

Abstract
The expression, processing, transport and activities of both coding and non-coding RNAs play critical roles in normal neuronal function and differentiation. Over the past decade, these same pathways have come under scrutiny as potential contributors to neurodegenerative disease. Here we focus broadly on the roles of RNA and RNA processing in neurodegeneration. We first discuss a set of “RNAopathies”, where non-coding repeat expansions drive pathogenesis through a surprisingly diverse set of mechanisms. We next explore an emerging class of “RNA binding proteinopathies” where redistribution and aggregation of the RNA binding proteins TDP-43 or FUS contribute to a potentially broad range of neurodegenerative disorders. Lastly, we delve into the potential contributions of alterations in both short and long non-coding RNAs to neurodegenerative illness.
Keywords: miRNA, TDP-43, Fragile X, Bidirectional transcription, CAG RNA, Repeat expansion disorders
1. Introduction
As the world population ages over the coming decades, the medical system will face an ever increasing incidence of neurodegenerative disorders. It is estimated that one in 7 people in the US will develop a neurodegenerative disorder in their lifetime, and dementia is now the 6th leading cause of death in the US (Dorsey et al., 2007; Thies and Bleiler, 2011). Neurodegeneration is the broad term used to describe the progressive loss first of neuronal function, then of the neurons themselves. As a central component of many forms of neurodegenerative disease is the presence of protein aggregates, much research to date has focused on protein quality control mechanisms in an effort to understand and possibly treat these disorders (Williams and Paulson, 2008; La Spada and Taylor, 2010; Selkoe, 2011). An emerging avenue of research in neurodegeneration focuses instead on the mechanisms by which RNA and RNA processing contribute to neuronal dysfunction and death. For a subset of diseases, direct mRNA toxicity via a gain of function mechanism has been proposed (Osborne and Thornton, 2006; O'Rourke and Swanson, 2009; Todd and Paulson, 2010). However, for a broader set of disorders, alterations in non-coding RNA, RNA splicing and RNA binding protein activity accompany or drive the neurodegenerative process in novel ways (Gallo et al., 2005; Cooper et al., 2009; Lagier-Tourenne et al., 2010). Moreover, the possibilities for transcriptional control mediated by long non-coding RNAs are just beginning to come to light, suggesting an even more complex mechanism by which disturbances at the RNA level can contribute to degeneration of the nervous system (Wapinski and Chang, 2011).
This review provides a broad summary of the myriad of ways by which both coding and noncoding RNAs and RNA binding proteins contribute to neurodegenerative disease. Our goal is to provide both a topical introduction for the uninitiated, as well as to facilitate cross-talk among researchers within the various subtopics we cover. We believe that there is likely significant and underappreciated overlap across different neurodegenerative disorders in terms of the mechanisms by which RNA contributes to disease pathogenesis, as evidenced by recent studies in ALS and frontotemporal dementia (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Given the breadth of this topic, aspects of our coverage of specific areas are at times limited or superficial. For each subtopic, we have therefore referenced numerous excellent recent reviews that should allow for more in-depth exploration.
2. Function and processing of RNAs in the central nervous system
To appreciate how alterations in the processing and expression of coding and non-coding RNAs contribute to neurodegeneration, it is important to review recent advances in our understanding of how RNA participates in the regulation of gene expression, RNA processing, and protein translation.
The human transcriptome is made up of both protein coding messenger RNAs (mRNAs) and multiple different classes of non-coding RNAs (ncRNAs), including ribosomal RNAs (rRNAs), transfer RNAs (tRNAs), small nuclear and nucleolar RNAs (snRNAs and snoRNAs, respectively), microRNAs (miRNAs), larger non-coding transcripts (e.g. lincRNAs and lncRNAs), and a host of recently described RNA species whose functions are less clear (e.g. vault RNAs, Y RNAs, and piRNA) (Cooper et al., 2009). Of the human genome, only about 1.2% of the sequence is protein-coding (Lander et al., 2001; Clark et al., 2011). Despite this, a much larger portion (up to 90%) of the genome is transcribed, indicating that a large number ncRNAs likely participate in numerous physiological processes in all cell types, including neurons (Lander et al., 2001; Birney et al., 2007; Wilhelm et al., 2008; Clark et al., 2011). Furthermore, many genes are transcribed in both the sense (i.e. in the direction that yields a protein encoding mRNA) and anti-sense (usually producing a shorter non-coding sequence) directions. The functions of antisense transcription are incompletely understood, but likely involve the regulation of transcription, stability, and translation of the sense transcripts with which they are associated (Katayama et al., 2005).
Messenger RNAs are initially transcribed as a pre-mRNA containing a 5′ untranslated region (5′UTR), a 3′ untranslated region (3′UTR) and numerous non-coding intronic regions (introns) between the protein coding regions of the mRNA. This pre-mRNA is spliced peri-transcriptionally, such that the intronic sequences are removed and variable combinations of the exons, including the 3′ and 5′ UTRs, are compiled into a complete coding sequence for translation (Pandit et al., 2008). For each pre-mRNA, there are usually multiple different potential alternatively spliced mature mRNAs produced, with different mature mRNAs being favored in specific tissue and cell types during distinct stages of development. Similarly, exonic combinations can be modulated in response to specific environmental cues (Poulos et al., 2011). The mature mRNA is then transported out of the nucleus and into the cytoplasm as part of a messenger ribonucleoprotein particle (mRNP). There, it associates with ribosomal machinery and is translated into its protein product in a regulated fashion. From initial transcription as a pre-mRNA and through its splicing, transport, translation, and degradation, the mRNA is associated with numerous RNA binding proteins and ncRNAs that regulate its processing, stability and translation.
Non-coding RNAs and their associated binding proteins are intimately involved in numerous cellular pathways. These pathways, which include RNA processing as well as regulation of transcription and translation, are critical determinants of neuronal differentiation and plasticity. Perhaps not surprisingly, alterations in these pathways have now been identified that contribute to a wide variety of disorders, including a number of neurodegenerative diseases (Cooper et al., 2009). For example, microRNAs (miRNAs) play a critical role in controlling gene expression post-transcriptionally. miRNAs are short sequences of between 20 and 30 nucleotides that are initially transcribed as longer primary miRNAs (pri-miRNAs) containing a hairpin structure, which must be enzymatically cleaved by Drosha, a nuclear RNaseIII (Zeng et al., 2005; Zeng and Cullen, 2006; Kim et al., 2009). The Drosha cleaved sequence, known as a precursor miRNA (pre-miRNA), is then transported to the cytoplasm, where it is cleaved by another RNase, Dicer, to produce the mature miRNA (Ketting et al., 2001; Yi et al., 2003; Murchison and Hannon, 2004; Kosik, 2006; Kim et al., 2009). These mature miRNAs then bind target sequences of mRNA transcripts to mediate either translational repression or mRNA degradation (Kosik, 2006). Interestingly, recent reports suggest that these miRNA–mRNA interactions can also lead to translational activation or transcriptional stimulation under certain circumstances (Kim et al., 2009).
