

Abstract
Despite a general repression of translation under hypoxia, cells selectively upregulate a set of hypoxia-inducible genes. Results from deep sequencing revealed that Let-7 and miR-103/107 are hypoxia-responsive microRNAs (HRMs) that are strongly induced in vascular endothelial cells. In silico bioinformatics and in vitro validation showed that these HRMs are induced by HIF1α and target argonaute 1 (AGO1), which anchors the microRNA-induced silencing complex (miRISC). HRM targeting of AGO1 resulted in the translational desuppression of VEGF mRNA. Inhibition of HRM or overexpression of AGO1 without the 3′ untranslated region decreased hypoxia-induced angiogenesis. Conversely, AGO1 knockdown increased angiogenesis under normoxia in vivo. In addition, data from tumor xenografts and human cancer specimens indicate that AGO1-mediated translational desuppression of VEGF may be associated with tumor angiogenesis and poor prognosis. These findings provide evidence for an angiogenic pathway involving HRMs that target AGO1 and suggest that this pathway may be a suitable target for anti- or proangiogenesis strategies.
Introduction
Hypoxic stress induces various physiological or pathophysiological responses in conditions including high-altitude adaptations, development, wound healing, ischemic heart disease, advanced atherosclerosis, stroke, and tumorigenesis. These multifaceted changes are controlled by a profound but coordinated gene expression profile, signified by a panel of hypoxia-inducible genes, despite a nominal inhibition of general protein synthesis. Among these genes, hypoxia-inducible factor 1α (HIF1α) is a key transcription factor (TF) robustly induced by oxygen deprivation to increase hundreds of proteins involved in angiogenesis, erythropoiesis, cell-cycle arrest, and metastasis (1, 2). Consensus hypoxia-responsive elements (HREs) in the promoter region are responsible for the HIF1α transcriptional induction of approximately 70 genes (3). However, mechanisms at the posttranscriptional level involving the 3′ UTR of mRNAs are also important for the hypoxia induction of genes, especially for genes lacking HREs. A synergism between transcriptional induction and posttranscriptional mechanisms seems crucial for the optimal induction of hypoxia-inducible genes (1, 4). For example, hypoxia can upregulate VEGF via HIF1α-dependent and -independent transcription (5, 6), 5′-cap–independent, internal ribosomal entry site–mediated (IRES-mediated) preferential translation (7), and increased mRNA stability (8, 9).
MicroRNAs (miRNAs) are noncoding small RNAs that regulate gene expression at the posttranscriptional level. After being processed from their hairpin precursors, miRNAs are loaded into the miRNA-induced silencing complex (miRISC). Consequently, Argonaute (AGO, also known as eukaryotic initiation factor 2C, eIF2C), the core protein in miRISC, tethers miRNA to the 3′ UTR of the target mRNAs, which results in mRNA degradation or translational repression (10, 11). Among AGO protein family members expressed in human cells, AGO1–AGO4 are involved in small RNA-induced gene silencing, and only AGO1 and AGO2 exhibit strand-dissociating activity of miRNA duplexes (12–14). Despite ample studies focusing on miRNAs responding to various stress conditions, little is known about the regulation of AGO expression under stress conditions and its functional consequences in the vasculature.
Several miRNAs are regulated by hypoxia and thus termed hypoxia-responsive miRNA (HRM) or hypoxamiRs (15–18). A subset of HRMs is considered proangiogenic (19). However, the molecular mechanisms by which HRMs promote angiogenesis remain elusive. Using sequencing-by-synthesis (SBS) deep sequencing, we generated a comprehensive and quantitative profile of HRMs in vascular endothelial cells (ECs). Bioinformatics in conjunction with experimental validation revealed that Let-7 and miR-103/107 are robustly induced in ECs and consequently target AGO1. Subsequently, a translational desuppression pathway upregulates VEGF and enhances angiogenesis in vitro and in vivo. In addition, results from clinical specimens provided evidence that this pathway may be implicated in tumor angiogenesis and progression. These findings suggest a novel role of HRMs in hypoxia-induced gene expression and the consequent angiogenesis via targeting AGO1.
Results
Hypoxia induces Let-7 and miR-103/107.
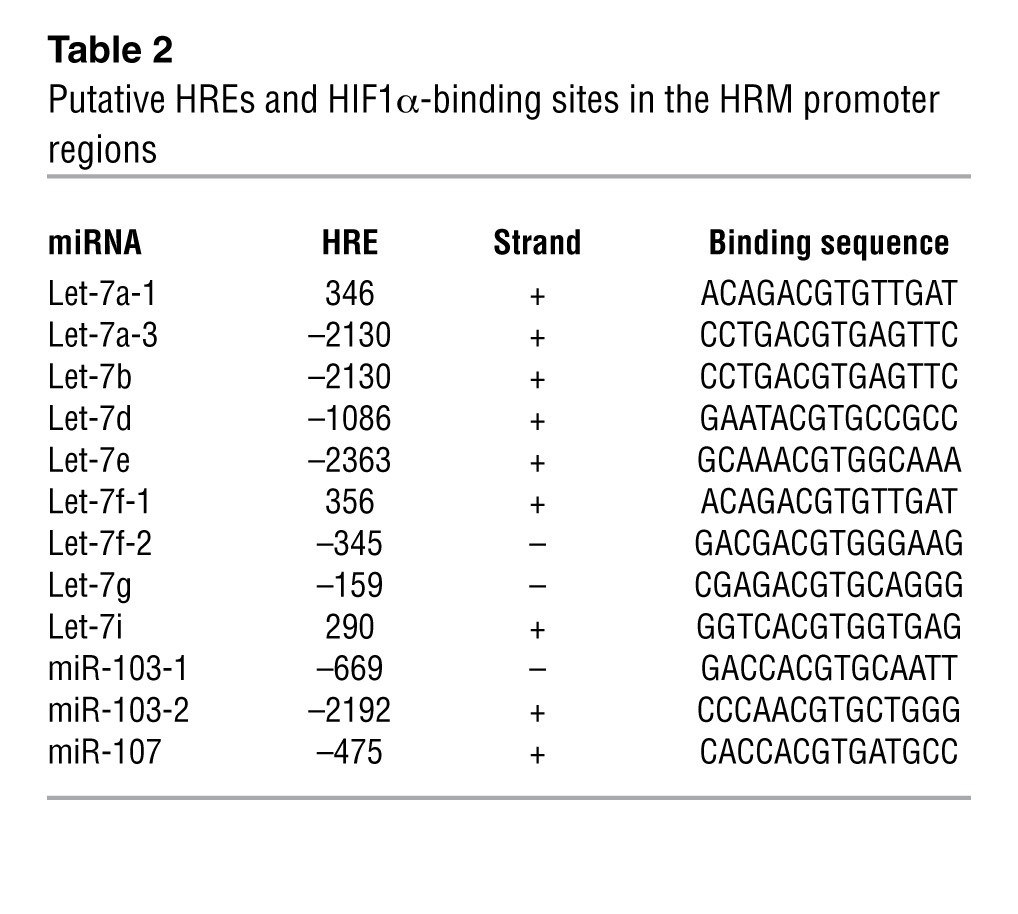
We performed SBS deep sequencing of small RNA libraries derived from HUVECs subjected to normoxia or hypoxia. Among all the miRNAs profiled, Let-7 and miR-103/107 were highly induced miRNA families (Table 1 and Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI65344DS1). Their induction by hypoxia was confirmed by Northern blotting (Figure 1A). The marked induction of specific miRNA paralogs, namely, Let-7a, Let-7e, miR-103, and miR-107, was validated by TaqMan quantitative PCR (qPCR) (Figure 1B). Because HIF1α is the key TF that mediates the induction of many hypoxia-inducible genes and was previously reported to induce miR-103 in cancer cells (18), we examined whether HIF1α can upregulate these HRMs in ECs. As depicted in Table 2 and Supplemental Table 2, Let-7 and miR-103/107 genes were all predicted to contain 1 or more conserved HREs in their promoter regions. Indeed, overexpression of HIF1α in HUVECs confirmed the induction of Let-7a, Let-7e, miR-103, and miR-107 (Figure 1C).
