

Abstract
After an initial response to chemotherapy, many patients with triple-negative breast cancer (TNBC) have recurrence of drug-resistant metastatic disease. Studies with TNBC cells suggest that chemotherapy-resistant populations of cancer stem-like cells (CSCs) with self-renewing and tumor-initiating capacities are responsible for these relapses. TGF-β has been shown to increase stem-like properties in human breast cancer cells. We analyzed RNA expression in matched pairs of primary breast cancer biopsies before and after chemotherapy. Biopsies after chemotherapy displayed increased RNA transcripts of genes associated with CSCs and TGF-β signaling. In TNBC cell lines and mouse xenografts, the chemotherapeutic drug paclitaxel increased autocrine TGF-β signaling and IL-8 expression and enriched for CSCs, as indicated by mammosphere formation and CSC markers. The TGF-β type I receptor kinase inhibitor LY2157299, a neutralizing TGF-β type II receptor antibody, and SMAD4 siRNA all blocked paclitaxel-induced IL8 transcription and CSC expansion. Moreover, treatment of TNBC xenografts with LY2157299 prevented reestablishment of tumors after paclitaxel treatment. These data suggest that chemotherapy-induced TGF-β signaling enhances tumor recurrence through IL-8–dependent expansion of CSCs and that TGF-β pathway inhibitors prevent the development of drug-resistant CSCs. These findings support testing a combination of TGF-β inhibitors and anticancer chemotherapy in patients with TNBC.
Introduction
Triple-negative breast cancers (TNBCs) lack detectable hormone receptors and ERBB2 gene amplification and represent the most virulent subtype of this malignancy (1). Cytotoxic chemotherapies such as taxanes are initially effective in most patients with metastatic TNBC; however, the majority of these tumors recur after chemotherapy (2). Metastatic tumor relapses are characterized by rapidly proliferating, drug-resistant cancers, associated with a high mortality rate. An increasing body of evidence suggests that survival of a small population of cells with stem-like properties may be responsible for these tumor recurrences after an initial response to chemotherapy (3–6). This population, interchangeably called cancer stem-like cells (CSCs) or tumor-initiating cells (TICs), retains the capacity to self-renew and regenerate the total bulk of a heterogeneous tumor comprised mostly of non–stem cells. In this study, we sought to identify clinically targetable molecules or pathways driving the survival of chemotherapy-resistant CSCs in TNBC.
Recent data suggest that the TGF-β family of cytokines plays a role in breast cancer stem cells. Shipitsin and colleagues showed that subpopulations with CSC features (CD44+) within breast tumors overexpress TGF-β1 and the TGF-β type I receptor (TGF-βR1). TGF-β is a potent inducer of an epithelial-to-mesenchymal transition (EMT) in mammary cells, and this transformation has been associated with acquisition of tumor stem-like properties (7). Indeed, a TGF-βR1/2 kinase inhibitor was shown to reverse EMT and induce a mesenchymal-to-epithelial differentiation in CD44+ mammary epithelial cells (8). TGF-β ligands are often enriched in the TNBC tumor microenvironment and can be produced by tumor cells or by tumor-associated stromal and immune cells (9, 10). These data suggest the possibility that the TGF-β pathway is involved in maintenance of CSCs in breast carcinomas.
TGF-β inhibitors have been proposed and are being developed as antimetastatic therapies in patients with cancer. However, the impact of these inhibitors on CSCs in breast cancer has not yet been explored. Using a small molecular weight TGF-βR1 kinase inhibitor and a neutralizing TGF-β type II receptor antibody currently in clinical development (11, 12), we determined the role of TGF-β signaling in chemotherapy-induced expansion of CSCs in TNBC cell lines and xenografts. We initially discovered enrichment of a TGF-β–responsive gene signature in chemotherapy-treated primary breast cancers. This signature correlated with TNBC cell lines with basal-like gene expression. In TNBC cell lines and xenografts, treatment with the chemotherapy agent paclitaxel expanded a population with CSC markers, high autocrine TGF-β signaling, and tumor-initiating capacity. These effects were abrogated by both TGF-β inhibitors as well as SMAD4 siRNA. Expression of IL-8 at the mRNA and protein level was also increased by chemotherapy. This induction required an intact TGF-β pathway, as it was blocked by the TGF-βR1 kinase inhibitor and SMAD4 siRNA. Finally, addition of the TGF-βR1 kinase inhibitor to paclitaxel abrogated expansion of the CSC fraction and IL-8 release in both cultured TNBC cell lines and xenografts established in athymic mice. These studies are the first to our knowledge to demonstrate the ability of TGF-β inhibitors to block the expansion of chemotherapy-resistant TICs in vivo. They provide a basis for future clinical studies testing their role in combination with chemotherapy in patients with TNBC.
Results
Chemotherapy-treated breast cancers display increased markers of TGF-β signaling and CSCs.
We first examined gene expression signatures enriched by chemotherapy in primary breast cancers (Table 1). RNA extracted from 17 matched breast tumor biopsies before and after neoadjuvant chemotherapy was quantitated using NanoString nCounter analysis (13). A TGF-β–responsive gene signature score (described in the Methods) was significantly enriched by chemotherapy in the biopsies after treatment (Figure 1A). When we applied the TGF-β signature to a second cohort of 21 patients treated with neoadjuvant systemic therapy (4–6 months), similar results were obtained with publically available gene expression in the pretreatment biopsy and the residual cancer (ref. 14 and Supplemental Figure 3B; supplemental material available online with this article; doi: 10.1172/JCI65416DS1). TGFBR2, TGFBR3, and SMAD4 genes were increased approximately 2 fold (Figure 1B), whereas TGFBR1, TGFB1, TGFB3, SMAD2, and SMAD7 were also increased but to a lesser degree (Supplemental Figure 3A). CD44 and ALDH1A1 (15), 2 CSC-related genes, were increased in samples after chemotherapy (Figure 1C). The TGF-β signature score was elevated in human breast cancer cell lines with basal-like gene expression but poorly represented among luminal cell lines, suggestive of an association with the basal-like subtype of breast cancer (Supplemental Figure 2). Further, several genes in the TGF-β pathway that were altered by chemotherapy in RNA from primary breast tumors (Figure 1D) were similarly altered in the human TNBC cell line SUM159 upon treatment with paclitaxel (Figure 1E), including the ligand TGFB1, the type II receptor TGF-βR2, and the transcription factors JUN, JUNB, FOS, and CDKN1A. These observations suggest that treatment with chemotherapy enriches for a population of cancer cells with basal-like gene expression, upregulated TGF-β signaling, and markers of tumor-initiating capacity.