In addition to short ncRNAs such as miRNAs, there are numerous classes of long non-coding RNAs (lncRNAs), including large intergenic non-coding RNAs (lincRNAs) that may meaningfully impact neurodegeneration. LincRNAs are non-protein coding genes which are highly conserved, transcriptionally regulated, spliced, and impart their function as RNA (Guttman et al., 2009). Between 2.3 and 17.2 kb, most lincRNAs studied to date function to modulate the transcription of target protein coding genes or participate in large scale shifts in the epigenetic landscape, as occurs in X-inactivation (Brannan et al., 1990; Brown et al., 1991; Mercer et al., 2009; Ponting et al., 2009; Huarte et al., 2010). Although the specific functions of most lncRNAs in the nervous system are not known, some evidence suggests further roles in neuronal differentiation and regulation of dendritic mRNA translation (Zalfa et al., 2003; Lin et al., 2008; Guttman et al., 2009; Huarte et al., 2010).
Longer variations of non-coding RNAs also include long interspersed nuclear elements (LINEs). LINE1 (or L1 elements) are typically about 6 kb in length, and may comprise up to 20% of the mammalian genome (Scott et al., 1987; Lander et al., 2001; Waterston et al., 2002; Gibbs et al., 2004). L1 elements are transcribed and exported from the nucleus, and contain two open reading frames which encode for a nucleic acid chaperone and a reverse transcriptase; however the majority of the transcribed sequence is not translated (Feng et al., 1996; Martin, 2006). The generation of these proteins allows L1 elements' autonomy for further retrotransposition (Feng et al., 1996; Martin, 2006; Singer et al., 2010). Once in the cytoplasm, L1 elements interact with RNA binding proteins, which includes their translated nucleic acid chaperone and reverse transcriptase which binds in cis, and are transported back into the nucleus (usually during mitosis) (Singer et al., 2010). In the nucleus, an endonuclease nicks target DNA sequences and allows reverse transcription of the L1 into the genome (Luan et al., 1993; Cost and Boeke, 1998). This process of L1 duplication and retrotransposition can alter the transcription of neighboring genes, and has recently been shown to play a role in neurodevelopment (Singer et al., 2010).
3. RNA dominant neurological disorders
3.1. RNA as a toxic species: the sequestration hypothesis
The concept that RNA itself acts as a primary toxic species in a neurological disorder was first proposed and established for myotonic dystrophy type 1 (DM1). DM1 is the most common adult onset muscular dystrophy and the third most common overall (Philips et al., 1998; Mankodi et al., 2000; Liquori et al., 2001; Kanadia et al., 2003; Wheeler and Thornton, 2007). In addition to skeletal muscle weakness and myotonia, patients with DM1 also have complications related to cardiac function, cataracts, and neuropsychiatric symptoms including cognitive dysfunction (Griggs and Wood, 1989; Jaspert et al., 1995; Modoni et al., 2004; Wheeler and Thornton, 2007). An autosomal dominant disorder, DM1 results from an expanded CTG repeat in the 3′ UTR of the DMPK gene (Brook et al., 1992; Fu et al., 1992; Mahadevan et al., 1992). Initial evaluations tested whether this repetitive sequence impaired production of the DMPK protein which contained the repeat, or if it may alter the expression of the genes surrounding the locus of the mutation. However, the expanded CTG repeats do not significantly alter DMPK protein expression and altered expression of neighboring genes did not recapitulate key features of the clinical disorder (Jansen et al., 1996; Reddy et al., 1996; Klesert et al., 2000; Sarkar et al., 2000; Filippova et al., 2001). However, expression of CUG repeat RNA in isolation was capable of eliciting cytotoxicity and recapitulating many of the central aspects of the human disease (Taneja et al., 1995; Philips et al., 1998; Mankodi et al., 2000). CUG repeat mRNAs form nuclear foci that co-localize with multiple members of the muscle blind-like (MBNL) splicing factor family in animal models and in affected tissues from patients (Miller et al., 2000; Mankodi et al., 2003). MBNL binds to CUG RNA repeats and redistributes from a diffuse pattern of nuclear staining to punctate foci in both patient tissue samples and when co-expressed with CUG RNA in cell culture models (Taneja et al., 1995; Mankodi et al., 2000; Miller et al., 2000). Overexpression of MBNL in mouse or Drosophila animal models of DM1 suppresses multiple aspects of the CUG RNA phenotype (Kanadia et al., 2006; Vicente-Crespo et al., 2008). MBNL knockout mice recapitulate many key features of the DM1 phenotype, including myotonia, dystrophic muscle changes, and some shared splicing abnormalities (Kanadia et al., 2003, 2006). Based on these findings, it was proposed that expanded repeats of CUG RNA might act to sequester MBNL and perhaps other key proteins as a primary disease mechanism (Mankodi et al., 2000, 2003; Miller et al., 2000).
There is significant evidence to support the hypothesis that MBNL and other proteins may contribute to DM1 pathology. In DM1 tissues, there is missplicing of a number of MBNL target messages in a fashion consistent with loss of MBNL function (Savkur et al., 2001; Mankodi et al., 2002; Wheeler et al., 2007; Osborne et al., 2009). These include missplicing of the chloride channel ClC1, which leads to myotonia; a splice variant of the glutamate receptor NMDAR1 that is predicted to impair proper dendritic targeting of the mRNA; and multiple splicing alterations in the microtubule-associated protein tau that may influence this protein's stability and aggregation properties (Sergeant et al., 2001; Mankodi et al., 2002; Jiang et al., 2004; Leroy et al., 2006; Wheeler et al., 2007; Dhaenens et al., 2008, 2011; Ghanem et al., 2009; Itoh et al., 2010). The best established of the MBNL dependent missplicing events in DM1 is the retention of a destabilizing splice variant in the chloride channel ClC1, which leads to myotonia(Mankodi et al., 2002; Wheeler et al., 2007). However, for most other identified splicing alterations, the evidence that these result from MBNL sequestration is associative rather than direct. This is especially important because of recent evidence indicating splicing alterations in several unrelated muscular dystrophy models, suggesting that muscle regeneration itself may lead to some of the observed alternative splicing changes (Orengo et al., 2011).