Table 1.
HRM profiles from SBS deep sequencing
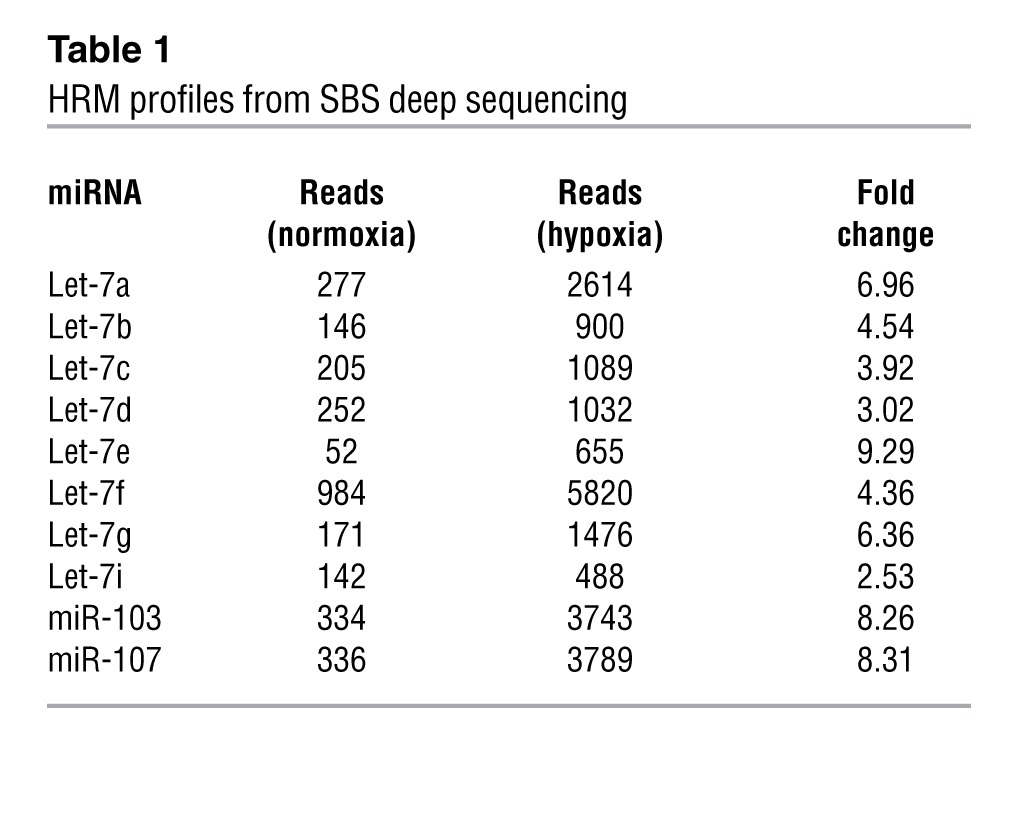
Figure 1. Hypoxia induces Let-7 and miR-103/107 in ECs.

(A and B) HUVECs were kept under normoxia (21% O2) or were subjected to hypoxia (2% O2) for 24 hours. (A) Northern blotting analysis of Let-7 and miR-103/107 expression. 18S rRNA was detected as loading control. (B) The indicated miRNAs were detected by TaqMan qPCR. The relative expression was normalized to that of U6 RNA, with that of normoxia set as 1. (C) HUVECs were infected with Ad-HIF1α for 72 hours, and miRNAs were quantified by TaqMan qPCR. The relative level of miRNA expression was compared with that of the mock-infected group (0 MOI), set as 1. Bar graphs represent mean ± SD from 3 independent experiments. *P < 0.05 compared with normoxia in B or mock-infected controls in C.
Table 2.
Putative HREs and HIF1α-binding sites in the HRM promoter regions
Let-7 and miR-103/107 target AGO1.
Given that Let-7 and miR-103/107 are greatly induced by hypoxia, we predicted the functional targets of these miRNAs by using miRanda (20), RNAhybrid (21), and TargetScan (22). Let-7 and miR-103/107 were predicted to target 169 and 148 mRNAs, respectively; these genes included AGO1, homeobox protein A1 (HOXA1), and apoptosis-antagonizing TF (AATF) (Supplemental Table 3, A and C). To verify whether the predicted genes are bona fide targets of HRMs, we first tested whether their expression was downregulated by hypoxia. The expression of AGO1 and HOXA1 but not AATF was decreased and that of HIF1α was increased in ECs under hypoxic stress (Figure 2A). Because AGO1 is a key protein anchoring miRISC and its mRNA 3′ UTR contains conserved seed sequences for all HRMs (Supplemental Figure 1), we further explored the HRM targeting of AGO1. Overexpression of the pre-miRs of Let-7 or miR-103/107 decreased the level of AGO1 in ECs under normoxia. In contrast, the inhibition of Let-7 or miR-103/107 with locked nucleic acid (LNA) increased the expression of AGO1 in hypoxic HUVECs (Figure 2, D and E, and Supplemental Figure 2B). Furthermore, exogenously expressed HIF1α, similar to hypoxia or pre-miRs, suppressed the level of AGO1 (Figure 2F). These data suggest that HIF1α-induced Let-7 and miR-103/107 suppressed AGO1 expression under hypoxia.