Table 1.
Clinicopathological characteristics of patients from whom biopsies were acquired before and after chemotherapy

Figure 1. Chemotherapy-treated breast cancers display increased markers of TGF-β signaling and CSCs.

(A) NanoString analysis of TGF-β pathway genes in RNA extracted from breast cancer biopsies before chemotherapy (Pre-Tx) and after chemotherapy (Post-Tx). A TGF-β gene expression score was generated in both groups, as described in Methods (n = 17; paired Student’s t test, P = 0.002). (B) Box plots of TGFBR2, TGFBR3, and SMAD4 gene expression between biopsies before and after chemotherapy (TGFBR2: P = 0.007, TGFBR3: P = 0.0026, SMAD4: P = 0.012). Symbols indicate individual tumors; horizontal bars represent the mean. (C) Box plots of gene expression of CSC genes CD44 and ALDH1A1 (CD44: P = 0.013; ALDH1A1: P = 0.0067). (D) Box plots of FOS, JUNB, JUN, CDKN1A gene expression in the 17 paired tumors (paired Student’s t test, P < 0.0008 for all 4 comparisons). (B–D) In box-and-whisker plots, horizontal bars indicate the medians, boxes indicate 25th to 75th percentiles, and whiskers indicate 10th and 90th percentiles. (E) TGFB1 and TGFBR2 mRNA from SUM159 cells treated with or without 10 nM paclitaxel for 6 days was measured by quantitative RT-PCR (*P = 0.03). Using the TGF-β–specific PCR array described in Methods, the expression of JUNB, JUN, FOS, and CDKN1A were assessed and quantified in both control (C) and paclitaxel-treated (P) cells. Error bars indicate SEM.
Paclitaxel enriches a CSC population with increased TGF-β signaling.
Paclitaxel is one of the most commonly used chemotherapies in patients with TNBC (16–19). IC50s for this drug in 4 TNBC cell lines were between 2 and 5 nM, respectively (Supplemental Table 1). These cells were treated with a concentration of paclitaxel (5 or 10 nM) that induced >50% cell death. CSCs, measured by flow cytometry of ALDEFLUOR+ SUM159 cells and CD44hiPROCR+ BT549 cells, were increased 7 to 9 fold, respectively, in response to paclitaxel (Figure 2A). Paclitaxel also increased the CSC fractions in SUM149 (ALDH+) and MDA231 (PROCR+/ESA+) cell lines 2 to 3 fold (Supplemental Figure 4). Mammosphere-forming efficiency, an operational surrogate of TICs, was also markedly increased by treatment with paclitaxel in BT549 cells and, more modestly, in SUM159 cells (Figure 2B). To confirm these findings in vivo, we established SUM159 xenografts in athymic mice. Mice with tumors measuring ≥100 mm3 were treated with paclitaxel for 5 days. After 10 days without additional treatment, the xenografts were harvested, dissociated into single cells, and tested in a mammosphere assay ex vivo. Mammosphere formation was markedly increased in a dose-dependent fashion in the paclitaxel-treated tumors compared with that in untreated controls (Figure 2C). Consistent with TGF-β pathway activation, SMAD2 phosphorylation was sustained in the paclitaxel-treated xenografts (Figure 2D). Similar results were observed in SUM159 and BT549 cells treated with paclitaxel (Supplemental Figure 5). In addition, treatment with paclitaxel induced TGF-β transcriptional reporter activity in dose-dependent fashion in both SUM159 and BT549 cells (Figure 2E). These results suggest that paclitaxel treatment expands a CSC population where autocrine TGF-β signaling is active.
Figure 2. Paclitaxel enriches a CSC population with increased TGF-β signaling.

(A and B) SUM159 and BT549 cells were treated with vehicle (control) or 10 nM paclitaxel for 4 days and allowed to recover in fresh media for another 4 days. Cells were trypsinized and analyzed by FACS for (A) ALDH activity and CD44hi/PROCR+ expression (*P < 0.01) or (B) assessed for their ability to form mammospheres (*P = 0.04). (C) SUM159 xenografts were treated with vehicle or 10 and 20 mg/kg/d paclitaxel for 5 consecutive days (treatment started at day 1 and ended at day 6 of x axis). Tumor diameters were measured every 3 days using calipers, and volume in mm3 was calculated as described in Methods. Xenografts were harvested on day 15, dissociated into single cells, and grown as mammospheres. Each bar represents the mean mammosphere number ± SEM (n = 3; *P < 0.05). Original magnification, ×100. (D) Whole lysates from the same SUM159 xenografts as in C were subjected to P-SMAD2, total SMAD2/3, and actin (control) immunoblot analysis. exp, exposure. (E) SUM159 and BT549 cells were treated with vehicle or 5 nM or 10 nM paclitaxel for 4 days and seeded in 24-well plates. Cells were transfected with pCAGA-Luc and Renilla plasmids; luciferase activity was determined 24 hours later using the Dual Luciferase Kit, as described in Methods (n = 3; *P < 0.05). Error bars indicate SEM.
TGF-β increases the CSC population in a SMAD4-dependent manner.
To determine whether TGF-β phenocopied the effect of paclitaxel on CSCs, we treated BT549 and SUM159 cells with recombinant TGF-β1 with or without the TGF-βR1 serine/threonine kinase small molecule inhibitor LY2157299 (11). Cotreatment with LY2157299 abrogated TGF-β–induced P-SMAD2 in both SUM159 and BT549 cells (Figure 3A). In SUM159 cells, exogenous TGF-β increased the proportion of ALDH+ cells by >3 fold and the mammosphere numbers by >2 fold. Both of these effects were blocked by LY2157299. In BT549 cells, TGF-β also increased the CD44hi/PROCR+ fraction, and this effect was abrogated by LY2157299 (Figure 3, B and C). Although TGF-β did not increase mammosphere formation in BT549 cells, treatment with LY2157299 alone markedly reduced baseline mammosphere number (Figure 3C), further suggesting that tumor-initiating properties are regulated by autocrine TGF-βR1 function.
Figure 3. TGF-β increases the CSC population in a SMAD4-dependent manner.