Identification of the gene responsible for myotonic dystrophy type 2 (DM2) further supports a critical role for MBNL sequestration in DM1 pathology (Liquori et al., 2001). DM2 has a clinical phenotype that is similar to DM1, although there is no congenital form of the disease and only minimal central nervous system involvement. DM2 results from a CCTG expansion in an intronic region of an unrelated gene, zinc finger nuclease 9 (Liquori et al., 2001). When transcribed, these CCUG repeats act very much like their counterparts in DM1, forming nuclear RNA foci that sequester MBNL proteins and are associated with similar splicing defects (Liquori et al., 2001; Mankodi et al., 2001). Taken together, DM1 and DM2 present the strongest case for a primary sequestration model of toxicity in RNA dominant disorders.
However, MBNL1 sequestration is only part of the DM1 story. In addition to causing splicing deficits, studies in patient derived tissues and multiple animal models of the disease demonstrate alterations in CUG binding protein 1 (CUGBP1) expression and activation (Philips et al., 1998; Timchenko et al., 2001). CUGBP1 is also a splicing factor, but it has numerous other roles in RNA processing, transport, and translation within neurons. Although CUGBP1 can bind to CUG RNA, it is not sequestered in RNA foci and instead appears to be activated by the presence of expanded CUG RNA expression. Importantly, overexpression of CUGBP1 recapitulates some critical features of DM1 pathology (Ward et al., 2010). Mice expressing 8 times the endogenous levels of CUGBP1 show significant skeletal muscle wasting, as evident by decreased muscle weight and functional stamina, histological evidence of degeneration, and alternative splicing (Ward et al., 2010). Together, these data suggest a concurrent role for MBNL and CUGBP1 in DM1 (Wang et al., 2007; Orengo et al., 2008).
In addition, the phenotypic differences between DM1 and DM2, especially in relation to cognitive decline and the congenital phenotype, indicate that further MBNL independent mechanisms are important in DM1. Indeed, comparison of transcriptional profiles from muscle taken from MBNL knockout and expanded CUG RNA transgenic mice reveal significant differences in transcriptional regulation that suggest roles for other factors (Osborne et al., 2009; Du et al., 2010). Some recent lines of evidence suggest roles for the sequestration or aberrant activation of MBNL independent pathways. In cardiac tissue, expression of a transgene containing the 3′ UTR of DMPK with an unexpanded repeat recapitulates many of the cardinal features of DM1 (Mahadevan et al., 2006). Both DM1 patients and these mice demonstrate upregulated expression of the cardiac specific transcription factor NKX2-5 in cardiac and non-cardiac tissues, leading to myocyte conduction defects due to aberrant transcription of other genes, including connexins 40 and 43 (Mahadevan et al., 2006; Yadava et al., 2008). These effects appear to be MBNL independent, as they are not associated with RNA foci formation or MBNL inclusions.
RNA binding protein sequestration is not unique to DM1 and DM2. In total, sequestration of specific RNA binding proteins is proposed as a primary pathogenic mechanism in at least five different RNA dominant disorders (see Table 1). One recently described example of this is spinocerebellar ataxia type 10 (SCA10). SCA10 is a rare cerebellar ataxia and epilepsy syndrome that results from an ATTCT repeat expansion in the 3′ UTR of the E46L gene (Matsuura et al., 2000; Lin and Ashizawa, 2003; White et al., 2010). In patient derived cells and in transfected cells, this RNA repeat drives formation of nuclear and cytoplasmic foci and cellular apoptosis (White et al., 2010). The RNA foci co-localize with the RNA binding protein hnRNP K and strongly bind to it in vitro and in vivo (White et al., 2010). Moreover, central aspects of the cellular RNA toxicity can be recapitulated by siRNA knockdown of hnRNP K (White et al., 2010). Future work will need to address whether interactions of AUUCU repeat RNA with hnRNP K are sufficient to explain the full clinical phenotype in animal models and whether overexpression of hnRNP K can suppress the cellular toxicity elicited by the AUUCU repeat RNA (White et al., 2010).
Table 1.
Potentially sequestered proteins in RNA dominant diseases.
| Disease | Repeat (antisense) | Bidirectional transcription? | Sequestered protein | Other pathogenesis factors |
|---|---|---|---|---|
| DM1 | CUG (CAG) | Yes | Muscleblind |
|
| DM2 | CCUG | Unknown | Muscleblind |
|
| SCA8 | CUG (CAG) | Yes | Muscleblind |
|
| HDL2 | CUG (CAG) | Yes | Muscleblind |
|
| SCA10 | AUUCU | Unknown | hnRNP K |
|
| SCA31 | UGGAA | Unknown | SFRS1 and 9 |
|
| FXTAS | CGG (CCG) | Yes | Muscleblind, hnRNPA2/B1, Pur α, Sam68, lamin A/C, others |
|
| c9FTD/ALS | GGGGCC | Unknown | Unknown |
|
Another form of spinocerebellar ataxia has recently been described, SCA31 (Sato et al., 2009). SCA31 arises from an inserted TGGAA repeat on chromosome 16. When this repeat is transcribed it forms nuclear inclusions and co-localizes with the serine/ arginine-rich splicing factors (SFRS) 1 and 9 (Sato et al., 2009). While this finding does not yet directly link SFRS1 and 9 inactivity to the pathology associated with SCA31, it does indicate yet another neurological disorder may be associated with nucleotide repeats capable of mediating neurodegeneration by way of protein sequestration.
