x axis), with the corresponding lipoprotein fractions (identified by human plasma standards) indicated at the top. Mean (thick line) and SEM (fine lines) are shown; n = 5 per group. (C) FPLC analysis of plasma lipoprotein profiles from DEREG/Ldlr–/– bone marrow chimeras treated for 8 weeks with PBS (gray line) or DT (black line). The triglyceride concentration in each fraction (y axis) is plotted against retention time (x axis), with the corresponding lipoprotein fractions (identified by human plasma standards) indicated at the top. Mean (thick line) and SEM (fine lines) are shown; n = 5 per group. (D) Spearman’s rank correlation analysis for VLDLs and atherosclerotic lesion size in the proximal aorta. Each dot represents an individual mouse and the curve is plotted with the corresponding correlation coefficient (r value) displayed. DT is represented by black circles; PBS is represented by gray circles.
Gel filtration analysis revealed a pronounced elevation of cholesterol in the VLDL/chylomicron remnant (CMR) fraction in DT-treated chimeric DEREG/Ldlr–/– mice (Figure 4B), whereas triglyceride content was lower in the VLDL/CMR particles (Figure 4C). Plasma VLDL/CMR cholesterol levels strongly correlated with lesion size (r = 0.956; P = 0.0006), as did total cholesterol and LDL cholesterol (Figure 4D and Supplemental Figure 4, A and B). Upon DT treatment, plasma apoB concentration was numerically increased, suggesting an increased number of VLDL particles rather than a larger amount of cholesterol per particle (Table 1). DT treatment affected neither body weight nor plasma markers, nor liver or kidney function (data not shown). DT-mediated elimination of Tregs from DEREG/Ldlr–/– mice was associated with markedly increased monocyte counts in peripheral blood (Table 1). No difference was detected in the distribution of monocyte subpopulations (Table 1). Interestingly, total monocyte counts in peripheral blood showed a positive correlation with lesion size (Supplemental Figure 4C), and an inverse correlation was found with GFP+ Tregs (Supplemental Figure 4D).
Plasma lipids were analyzed in 4 treatment groups of chimeric mice: recipient Ldlr–/– mice receiving wild-type bone marrow and treated with (a) PBS or (b) DT; or Ldlr–/– mice receiving DEREG bone marrow and treated with (c) PBS or (d) DT. Of note, DT treatment had no significant effect on atherosclerotic lesions (Figure 2C), total cholesterol, triglycerides, body weight, or hematological parameters (Table 1). This corroborates previous work showing that lipid levels in Apoe–/– mice are not affected by DT administration (23).
Treg depletion impairs lipoprotein catabolism.
To determine the effects of Treg depletion on VLDL biosynthesis in the liver, we analyzed plasma lipids after administering Triton WR-1339, which inhibits lipoprotein lipase–dependent (LPL-dependent) lipoprotein catabolism. Cholesterol levels in plasma rose to a similar extent in both groups (Figure 5A), suggesting that the elevated VLDL levels were not due to increased biosynthesis in the liver.
Figure 5. VLDL/CMR lipoprotein catabolism is impaired in Treg-depleted mice.

(A) Biosynthesis of apoB-containing plasma lipoproteins. Data show plasma cholesterol levels in chimeric DEREG/Ldlr–/– mice treated for 8 weeks with DT or PBS followed by i.v. administration of Triton WR-1339 to inhibit lipoprotein lipase–dependent VLDL catabolism; n = 9 (PBS) and n = 7 (DT), respectively. (B) Clearance of injected FITC-VLDL in chimeric DEREG/Ldlr–/– mice treated for 8 weeks with DT or PBS. FITC-derived fluorescence was analyzed in plasma samples at the indicated time points. Data for each individual were normalized to the fluorescence of plasma taken 1 minute after injection; n = 4 per group. (C) In vivo turnover of CM particles injected into chimeric DEREG/Ldlr–/– mice treated for 8 weeks with DT or PBS. Data show kinetics of the CM [14C] retinol core particle clearance from blood and are expressed as radiolabeled moieties corrected for weight; n = 5 (PBS); n = 7 (DT). (D) CM [14C] retinol uptake in the liver. Data expressed as radiolabeled moieties corrected for weight; n = 5 (PBS); n = 7 (DT). *P < 0.05.
VLDL catabolism was assessed after i.v. injections of FITC-labeled mouse VLDLs. This analysis revealed significantly impaired clearance of VLDL particles in Treg-depleted DEREG/Ldlr–/– mice (Figure 5B). The gel filtration approach cannot resolve VLDLs from CMR particles, and we therefore analyzed chylomicron (CM) catabolism by using a double-isotope technique that permitted evaluation of triglyceride and particle elimination. Rats received [14C]-oleate and [3H]-retinol by intragastric infusion; CMs containing [14C]-triglyceride and [3H]-retinol were isolated and injected into DEREG/Ldlr–/– mice treated with PBS or DT. Analysis at different time points after injection showed delayed clearance of retinol without concomitant changes in triglycerides, reflecting a putative defect in core particle turnover (Figure 5C and Supplemental Figure 5). Liver uptake of [3H]-retinol was significantly reduced, indicating hampered uptake of CM particles in DT-treated animals (Figure 5D). Together, our studies of lipoprotein metabolism show that the elevated plasma cholesterol in DT-treated DEREG/Ldlr–/– mice was a result of reduced clearance of VLDL and CM remnants.