(A) SUM159 and BT549 cells were preincubated with 1 to 5 μM LY2157299 for 24 hours, followed by treatment with 2.5 ng/ml TGF-β1 for 2 hours in media supplemented with 0.5% FBS. Cell lysates were prepared and subjected to immunoblot analysis with total SMAD2/3 and P-SMAD2 antibodies. (B) SUM159 and BT549 cells were treated with 2.5 ng/ml TGF-β1 with or without 5 μM LY2157299 for 6 days. TGF-β1 and inhibitor were replenished every 3 days. ALDEFLUOR assay was performed in SUM159 cells as described in Methods (n = 3; *P < 0.01). BT549 cells were stained with CD44 and PROCR antibodies, followed by FACS analysis (n = 3; #P < 0.002). Error bars indicate SEM. (C) SUM159 and BT549 cells were seeded in low-adherent dishes and treated with 2.5 ng/ml TGF-β1 with or without 5 μM LY2157299 for 6 days. TGF-β and inhibitor were replenished every 3 days. Mammosphere number was calculated using a GelCount reader and software. Each bar represents the mean number ± SEM (*P < 0.02). (D) SUM159 and BT549 cells were transfected with nontargeting control (CTL) or 2 different SMAD4 siRNA oligonucleotides for 72 hours; lysates of these cells were separated by SDS-PAGE and subjected to immunoblot analysis with SMAD4 and actin antibodies. (E) Control and SMAD4 siRNA-transfected SUM159 and BT549 cells were cultured as mammospheres in the presence or absence of 2.5 ng/ml TGF-β1. Mammosphere number was calculated using a Gel Count reader and software after 6 days. Each bar represents the mammosphere number ± SEM (n = 3; *P < 0.04).
The type I TGF-β receptor ALK5 phosphorylates SMAD2/3, which, in turn, forms a complex with SMAD4. This complex translocates to the nucleus, in which it binds DNA to induce the transcription of TGF-β–dependent genes (20). Thus, we examined whether the effects of LY2157299 were mirrored by downregulation of SMAD4. Transfection with SMAD4 siRNA oligonucleotides downregulated SMAD4 protein levels (Figure 3D and Supplemental Figure 6B) and reduced both baseline and TGF-β–induced mammosphere formation (Figure 3E and Supplemental Figure 8) in SUM159, BT549, and MDA231 cell lines. These data further suggest that tumor-initiating properties in TNBC are regulated by autocrine TGF-β/SMAD4 signaling.
Paclitaxel and TGF-β induce SMAD4-dependent expression of IL-8.
Like TGF-β, the cytokine IL-8 has been shown to regulate CSCs (21). Therefore, we sought to determine whether there was a link among paclitaxel, TGF-β, and IL-8 in regulating CSCs. Paclitaxel significantly induced IL8 mRNA expression in SUM159 and BT549 cells (Figure 4A). Exogenous TGF-β also induced IL8 mRNA expression in SUM159 and MDA231 cells (Supplemental Figure 6A). Further, TGF-β–induced secretion of IL-8 was completely abrogated by LY2157299 (Figure 4B). Knockdown of SMAD4 by siRNA also reduced >50% of basal IL8 mRNA levels (Supplemental Figure 6B) and TGF-β–induced IL-8–secreted protein levels (Figure 4C). To determine the importance of TGF-β–induced IL-8 on CSCs, we targeted IL-8 function using 2 methods: (a) RNAi targeting the IL-8 receptors CXCR1 and CXCR2 and (b) an IL-8–neutralizing antibody. Knockdown of CXCR1 and CXCR2 inhibited basal and TGF-β–induced SUM159 mammosphere formation (Figure 4D and Supplemental Figure 7A). However, treatment with the neutralizing IL-8 antibody only reduced basal but not TGF-β–induced mammosphere formation (Supplemental Figure 7B). Of note, addition of recombinant human IL-8 only modestly rescued the inhibition of mammosphere formation by MDA231 cells stably expressing SMAD4 shRNA (Supplemental Figure 8). These data imply that (a) the effects of TGF-β on TICs are at least in part mediated by the IL-8/CXCR1/2 axis; (b) the ability of IL-8 to induce mammosphere formation still requires TGF-β signaling, which is disabled upon stable SMAD4 knockdown; and (c) IL-8 is not the only mediator of TGF-β–induced CSCs.
Figure 4. Paclitaxel and TGF-β induce SMAD4-dependent expression of IL-8.

(A) SUM159 and BT549 cells were treated with 5 to 10 nM paclitaxel for 4 days. RT-qPCR analysis was performed to assess IL8 and GAPDH RNA levels (*P < 0.05, **P < 0.003). (B) SUM159 cells were treated with 2.5 ng/ml TGF-β with or without 5 μM LY2157299 and grown as mammospheres. Media was collected, and IL-8 protein levels were measured by ELISA; IL-8 levels were normalized to total protein (*P = 0.02). (C) SUM159 cells were transfected with control or SMAD4 siRNA and plated as mammospheres with or without TGF-β1. After 6 days, mammospheres and media were collected and analyzed for IL-8 levels by ELISA. IL-8 levels were normalized to total protein (*P < 0.001). (D) SUM159 cells were transfected with control or both CXCR1 and CXCR2 siRNA and plated as mammospheres with or without 2.5 ng/ml TGF-β1 for 6 days. Mammosphere number was then quantitated, as described in Methods (*P = 0.016). (E) SUM159 cells were transfected with control or SMAD4 siRNA. Forty-eight hours later, 10 nM paclitaxel was added for 24 hours before mRNA extraction and RT-qPCR using IL-8–specific primers (*P < 0.002, **P = 0.002). (F) RT-qPCR analysis of IL8 mRNA levels in SUM159 and BT549 cells treated with 5 μM LY2157299 and 5 nM paclitaxel (BT549 cells) or 10 nM paclitaxel (SUM159 cells) as indicated for 6 days (*P < 0.007, **P < 0.001). (G) Media from cells treated with paclitaxel with or without LY2157299 was collected and subjected to IL-8 ELISA assay. Raw IL-8 levels were normalized to cell number (*P < 0.001). Error bars indicate SEM.
Finally, we determined the effect of TGF-β inhibition on paclitaxel-induced IL-8 expression. Both transfection of SMAD4 siRNA (Figure 4E) and treatment with LY2157299 (Figure 4, F and G) reduced paclitaxel-induced IL8 mRNA and protein levels. These findings suggest that paclitaxel-induced IL-8 levels are regulated by the TGF-β pathway.
Paclitaxel requires TGF-β signaling to expand the CSC population.
In SUM159 and BT549 cells, treatment with LY2157299 abrogated paclitaxel-induced phosphorylation of SMAD2 and the concurrent expansion of CSCs, as measured by flow cytometry for ALDH or CSC surface markers (Figure 5, A and B, and Supplemental Figure 9A). In addition, LY2157299 reduced the paclitaxel-enriched MDA231 CSC population measured by FACS analysis (Supplemental Figure 9B). Similarly, primary mammosphere formation by paclitaxel-treated cells was inhibited by treatment with LY2157299 (Figure 5C) and SMAD4 siRNA oligonucleotides (Figure 5D).