3.2. Many roads lead to neuronal dysfunction and death in FXTAS
Fragile X-associated Tremor/Ataxia Syndrome (FXTAS) is a neurodegenerative disorder which primarily affects the grandfathers of children with Fragile X Syndrome (FXS). FXS, the most common known inherited form of mental retardation, results from a large expansion of a CGG trinucleotide repeat in the 5′ UTR of the FMR1 gene on the X chromosome (Oberle et al., 1991; Verkerk et al., 1991; Yu et al., 1991). CGG expansion to greater than 200 repeats induces hypermethylation of the repeat sequence and a neighboring CpG island within the FMR1 promoter, preventing transcription of FMR1 mRNA and leading to the absence of the FMR1 protein product, FMRP (Bell et al., 1991; Pieretti et al., 1991; Feng et al., 1995). This expansion from a normal repeat size of 6–45 CGGs to a full mutation range repeat takes at least two generations (Fu et al., 1991). While initially thought to be asymptomatic, carriers of a “pre-mutation” range repeat of 55–200 CGGs have a distinct clinical and molecular phenotype (Hagerman et al., 2001). Specifically, FXTAS patients acquire a progressive neurodegenerative disorder characterized by action tremor, gait difficulties, neuropsychiatric symptoms, peripheral neuropathy and dementia after the age of 50 (Hagerman et al., 2001; Basuta et al., 2011).
Pathologically, FXTAS patients display widespread neurodegeneration and brain atrophy with intranuclear ubiquitin positive inclusions in neurons and astrocytes (Greco et al., 2002; Iwahashi et al., 2006). Further, pre-mutation CGG repeat expansions lead paradoxically to increased expression of FMR1 mRNA and decreased expression of FMRP, depending on the size of the repeat expansion and the tissue type sampled (Tassone et al., 2000a, b; Kenneson et al., 2001). The increase in expression of the CGG repeat mRNA is significant, often as high as 5-fold greater in patient derived cell lines or in animal models of the disease. This increased FMR1 RNA expression results from greater FMR1 transcription and not from an increase in FMR1 mRNA stability associated with the enlarged repeat (Tassone et al., 2000a, b, 2007). Evidence now suggests this augmented transcription is triggered by epigenetic alterations induced by the CGG repeat expansion itself as DNA (Todd et al., 2010). Specifically, CGG repeat expansions elicit chromatin changes in vitro, in a Drosophila model of the disease, and in patient derived cell lines. Interestingly, these alterations in local chromatin structure are dynamic and modifiable by genetic and pharmacologic means, suggesting that agents aimed at chromatin remodeling might have therapeutic potential in FXTAS (Todd et al., 2010).
FXTAS pathogenesis is hypothesized to result from a primary mRNA toxic gain of function mechanism akin to DM1 (Hagerman et al., 2001; Todd and Paulson, 2010). FMR1 mRNA but not FMR protein was found in FXTAS patient derived nuclear inclusions (Tassone et al., 2004). Further, ectopic expression of an expanded CGG repeat in the context of a heterologous transcript was sufficient to cause neurodegeneration and intranuclear inclusions in Drosophila (Jin et al., 2003), transfected cells (Hashem et al., 2009), and in murine purkinje cells (Hashem et al., 2009). However, unlike DM1, the primary sequestered RNA binding protein in FXTAS remains unclear. A proteomic analysis of the nuclear inclusions in FXTAS revealed a large number of proteins in the aggregates, including the RNA binding protein hnRNP A2/B1, the nuclear envelope protein lamin A/C and the small heat shock protein αB-crystallin (Iwahashi et al., 2006). Importantly, a number of other potential toxic aggregating proteins were not seen in the inclusions, including α-synuclein and tau, although TDP-43 was not specifically tested (see Section 4). Subsequently, Jin and colleagues identified two CGG repeat RNA binding proteins, Pur α and hnRNPA2/B1, by affinity chromatography (Jin et al., 2007). Both are present in FXTAS patient inclusions and co-expression of either of these proteins suppresses CGG repeat mediated toxicity in a Drosophila model (Jin et al., 2007; Sofola et al., 2007a, b). Interestingly, hnRNPA2/B1 binds to CUGBP1 and overexpression of CUGBP1 suppresses the CGG repeat phenotype in Drosophila (Sofola et al., 2007a, b). However, as the changes in CUGBP1 are in the opposite direction of those seen in DM1, the relevance of this finding to FXTAS pathogenesis remains unclear (Sofola et al., 2007a, b).
More recent evidence suggests that altered splicing is important in FXTAS pathogenesis. Sam68 is another RNA binding protein identified within FXTAS human patient brain inclusions (Sellier et al., 2010). Sam 68 binds to both RNA and DNA and is a known splicing factor (Richard, 2010). In cell culture, Sam 68 is recruited to CGG repeat aggregates as an early event in pathogenesis. This sequestration leads to altered splicing of a number of Sam68 target mRNAs, which are also misspliced in FXTAS model mice and human patient brain samples (Sellier et al., 2010). Moreover, knockdown of Sam68 prevented CGG RNA induced aggregate formation. Interestingly, Sam68 does not bind directly to the CGG RNA, suggesting that an intermediate CGG binding protein is required to facilitate this initial interaction (Sellier et al., 2010).
Although sequestration of Sam68 and other RNA binding proteins by CGG repeat RNA may be an important aspect of FXTAS pathogenesis, other mechanisms likely contribute to neuronal dysfunction and neurodegeneration in this disorder. There is evidence for a failure of protein quality control pathways and altered cellular homeostasis in FXTAS model systems and in patient samples as evidenced by the presence of large proteinaceous ubiquitinated intranuclear inclusions (Greco et al., 2002; Willemsen et al., 2003). In a Drosophila model of the disease, co-expression of the chaperone heat shock protein 70 (Hsp70) suppresses CGG repeat induced neurodegeneration (Jin et al., 2003). Similarly, in patient derived fibroblast cultures and in neuronal cultures from female heterozygous FXTAS model mice, there are significant markers of cellular stress, including elevated expression of multiple heat shock proteins, increased nuclear heterochromatin formation and decreased cellular viability (Garcia-Arocena et al., 2010; Chen et al., 2009). In HEK cells stably transfected with 120 CGG repeats, gene activation triggers reduced cell viability and an increased sensitivity to drugs that impair protein quality control pathways (Handa et al., 2005). In transfected human neuroblastomas containing the FMR1 5′ UTR with 88 repeats in front of eGFP, there was a similar reduction in viability associated with gene activation and αB-crystallin positive (but ubiquitin negative) intranuclear inclusion formation at 72 h after gene transfection (Arocena et al., 2005). Interestingly, in both patient derived fibroblasts as well as transfected cells, CGG repeat expression leads to alterations in the expression and distribution of lamin A/C within the nuclear membrane (Garcia-Arocena et al., 2010; Arocena et al., 2005). Given that lamin A/C is found as a component of the intranuclear inclusions, the authors proposed that one component of FXTAS pathogenesis could be as a “laminopathy”, as occurs in Emory-Dreifuss muscular dystrophy and the premature aging disorder Progeria (Arocena et al., 2005; Capell and Collins, 2006; Garcia-Arocena et al., 2010; Zaremba-Czogalla et al., 2011).