Figure 2. Let-7 and miR-103/107 target AGO1.

(A) HUVECs were subjected to normoxia (time 0) or hypoxia for the indicated times. Cell lysates underwent SDS-PAGE, then Western blot analysis with various antibodies. (B and C) HUVECs were transfected under normoxia for 72 hours with control RNA, pre–Let-7a and pre–Let-7e, or pre-103 and pre-107, at 20 or 40 nM each. Western blot analysis was performed to assess the AGO1 level. (D and E) HUVECs were transfected with LNAs inhibiting Let-7a and Let-7e or miR-103 and miR-107 at indicated concentrations for 48 hours before 24-hour hypoxic stress. Western blot analysis was performed to assess the level of AGO1. (F) Western blot analysis of AGO1 level in HUVECs infected with Ad-HIF1α. Relative protein expression was obtained by normalizing to that of β-actin or α-tubulin. Bar graphs represent mean ± SD from 3 independent experiments. *P < 0.05 versus respective controls set to 1.
HRMs targeting AGO1 mRNA in miRISC.
To demonstrate a direct targeting of AGO1 3′ UTR by Let-7 and miR-103/107, we created reporter constructs containing luciferase fused to the WT AGO1 3′ UTR (AGO1–3′ UTR). Human embryonic kidney 293 (HEK293) cells cotransfected with the AGO1–3′ UTR reporter together with pre–Let-7e, pre–miR-103, or a combination of these 2 pre-miRs conferred lower luciferase activity as compared with cells cotransfected with control RNA (Figure 3A). However, pre–Let-7e or pre–miR-103 induction was unable to decrease the luciferase activity in cells cotransfected with AGO1–3′ UTR mutant constructs with mutated Let-7 or miR-103/107 binding sites. We also constructed an AGO2–3′ UTR reporter. The luciferase reporter activity of the constructed AGO2–3′ UTR was not affected by the cotransfected pre–Let-7 or miR-103, which is consistent with the lack of Let-7 and miR-103/107 target sequences at the AGO2–3′ UTR. Furthermore, hypoxia, like pre–Let-7 or miR-103, decreased the luciferase activity of AGO1–3′ UTR, but not that of AGO2–3′ UTR in ECs (Figure 3B).
Figure 3. Posttranscriptional targeting of AGO1 mRNA in AGO1-mediated miRISC.

(A) HEK293 cells were transfected with the WT Luc-AGO1–3′ UTR (WT), Luc-AGO1–3′ UTR with miR-103/107 or Let-7 target sites mutated (mut), or Luc-AGO2–3′ UTR, together with pre–Let-7e (40 nM), pre-103 (40 nM), pre–Let-7e and pre-103 (20 nM each), or control RNA (40 nM). (B) Bovine aortic ECs (BAECs) transfected with Luc-AGO1–3′ UTR or -AGO2–3′ UTR were subjected to normoxia (Nx) or hypoxia (Hx). CMV–β-gal was cotransfected in all groups as a transfection control. Luciferase activity was normalized to that of β-gal. (C–G) HUVECs were subjected to normoxia or hypoxia. (C and D) Western blot and qPCR analyses of protein and mRNA levels of AGO1–3. (E–G) AGO1 was immunoprecipitated from cell lysates. The immunoprecipitates were subjected to AGO1 immunoblotting (F) and the AGO1-associated miRNAs and AGO1 mRNA were quantified by qPCR (E and G). Data represent mean ± SD from 3 independent experiments. *P < 0.05 compared with control RNA group in A or normoxia group in B–G.
miRNA/mRNA complexes associate with AGO proteins to form miRISC. For AGO1–4 found in human cells (13), hypoxia decreased only AGO1 levels in HUVECs (Figure 3C; AGO4 protein level was under the detection limit), but the mRNA level of AGO1–3 remained unchanged (Figure 3D). These results suggest that HRMs targeting AGO1 was due mainly to translational suppression but not AGO1 mRNA degradation, possibly because of imperfect complementarities between the Let-7 and miR-103/107 and AGO1 mRNA (refs. 23, 24 and Supplemental Figure 1).
Let-7 and miR-103/107 should target AGO1 mRNA within the AGO1-associated miRISC. We therefore compared the amount of AGO1-associated Let-7, miR-103/107, and AGO1 mRNA in normoxic HUVECs and that in hypoxic cells. IP of AGO1 and subsequent qPCR analysis revealed that Let-7a/e and miR-103/107 were significantly enriched in AGO1-miRISC in hypoxic ECs, even though the level of AGO1 was lower (Figure 3, E and F, and Supplemental Figure 3). Consistently, the level of AGO1 mRNA, as an HRM target, was also increased in the AGO1-miRISC in hypoxic cells (Figure 3G). However, we detected neither miRNA nor AGO1 mRNA in IgG isotype controls (Supplemental Figure 3).
Translational desuppression of VEGF mediated by HRMs targeting AGO1.
The sequential events of HIF1α upregulation, Let-7 and miR-103/107 induction, and AGO1 suppression (Figure 4, A–C) led us to propose an HRM-induced translational desuppression pathway: during hypoxia, the HIF1α-induced HRMs, through repressing AGO1-miRISC, release mRNAs that are translationally suppressed under normoxia, which results in the upregulation of the encoded proteins (Figure 4D). To validate this pathway, we searched for mRNA that is decreased in AGO1-miRISC under hypoxia. We tested VEGF as a prototypic gene because it can be induced by hypoxia due to increased mRNA stability or translational efficiency, possibly mediated by AGO1-miRISC. Indeed, we detected a marked decrease in mRNA level of AGO1-associated VEGF in ECs under hypoxia as compared with the normoxic control (Figure 4E). We then tested whether HRM targeting AGO1 mediates hypoxia-induced VEGF. Inhibition of HRMs or overexpression of AGO1 by using a construct encoding the AGO1 ORF without its 3′ UTR attenuated the hypoxia-induced VEGF in ECs (Figure 4, F and G). However, when AGO1 was knocked down to mimic hypoxia suppression, VEGF expression was increased in normoxic ECs. Furthermore, when HUVECs were treated with cycloheximide, a translation inhibitor, the effect of AGO1 knockdown on VEGF induction was abolished (Figure 4H). In a control experiment, VEGF levels were not altered by siRNA against AGO2, whose expression was not repressed under hypoxia (Supplemental Figure 4A). These results suggest that hypoxia-induced VEGF requires translational desuppression through HRM-specific targeting of AGO1.
Figure 4. Translational desuppression of VEGF caused by AGO1 targeting.