Figure 5. Paclitaxel requires TGF-β signaling to expand CSCs.

(A) SUM159 and BT549 cells were treated with 5 μM LY2157299 and 5 nM paclitaxel (BT549 cells) or 10 nM paclitaxel (SUM159 cells) for 6 days. The proportion of ALDH+ and CD44hi/PROCR+ cells was determined by flow cytometry (SUM159: *P < 0.03; BT549: #P < 0.001; n = 3). (B) P-SMAD2 levels were assessed by immunoblot analysis of cell lysates. (C) SUM159 and BT549 cells were treated with 5 μM LY2157299 and 5 nM paclitaxel for 6 days. Cells were then seeded in either 6- or 24-well ultra-low adherent plates for mammosphere formation. Mammosphere number was calculated using the GelCount reader and software (BT549: *P < 0.002; n = 3). (D) SUM159 cells were treated with 10 nM paclitaxel for 3 days, followed by transfection with either CTL or SMAD4 siRNA. Transfected cells were seeded as mammospheres for 6 days. Mammospheres were imaged and their total number was quantitated (*P < 0.001). Original magnification, ×100. (E) SUM159 ALDH+ and ALDH– cells were flow sorted; an equal number of ALDH+ and ALDH– cells were seeded in a 3D Matrigel morphogenesis assay, as described in Methods. Three days later, cells were treated with LY2157299 (5 μM) or paclitaxel (5 nM) or both for 9 days. Fresh medium and inhibitors were added every 3 days. Original magnification, ×100. Error bars indicate SEM.
SMAD4 also mediates the transcriptional effects of BMP and activin receptors (22). Further, type I TGF-β receptor (ALK5) inhibitors like LY2157299 have been shown to inhibit the kinase activity of ALK4 and ALK7, the type I receptors for activin (23–25). Therefore, to determine whether the effects observed with SMAD4 siRNA and LY2157299 were specific to inhibition of ALK5, we used a monoclonal antibody (TR1) that specifically neutralizes the type II TGF-β receptor (TGF-βR2) (12). TR1 functions by preventing the binding of TGF-β to TGF-βR2 and induces the internalization of TGF-βR2. Cotreatment with TR1 also decreased the paclitaxel-induced expansion of the ALDH+ SUM159 cells (Supplemental Figure 9C), suggesting that this expansion is the result of drug-induced autocrine activation of TGF-βR2. Finally, we flow sorted ALDH+ and ALDH– SUM159 cells prior to plating them in a 3D-Morphogenesis (Matrigel) assay. Under these conditions, ALDH+ cells but not ALDH– cells continued to grow in the presence of paclitaxel. However, addition of LY2157299 eliminated the 3D growth of ALDH+ cells treated with paclitaxel (Figure 5E). These results suggest that TGF-β inhibition can reduce the chemotherapy-resistant CSC populations in TNBC.
Pharmacological inhibition of TGF-β in vivo reduces the paclitaxel-resistant CSC population.
We next established SUM159 xenografts in female athymic mice. Mice (n = 10 per group) with tumors measuring ≥75 mm3 were randomized to treatment with vehicle, paclitaxel (20 mg/kg/d i.p. 5 times), LY2157299 (100 mg/kg twice a day p.o. 14 times), and a combination both drugs (Figure 6A). Some xenografts were harvested 5 days immediately after completing treatment with paclitaxel. Immunoblot analysis of these tumor lysates showed higher levels of P-SMAD2 in paclitaxel-treated xenografts compared with those in untreated xenografts. P-SMAD2 was undetectable in tumors from mice treated with LY2157299 (Figure 6B). Interestingly, P-SMAD2 levels were maintained in paclitaxel-treated and control tumors harvested on day 14, even though treatment with the taxane had been discontinued on day 5. Tumors previously treated with LY2157299 exhibited very low or undetectable levels of P-SMAD2 (Figure 6C). On day 14, tumor volume was significantly lower in the group treated with LY2157299 and paclitaxel compared with that of tumors treated with paclitaxel alone (Figure 6D). In all 4 treatment arms, mice maintained their body weight, and there were no signs of heart or liver toxicity upon histopathological evaluation (data not shown). Dissociated cell suspensions from freshly harvested tumors were assessed for CSCs by flow cytometry. The ALDH+ fraction of cell suspensions from xenografts harvested on day 14 was significantly lower in tumors treated with LY2157299 compared with that of paclitaxel-treated and control xenografts (Figure 6E). Finally, mice treated with paclitaxel alone exhibited >5-fold higher serum levels of tumor-derived IL-8 compared with untreated controls; these levels were reduced by >50% in mice treated with the combination of LY2157299 and paclitaxel (Figure 6F).
Figure 6. TGF-β inhibition in vivo abrogates tumor-initiating potential after chemotherapy.

(A–C) Female athymic mice were injected with SUM159 cells in the number 4 mammary fat pad. (A) Once tumors reached a volume of ≥75 mm3, mice were randomized to 4 treatment groups (n = 10 mice per group): (a) vehicle (control), (b) LY2157299 (100 mg/kg/p.o. twice daily), (c) paclitaxel (20 mg/kg/d i.p. 5 times), and (d) both drugs. The arrow at day 5 represents the cessation of paclitaxel treatment. The arrow and star at day 15 indicate the cessation of LY2157299 treatment and the procurement of xenografts, respectively. The arrow at day 23 represents the cessation of monitoring for tumor growth. Lysates from xenografts harvested after (B) 5 days and (C) 14 days were assessed for total SMAD2/3 and P-SMAD2 levels by immunoblot analysis. (D) Tumor volumes calculated on day 14 (*P = 0.035; Mann-Whitney). Symbols indicate individual tumors; horizontal bars represent the mean. (E) Single cell suspensions derived from xenografts harvested on day 14 were analyzed for ALDH activity using the ALDEFLUOR assay (*P = 0.002). (F) Serum was collected from mice at the time of tumor harvesting and sacrifice and analyzed for IL-8 protein levels by ELISA. Each bar represents mean IL-8 levels over control ± SEM (n = 3). ns, not significant. (G) Progression of tumor volumes during days 14–23 in mice treated with paclitaxel with or without LY2157299 during days 1–14 (n = 5). Tumors treated with vehicle (controls) or LY2157299 alone are not included, as they had reached a volume of ≥1 cm3 on day 14 (start of x axis). Because of the large tumor burden, these mice had to be sacrificed as per institutional guidelines. Error bars indicate SEM.