Although FXTAS is a degenerative disease that affects primarily a geriatric population, a number of recent studies suggest that the CGG pre-mutation also alters neuronal and synaptic development. To understand the relevance of these findings, it is important to appreciate the normal functions of FMRP, the dynamics of FMR1 translation under normal circumstances and the potential effect of the expanded CGG repeat on translational inefficiency. FMRP itself is an RNA binding protein that regulates dendritic synthesis of proteins in response to synaptic stimulation (Ronesi and Huber, 2008). Interestingly, one of the messages FMRP likely regulates at synapses is FMR1, which is rapidly translated in response to metabotropic glutamate receptor stimulation (Weiler et al., 1997; Todd et al., 2003; Hou et al., 2006). Thus, efficient translation of FMRP at synapses is an important part of the protein's basal function. The CGG repeat in FMR1 acts to impair translation by interfering with ribosomal scanning through the 5′ UTR, preventing appropriate loading of CGG expanded FMR1 mRNA into polyribosomal complexes (Primerano et al., 2002; Ludwig et al., 2009, 2011). Although FMRP expression levels in patients with FXTAS have been widely reported as only slightly decreased, most of these measurements have been done on lymphocytes or whole blood samples rather than on brain tissue, where changes might be expected to be most robust (Tassone et al., 2000a, b; Kenneson et al., 2001; Primerano et al., 2002; Singh et al., 2007). Given evidence from mice that FMRP expression declines with age (Singh et al., 2007), a role for FMRP insufficiency in FXTAS, especially in patients with large CGG repeat expansions, deserves further consideration (Hessl et al., 2011).
Most of the studies addressing a potential role for FMRP insufficiency have utilized one of two knock-in (KI) mouse models where a portion of the human 5′ UTR from a pre-mutation carrier has been inserted into the FMR1 mouse locus (Bontekoe et al., 2001; Peier and Nelson, 2002; Entezam et al., 2007; Hashem et al., 2009). Both mice, as well as a YAC transgenic model mouse, demonstrate intra-generational instability, elevated FMR1 mRNA expression and variably decreased FMRP expression at larger premutation repeat lengths (Peier and Nelson, 2002; Willemsen et al., 2003; Entezam et al., 2007; Brouwer et al., 2008; Hashem et al., 2009).
In the best characterized of the models (Willemsen et al., 2003; Berman and Willemsen, 2009), CGG KI mice show signs of hippocampal-dependent cognitive impairment at an early age that precedes frank neurodegeneration or inclusion formation (Hunsaker et al., 2009). These same mice also show signs of age dependent cognitive decline in later life as measured by impaired visual-spatial learning and performance on the Morris water maze (Van Dam et al., 2005). Interestingly, cultured hippocampal neurons from heterozygous female CGG KI mice demonstrate early developmental defects including delayed dendritic complexity and outgrowth (Chen et al., 2010). As a follow-up to this study, Cunningham and colleagues identified significant delays in cortical development in CGG KI mice during embryogenesis characterized by migrational defects in the neocortex and altered expression of neuronal lineage markers (Cunningham et al., 2010). As these mice have relatively large CGG repeat expansions (∼150 CGGs), they express only about 50% of basal FMRP levels in embryonic tissues. Importantly, defects in neural stem cell proliferation, which have been reported in Fmr1 knockout mouse models (Tervonen et al., 2009), were not present in these animals, suggesting the etiology of the developmental defects in the CGG KI mice may be more than just FMRP insufficiency (Cunningham et al., 2010).
In the more recently described of the two knock-in models, the inefficiency of CGG-Fmr1 translation for a given repeat size appears to be greater (Entezam et al., 2007). This translational inefficiency leads to lower FMRP levels for a given CGG repeat size, especially in certain brain regions, though greater basal CGG Fmr1 RNA expression (Entezam et al., 2007). Despite the increased amount of CGG-containing RNA produced, these mice generate few intranuclear inclusions, although they do demonstrate mild cerebellar and cortical neurodegeneration with age (Entezam et al., 2007). In a recent study, these mice were found to exhibit some features also seen in Fmr1 KO mice. Most notably, they had decreased dendritic arborization and longer thin spines on histological analysis, along with increased regional brain protein synthesis rates (Qin et al., 2011). Taken together, these data suggest that therapeutic strategies aimed selectively at eliminating the toxic CGG repeat mRNA run a significant risk or exacerbating symptoms of FXTAS that may relate to decreased basal or activity dependent FMRP expression (Todd et al., 2010).
3.3. Bidirectional transcription of nucleotide repeats
Spinocerebellar ataxia type 8 (SCA8) is an autosomal dominant disorder characterized by cerebellar ataxia and progressive cerebellar neurodegeneration. Early work on SCA8 suggested yet another example of an RNA binding protein sequestration mechanism (Koob et al., 1999). SCA8 arises from a CTG expansion in the 3′ end of a non-protein coding RNA, Ataxin 8OS. As in DM1, this CUG RNA expansion leads to nuclear RNA foci formation and MBNL sequestration in the cerebellum, with associated splicing changes in the GABA-A transporter 4 (GAT4) in patient tissue samples and animal models (Daughters et al., 2009). Additionally, bidirectional transcription at the SCA8 locus through the expanded repeat mutation has been observed (Moseley et al., 2006). The antisense transcript, Ataxin8, includes a short cryptic open reading frame that includes the CAG repeat in the glutamine reading frame (Moseley et al., 2006). Furthermore, the identification of poly-glutamine inclusions in both animal models and human brain tissues suggests that SCA8 may also be a polyglutamine disorder, akin to numerous other spinocerebellar ataxias (Moseley et al., 2006).