Treg depletion modulates expression of liver genes involved in lipoprotein metabolism.
To assess the effect of Treg depletion on genes controlling metabolism, a global gene expression array was performed on mRNA isolated from the 2 major organs involved in lipoprotein metabolism — liver and intestine — comparing DT- versus PBS-treated chimeric DEREG/Ldlr–/– mice. It revealed that Treg elimination caused substantial changes in the expression pattern of genes regulating metabolism as well as inflammation and immunity in liver (Figure 6), whereas no significant changes were detected in intestine (data not shown). In the 2 latter groups of genes (clusters 2, 3, 5, 6, and 7), expression was increased for virtually all genes in DT-treated animals, whereas a more complex pattern was observed for metabolic genes (clusters 1 and 4).
Figure 6. Gene expression array analysis shows changes in liver transcriptomes upon Treg depletion.

Shown here are gene expression heat maps of global Affymetrix gene expression array analysis of liver RNA from DEREG/Ldlr–/– mice. The rows correspond to genes and the columns to individual mice treated with DT or PBS, respectively. Relative gene expression is shown based on normalization for each gene in the DT- and PBS-treated groups. Color coding indicates increased gene expression in red, and decreased expression in blue compared with the other group, respectively. Sets of genes involved in functional annotation clusters as defined by the DAVID annotation analysis are grouped accordingly. Arbitrary titles that summarize the functional role of displayed genes in a cluster are shown in parentheses. Clusters show differentially expressed transcripts based on an FDR less than 0.1 and an absolute linear fold change greater than 1.5.
Quantitative PCR analysis of specific mRNA species showed increased expression of the 2 proinflammatory cytokines IFN-γ and TNF-α and of the T cell marker CD3, implying increased Th1-type inflammatory activity in the liver of DT-treated chimeric mice (Figure 7). In contrast, no effects were registered for genes involved in NKT cell control of lipase activity (Supplemental Figure 6), although this pathway was previously reported to modulate lipoprotein metabolism (24).
Figure 7. Treg depletion modulates expression of genes controlling inflammation and lipid metabolism in the liver.

Quantitative real-time RT-PCR analysis of liver mRNA from DEREG/Ldlr–/– mice treated for 8 weeks with DT or PBS. Signals were normalized to Hprt. *P < 0.05; n = 6 per group. (A) Ifng, (B) Tnfa, (C) Cd3, (D) Apoc3, (E) Apob, (F) Dgat, (G) Lpl, (H) Lipc, (I) Vldlr, (J) Pltp, (K) Sort1, and (L) Atf3.
No changes were observed in the mRNA levels of a set of genes involved in lipoprotein biosynthesis, including Apo3, Apob, and diacylglycerol acyltransferase (Dgat) (Figure 7). In contrast, several genes involved in lipoprotein catabolism showed differential expression upon Treg depletion (Figure 7). Lpl and hepatic lipase (Lipc) were both upregulated, as was the VLDL receptor. mRNA for phospholipid transfer protein (Pltp) was also significantly increased. In contrast, mRNA for the VLDL binding protein, sortilin-1, was significantly reduced and activating transcription factor-3 (Atf3), a transcription factor that represses sortilin-1 expression, was robustly enhanced in the livers of Treg-depleted mice (Figure 7). Western blot analysis confirmed that sortilin-1 protein was significantly reduced under these conditions, indicating that sortilin-1 is a target of Treg depletion (Figure 8, A and B).
Figure 8. Effects of Treg depletion on sortilin-1, LPL, HL, and PLTP.

(A) Immunoblot of liver tissue showing sortilin-1 protein in DT- or PBS-treated DEREG/Ldlr–/– mice. Full uncut images of sortilin-1 and tubulin immunoblots are shown in Supplemental Figure 7. (B) Scattergram shows individual values expressed as a ratio of sortilin-1 protein expression to α–tubulin protein expression. (C) LPL activity in postheparin plasma was measured using [3H]-labeled triolein in an emulsion with Intralipid 10% composition as the enzyme substrate; n = 10 (PBS); n = 9 (DT). (D) HL activity in postheparin plasma was measured using a gum arabic–stabilized emulsion; n = 10 (PBS); n = 9 (DT). (E) PLTP activity was measured using an exogenous lipoprotein-independent phospholipid transfer assay; n = 10 (PBS); n = 10 (DT). All data are from DEREG/Ldlr–/– mice treated for 8 weeks with DT or PBS. Mean ± SEM is shown. *P < 0.05; **P < 0.01.
Treg depletion increases activity of LPL, hepatic lipase, and PLTP in plasma.
To further evaluate factors involved in VLDL/CMR metabolism, we analyzed LPL and hepatic lipase (HL) activity in postheparin plasma (Figure 8, C and D). The lipolytic activities of these 2 enzymes were significantly increased in DT-treated chimeric DEREG/Ldlr–/– mice. Plasma PLTP activity was also significantly increased in Treg-depleted bone marrow chimeras (Figure 8E). In contrast, lecithin:cholesterol acyl transferase (LCAT) activity was not affected (data not shown). Together, these alterations may explain the difference in clearance between triglycerides and core particle labels resulting in triglyceride-poor VLDL particles in the circulation of Treg-depleted mice.