TGF-β inhibition in vivo abrogates tumor-initiating potential after chemotherapy.
Upon cessation of LY2157299 treatment at day 14, mice (n = 5) previously receiving paclitaxel (for 5 days) with or without LY2157299 were monitored for an additional 9 days to assess tumor growth. During this follow up, tumors in mice previously treated with LY2157299 and paclitaxel exhibited a slower rate of growth compared with that of tumors in mice treated with paclitaxel alone (Figure 6G). Tumors from all 4 groups were harvested on day 14; single tumor cell suspensions were prepared and injected in limiting dilutions (102–104) into tumor-free mice, which were monitored for subsequent tumor formation for 25 days. This limiting dilution assay is considered a rigorous test of cells with tumor-initiating capacity within a cancer population. SUM159 cells from tumors previously treated with LY2157299 alone or in combination with paclitaxel exhibited reduced tumor-forming potential (Table 2; P < 0.01). The combination of LY2157299 and paclitaxel significantly reduced the CSC frequency (Table 2). Taken together, these data suggest that TGF-β signaling sustains the tumor-initiating capacity of TNBC stem cells, at least in part, through regulation of IL-8. Thus, we propose the use of TGF-β pathway inhibitors in combination with chemotherapy to target drug-resistant TICs in TNBC.
Table 2.
Tumor incidence in limiting dilution assay

Discussion
It has been proposed that tumor recurrences following treatment with anticancer chemotherapy are driven by a subpopulation of CSCs (26, 27). Compared with the bulk tumor cell population, CSCs are thought to possess intrinsic resistance to chemotherapy. Indeed, gene expression data from tumors that remain in the breast after neoadjuvant chemotherapy for invasive breast cancer have revealed enrichment of a cancer stem cell-like signature (26).Consistent with this notion, persistence of tumor cells in the breast after chemotherapy is associated with a high rate of metastatic recurrences and shorter survival compared with that in patients that exhibit a pathological complete response (2, 26). In an effort to discover targetable signaling pathways present in drug-resistant cells, we examined gene expression signatures enriched by chemotherapy in primary breast cancers. RNA extracted from primary breast tumors before and after chemotherapy showed enrichment of CSC-related genes and a TGF-β–responsive gene signature that was also associated with the more aggressive basal-like subtype of breast cancer. We also examined levels of phosphorylated SMAD2 in tumor cell nuclei by IHC. Staining intensity and percentage of P-SMAD2–positive nuclei were not different in archival tumor sections before and after treatment (data not shown). These results concur with recent findings that a TGF-β–responsive signature was a better predictor of recurrence than P-SMAD2 tumor levels in a large cohort of patients with breast cancer (28).
In this study, treatment of TNBC cells with the microtubule inhibitor paclitaxel induced TGF-β reporter activity and upregulated genes in the TGF-β pathway. In some cases this was associated with increased phosphorylation of SMAD2. These data suggest an effect of paclitaxel on TGF-β signaling, which could be direct and/or indirect through negative selection, that is by sparing those CSCs with high TGF-β signaling. However, data in Figure 5 argue for a direct effect, albeit by molecular mechanisms that require further investigation: treatment with the type I TGF-β receptor kinase inhibitor LY2157299 and RNAi-mediated knockdown of SMAD4 inhibited paclitaxel-induced SUM159 and BT549 mammosphere formation and ALDH+ or CD44hi/PROCR+ cells, respectively. Our data do not exclude a simultaneous effect through negative selection. Nonetheless, data in Figure 5E suggest that might be the case: paclitaxel eliminates ALDH– cells but spares ALDH+ cells. Addition of LY2157299 eliminates paclitaxel-treated ALDH+, suggesting that these cells harbor autocrine TGF-β signaling causally associated with resistance to paclitaxel.
We also showed that paclitaxel induces transcription and secretion of the CSC-associated cytokine IL-8 to drive CSC expansion. Fas ligand and IL-8 are released by tumors upon treatment with chemotherapy (29). Of note, an IL8 gene expression signature correlates with poor prognosis in patients with basal-like breast cancer (30), and IL-8 has been shown to expand the CSC fraction in breast cancers (21, 31). Blockade of IL-8 has also been reported to abrogate chemotherapy-induced enrichment of CSCs (21). Further, like TGF-β, IL-8 is an osteolytic factor that can facilitate bone metastasis (32, 33). Both cytokines can expand the CSC population, which may explain the high recurrence rates observed in patients with TNBC. Taken together, these data suggest that following anticancer therapy, upregulation of autocrine/paracrine TGF-β signaling at tumor sites and in situ levels of IL-8 contribute to a tumor microenvironment that sustains CSCs in the primary cancer and also facilitates progression at metastatic sites.
We also showed a causal association between TGF-β signaling and IL-8 expression, since the latter was blocked by treatment with the TGF-βR1 kinase inhibitor LY2157299, a TGF-β type II receptor–neutralizing antibody and SMAD4 siRNA. However, we acknowledge the inconsistency between the efficacy of LY2157299 observed in vitro and in vivo in this article. We would like to point out that the variable monotherapeutic efficacy of TGF-β inhibitors in 2-dimensional and 3-dimensional culture systems has been observed and reported (34). Phase I safety and dose-finding clinical trials with LY2157299 have shown that it is well tolerated (35, 36). Efficacy studies using an intermittent schedule of this small molecule are currently ongoing in patients with hepatocellular carcinoma and multiple myeloma (37). In the xenograft studies shown herein, mouse body weight and histological examination of organs did not reveal any evidence of toxicity related to the TGF-β inhibitor. Further, the combination of LY2157299 with paclitaxel significantly reduced IL-8 expression, stem cell markers, and the tumor-initiating potential of SUM159 cancer cells compared with either drug alone.
The TGF-β type II receptor–neutralizing antibody, TR1 (12), also blocked chemotherapy-induced expansion of CSCs (Supplemental Figure 9C) by FACS analysis. Since this antibody blocks access of endogenous ligands to TGF-βRII (12), this result implies that chemotherapy-mediated induction of IL-8 follows autocrine activation of the type II receptor. This is also consistent with previous reports showing that TGF-βRII can activate MAPK (38) and PI3K (39) independently of TGF-βR1 and that RNA interference of p38MAPK abrogates IL-8 expression in a mouse model of MDA231 bone metastasis (40). Further, TGF-β can still induce p38MAPK-dependent invasion and IL-11 secretion in SMAD4-null MDA468 cells (40). In data not shown herein, treatment with LY2157299 abrogated paclitaxel-induced enrichment of CSCs in MDA468 cells. Because of the potential pharmacological advantages of antibodies and the high specificity of TR1 to TGF-βRII and the TGF-β pathway, this monoclonal antibody should also be pursued in the clinic in parallel to LY2157299 for the treatment of patients with TNBC.