Huntington disease-like 2 (HDL2) provides another example of a toxic effect possibly due to antisense gene transcription. HDL2 is a neurodegenerative disorder that is clinically and pathologically similar to Huntington's disease, including the presence of 1C2 positive (presumably polyglutamine) aggregates (Rudnicki et al., 2008). However, HDL2 results from a CTG expansion in the 3′ UTR of the Junctophilin gene. As in SCA8, initial research supported a mechanism related to RNA foci, MBNL recruitment and splicing defects (Rudnicki et al., 2007, 2008). In recently published work using a BAC transgenic mouse model of the disease, Wilburn and colleagues established that there is an antisense CAG transcript generated which is translated to produce a polyglutamine peptide (Wilburn et al., 2011). They further demonstrated that even when the CUG sense transcript is silenced, the CAG antisense transcript is still generated and is sufficient to drive pathogenicity and the behavioral phenotypes observed. These findings suggest a concurrent mechanism in which a toxic gain-of-function RNA and a deleterious polyglutamine protein can be generated from the same repeat expansion, but that, in the case of HDL2, protein derived toxicity appears to be the most relevant (see Fig. 1).
Fig. 1.
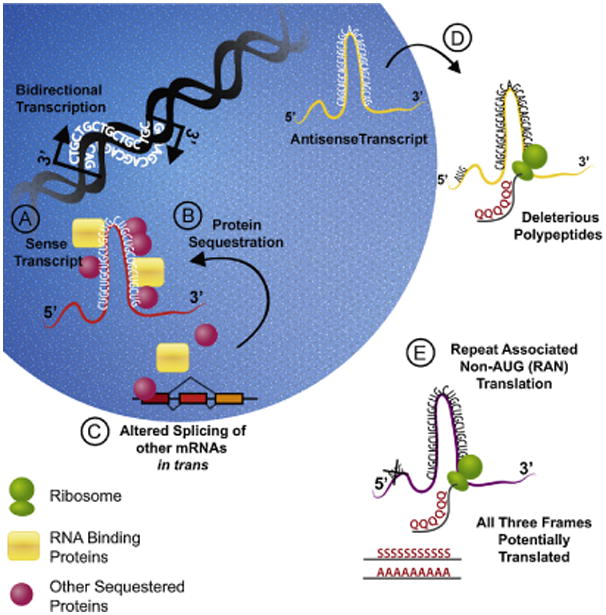
Mechanisms underlying RNA mediated neurodegeneration in nucleotide repeat disorders. Bidirectional transcription atloci with expanded nucleotide repeats leads to the generation of multiple potentially toxic products. (A) Sense transcripts of tri-nucleotide CUG (shown here), CAG, or CGG repeats form secondary mRNA hairpin structures that can bind to and sequester RNA-binding proteins. (B and C) The decreased availability of RNA binding proteins such as splicing factors leads to alterations in the splicing and expression of other mRNAs in trans. Transcription factors and other nucleotide associated proteins may also be sequestered with effects on neuronal homeostasis. (D) Antisense transcription through the repeats leads to the generation of other potentially toxic mRNA sequences. These antisense transcripts sometimes contain open reading frames through the repeat, leading to translation of potentially toxic amino acid homopolymer production (e.g. polyglutamine containing proteins). (E) Homopolymer containing proteins may also be generated through a novel process known as repeat-associated non-ATG initiated (RAN) translation. Shown is the production of a polyglutamine peptide; however, alternative homopolymers in all three potential reading frames may be generated from each transcript.
Bidirectional transcription at the FMR1 locus may also play a role in pathology of FXTAS. One antisense transcript has been discovered in human lymphoblastoma cells which overlaps FMR1 in exon 1,and is transcribed though the CGG repeats in the 5′ UTR (Ladd et al., 2007). This antisense transcript, deemed ASFMR1, is spliced, processed, and exported from the nucleus (Ladd et al., 2007). ASFMR1 contains an open reading frame that, if utilized, would encode for a polyproline tract through the CGG/CCG repeat. In addition to evidence of an alternative splice site in FXTAS patient ASFMR1, these data suggest an alternate role for aberrant protein generation in FXTAS pathology(Ladd et al.,2007).However, ASFMR1 is not the only antisense transcript of FMR1; an additional transcript, FMR4, has been described that overlaps only the very 5′ end of the FMR1 gene (Khalil et al., 2008). FMR4 does not regulate FMR1 expression, though as with ASFMR1, its expression is enhanced in XTAS patient tissues (Khalil et al.,2008).Interestingly,FMR4 ncRNA functions in cell cycle regulation and over expression of FMR4 lead to increased cell proliferation in vitro (Khalil et al., 2008). These data suggest yet another potential component contributing the pathology of FXTAS, especially in light of transcriptional upregulation at the FMR1 locus.
In addition to these three disorders, there is evidence of bidirectional transcription through the nucleotide repeat sequence in other nucleotide repeat diseases, including DM1 and spinocerebellar ataxia type 7 (Cho et al., 2005; Sopher et al., 2011). These antisense RNAs clearly influence transcription of the sense transcript and perhaps the epigenetic landscape of the associated genes, but they may also have important potential roles in toxicity. Future studies will be needed to define how each of these transcripts influences disease pathogenesis.
3.4. CAG repeats and RAN translation: novel disease mechanisms for nucleotide repeat disorders
The potential toxicity of CAG RNA repeats is relevant to a number of neurodegenerative diseases (some of which are discussed above), and as such a number of recent studies focused on CAG toxicity both within disease context and in isolate. Early work demonstrated the ability of proteins to bind to cytoplasmic CAG RNA repeats in a length-dependent fashion (McLaughlin et al., 1996). In more recent work, MBNL is capable of binding to ectopically expressed CAG repeat RNA in Caenorhabditis elegans (L.C. Wang et al., 2011). Similarly, a Drosophila model of spinocerebellar ataxia 3 expressing a non-coding expanded CAG repeat elicited neurodegeneration (Li et al., 2008). Interestingly, up-regulating the fly MBNL homolog caused a marked increase in polyglutamine-containing ataxin3 neurodegeneration, which is the opposite direction of interaction to that seen with the CUG repeat expression (Houseley et al., 2005; Li et al., 2008; Braida et al., 2010; Wojciechowska and Krzyzosiak, 2011). This effect was also investigated in a mouse model which expressed CAG repeats in the 3′ UTR of a green fluorescence protein reporter (Hsu et al., 2011). In this model, longer CAG repeats formed RNA foci and led to functional abnormalities in skeletal muscle and sperm which expressed the transgene (Hsu et al., 2011).