Discussion
Regulatory immunity has been implicated in atherosclerosis, but the mechanism and target organ(s) of atheroprotection remain largely unknown. We now show in the chimeric DEREG/Ldlr–/– model that: (a) depletion of FOXP3+ Treg leads to substantially increased atherosclerosis; (b) elimination of FOXP3+ Tregs aggravates hypercholesterolemia; and (c) VLDLs and CMRs are dramatically elevated in plasma due to reduced clearance in the absence of the LDL receptor. Together, these findings establish that FOXP3+ Tregs exert an important atheroprotective effect, and show that plasma lipoprotein metabolism is modulated in part by regulatory immunity.
Previous studies demonstrated that regulatory immunity affects atherosclerosis progression. Elimination of CD4+CD25+ cells increases disease severity, and the transfer of T cells from Cd28–/– mice is associated with reduced numbers of CD4+CD25+ natural Tregs accelerating atherosclerosis in Ldlr–/– mice (16). Similarly, Ldlr–/– mice deficient in ICOS displayed decreased FOXP3+ T cells and increased atherosclerosis (25), while expansion of the circulating Treg pool in Apoe–/– mice by CD31 administration reduced atherosclerosis (26), as did the adoptive transfer of CD4+CD25+ cells from atherosclerotic to disease-prone mice (27). Furthermore, expansion of another regulatory T cell pool, the Tr1 cell population, by global or antigen-specific methods, also reduces atherosclerosis in Apoe–/– mice (28, 29). While all these studies highlight the role of regulatory cellular immunity in atherosclerosis, direct evidence for atheroprotective actions of FOXP3-driven immunity is lacking.
We now provide such evidence by using the DEREG model. We had expected that elimination of Tregs would unleash plaque inflammation, causing enhanced production of several proinflammatory mediators. Instead, immune cells and local inflammatory markers in the lesions were either unchanged or reduced when Tregs were depleted, which is likely attributable to the advanced stage of lesions with low cellularity in DT-treated mice. Circulating levels of the inflammatory marker SAA remained unaltered, and while monocyte counts were elevated, no changes were registered between monocyte subclasses. Together, these data suggest that modulation of vascular inflammation may not be the only, and perhaps not even the major, target of Treg control in atherosclerosis.
DT treatment of DEREG/Ldlr–/– mice profoundly reduced the population of GFP+FOXP3+ Tregs. This led to a more than 3-fold increase in effector T cell proliferation due to loss of suppressor activity. However, it did not cause any substantial reduction in the total number of FOXP3+ T cells. Bone marrow transplantation (BMT) experiments using CD45 isotypic markers clarified that this was due to an expansion of FOXP3+ T cells derived from the recipient. This, in turn, may reflect that FOXP3+ cells are less sensitive to irradiation used in the BMT protocol than other T cell types. In line with previous data (22), the robust effect of DT treatment on T effector cell proliferation shows that the recipient-derived FOXP3+ T cells were not effective as suppressor cells, possibly reflecting that they were not fully differentiated Tregs.
In addition to these changes in FOXP3+ cell populations, it was apparent that the depletion of transgenic FOXP3+ Tregs (GFP+FOXP3+) waned over time despite continued DT treatment. This may reflect genetic alterations in the artificial Foxp3 promoter, an increasingly efficient DT metabolism in the recipients, or immune reactions to DT. At any rate, our data show that selective depletion of Tregs over an extended period is possible by using the DEREG/DT strategy; this permits studies of chronic pathological processes such as atherosclerosis.
The phenotypic screen of DEREG/Ldlr–/– chimeras revealed increased hypercholesterolemia upon Treg depletion, with a 1.7-fold increase in total plasma cholesterol. This was due to a dramatic increase in large, cholesterol-rich, VLDL/CMR–sized particles. Since such particles are highly atherogenic in Ldlr–/– mice (30), it is likely that their accumulation in blood crucially contributed to the accelerated atherosclerosis observed when Tregs were depleted. This notion is supported by the strong correlation between VLDL cholesterol concentration in plasma and lesion size. This, in turn, suggests that Tregs under normal conditions control VLDL cholesterol levels and that this contributes to their atheroprotective role.
The clearance of injected VLDLs and CMs was reduced in Treg-ablated DEREG/Ldlr–/– mice. In contrast, neither lipoprotein secretion nor mRNA expression of enzymes involved in the VLDL biosynthesis pathway was affected by Treg depletion. Therefore, Tregs modulate blood lipid levels by regulating lipoprotein catabolism.
VLDLs and CM particles are catabolized in a complex process that involves hydrolysis of triglycerides by several lipases including LPL and HL, and uptake of the remaining cholesterol-rich particles through several receptors, mainly in the liver. Among them, Vldlr mRNA was increased, Lrp1 mRNA was unchanged, and Ldlr was eliminated by gene targeting in the present study. However, sortilin-1, a receptor involved both in intracellular processing and cell surface uptake of VLDLs, was significantly reduced, both in mRNA and protein levels. Furthermore, mRNA for ATF3, a sortilin-inhibiting transcription factor, was increased, supporting the notion that sortilin-1 expression was attenuated in Treg-depleted mice.