(A–C) HUVECs were subjected to normoxia or hypoxia for the indicated times. Western blot analysis of protein levels of HIF1α (A) and AGO1 (C) and TaqMan qPCR analysis of miRNAs (B). (D) Model of translational suppression under normoxia and desuppression under hypoxia. Red arrows indicate up- or downregulation of various molecules. (E) AGO1-associated mRNAs were enriched by IP. Level of VEGF was detected by qPCR. (F) HUVECs were transfected with a mixture of anti–Let-7a/e/miR-103 (10 nM each) or control RNA (30 nM). (G) HUVECs were transfected with a control vector expressing HA or HA-AGO1-ORF plasmid containing AGO1 ORF without 3′ UTR. (H) HUVECs were transfected with AGO1 siRNA or control RNA for 48 hours, then treated with vehicle control or cycloheximide (CHX) (5 μg/ml) for 4 hours. Cells in F–H were then subjected to hypoxia or kept as normoxia controls, and various proteins were detected by Western blot analysis. *P < 0.05 compared with normoxia group.
HRMs targeting AGO1 enhances angiogenesis.
To investigate the functional consequence of translational desuppressed VEGF, we assessed the role of HRMs targeting of AGO1 in in vitro angiogenesis. Consistent with results presented in Figure 4, F–H, blockage of HRMs abolished the hypoxia-induced endothelial tube formation in Matrigel (Figure 5A), which was simulated by AGO1 overexpression (Figure 5B). In contrast, knockdown of AGO1 in normoxic ECs resulted in enhanced tube formation (Figure 5C). These findings indicate that HRMs targeting AGO1 is functionally important for angiogenesis in vitro.
Figure 5. HRM/AGO1 pathway mediates hypoxia-induced angiogenesis in vitro and responds to hypoxia in vivo.

(A–C) HUVECs were prepared as described in Figure 4, F–H, and underwent in vitro angiogenesis assays. The tube/joint numbers were counted, with normoxia/control RNA group (A), normoxia/control plasmid group (B), or control RNA group (C) set to 1. Scale bar: 100 μm. (D and E) C57BL/6 mice were subjected to hypoxia (10% O2) or normoxia (21% O2) for 5 days. Let-7 and miR-103/107 expression in multi-organs/tissues was detected by TaqMan miRNA qPCR (D) and AGO1 protein level by Western blot analysis (E). The levels of miRNAs in normoxia were set to 1. (F and G) Control RNA or antagomirs (antmiR) were delivered with pluronic gel to hind-limb muscles of C57BL/6 mice and then kept under normoxia or hypoxia for 5 days. The miRNAs in the hind limbs were quantified by qPCR (F) and VEGF by Western blot analysis (G). (D–G) Data represent results from at least 6 animals per group. *P < 0.05 compared with controls or between indicated groups.
To validate these findings in vivo, we first examined the expression of HRMs and AGO1 in C57BL/6 mice subjected to 5-day 10% O2 treatment, which mimics the partial pressure of oxygen at approximately 5000 m altitude (25). Let-7a, Let-7e, and miR-103 expression were abundant and robustly induced in multiple tissues/organs in mice exposed to such hypoxia, whereas miR-107 expression was less abundant and not significantly changed (Figure 5D). In accordance with the miRNA induction, AGO1 expression was suppressed (Figure 5E). Similar results were obtained in mice subjected to hypoxia with lower oxygen content or longer duration (8% O2 for 3 days or 10% O2 for 7 days; Supplemental Figure 5, A–C). We next delivered antagomirs against Let-7a, Let-7e, and miR-103 to the hind limbs of C57BL/6 mice (Figure 5F): Hypoxia-induced VEGF expression was attenuated in antagomir-administered muscles as compared with muscles of mice receiving control RNA (Figure 5G).
To evaluate the effect of miRNA targeting of AGO1 on angiogenesis in vivo, we implanted Matrigel mixed with control RNA or antagomirs against Let-7a, -7e, and miR-103 in C57BL/6 mice. Hypoxia-induced angiogenesis was apparent in Matrigel plugs mixed with control RNA, as signified by the increased hemoglobin (Hb) content, infiltrated red blood cells, and staining for vWF and CD31 (Figure 6, A–C). Notably, these angiogenic responses were greatly decreased in Matrigel plugs mixed with antagomirs, which abolished the hypoxia-induced Let-7a, -7e, and miR-103 (Figure 6, A–C, and Supplemental Figure 6). In normoxic mice, little angiogenesis was seen in Matrigel plugs mixed with control RNA or antagomirs. We also tested the role of AGO1 by using SCID mice. Matrigel mixed with HUVECs was s.c. injected into the dorsal sides of SCID mice. As compared with HUVECs transfected with a control vector, cells overexpressing AGO1 resulted in a lower angiogenic response in hypoxic mice (Figure 6, D and E). In the complementary experiment, we administered Matrigel mixed with ECs transfected with control RNA or AGO1 siRNA to SCID mice. Under normoxia, angiogenesis was increased in the implanted Matrigel plugs, which depended on the dose of AGO1 siRNA (Figure 6, F–H). Thus, angiogenesis could be induced by HRMs targeting AGO1, leading to translationally desuppressed VEGF.
Figure 6. HRM/AGO1 pathway regulates hypoxia-induced angiogenesis in vivo.

(A–C) Matrigel mixed with control RNA (15 μg/plug) or antagomirs against Let-7a, Let-7e, and miR-103 (5 μg/plug each) was injected s.c. into C57BL/6 mice before 5-day normoxia or hypoxia. Gross morphology (A) and Hb quantification (B) as well as H&E, vWF, and CD31 staining of Matrigel plugs (C). (D and E) HUVECs transfected with control or HA-AGO1-ORF plasmid were mixed with Matrigel and s.c. injected into SCID mice, which were then kept under normoxia or hypoxia for 3 days. Gross morphology (D) was photographed and Hb quantified (E). (F–H) HUVECs transfected with control RNA (40 nM) or AGO1 siRNA (20 or 40 nM) were mixed with Matrigel and s.c. injected into SCID mice. Five days after injection, the plugs were harvested for gross morphology (F), Hb quantification (G), histology, and IHC (H). In C, E, and G, the Hb content in normoxia/control RNA or control plasmid group was set to 1. Bar graphs represent mean ± SEM. Results are from 5–10 animals per group. Scale bar: 100 μm. *P < 0.05 compared with controls or between indicated groups.
The HRM/AGO1/VEGF pathway is correlated with tumorigenesis.
Because hypoxia-induced angiogenesis is critical in tumorigenesis, we postulated that HRM-mediated desuppression is also present in solid tumors. We therefore examined the levels of miRNAs and AGO1 in breast tumor xenografts in SCID mice. In these expanded xenografts, hypoxic cores were formed (Figure 7A). As compared with oxygenated distal regions, the hypoxic core showed increased HRM levels but decreased AGO1 expression (Figure 7, B and C).