In summary, results presented herein link anticancer chemotherapy, autocrine TGF-β signaling, IL-8 expression, and the expansion of cells with phenotypic markers of stem cells with tumor-initiating capacity. We surmise that, in the clinic, this phenomenon is associated with metastatic cancer recurrences and poor patient outcome. These data also imply that anticancer chemotherapy in combination with systemic antagonists of TGF-β signaling, either small molecule kinase inhibitors or therapeutic antibodies, should limit basal-like breast tumor recurrences following chemotherapy and, as a result, improve the outcome of patients with this subtype of breast cancer.
Methods
Analysis of TGF-β gene expression signature.
Raw Affymetrix U133A array CEL files for the 51 breast cancer cell lines were downloaded from ArrayExpress (E-TABM-157) and RMA normalized in R ( http://cran.r-project.org). Data were collapsed to the gene level by filtering duplicate Entrez IDs and by retaining only the most variable probe set for each gene. Genes present in a published TGF-β–induced signature in breast cancer cell lines (41) (TGF-β–responsive score [TBRS]) were extracted from the data and used for unsupervised hierarchical clustering (Spearman’s correlation with complete clustering) in R ( http://cran.r-project.org). The molecular subtype of the cell lines was defined as described previously (42). For plotting, the signature score was normalized across all cell lines to a Z score by subtracting the mean score and dividing by the standard deviation.
NanoString gene expression analysis.
Genes (n = 152) significantly regulated by TGF-β in 4 individual breast cell lines (41) were used to construct a TGF-β gene expression signature (TBRS). Genes upregulated (n = 101) and downregulated (n = 51) in this signature were quantified by NanoString in 17 paired breast tumors biopsied before and after neoadjuvant chemotherapy. Data from 96 upregulated genes and 49 downregulated genes met quality controls and were available for analysis. The TGF-β signature score “s” was calculated as:
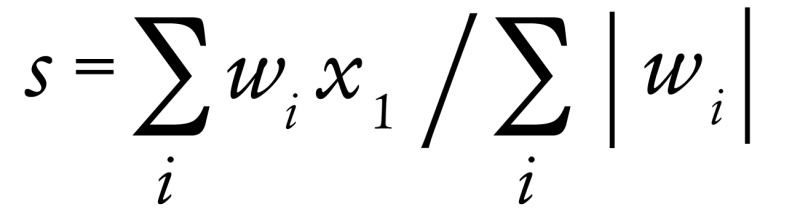
(Equation 1)
where w is the weight +1 or –1, depending on whether the gene was upregulated or downregulated in the signature (41), and x is the normalized gene expression level.
Cell lines and inhibitors.
SUM159, BT549, SUM149, and MDA231 cells were from ATCC. SUM159 and SUM149 cells were cultured in Ham’s F-12/5% FΒS and 500 ng/ml hydrocortisone; BT549 cells were cultured in RPMI1640/10% FΒS; and MDA231 cells were cultured in DMEM/10% FΒS. All cells were maintained at 37°C in 5% CO2. LY2157299 and TR1 were provided by Eli Lilly and ImClone, respectively. Paclitaxel for in vitro studies was obtained from Sigma-Aldrich. Paclitaxel for in vivo studies was obtained from the Vanderbilt Hospital Outpatient Pharmacy. The IL-8–neutralizing antibody was from R&D Systems.
Quantitative RT-PCR.
RNA was harvested using the RNAeasy Kit (Qiagen) according to the manufacturer’s instructions and used to synthesize cDNA (iSCRIPT cDNA Synthesis Kit, Bio-Rad) also according to the manufacturer’s instructions. PCR reactions were performed using SYBR Green Master Mix (Bio-Rad) and the Bio-Rad IQ5 cycler. Relative mRNA levels were standardized to GAPDH mRNA levels. The TGF-β/BMP PCR Array (SA Biosciences), containing primers for 84 genes, was performed according to the manufacturer’s instructions. Gene expression was verified in cells independently treated with paclitaxel and using primer sets different than the ones used in the TGF-β/BMP Array.
Flow cytometry.
The ALDEFLUOR assay (Stemcell Technologies) was performed according to the manufacturer’s guidelines to identify cells with high ALDH activity. Cells were passed through a 35-μm filter, suspended in ALDEFLUOR assay buffer plus BODIPY aminoacetaldehyde (BAAA), and incubated for 45 minutes at 37°C in the presence or absence of the ALDH inhibitor diethylaminobenzaldehyde (DEAB). CD44 (BD Biosciences, 1:25) and PROCR (BD Biosciences, 1:5) were incubated with single cells in PBS/1% FΒS for 30 minutes at 4°C. Cells were stained with propidium iodide to exclude nonviable cells. CSC populations were identified as ALDH+ (31) in SUM159 or SUM149 cells and as CD44hi/PROCR+ (43, 44) in BT549 cells (Supplemental Figure 1). In MDA231 cells, PROCR+/ESA+ (43) positive cells were used to identify CSCs.
Mammosphere assays.
Single cell suspensions were seeded in 6-well or 24-well ultra-low attachment plates (Corning) in serum-free DMEM/F12/20 ng/ml EGF (R&D Systems) and 1× B27 (Invitrogen). Monoclonality of mammospheres was verified using methods described previously (45). Mammosphere number and volume was determined using the GelCount mammalian cell colony counter (Oxford Optronix). Secondary mammospheres were obtained following the mechanical and enzymatic dissociation of primary mammospheres as described previously (3).
Xenograft studies and limiting dilution assay.
Athymic female mice (Harlan Sprague Dawley) were inoculated with 5 × 106 SUM159PT cells mixed with Matrigel (1:1) subcutaneously or orthotopically in the number 4 mammary gland. Six days later, tumor-bearing mice were randomized to treatment with saline, LY2157299 (100 mg/kg bid via orogastric gavage), paclitaxel (20 mg/kg/d 5 times i.p.), or both drugs. Tumor diameters were serially measured with calipers, and mouse weight was determined 3 times weekly. Tumor volume (in mm3) was calculated by the following formula: volume = length/2 × width2. Blood was collected from anesthetized mice and centrifuged at 200 g for collection of serum, which was stored at –80°C in 50 μl aliquots. Hearts, livers, and xenografts were flash frozen or fixed in 10% formalin, followed by embedding in paraffin. Xenografts were harvested and rinsed in PBS, mechanically minced with sterile blades in DMEM/F12/5% FBS and antibiotics/antimycotics in C-tubes (Miltenyi Biotech), and incubated with 1x collagenase/hyaluronidase plus 0.2 mg/ml DNAseI for 1 to 2 hours at 37°C. The dissociated cells were passed through 70- and 35-μm filters and separated from debris using a Ficoll-Paque gradient. Cell viability was verified by trypan blue exclusion before inoculation into tumor-free mice, FACS analysis, or mammosphere assays.