Sortilin-1 was recently identified as a major regulator of plasma cholesterol in a genome-wide association study (31). It acts intracellularly to modulate VLDL secretion by binding apoB-containing particles in the Golgi apparatus and targeting them to a lysosomal compartment (32). In addition, sortilin-1 operates on the hepatic cell surface, where it binds apoB-containing particles and guides them to lysosomal degradation (32). Our current data showing that the hypercholesterolemic phenotype was associated with reduced sortilin-1 expression is in line with these actions of sortilin-1 as a lipoprotein-regulating receptor. It is likely that the lack of functional LDL receptors in the chimeric mice augmented the lipoprotein-clearing action of sortilin-1. The fact that the reduction in VLDL secretion did not reach significance may reflect limitations in the assay, or it may mean that the effect on particle clearance was more important in the present model.
Nascent VLDL particles are modified by the lipolytic enzymes, HL and LPL. Both lipases were upregulated, offering an explanation for the accumulation of cholesterol-rich and relatively triglyceride-poor particles in Treg-depleted mice. Since sortilin-1–dependent clearance was reduced, these lipoprotein particles continued to circulate while triglycerides were persistently catabolized by the lipases, which showed elevated expression and activity.
Increased PLTP activity may also contribute to the observed phenotype. PLTP transfers phospholipids between lipoproteins, modulates HDL composition, and can increase VLDL levels (33–35). This results in an atherogenic lipoprotein profile with elevated VLDL and reduced HDL, increased plasma cholesterol, and impaired reverse cholesterol transport. Accordingly, hypercholesterolemic mice carrying a PLTP transgene develop accelerated atherosclerosis (36, 37), whereas PLTP deficiency protects from disease (38).
In summary, we provide evidence that Treg depletion results in significantly accelerated atherosclerosis and an atherogenic lipoprotein profile that is mediated by reduced clearance of large lipoprotein particles. Our findings highlight that immune mechanisms have an impact on metabolic events and show that chronic inflammation can promote cardiovascular disease by instigating metabolic disturbances.
Methods
Animals and treatment protocols.
DEREG mice on a C57BL/6 background were previously generated (13) and were maintained and used as hemizygous transgenics. Offspring were typed by flow cytometry for the presence of an eGFP+ cell population within the CD3+CD4+ population of peripheral blood cells. Bone marrow cells, isolated from the femur and tibia of male 8- to 10-week-old DEREG– or DEREG+ littermates were i.v. injected (7.5 × 106 cells per mouse) into lethally irradiated (14 Gy) 6- to 8-week-old Ldlr–/– mice (The Jackson Laboratory). After recovery for 6 weeks, Ldlr–/– mice were fed a high-cholesterol diet (1.25% cholesterol, 0% cholate; Research Diets). Mice were randomly assigned to treatment with DT (6.25 mg/kg body weight, i.p. injection 2 times per week; Merck, no. 322326) or control (100 μl of endotoxin-free PBS containing 0.1% BSA). Following 8 weeks of treatment, the mice were anesthetized by CO2 asphyxiation and their organs were harvested. The treatment groups comprised:
Group A: Wild-type bone marrow injected into Ldlr–/– mice treated for 8 weeks with PBS; i.p. injection.
Group B: Wild-type bone marrow injected into Ldlr–/– mice treated for 8 weeks with DT; i.p. injection.
Group C: DEREG bone marrow injected into Ldlr–/– mice treated for 8 weeks with PBS; i.p. injection.
Group D: DEREG bone marrow injected into Ldlr–/– mice treated for 8 weeks with DT; i.p. injection.
For certain studies regarding the origin of FOXP3+ cells, Ldlr–/– mice were crossed with CD451/1 mice (B6.SJL-Ptprca Pepcb/BoyJ, stock-nr: 002014, The Jackson Laboratory) to generate Ldlr–/–/CD451/1 mice which then received bone marrow from DEREG mice on a regular C57BL/6 background (CD452/2). Staining for CD45.1/CD45.2 was performed as with other cell surface markers using antibodies directed against CD45.1 and CD45.2 (eBioscience).
Tissue preparation and histological analysis.
To evaluate atherosclerotic lesion size, a total of 8 cryosections (10 μm) extending cranially from the aortic valves at 100-μm intervals were prepared, stained with Oil Red O and hematoxylin, and analyzed using Leica Q500MC image analysis software (19). Lesion area per cross-section and relative area of lesions were quantified and the results expressed as the mean of 4 consecutive sections per mouse available for all individual mice. In brief, relative lesion area was calculated for each section as F% = (100 × L/A), where L is lesion area and A is area inside the external elastic lamina. F% is averaged over all levels analyzed (300–600 μm2 above aortic cusps), and the mean is calculated for each treatment group. This method eliminates artifacts caused by oblique sections. Necrotic core was expressed as acellular area per lesion area.
Immunolocalization studies.
Primary antibodies used for immunohistochemistry comprised anti-CD68 (AbD Serotec), anti–VCAM-1 and anti–I-Ab (both BD Biosciences), anti-CD3 (Southern Biotechnology), and anti-FOXP3 (eBioscience); all of them were rat IgG to mouse antigens. Isotype-matched antibodies were used as controls. Antibodies were titrated to optimum performance and applied to acetone-fixed cryosections followed by detection using an ABC peroxidase kit and DAB substrate (both from Vector Laboratories). Immunofluorescence was performed on aortic sections as described (13). Quantification of stainings was documented as the ratio of thresholded chromogen to fluorogen area or number of stained cells per intimal lesion area and nucleated-positive (DAPI) area. Quantitative image analysis was performed independently by 2 investigators blinded to the study protocol using either Quantimed (Leica) or analySIS (Olympus) software.