Figure 7. Implication of HRM-mediated VEGF desuppression in tumorigenesis.

(A–C) Mammary fat pads of SCID mice were implanted with 5 × 105 MDA-MB-231 human breast cancer cells. After 4 weeks, mice were killed and tumors were harvested. Core regions of necrotic tumors and distal areas shown in A were collected for detection of miRNAs (B) and AGO1 protein (C). Scale bar: 2 mm. (B) Specimens collected from 10 animals were pooled into 4 samples (each containing 2 or 3 specimens as indicated) for miRNA qPCR. Bars represent the ratio of miRNA level in the core relative to that in the distal region. (C) Representative images of Western blot analysis with samples from individual mice. (D) Levels of Let-7e and miR-103 was detected in paired normal (N) and tumor (T) tissues, and the T/N expression ratio was calculated. (E and F) Representative IHC staining of AGO1, VEGF, CD31, and ISH of Let-7e and miR-103 in serial sections from patients with AGO1 high/VEGF low (E) and AGO1 low/VEGF high expression patterns (F). Scale bar: 200 μm (×100); 100 μm (×400). Yellow arrowheads indicate the location of microvessels. (G and H) Kaplan-Meier analysis of disease-free (G) and overall survival (H) for 173 HCC patients, stratified by AGO1 and VEGF expression.
Given that hypoxic stress is associated with the majority of human tumors (26), we compared HRM levels in paired tumor and normal tissues from patients with breast, colon, or liver cancers. Let-7e and/or miR-103 levels were elevated in tumor tissues in 11 of 16 pairs (Figure 7D). To evaluate the clinical relevance of the AGO1-mediated translational desuppression of VEGF, we performed in situ hybridization (ISH) and immunohistochemistry (IHC), and correlated the VEGF and AGO1 expression in 173 human hepatocellular carcinoma (HCC) specimens. We found inverse correlations between HRM and AGO1 expression, AGO1 and VEGF expression, and AGO1 and microvessel density (revealed by CD31 staining) (Figure 7, E and F, and Supplemental Table 4). Furthermore, a high level of AGO1 but low level of VEGF was associated with both increased disease-free (P = 0.009) and overall survival (P = 0.055). In contrast, poor cancer survival was correlated with low levels of AGO1 but high levels of VEGF (Figure 7, G and H). These findings suggest that the HRM-targeted AGO1 and the subsequent desuppressed VEGF expression may be involved in tumor progression and is indicative of cancer prognosis.
Discussion
Under hypoxia, cells adjust their transcription and translation to adapt to the deficit in O2 and energy. Hypoxia decreases global protein synthesis, mainly by phosphorylation of eukaryotic initiation factor 2 α and inhibition of mammalian target of rapamycin activity, thus causing suppressed 5′-cap-dependent mRNA translation (27–29). However, many genes involved in glycolysis, cell cycle arrest, and angiogenesis are highly expressed. The transcriptional induction by TFs such as HIF ensures an abundant level of mRNAs that enables the translation of some of these genes under the suppressed translational machinery. In addition to direct transcriptional induction, noncanonical IRES-initiated translation and decreased proteosome-mediated protein degradation are involved in the induction of some of these hypoxia-inducible genes (7, 30, 31). Our current study suggests that HRMs such as Let-7 and miR-103 targeting AGO1 results in translational desuppression under hypoxia. This newly defined mechanism may allow for a timely release of sequestered mRNAs for translation, which synergizes with those previously defined, thus leading to an optimal concentration of mRNAs to overcome the suppressed translational machinery.
In the current study, we identified VEGF, a key mediator of hypoxic response, as a target gene regulated by such machinery. We provided evidence that HRMs, by targeting AGO1, release VEGF mRNA sequestered in miRISC, thus contributing to VEGF induction (Figure 4, E–H). Because the silencing effect of miRISC requires AGO1 protein as well as the miRNA-mRNA duplex, hypoxia-suppressed AGO1 expression should also release miRNAs that would otherwise target VEGF mRNAs under normoxia. Indeed, hypoxia greatly decreased AGO1-associated miR-15 and miR-29 levels, previously reported to target VEGF (refs. 32, 33, and Supplemental Figure 7A). In contrast, the mRNAs encoding hypoxia-suppressed genes and their cognate miRNAs would still be enriched in hypoxic AGO1-miRISC. For example, the AGO1 protein-associated AGO1 mRNA and HRMs were concomitantly higher under hypoxia, despite the reduced expression of AGO1 (Figure 3, E–G). Of note, HIF2α overexpression can also induce HRMs, probably through the putative HREs in the promoter regions of these HRMs (Supplemental Figure 8). Therefore, both HIF1α and HIF2α may upregulate HRMs under hypoxia. Given the induction of HRMs by HIF (Figure 1C and Supplemental Figure 8) and the time course of HIF, HRM, and AGO1 expression (Figure 2A and Figure 4, A–C), the translational desuppression may also be considered an HIF-integrated network. Such a mode of action, combined with enhanced transcription and translation, may confer maximal upregulation of VEGF and serve as an essential mechanism, especially in chronic hypoxia. In line with this, the hypoxia-activated HRM/AGO1 pathway was detected under both physiological and pathophysiological conditions in vivo (Figure 5, D and E, and Figure 7, A–C).
Because of the multitude of genes regulated by miRNAs and miRISC, other hypoxia-inducible genes may also be translationally desuppressed in this manner. To test this likelihood, we performed microarray experiments to compare mRNAs associated with AGO1-miRISC under normoxia and hypoxia. In addition to VEGF, multiple genes involved in glycolysis and angiogenesis may be desuppressed under hypoxia (Supplemental Tables 5 and 6). Among these, mRNAs encoding PDGF and placental growth factor (PlGF) were verified by qPCR (Supplemental Figure 7B). Belonging to the same family as VEGF, PDGF and PlGF are highly induced by hypoxia and are potent angiogenic factors under various physiological and pathological conditions (34–36). Interestingly, consensus HREs were not found in the promoter regions of PDGF and PlGF. Hence, their induction by hypoxia seems independent of HIF direct transactivation (37). An intriguing issue is the specificity of AGO1-mediated desuppression because not all mRNA transcripts were released from the AGO1-miRISC under hypoxia (Figure 3G). This finding suggests that the desuppression mechanism is not a generic effect resulting from global AGO1 decrease.