CAG repeats form hairpin structures and RNA foci with MBNL in human Huntington patient fibroblasts (de Mezer et al., 2011). Interestingly, in vitro evidence also suggests that CAG repeat RNA hairpins can be cleaved by Dicer and made into functional miRNAs which could have broad-scale effects on other target gene regulation (Krol et al., 2007). Similarly, expression of bidirectional transcripts may also contribute to toxicity via this mechanism. In Drosophila, expressing both CAG and complementary CUG expanded repeats results in increased toxicity as a result of dysregulated transcription of numerous genes (Krol et al., 2007). These complementary CTG expanded repeats result in the generation of miRNA-like sequences (Yu et al., 2011). Surprisingly, this increased toxicity with co-expression of CUG and CAG transcripts is the exact opposite of effects seen when CGG and CCGRN As are co-expressed in Drosophila, where dsRNA duplex formation triggers transcript degradation through an Argonaute dependent pathway, leading to phenotypic rescue (Sofola et al., 2007a, b). Thus, it appears that CAG RNA repeats are capable of eliciting toxicity in a variety of systems, either in isolation, or as a duplex with CUG RNA. However, given the overwhelming evidence for toxicity of polyglutamine containing proteins in neurons (Williams and Paulson, 2008), it remains unclear whether CAG RNA toxicity in these disorders will emerge as a significant or minor contributor to pathogenesis.
Even more provocative is a recent study suggesting a novel mechanism by which non-coding RNA repeats might lead to toxicity and neurodegeneration. Zu et al. describe an aberrant repeat-associated non-ATG (RAN) translation which generates polyglutamine, polyalanine, and polyserine tracts from otherwise non-coding CAG repeat mRNA messages (Zu et al., 2011). This was initially identified when transfection of a CAG repeat-containing Ataxin8OS construct lacking the ATG start site failed to prevent translation of the polyglutamine tract. Further experiments using C-terminal epitope tags in differing reading frames demonstrated that this aberrant translation occurred in all three possible reading frames of the CAG repeat. The effect of RAN translation was repeat length-dependent, in that constructs with 15 CAG repeats did not trigger AUG independent translation, but those with 45 or greater did. This RAN translation only occurs when the repeat tract is complementary and forms a hairpin structure. Zu et al. further identified staining for these novel proteins in a SCA8 mouse model, and in human SCA8 cerebellum using an antibody generated against the putative SCA8-GCA frame (alanine) peptide. A similar approach demonstrated a novel polyglutamine protein in DM1, presumably as a result of RAN translation from an antisense transcript though the CTG/CAG repeat (Cho et al., 2005; Zu et al., 2011). These findings suggest a novel mechanism for aberrant protein synthesis from repetitive nucleotide sequences in the absence of an obvious open reading frame. While further studies are required to establish whether RAN translation significantly contributes to pathogenesis in DM1 and SCA8, the potential extension of this mechanism to other nucleotide repeat disorders could dramatically change our understanding of what drives neurodegeneration in this class of diseases (Pearson, 2011).
4. TDP-43 and FUS as “RNA binding-proteinopathies”
A major area of advancement in neurodegenerative disease research over the past five years involves the identification of two proteins commonly found in neuronal inclusions: the Tar DNA binding protein of 43 kD (TDP-43) and the Fused in Sarcoma/ Translocated in Liposarcoma protein (FUS/TLS) (Lagier-Tourenne and Cleveland, 2009; Buratti and Baralle, 2010; Chen-Plotkin et al., 2010; Gendron et al., 2010; Lagier-Tourenne et al., 2010; Mackenzie et al., 2010). TDP-43 was identified as a component of ubiquitin positive but tau negative inclusions in patients with frontotemporal lobar dementia (FTLD) and in neuronal inclusions in the vast majority of sporadic cases of amyotrophic lateral sclerosis (ALS) (Neumann et al., 2006). Subsequent analysis of familial and sporadic cases of ALS revealed mutations in TDP-43 as a rare cause of the disease (Gitcho et al., 2008; Kabashi et al., 2008; Sreedharan et al., 2008; Yokoseki et al., 2008). TDP-43 pathology has now been identified in numerous neurodegenerative conditions associated with proteinaceous neuronal and non-neuronal inclusion formation, including Alzheimer's disease, Parkinson's disease, polyglutamine disorders suchasHuntington's disease, and Inclusion Body Myopathy (Amador-Ortiz et al., 2007; Guinto et al., 2007; Higashi et al., 2007; Nakashima-Yasuda et al., 2007; Neumann et al., 2007; Hu et al., 2008; Lin and Dickson, 2008; Schwab et al., 2008; Uryu et al., 2008; Weihl et al., 2008; Arai et al., 2009; Finch et al., 2009; Chanson et al., 2010; Lagier-Tourenne et al., 2010; Haraguchi et al., 2011; Pao et al., 2011).
TDP-43 is a ubiquitously expressed 414 amino acid protein with two N-terminal RNA recognition motifs and a flexible glycine rich region in the C terminus that mediates interactions with other proteins (Kuo et al., 2009; Lagier-Tourenne and Cleveland, 2009). Under normal circumstances, TDP-43is predominantly localized in the nucleus associated with a large number of proteins and mRNAs in heterogeneous nuclear ribonucleoprotein particles (hnRNPs) (Buratti et al., 2005; Freibaum et al., 2010). As a component of these hnRNPs, it binds to both RNA and DNA in vitro and in vivo (Buratti and Baralle, 2001; Casafont et al., 2009; Lalmansingh et al., 2011). The exact critical roles of TDP-43 within these complexes are still being elucidated, but evidence to date supports functional roles at multiple steps in RNA expression and processing, including transcriptional regulation, mRNA splicing, RNA transport/stabilization and translation (Ou et al., 1995; Buratti et al., 2001, 2004, 2005; I.F. Wang et al., 2008; Freibaum et al., 2010; McDonald et al., 2011).