Flow cytometry.
Flow cytometry was performed on leukocytes isolated from blood, spleen, or inguinal lymph nodes. Primary labeled antibodies were used for extracellular staining against CD3, CD4, and CD45 isoforms CD45.1 and CD45.2 (all from BD Biosciences) and CD25 (eBioscience). Intracellular staining was performed using the anti–mouse/rat FOXP3 Staining Set (eBioscience), rabbit anti-GFP (Invitrogen), and anti-FOXP3 (eBioscience). The staining procedure to detect monocyte subclasses was performed essentially as described (39). Briefly, cells were incubated for 15 minutes at room temperature with FcBlock (anti–CD16/CD32; eBioscience) and fixable Aqua LIVE/DEAD stain (Invitrogen). Subsequently, the cells were washed and incubated for 45 minutes at room temperature with the following antibodies: anti-Ly6G, anti-CD49b, anti-CD19, anti-CD3, anti NK1.1 (all PE-conjugated), anti–CD11b-PECy7, and anti–Ly6C-FITC (all from eBioscience). All samples were washed, fixed in 1% formalin, and analyzed on a CyAn ADP Analyzer using Summit version 4.3 software (both Beckman Coulter).
Functional immune assays.
Inguinal lymph node cells were isolated by meshing lymph nodes from individual mice through a 100 μm nylon mesh followed by osmotic lysis of red blood cells (EL buffer; QIAGEN). Cells were washed in PBS, cultured in duplicate in a 96-well plate at 5 × 105 cells per well in IMDM containing 5% FCS, and incubated in the absence or presence of rat anti-mouse CD3 antibody (1 μg/ml; SouthernBiotech) for 72 hours. During the last 18 hours, 3H-thymidine (10 μCi/ml; GE Healthcare) was added to the cultures for evaluation of cell proliferation using a scintillation counter (PerkinElmer).
Hematology and plasma biochemistry.
Whole blood (EDTA) was analyzed by a Scil Vet abc hemocounter (Scil). Clinical biochemistry profiling of plasma was made on a Vitros DT60II Chemistry System (Ortho Clinical Diagnostics, Johnson & Johnson Co.) as described previously (40).
Lipoprotein analysis.
Lipoprotein profiles were analyzed using a micro-FPLC column (30 × 0.32 cm Superose 6B; GE Healthcare) coupled to a system for online separation and subsequent detection of cholesterol (41). Plasma cholesterol and triglycerides were measured using enzymatic colorimetric kits (Randox Laboratories) according to the manufacturer’s protocol. ApoB concentrations were measured by a commercial ELISA following the manufacturer’s instructions (ALerCHEK).
Analysis of lipoprotein catabolism and biosynthesis.
For in vivo clearance, VLDLs (d = 1.006 – 1.019 g/ml) were isolated by ultracentrifugation from pooled plasma of Apoe–/– mice in the presence of 2 mM benzamidine, 0.5 mM PMSF, and 0.1 U/ml aprotinin. After isolation, VLDLs were dialyzed against PBS containing 1 mM EDTA and labeled using a modified version of a previously described method (42). In brief, 2 mg/ml VLDL was dialyzed overnight against 0.5 M NaHCO3 pH 9.5. Next, 50 μg of FITC (Sigma-Aldrich) dissolved in DMSO (1 mg/ml) was added for each milligram of protein in VLDLs and incubated at room temperature for 2 hours. Conjugates were separated from free fluorochrome by gel filtration using a PD10 column (GE Healthcare) eluted with PBS. FITC conjugation was evaluated by absorption spectroscopy at 495 nm against an FITC standard curve. Protein concentration was determined by the Bradford assay (Bio-Rad) and adjusted to 2 mg/ml. Of this preparation, 100 μl was injected into the tail vein and blood samples were collected into EDTA-coated tubes 1, 5, 15, 30, and 60 minutes after injection. After centrifugation (1,500 g for 5 minutes), plasma samples were diluted 1:20 in PBS and fluorescence (excitation 485 nm, emission 535 nm) was analyzed using a Fluoroskan Ascent FL plate fluorescence reader (Thermo Scientific). Data presented are normalized to the fluorescence measured in samples obtained at 1 minute (= 100 %).
VLDL synthesis was assessed after irreversibly blocking VLDL catabolism (43). Briefly, fasted mice received Triton WR-1339 (500 mg/kg i.v. tyloxapol; Sigma-Aldrich) using a 10% (w/v) solution and were bled from the tail into EDTA-coated tubes at the indicated time points. Triglycerides and cholesterol were measured in plasma as described above.
For in vivo measurements of CM clearance, uncirculated double-labeled ([3H]-retinol ester and [14C]-triacylglycerol) CMs were isolated from rat intestinal lymph through cannulation of the thoracic duct and handled as described elsewhere (44). Chimeric DEREG/Ldlr–/– mice treated for 8 weeks with DT or PBS and fed a high-cholesterol diet were injected with CMs corresponding to 2 mg triglycerides through the tail vein, and blood was collected after 1, 3, 7, and 15 minutes from the retro-orbital vein plexus in temporarily isoflurane-anesthetized animals. After the final bleeding, the mice were sacrificed and their tissues were collected and rinsed in PBS. Blood samples were extracted for esterified lipids as previously described (44), traced for the radiolabeled moieties, and corrected for weight. Tissue samples were homogenized in chloroform/methanol (2:1) using tubes with ceramic beads, total lipids were extracted, and the tissues were adjusted for weight.