Immunoblot analysis.
Cells in monolayer and mammospheres were lysed with NP40 lysis buffer (150 mM Tris, pH 7.4, 100 mM NaF, 120 mM NaCl, 0.5% NP-40, 100 μM sodium vanadate, and 1× protease inhibitor cocktail [Roche]). Lysates (40 μg) were resolved by SDS-PAGE and transferred to nitrocellulose membranes; these were first incubated with primary antibodies at 4°C overnight or at room temperature for 2 hours, followed by incubation with HRP-conjugated anti-rabbit and anti-mouse secondary antibodies (Santa Cruz Biotechnology) for 1 hour at room temperature. Immunoreactive bands were visualized by enhanced chemiluminescence (Thermo Scientific). The following primary antibodies were used: SMAD4 (Santa Cruz Biotechnology, 1:1,000), P-SMAD2 (Cell Signaling, 1:500), total SMAD2/3 (Cell Signaling, 1:1,000), β-actin (Cell Signaling, 1:1,000), and IL8RA and IL8RB (Santa Cruz Biotechnology, 1:500).
Transcriptional reporter assays.
Following treatment with paclitaxel, cells were seeded in 24-well dishes. Twenty-four hours later, cells were transfected with pCAGA-Luc and CMV-Renilla (Promega) using Fugene 6 transfection reagent (Roche). pCAGA-luciferase consists of 12 Smad3/Smad4-binding sequences (CAGA boxes) and the luciferase-coding sequence. Twenty-four hours later, firefly and Renilla reniformis luciferase activity was measured using the Dual Luciferase Kit (Promega) according to the manufacturer’s instructions in a Moonlight 3010 luminometer (Analytical Luminescence Laboratory).
RNAi experiments.
SMAD4 and CXCR1/2 siRNA (Dharmacon) was transiently transfected using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. SMAD4 shRNA retroviral vectors were provided by Yibin Kang (Princeton University, Princeton, New Jersey, USA) and transfected into Phoenix cells (ATCC) to generate retroviral particles. MDA-231 cells were transduced with retrovirus-containing media and selected in puromycin. Single cell clones were obtained and expanded.
IL-8 ELISA assay.
Cells in 6-well plates were treated with LY2157299, paclitaxel, or both for 3 days in growth medium; after this time, the cell medium was collected and centrifuged to remove cellular debris. For each treatment group cell number was determined. IL-8 ELISA (R&D Systems) was performed according to the manufacturer’s instructions. IL-8 readings were normalized to cell number, and the fold change compared with control group was calculated and graphed using GraphPad Prism Software. For in vivo serum IL-8 analysis, IL-8 levels were normalized to tumor volume and the fold change compared with the control group.
Statistics.
Statistical differences were determined using the Student’s t test. For the paired tumor NanoString analyses (before and after chemotherapy), paired t tests were used. For animal studies, the differences among treatment groups were determined by Mann-Whitney test with Bonferroni post-hoc corrections. Extreme limiting dilution assay analysis was performed to determine the statistical differences in stem cell-like frequency among the treatment groups as described previously (46). A P value of less than 0.05 was considered to be statistically significant.
Study approval.
Animal studies were approved and performed in accordance with the Vanderbilt Institutional Animal Care and Use Committee. Breast tumor blocks were identified retrospectively from a cohort of patients who had consented to use of any deidentified tissues for research purposes under the auspices of an IRB-approved protocol (Vanderbilt IRB no. 030747). Selection was based on the following criteria: (a) the patient received neoadjuvant chemotherapy and had residual cancer in the breast in the mastectomy specimen; (b) sufficient formalin-fixed paraffin-embedded (FFPE) tissue was available for nucleic acid purification; and (c) pretreatment biopsies were available for comparison. Samples were included only if they were deemed by an expert pathologist to contain ≥20% tumor cells in the entire section or if they contained a region with ≥20% tumor cells that could be macrodissected. In total, 17 patients were included. Nucleic acid purification was performed using the RNAEasy FFPE Kit (Qiagen) according to the manufacturer’s instructions. RNA samples were provided to NanoString for analysis. The raw transcript counts were normalized by dividing by the geometric mean of 7 preselected normalization “housekeeper” genes (WAS, CD40, B2M, NAGA, TUBB, NPAS2, and POLR1B), which cover a range of levels of constitutive expression.
Supplementary Material
Acknowledgments
This work was supported by ACS Clinical Research Professorship Grant CRP-07-234 (to C.L. Arteaga), The Lee Jeans Translational Breast Cancer Research Program (to C.L. Arteaga), Breast Cancer Specialized Program of Research Excellence (SPORE) P50 CA98131, Vanderbilt-Ingram Cancer Center Support Grant P30 CA68485, Stand Up to Cancer Dream Team Translational Research Grant, a Program of the Entertainment Industry Foundation (SU2C-AACR-DT0209) (to C.L. Arteaga), Susan G. Komen for the Cure Foundation Grant SAC100013 (to C.L. Arteaga), NIH R01 CA143126 (to R.S. Cook), and Susan G. Komen for the Cure Foundation Career Catalyst Grant KG100677 (to R.S. Cook).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(3):1348–1358. doi:10.1172/JCI65416.