Among all the AGO family members, only AGO1 is suppressed by hypoxia in ECs. A recent study reported that hypoxia increases AGO2 expression through posttranslational hydroxylation in cultured SMCs, which enhances its stability (38). In contrast, Ho et al. reported that hypoxia slightly decreases AGO2 in ECs (39). However, none of our newly identified HRMs were predicted to target AGO2, nor did hypoxia significantly alter AGO2 expression in cultured ECs or various tissues in vivo (Figure 3, A–D, and Supplemental Figure 5D). Because AGO1 expression is decreased under hypoxia, AGO2 is likely involved in mediating miRNA targeting. Indeed, AGO2-associated HRMs were also increased under hypoxia (Supplemental Figure 4B). In addition, AGO2 would be required for stabilizing mature miRNAs under hypoxia (40). Because levels of AGO2-associated VEGF, PlGF, and PDGF mRNAs in normoxic cells were similar to those in hypoxic cells (Supplemental Figure 4C) and AGO2 silencing did not enhance VEGF expression in ECs (Supplemental Figure 4A), AGO2 may not play a major role in the hypoxia-induced translational desuppression.
Results presented in Figure 7 suggest that HRM/AGO1-mediated translational desuppression may contribute to hypoxia-induced angiogenesis during tumorigenesis. Antagonists against this angiogenic pathway may disrupt the tumorigenesis by targeting VEGF and other proangiogenic factors. Because of the heterogeneity of solid tumors and the distinct tumor origin, the induced HRMs and the involved mechanisms may differ. For example, although hypoxia can decrease AGO1 expression via Let-7 targeting, Let-7 may also target oncogenes (41, 42). The overall functional outcome of Let-7 would depend on integrated targeting of genes. In contrast to Let-7’s role as a tumor suppressor, miR-103/107 expression was found inversely correlated with breast cancer survival (43). We and our collaborators also demonstrated that miR-103/107 levels are negatively correlated with survival rates for colon cancer patients (44). Thus, during the development of certain types of cancer, miR-103/107 but not Let-7 may be protumorigenic.
Interestingly, Let-7 and miR-103/107 also target human Dicer (43, 45). As well, recent work by Ho et al. demonstrated that full expression of hypoxia-responsive genes such as VEGF in chronic hypoxia depends on hypoxia-repressed Dicer and changes in posttranscriptional gene regulation (39). Together with Dicer-mediated suppression of miRNA targeting, HRM/AGO1-mediated translational desuppression adds a new mode of regulation of hypoxia-induced gene expression in concert with the HIF-orchestrated network. These pathways highlight the importance of miRNAs and their machinery in modulating the cellular response to hypoxic stresses. Collectively, our findings demonstrate the crucial role of HRM/AGO1-mediated translational desuppression in the hypoxic response (Figure 4D and Figures 5 and 6) and the therapeutic potential of targeting this pathway in pathological angiogenesis.
Methods
Small RNA library and SBS deep sequencing.
Total RNA was extracted by a standard Trizol protocol (Invitrogen). The small RNA library was constructed as described (46). Briefly, 20- to 30-nt small RNAs were gel isolated; then the 5′ and 3′ ends were ligated to single-stranded oligonucleotides (5′ tags and 3′ adaptor) (for oligo sequences, see Supplemental Methods). The ligates then underwent RT-PCR, which resulted in a library of small RNA cDNA molecules. After performing quality control, we used the cDNA library for deep sequencing by Illunima analyzer. miRNA profiles are demonstrated by their reads in the libraries derived from normoxic or hypoxic cells, and fold change under hypoxia relative to normoxic controls was thus determined.
In silico bioinformatics analysis.
Reads from SBS deep sequencing were analyzed as follows: (a) sorting of normoxia and hypoxia libraries based on 5′ tag sequences, (b) trimming the 3′ adaptor sequences, and (c) eliminating sequences of less than 18 or more than 28 nt. All obtained sequences were searched against the human genome by use of a short oligonucleotide alignment program for completely matched sequences. After alignment to the miRNA registry (miRBase 13.0), the miRNA expression profiles were generated and presented as specific miRNA with copy numbers normalized by total small RNA reads. HREs in miRNA promoter regions were searched as described (47). Briefly, 3 binding consensus sequences from proTransFac 7.0 (48) were used to scan the putative promoter regions, and potential HREs were calculated by the position-weight matrix approach. To predict the miRNA targets, we used miRanda, RNAhybrid, and TargetScan to scan cross-species–conserved 3′ UTRs of mRNAs.
Plasmids and luciferase assay.
Human AGO1 or AGO2 3′ UTR containing miR-103/107 and Let-7 target sequences were subcloned into pMIR-REPORT vector (Ambion) to generate pMIR-Luc-AGO1/2–3′ UTR. To create the pMIR-Luc-AGO1–3′ UTR-103/7-mut, a GC to CG mutation was introduced into the miR-103/107 target sequence by using PfuTurbo (Stratagene). To create the pMIR-Luc-AGO1–3′ UTR–Let-7-mut, approximately 50-bp flanking Let-7 target sites were deleted using a 2-step PCR deletion strategy (49). N-terminal HA-AGO1-ORF (plasmid 21535; Addgene) was originally generated by Lian et al. (50). Luciferase reporter constructs were cotransfected with β-gal with or without pre-miR or control RNA. Luciferase activity was measured by use of the Dual-Glo Luciferase Reporter Assay Kit (Promega). The luciferase activity was normalized to that of β-gal.
Immunoprecipitation of AGO-miRISC.
HUVECs were lysed with a buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% NP-40, 1 mM EDTA, and 100 units/μl RNase inhibitor. The lysates were incubated with anti-AGO1 (clone 2A7; Wako Chemicals) or anti-AGO2 (clone 4G8; Wako Chemicals) at 4°C overnight and then protein G Dynabeads for 4 hours. The beads were then spun down, and the immunoprecipitated proteins and RNAs were extracted with lysis buffer and Trizol, respectively. The microarray data of AGO1-associated mRNAs have been deposited in Gene Expression Omnibus (GEO GSE43165), with an assigned accession number upon approval of GEO.
Animal experiments.
We used 8- to 12-week-old male C57BL/6 or female SCID mice. The mice subjected to hypoxia were placed in a sealed chamber infused with 10% O2 and 90% N2 that was kept at room temperature for 5 days. Age- and sex-matched mice were maintained at room temperature, which represented the normoxic control.
AntagomiRs against Let-7a, -7e, and miR-103 were designed as described (51) (see Supplemental Methods for details). On day 1, mice were anesthetized. Then 30 μg control RNA or antagomirs (10 μg of Let-7a, Let-7e, and miR-103 each) mixed with 25% pluronic gel in a total volume of 80 μl was administered into each designated hind limb. After immediate gelification, the skin was reapproximated. The animals were kept for 2 days for recovery before subsequent procedures.