LPL and HL were released into the plasma with heparin injections (50 U/kg), and postheparin blood was collected from the tail vein after 15 minutes into heparin-coated tubes. Plasma was collected by ultracentrifugation and stored at –80°C. LPL activity was assessed in a system using [3H]-labeled triolein in an emulsion with Intralipid 10% composition as the enzyme substrate. Rat serum was added as apoCII donor (45). HL was inhibited using antibodies against rat HL (46). Specific HL activity was measured using a gum arabic–stabilized emulsion (47). Postheparin plasma lipase activity was determined as the mean of triplicate readings. Lipase activity is expressed as mU/ml, where 1 mU corresponds to 1 nmol of fatty acid released per minute at 25°C (48). Phospholipid transfer activity of PLTP was measured with a radiometric activity assay as described by Jauhiainen et al. (49) using an exogenous, lipoprotein-independent phospholipid transfer assay with labeled phospholipid vesicles as a substrate.
RNA isolation.
mRNA was isolated from liver, intestine, heart, white adipose tissue, skeletal muscle, spleen, and lung using the RNeasy kit from QIAGEN. Total RNA quality was analyzed by an Agilent 2100 BioAnalyzer (Agilent Technologies) and quantified using a Nanodrop 1000 (Thermo Fisher Scientific).
Gene expression array.
mRNA of liver and intestine was submitted to a core facility (Affymetrix Core Facility, Novum, Huddinge, Sweden and Uppsala Array Platform, Uppsala University, Sweden) for hybridization to GeneChip Mouse Gene 1.0 ST Arrays (Affymetrix). The microarray expression data were preprocessed with R statistical software (50) using the RMA method as implemented in the Affymetrix package. Differential expression analysis was performed with the moderated t statistic and false discovery rate (FDR) multiple test correction methods from the limma package. Transcripts with an FDR less than 0.1 and an absolute linear fold change greater than 1.5 were considered significantly differentially expressed. The raw microarray expression data have been deposited in NCBI’s Gene Expression Omnibus (Gene Expression Omnibus accession numbers GSE36279 for liver and GSE39351 for intestine; http://www.ncbi.nlm.nih.gov/geo). The differentially expressed genes were clustered according to their annotation using the DAVID Bioinformatics Resource to identify significantly enriched functional annotation terms in the categories of gene ontology and pathways (51).
Real-time PCR.
Superscript-II and random hexamers (both from Invitrogen) were used for reverse transcription. Real-time PCR used TaqMan Universal Master Mix and premanufactured primers and probes (Assay-on-Demand) for genes of interest, with hypoxanthine guanidine ribonucleosyltransferase (Hprt) used as the housekeeping gene, and was run on an ABI 7900 HT Fast RT-PCR system (all Applied Biosystems). Data were analyzed on the basis of the relative expression method with the formula: 2–ΔΔCT, where ΔΔCT = ΔCT (sample) – ΔCT (calibrator = average CT values of all samples), and ΔCT is the CT of the Hprt housekeeping gene subtracted from the CT of the target gene (52).
Western blot analysis.
Total proteins were isolated from a 25-mg liver sample using T-PER (Thermo Scientific) and homogenized with glass homogenizers. Protein concentration was determined by the Bradford assay and adjusted to 1.5 mg/ml. Thirty-microgram samples were diluted with 2× Laemmli sample buffer, boiled, separated by electrophoresis using 4%–15% Mini-PROTEAN TGX Precast Gels, and transferred to a PVDF membrane (all Bio-Rad). Sortilin-1 was detected using a rabbit polyclonal anti–sortilin antibody (ab16640; Abcam), followed by peroxidase-labeled goat anti-rabbit IgG (Dako), and visualized using an ECL system (GE Healthcare). Rabbit anti-tubulin antibody (ab4074, Abcam) was used as a loading control. Quantification was performed using the ImageJ software (NIH) gel analyzer tool.
Statistics.
Values are expressed as mean ± SEM unless otherwise indicated. Groupwise comparisons (n >2) were performed using a nonparametric Kruskal-Wallis test followed by a Mann-Whitney U test as appropriate. Pairwise comparisons were performed using a nonparametric Mann-Whitney test unless otherwise indicated. Spearman’s rank correlation test was performed to assess correlations. Statistics performed for the arrays are described in that section. A P value less than 0.05 was considered significant. All experiments reported here were repeated at least twice with similar results.
Study approval.
All animal experiments were approved by the Stockholm Regional Board for Animal Ethics.