References
- 1.Dent R, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 pt 1):4429–34. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 2.Liedtke C, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–1281. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- 3.Al-Hajj M, Wicha M, Benito-Hernandez A, Morrison S, Clarke M. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: Models and concepts. Annu Rev Med. 2007;58:267–284. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 5.McDermott SP, Wicha MS. Targeting breast cancer stem cells. Mol Oncol. 2010;4(5):404–419. doi: 10.1016/j.molonc.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100(9):672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 7.Mani S, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shipitsin M, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11(3):259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 9.Bierie B, Moses HL. Tumour microenvironment: TGF[beta]: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6(7):506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 10.Bierie B, et al. Transforming growth factor–β regulates mammary carcinoma cell survival and interaction with the adjacent microenvironment. Cancer Res. 2008;68(6):1809–1819. doi: 10.1158/0008-5472.CAN-07-5597. [DOI] [PubMed] [Google Scholar]
- 11.Bueno L, et al. Semi-mechanistic modelling of the tumour growth inhibitory effects of LY2157299, a new type I receptor TGF-[beta] kinase antagonist, in mice. Eur J Cancer. 2008;44(1):142–150. doi: 10.1016/j.ejca.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 12.Zhong Z, et al. Anti-transforming growth factor β receptor II antibody has therapeutic efficacy against primary tumor growth and metastasis through multieffects on cancer, stroma, and immune cells. Clin Cancer Res. 2010;16(4):1191–1205. doi: 10.1158/1078-0432.CCR-09-1634. [DOI] [PubMed] [Google Scholar]
- 13.Geiss GK, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotech. 2008;26(3):317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez-Angulo AM, et al. Gene expression, molecular class changes, and pathway analysis after neoadjuvant systemic therapy for breast cancer. Clin Cancer Res. 2012;18(4):1109–1119. doi: 10.1158/1078-0432.CCR-11-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ginestier C, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conlin A, Seidman A. Beyond cytotoxic chemotherapy for the first-line treatment of HER2-negative, hormone-insensitive metastatic breast cancer: current status and future opportunities. Clin Breast Cancer. 2008;8(3):215–223. doi: 10.3816/CBC.2008.n.024. [DOI] [PubMed] [Google Scholar]
- 17.Hortobagyi G. Docetaxel in breast cancer and a rationale for combination therapy. Oncology (Williston Park). 1997;11(6 suppl 6):11–15. [PubMed] [Google Scholar]
- 18.Carey LA, et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13(8):2329–2334. doi: 10.1158/1078-0432.CCR-06-1109. [DOI] [PubMed] [Google Scholar]
- 19.Torrisi R, et al. Tailored preoperative treatment of locally advanced triple negative (hormone receptor negative and HER2 negative) breast cancer with epirubicin, cisplatin, and infusional fluorouracil followed by weekly paclitaxel. Cancer Chemother Pharmacol. 2008;62(4):667–672. doi: 10.1007/s00280-007-0652-z. [DOI] [PubMed] [Google Scholar]
- 20.Derynck R, Akhurst RJ, Balmain A. TGF-[beta] signaling in tumor suppression and cancer progression. Nat Genet. 2001;29(2):117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 21.Ginestier C, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010;120(2):485–497. doi: 10.1172/JCI39397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu R-H, et al. NANOG is a direct target of TGFbeta/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell. 2008;3(2):196–206. doi: 10.1016/j.stem.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DaCosta Byfield S, Major C, Laping NJ, Roberts AB. SB-505124 is a selective inhibitor of transforming growth factor-beta type I receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2004;65(3):744–752. doi: 10.1124/mol.65.3.744. [DOI] [PubMed] [Google Scholar]
- 24.Ganapathy V, et al. Targeting the Transforming Growth Factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol Cancer. 2010;9:122. doi: 10.1186/1476-4598-9-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inman GJ, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62(1):65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 26.Creighton C, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106(33):13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 28.Bruna A, et al. TGF-beta induces the formation of tumour-initiating cells in claudinlow breast cancer. Nat Commun. 2012;3:1055. doi: 10.1038/ncomms2039. [DOI] [PubMed] [Google Scholar]
- 29.Chhipa RR, Bhat MK. Bystander killing of breast cancer MCF-7 cells by MDA-MB-231 cells exposed to 5-fluorouracil is mediated via Fas. J Cell Biochem. 2007;101(1):68–79. doi: 10.1002/jcb.21153. [DOI] [PubMed] [Google Scholar]
- 30.Rody A, et al. A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res. 2011;13(5):R97. doi: 10.1186/bcr3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charafe-Jauffret E, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009;69(4):1302–1313. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bendre MS, et al. Tumor-derived interleukin-8 stimulates osteolysis independent of the receptor activator of nuclear factor-kappaB ligand pathway. Cancer Res. 2005;65(23):11001–11009. doi: 10.1158/0008-5472.CAN-05-2630. [DOI] [PubMed] [Google Scholar]
- 33.Kang Y, et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci U S A. 2005;102(39):13909–13914. doi: 10.1073/pnas.0506517102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bauer J, et al. RNA interference (RNAi) screening approach identifies agents that enhance paclitaxel activity in breast cancer cells. Breast Cancer Res. 2010;12(3):R41. doi: 10.1186/bcr2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azaro A, et al. The oral transforming growth factor-beta (TGF-ss) receptor I kinase inhibitor LY2157299 plus lomustine in patients with treatment-refractory malignant glioma: The first human dose study. ASCO Meeting Abstracts. 2012;30:2042. [Google Scholar]
- 36.Rodon Ahnert J, et al. First human dose (FHD) study of the oral transforming growth factor-beta receptor I kinase inhibitor LY2157299 in patients with treatment-refractory malignant glioma. ASCO Meeting Abstracts. 2011;29:3011. [Google Scholar]
- 37.Hawinkels LJAC, ten Dijke P. Exploring anti-TGF-β therapies in cancer and fibrosis. Growth Factors. 2011;29(4):140–152. doi: 10.3109/08977194.2011.595411. [DOI] [PubMed] [Google Scholar]
- 38.Janda E, et al. Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis. J Cell Biol. 2002;156(2):299–314. doi: 10.1083/jcb.200109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275(47):36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- 40.Gupta J, Robbins J, Jilling T, Seth P. TGFbeta-dependent induction of Interleukin-11 and Interleukin-8 involves SMAD and p38 MAPK pathways in breast tumor models with varied bone metastases potential. Cancer Biol Ther. 2011;11(3):311–316. doi: 10.4161/cbt.11.3.14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Padua D, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133(1):66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neve R, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10(6):515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hwang-Verslues WW, et al. Multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem cell markers. PLoS One. 2009;4(12):e8377. doi: 10.1371/journal.pone.0008377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoon C-H, et al. PTTG1 oncogene promotes tumor malignancy via epithelial to mesenchymal transition and expansion of cancer stem cell population. J Biol Chem. 2012;287(23):19516–19527. doi: 10.1074/jbc.M111.337428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, et al. Transforming growth factor-[beta] regulates the sphere-initiating stem cell-like feature in breast cancer through miRNA-181 and ATM. Oncogene. 2011;30(12):1470–1480. doi: 10.1038/onc.2010.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu Y, Smyth GK. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1–2):70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.