In vivo Matrigel plug assay followed a published protocol (52) with modification. First, 2 Matrigel plugs premixed with 60 U/ml heparin and 40 ng/ml basic fibroblast growth factor, together with 15 μg control RNA or antagomirs (5 μg each of Let-7a, Let-7e, and miR-103) were s.c. injected (500 μl) into the dorsal surface of C57BL/6 mice. After 24-hour recovery, animals were subjected to normoxia or hypoxia for 5 days. The mice were sacrificed, and Matrigel plugs were photographed and harvested for Hb quantification and histology. For Hb quantification, Matrigel plugs were homogenized in 200 μl distilled deionized water. The supernatant after centrifugation was used for Drabkin’s assay (Sigma-Aldrich). For IHC, Matrigel plugs were fixed with formaldehyde and processed as described in Supplemental Methods.
The experiments involving SCID mice were conducted as described (53), with modification. Briefly, HUVECs were transfected with siRNA or DNA plasmids. At 18 hours after transfection, 1 × 106 cells were mixed with 400 μl Matrigel containing 30 units/ml heparin and s.c. injected into the dorsal surface of 8-week-old female SCID mice. On day 3 (for AGO1 rescue experiments) or 5 (for AGO1 siRNA experiments), mice were sacrificed and Matrigel plugs were harvested and photographed.
Female SCID mice (6 to 8 weeks old) were used as recipients in the tumor xenograft model. Specifically, MDA-MB-231 human breast adenocarcinoma cells (5 × 105) were injected into the mammary fat pad. Body weight and tumor volume were monitored weekly for 4 weeks. Tumors of 10–15 mm in diameter were collected at autopsy. Core regions of necrotic tumors and distal areas were separated for detection of miRNA and AGO1 protein.
Clinical samples, ISH, and IHC.
The surgical specimens used for ISH and IHC were formalin-fixed and paraffin-embedded (FFPE) before being archived. ISH and IHC were performed following protocols previously described (44) with minor modification. For ISH, 50 nM 3′-DIG–labeled Let-7e or miR-103 LNA were used as probes. Upon reaction with alkaline phosphatase-linked antibody against DIG (Roche), ISH signals were visualized by use of bromochloroindolyl phosphate-nitro blue tetrazolium substrates. For IHC, rabbit antibodies against human AGO1 (Cell Signaling), VEGF (Santa Cruz Biotechnology Inc.), or CD31 (Santa Cruz Biotechnology Inc.) were used as primary antibodies, followed by sequential incubation with biotinylated secondary antibody, and horseradish peroxidase-conjugated streptavidin. The staining was visualized by use of 3-3′ diaminobenzidine tetrahydrochloride (DAB), and counterstaining was with light hematoxylin. All samples were viewed and photographed under an Olympus microscope. A 4-point scoring system based on staining intensity (range from 0 to 3, with 0 being no expression and 3 maximal expression) was used to evaluate the relative expression of various molecules. All IHC images were reviewed and scored independently by 2 pathologists.
Statistics.
The significance of variability was determined by 2-tailed Student’s t test for 2-group comparison. For multi-group comparison, ANOVA was performed, followed by post-hoc testing to compare differences among groups. Unless otherwise indicated, all results are presented as mean ± SD or mean ± SEM from at least 3 independent experiments. P < 0.05 was considered statistically significant. Correlation between prognostic molecules was determined by Spearman’s nonparametric correlation test. The Kaplan-Meier estimate was used for analysis of disease-free and overall survival, with the log-rank test used to compare the difference.
Study approval.
The animal experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California, Riverside, and the University of California, San Diego. The normal and tumor (N/T) paired samples and human tissues used for miRNA ISH and IHC were from patients undergoing tumor resection or surgical procedures at Taipei Veterans General Hospital. All clinical samples were obtained with informed consent and institutional review board approval (protocol number: 201010021IC) and paired based on patient sex, age, and histopathological diagnoses.
Supplemental methods.
Additional experimental procedures are described in Supplemental Methods.
Supplementary Material
Acknowledgments
This work is supported in part by the NIH Heart, Lung, and Blood Institute (grants HL89940, HL105318, and HL106567 to J.Y-J. Shyy), the American Heart Association (grant 11POST7590128 to Z. Chen), and the National Science Council of the Republic of China (grant NSC 101-2911-I-009-101 to H. Huang). We thank Edward Chan (Department of Oral Biology, University of Florida College of Dentistry Health Science Center) for sharing the HA-AGO1 plasmid in the Addgene repository. The authors are grateful for useful comments from David Johnson, Craig Byus, Amy Pasquinelli, and Traci Marin.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(3):1057–1067. doi:10.1172/JCI65344.
References
- 1.Manalo DJ, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105(2):659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 2.Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007;2007(407):cm8. doi: 10.1126/stke.4072007cm8. [DOI] [PubMed] [Google Scholar]
- 3.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005(306):re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 4.Kenneth NS, Rocha S. Regulation of gene expression by hypoxia. Biochem J. 2008;414(1):19–29. doi: 10.1042/BJ20081055. [DOI] [PubMed] [Google Scholar]
- 5.Arany Z, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451(7181):1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 6.Forsythe JA, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16(9):4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stein I, Itin A, Einat P, Skaliter R, Grossman Z, Keshet E. Translation of vascular endothelial growth factor mRNA by internal ribosome entry: implications for translation under hypoxia. Mol Cell Biol. 1998;18(6):3112–3119. doi: 10.1128/mcb.18.6.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikeda E, Achen MG, Breier G, Risau W. Hypoxia-induced transcriptional activation and increased mRNA stability of vascular endothelial growth factor in C6 glioma cells. J Biol Chem. 1995;270(34):19761–19766. doi: 10.1074/jbc.270.34.19761. [DOI] [PubMed] [Google Scholar]
- 9.Ray PS, Jia J, Yao P, Majumder M, Hatzoglou M, Fox PL. A stress-responsive RNA switch regulates VEGFA expression. Nature. 2009;457(7231):915–919. doi: 10.1038/nature07598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 11.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki T, Shiohama A, Minoshima S, Shimizu N. Identification of eight members of the Argonaute family in the human genome small star, filled. Genomics. 2003;82(3):323–330. doi: 10.1016/S0888-7543(03)00129-0. [DOI] [PubMed] [Google Scholar]
- 14.Wang B, Li S, Qi HH, Chowdhury D, Shi Y, Novina CD. Distinct passenger strand and mRNA cleavage activities of human Argonaute proteins. Nat Struct Mol Biol. 2009;16(12):1259–1266. doi: 10.1038/nsmb.1712. [DOI] [PubMed] [Google Scholar]