Supplementary Material
Acknowledgments
We are grateful for the excellent technical assistance of Kristina Edwardsson, Leo Johansson, Anneli Olsson, André Strodthoff, and Ingrid Törnberg. This work was supported by grants from the Swedish Research Council, Swedish Heart-Lung Foundation, Stockholm County Council (ALF), the Foundation for Strategic Research, Loo and Hans Osterman Foundation, Karolinska University Hospital, The Leducq Foundation, and the European Union (AtheroRemo project). R. Klingenberg was supported by a fellowship from the Deutsche Forschungsgemeinschaft (KL1398/2-1, 2-2), by the Swedish Heart-Lung Foundation, Swiss Heart Foundation, and the Roche Research Foundation, Switzerland. The DEREG mouse model, its progeny, and modifications thereof are protected by a patent owned by the Technical University of Munich (TUM), Germany.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(3):1323–1334. doi:10.1172/JCI63891.
References
- 1.Baigent C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366(9493):1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 2.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352(16):1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 3.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12(3):204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 4.Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 5.Hermansson A, et al. Inhibition of T cell response to native low-density lipoprotein reduces atherosclerosis. J Exp Med. 2010;207(5):1081–1093. doi: 10.1084/jem.20092243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6(7):508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 7.Klingenberg R, Hansson GK. Treating inflammation in atherosclerotic cardiovascular disease: emerging therapies. Eur Heart J. 2009;30(23):2838–2844. doi: 10.1093/eurheartj/ehp477. [DOI] [PubMed] [Google Scholar]
- 8.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 9.Shevach EM. From vanilla to 28 flavors: multiple varieties of T regulatory cells. Immunity. 2006;25(2):195–201. doi: 10.1016/j.immuni.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 11.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. . Nat Immunol. 2003;4(4):330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 12.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. . Nat Immunol. 2003;4(4):337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 13.Lahl K, et al. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. . J Exp Med. 2007;204(1):57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taguchi O, Takahashi T. Administration of anti-interleukin-2 receptor alpha antibody in vivo induces localized autoimmune disease. Eur J Immunol. 1996;26(7):1608–1612. doi: 10.1002/eji.1830260730. [DOI] [PubMed] [Google Scholar]
- 15.McHugh RS, Shevach EM. Cutting edge: depletion of CD4+CD25+ regulatory T cells is necessary, but not sufficient, for induction of organ-specific autoimmune disease. . J Immunol. 2002;168(12):5979–5983. doi: 10.4049/jimmunol.168.12.5979. [DOI] [PubMed] [Google Scholar]
- 16.Ait-Oufella H, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12(2):178–180. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 17.Mallat Z, et al. Protective role of interleukin-10 in atherosclerosis. Circ Res. 1999;85(8):e17–e24. doi: 10.1161/01.RES.85.8.e17. [DOI] [PubMed] [Google Scholar]
- 18.Pinderski LJ, et al. Overexpression of interleukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL receptor-deficient mice by altering lymphocyte and macrophage phenotypes. Circ Res. 2002;90(10):1064–1071. doi: 10.1161/01.RES.0000018941.10726.FA. [DOI] [PubMed] [Google Scholar]
- 19.Robertson AK, Rudling M, Zhou X, Gorelik L, Flavell RA, Hansson GK. Disruption of TGF-beta signaling in T cells accelerates atherosclerosis. J Clin Invest. 2003;112(9):1342–1350. doi: 10.1172/JCI18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gojova A, et al. Specific abrogation of transforming growth factor-beta signaling in T cells alters atherosclerotic lesion size and composition in mice. Blood. 2003;102(12):4052–4058. doi: 10.1182/blood-2003-05-1729. [DOI] [PubMed] [Google Scholar]
- 21.George J. Mechanisms of disease: the evolving role of regulatory T cells in atherosclerosis. Nat Clin Pract Cardiovasc Med. 2008;5(9):531–540. doi: 10.1038/ncpcardio1279. [DOI] [PubMed] [Google Scholar]
- 22.Baru AM, Untucht C, Ganesh V, Hesse C, Mayer CT, Sparwasser T. Optimal isolation of functional Foxp3+ induced regulatory T cells using DEREG mice. . PLoS One. 2012;7(9):e44760. doi: 10.1371/journal.pone.0044760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stoneman V, et al. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ Res. 2007;100(6):884–893. doi: 10.1161/01.RES.0000260802.75766.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lo JC, et al. Lymphotoxin beta receptor-dependent control of lipid homeostasis. Science. 2007;316(5822):285–288. doi: 10.1126/science.1137221. [DOI] [PubMed] [Google Scholar]
- 25.Gotsman I, et al. Impaired regulatory T-cell response and enhanced atherosclerosis in the absence of inducible costimulatory molecule. Circulation. 2006;114(19):2047–2055. doi: 10.1161/CIRCULATIONAHA.106.633263. [DOI] [PubMed] [Google Scholar]
- 26.Groyer E, et al. Atheroprotective effect of CD31 receptor globulin through enrichment of circulating regulatory T-cells. J Am Coll Cardiol. 2007;50(4):344–350. doi: 10.1016/j.jacc.2007.04.040. [DOI] [PubMed] [Google Scholar]
- 27.Mor A, et al. Role of naturally occurring CD4+ CD25+ regulatory T cells in experimental atherosclerosis. . Arterioscler Thromb Vasc Biol. 2007;27(4):893–900. doi: 10.1161/01.ATV.0000259365.31469.89. [DOI] [PubMed] [Google Scholar]
- 28.Mallat Z, et al. Induction of a regulatory T cell type 1 response reduces the development of atherosclerosis in apolipoprotein E-knockout mice. Circulation. 2003;108(10):1232–1237. doi: 10.1161/01.CIR.0000089083.61317.A1. [DOI] [PubMed] [Google Scholar]
- 29.Klingenberg R, et al. Intranasal immunization with an apolipoprotein B-100 fusion protein induces antigen-specific regulatory T cells and reduces atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30(5):946–952. doi: 10.1161/ATVBAHA.109.202671. [DOI] [PubMed] [Google Scholar]
- 30.VanderLaan PA, Reardon CA, Thisted RA, Getz GS. VLDL best predicts aortic root atherosclerosis in LDL receptor deficient mice. J Lipid Res. 2009;50(3):376–385. doi: 10.1194/jlr.M800284-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Musunuru K, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466(7307):714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strong A, et al. Hepatic sortilin regulates both apolipoprotein B secretion and LDL catabolism. J Clin Invest. 2012;122(8):2807–2816. doi: 10.1172/JCI63563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samyn H, et al. Plasma phospholipid transfer activity is essential for increased atherogenesis in PLTP transgenic mice: a mutation-inactivation study. J Lipid Res. 2008;49(12):2504–2512. doi: 10.1194/jlr.M800080-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.van Haperen R, et al. Elevated expression of phospholipid transfer protein in bone marrow derived cells causes atherosclerosis. PLoS One. 2008;3(5):e2255. doi: 10.1371/journal.pone.0002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samyn H, et al. Elevation of systemic PLTP, but not macrophage-PLTP, impairs macrophage reverse cholesterol transport in transgenic mice. Atherosclerosis. 2009;204(2):429–434. doi: 10.1016/j.atherosclerosis.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 36.Jiang XC, et al. Targeted mutation of plasma phospholipid transfer protein gene markedly reduces high-density lipoprotein levels. J Clin Invest. 1999;103(6):907–914. doi: 10.1172/JCI5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moerland M, et al. Acute elevation of plasma PLTP activity strongly increases pre-existing atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28(7):1277–1282. doi: 10.1161/ATVBAHA.108.165084. [DOI] [PubMed] [Google Scholar]
- 38.Jiang XC, et al. Apolipoprotein B secretion and atherosclerosis are decreased in mice with phospholipid-transfer protein deficiency. Nat Med. 2001;7(7):847–852. doi: 10.1038/89977. [DOI] [PubMed] [Google Scholar]
- 39.Swirski FK, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117(1):195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou X, Hansson GK. Effect of sex and age on serum biochemical reference ranges in C57BL/6J mice. Comp Med. 2004;54(2):176–178. [PubMed] [Google Scholar]
- 41.Parini P, Johansson L, Broijersen A, Angelin B, Rudling M. Lipoprotein profiles in plasma and interstitial fluid analyzed with an automated gel-filtration system. Eur J Clin Invest. 2006;36(2):98–104. doi: 10.1111/j.1365-2362.2006.01597.x. [DOI] [PubMed] [Google Scholar]
- 42.Schmitz G, Wulf G, Bruning T, Assmann G. Flow-cytometric determination of high-density-lipoprotein binding sites on human leukocytes. Clin Chem. 1987;33(12):2195–2203. [PubMed] [Google Scholar]
- 43.Merkel M, et al. Apolipoprotein AV accelerates plasma hydrolysis of triglyceride-rich lipoproteins by interaction with proteoglycan-bound lipoprotein lipase. J Biol Chem. 2005;280(22):21553–21560. doi: 10.1074/jbc.M411412200. [DOI] [PubMed] [Google Scholar]
- 44.Hultin M, Olivecrona T. Conversion of chylomicrons into remnants. Atherosclerosis. 1998;141(Suppl 1):S25–S29. doi: 10.1016/s0021-9150(98)00229-9. [DOI] [PubMed] [Google Scholar]
- 45.Bergo M, Olivecrona G, Olivecrona T. Forms of lipoprotein lipase in rat tissues: in adipose tissue the proportion of inactive lipase increases on fasting. Biochem J. 1996;3:893–898. doi: 10.1042/bj3130893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruge T, Wu G, Olivecrona T, Olivecrona G. Nutritional regulation of lipoprotein lipase in mice. Int J Biochem Cell Biol. 2004;36(2):320–329. doi: 10.1016/S1357-2725(03)00256-5. [DOI] [PubMed] [Google Scholar]
- 47.Hultin M, Olivecrona G, Olivecrona T. Effect of protamine on lipoprotein lipase and hepatic lipase in rats. Biochem J. 1994;3:959–966. doi: 10.1042/bj3040959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bengtsson-Olivecrona G, Olivecrona T. Phospholipase activity of milk lipoprotein lipase. Methods Enzymol. 1991;197:345–356. doi: 10.1016/0076-6879(91)97160-z. [DOI] [PubMed] [Google Scholar]
- 49.Jauhiainen M, Ehnholm C. Determination of human plasma phospholipid transfer protein mass and activity. Methods. 2005;36(2):97–101. doi: 10.1016/j.ymeth.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 50. R Development Core Team.R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2008. [Google Scholar]
- 51.Dennis G, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(5):P3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- 52.Klingenberg R, et al. Sphingosine-1-phosphate analogue FTY720 causes lymphocyte redistribution and hypercholesterolemia in apoE-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27(11):2392–2399. doi: 10.1161/ATVBAHA.107.149476. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.