- 15.Fasanaro P, et al. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem. 2008;283(23):15878–15883. doi: 10.1074/jbc.M800731200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh G, et al. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-alpha isoforms and promotes angiogenesis. J Clin Invest. 2010;120(11):4141–4154. doi: 10.1172/JCI42980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang X, et al. Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol Cell. 2009;35(6):856–867. doi: 10.1016/j.molcel.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kulshreshtha R, et al. A microRNA signature of hypoxia. Mol Cell Biol. 2007;27(5):1859–1867. doi: 10.1128/MCB.01395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Olson EN. AngiomiRs--key regulators of angiogenesis. Curr Opin Genet Dev. 2009;19(3):205–211. doi: 10.1016/j.gde.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2(11):e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krek A, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37(5):495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 22.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787–798. doi: 10.1016/S0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 23.Hutvagner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science. 2002;297(5589):2056–2060. doi: 10.1126/science.1073827. [DOI] [PubMed] [Google Scholar]
- 24.Pillai RS, et al. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309(5740):1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- 25.Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58(7):1408–1416. [PubMed] [Google Scholar]
- 26.Schoch HJ, Fischer S, Marti HH. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 2002;125(Pt 11):2549–2557. doi: 10.1093/brain/awf257. [DOI] [PubMed] [Google Scholar]
- 27.Connolly E, Braunstein S, Formenti S, Schneider RJ. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol Cell Biol. 2006;26(10):3955–3965. doi: 10.1128/MCB.26.10.3955-3965.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fahling M. Cellular oxygen sensing, signalling and how to survive translational arrest in hypoxia. Acta Physiol (Oxf). 2009;195(2):205–230. doi: 10.1111/j.1748-1716.2008.01894.x. [DOI] [PubMed] [Google Scholar]
- 29.Thomas JD, Johannes GJ. Identification of mRNAs that continue to associate with polysomes during hypoxia. RNA. 2007;13(7):1116–1131. doi: 10.1261/rna.534807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lang KJ, Kappel A, Goodall GJ. Hypoxia-inducible factor-1alpha mRNA contains an internal ribosome entry site that allows efficient translation during normoxia and hypoxia. Mol Biol Cell. 2002;13(5):1792–1801. doi: 10.1091/mbc.02-02-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Semenza GL. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev. 1998;8(5):588–594. doi: 10.1016/S0959-437X(98)80016-6. [DOI] [PubMed] [Google Scholar]
- 32.Kwiecinski M, et al. Expression of platelet-derived growth factor-C and insulin-like growth factor I in hepatic stellate cells is inhibited by miR-29. Lab Invest. 2012;92(7):978–987. doi: 10.1038/labinvest.2012.70. [DOI] [PubMed] [Google Scholar]
- 33.Yin KJ, Olsen K, Hamblin M, Zhang J, Schwendeman SP, Chen YE. Vascular endothelial cell-specific microRNA-15a inhibits angiogenesis in hindlimb ischemia. J Biol Chem. 2012;287(32):27055–27064. doi: 10.1074/jbc.M112.364414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carmeliet P, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001;7(5):575–583. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]
- 35.Green CJ, et al. Placenta growth factor gene expression is induced by hypoxia in fibroblasts: a central role for metal transcription factor-1. Cancer Res. 2001;61(6):2696–2703. [PubMed] [Google Scholar]
- 36.Kourembanas S, Hannan RL, Faller DV. Oxygen tension regulates the expression of the platelet-derived growth factor-B chain gene in human endothelial cells. J Clin Invest. 1990;86(2):670–674. doi: 10.1172/JCI114759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Semenza GL. Vascular responses to hypoxia and ischemia. Arterioscler Thromb Vasc Biol. 2010;30(4):648–652. doi: 10.1161/ATVBAHA.108.181644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu C, et al. Hypoxia potentiates microRNA-mediated gene silencing through posttranslational modification of Argonaute2. Mol Cell Biol. 2011;31(23):4760–4774. doi: 10.1128/MCB.05776-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ho JJ, et al. Functional importance of Dicer protein in the adaptive cellular response to hypoxia. J Biol Chem. 2012;287(34):29003–29020. doi: 10.1074/jbc.M112.373365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diederichs S, Haber DA. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell. 2007;131(6):1097–1108. doi: 10.1016/j.cell.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 41.Johnson CD, et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007;67(16):7713–7722. doi: 10.1158/0008-5472.CAN-07-1083. [DOI] [PubMed] [Google Scholar]
- 42.Shell S, et al. Let-7 expression defines two differentiation stages of cancer. Proc Natl Acad Sci U S A. 2007;104(27):11400–11405. doi: 10.1073/pnas.0704372104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martello G, et al. A MicroRNA targeting dicer for metastasis control. Cell. 2010;141(7):1195–1207. doi: 10.1016/j.cell.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 44.Chen HY, et al. miR-103/107 promote metastasis of colorectal cancer by targeting the metastasis suppressors DAPK and KLF4. Cancer Res. 2012;72(14):3631–3641. doi: 10.1158/0008-5472.CAN-12-0667. [DOI] [PubMed] [Google Scholar]
- 45.Tokumaru S, Suzuki M, Yamada H, Nagino M, Takahashi T. let-7 regulates Dicer expression and constitutes a negative feedback loop. Carcinogenesis. 2008;29(11):2073–2077. doi: 10.1093/carcin/bgn187. [DOI] [PubMed] [Google Scholar]
- 46.Lu C, Meyers BC, Green PJ. Construction of small RNA cDNA libraries for deep sequencing. Methods. 2007;43(2):110–117. doi: 10.1016/j.ymeth.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 47.Kulshreshtha R, Davuluri RV, Calin GA, Ivan M. A microRNA component of the hypoxic response. Cell Death Differ. 2008;15(4):667–671. doi: 10.1038/sj.cdd.4402310. [DOI] [PubMed] [Google Scholar]
- 48.Wingender E, Karas H, Knüppel R. TRANSFAC database as a bridge between sequence data libraries and biological function. Pac Symp Biocomput. 1997:477–485. [PubMed] [Google Scholar]
- 49.Ogel ZB, McPherson MJ. Efficient deletion mutagenesis by PCR. Protein Eng. 1992;5(5):467–468. doi: 10.1093/protein/5.5.467. [DOI] [PubMed] [Google Scholar]
- 50.Lian SL, Li S, Abadal GX, Pauley BA, Fritzler MJ, Chan EK. The C-terminal half of human Ago2 binds to multiple GW-rich regions of GW182 and requires GW182 to mediate silencing. RNA. 2009;15(5):804–813. doi: 10.1261/rna.1229409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krutzfeldt J, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438(7068):685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 52.Passaniti A, et al. A simple, quantitative method for assessing angiogenesis and antiangiogenic agents using reconstituted basement membrane, heparin, and fibroblast growth factor. Lab Invest. 1992;67(4):519–528. [PubMed] [Google Scholar]
- 53.Bonauer A, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324(5935):1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.