In 2009, a second RNA and DNA binding protein, FUS, was identified as a cause of familial ALS (Kwiatkowski et al., 2009; Vance et al., 2009). As with TDP-43, subsequent reports identified it as a component of inclusions in other neurodegenerative disorders, including some forms of frontotemporal dementia (FTD), Basophilic inclusion body disease, and neuronal intermediate filament inclusion disease (Munoz et al., 2009; Neumann et al., 2009; Doi et al., 2010). As with TDP-43, FUS was also found in association with the neuronal intranuclear inclusions seen in a number of polyglutamine disorders (Doi et al., 2010). Importantly, FUS is not a component of inclusions in sporadic ALS and does not co-localize with TDP-43 inclusions in most other disorders (Kwiatkowski et al., 2009; Vance et al., 2009; Lagier-Tourenne et al., 2010). However, there are significant functional similarities between FUS and TDP-43 that have led investigators to consider them as a new class of proteins involved in neuronal inclusion formation rather than solely as distinct entities (Lagier-Tourenne et al., 2010). As with TDP-43, FUS binds avidly to RNA and DNA and is found largely in nuclear RNP complexes, albeit with different protein and RNA partners and a slightly different localization pattern (Zinszner et al., 1997; Hicks et al., 2000). Similarly, FUS is thought to participate in a number of stages RNA transcription and processing, including RNA splicing, transport and stability (Zinszner et al., 1997; Hicks et al., 2000; Gal et al., 2010; Ito et al., 2011).
One of the pathological hallmarks of the ALS and FTLD associated with TDP-43 inclusions is the redistribution of TDP-43 from the nucleus and into the cytoplasm (Neumann et al., 2006). This appears to be an early event in the disease process, and can occur in the absence of frank intranuclear or cytoplasmic inclusions in both patients and mouse models (Giordana et al., 2010; Wils et al., 2010). This cytoplasmic redistribution is associated with a significant decrease in nuclear TDP-43 staining (Kwiatkowski et al., 2009; Vance et al., 2009). Interestingly, the redistribution of endogenous TDP-43 can be triggered by expression of mutated forms of TDP-43 lacking a nuclear localization signal (NLS), leading to recapitulation of central features of disease pathology (Winton et al., 2008). Importantly, FUS also redistributes to the cytoplasm in ALS cases resultant from FUS mutations as well as in other degenerative disorders where it aggregates (Kwiatkowski et al., 2009; Vance et al., 2009).
A number of lines of evidence suggest that the cytoplasmic redistribution of TDP-43 or FUS is a fundamental component of disease pathogenesis. In neuronal cultures, the cytoplasmic redistribution of fluorescently labeled wildtype or mutant TDP-43 was predictive of neuronal death (Barmada et al., 2010). Interestingly, death was independent of nuclear or cytoplasmic inclusion formation. In Drosophila models, human TDP-43 expression is toxic when expressed in any of a number of model systems, with generally greater toxicity when pathogenic mutants of TDP-43 are expressed (Hanson et al., 2010; Li et al., 2010; Ritson et al., 2010; Estes et al., 2011). This toxicity is enhanced by limiting expression to the cytoplasm and largely alleviated by limiting expression of the protein to the nucleus by eliminating the nuclear localization or exportation sequence, respectively. Recent work using a mouse model expressing human TDP-43 with a mutated NLS also showed greater toxicity compared to expression of wildtype human TDP-43 alone, although this analysis was complicated by dosage issues related to increased hTDP-43 in the NLS mutants compared to wildtype TDP-43 controls (Igaz et al., 2011). Interestingly, these NLS mutants were associated with relatively few inclusions and elicited at least a portion of their toxicity by triggering transcriptional down regulation of the endogenous TDP-43 gene, effectively creating a nuclear TDP-43 null mouse in affected neurons. Neurons expressing the human TDP-43 NLS mutants thus exhibit significant transcriptional abnormalities that mirror those reported in association with small interfering RNA (siRNA) knockdown of TDP-43 (Igaz et al., 2011).
Like TDP-43, overexpression of human FUS is toxic in multiple model systems and is associated with cytoplasmic redistribution and aggregate formation (Chen et al., 2011; Igaz et al., 2011; Ju et al., 2011; Kryndushkin et al., 2011; Lanson et al., 2011; Sun et al., 2011). This toxicity is increased with pathogenic mutations in some but not all model systems, and pathogenic mutations do not appear to alter its intrinsic aggregation properties (Fushimi et al., 2011; Igaz et al., 2011; Lanson et al., 2011; Sun et al., 2011). As with TDP-43, cytoplasmic distribution of FUS is associated with greater toxicity, and blocking cytoplasmic redistribution of the protein effectively abrogates toxicity (Lanson et al., 2011; Sun et al., 2011).
In addition to changes in subcellular localization, the ability of TDP-43 and FUS to bind to RNA appears critical to both their normal functions as well as their ability to induce neurodegeneration. In C. elegans, a mutation in either of the two major RNA binding domains in TDP-43 completely blocks its neurotoxic effects (Ash et al., 2010). Similarly, in a yeast model of FUS related toxicity, mutations in the RNA binding protein block toxicity and aggregation (Sun et al., 2011). Moreover, genetic suppressors of FUS toxicity included numerous RNA binding proteins normally found in mRNPs and RNA stress granules (Ju et al., 2011; Sun et al., 2011), further supporting a role for FUS interactions with RNA in pathogenicity.
What do TDP-43 and FUS do in the cytoplasm? Under physiological conditions, both proteins are found in association with RNA granules that transport mRNAs into dendrites (Elvira et al., 2006). For FUS in particular, there is evidence that these proteins play an active role in activity dependent transport and translational regulation at synapses (Belly et al., 2005; Fujii et al., 2005). Consistent with this finding, in mice lacking FUS, there are significant dendritic spine abnormalities, with a decrease in total spine number and an increase in long thin filopodia-like spines (Fujii et al., 2005). Abnormalities in spine development are also seen with decreased TDP-43 expression in Drosophila neurons, where TDP-43 has been shown to play an active role in the regulated translation of Futch/MAP1B mRNA (Godena et al., 2011).
There is also an emerging role for both TDP-43 and FUS in stress granule formation and stability (Colombrita et al., 2009; Bosco et al., 2010). Stress granules are microscopically visible cytoplasmic domains that house RNA binding proteins and their cognate translationally arrested mRNAs (Thomas et al., 2011). Their formation is triggered by a host of cellular insults (oxidative stress, heat shock, toxins, axotomy) and is associated with a rapid redistribution of certain hnRNP proteins from the nucleus into the cytoplasm. Normally their formation is transient, allowing for redistribution of mRNAs for storage, decay or translation after the insult. In response to a variety of cellular insults, TDP-43 and FUS both translocate from the nucleus into stress granules (Colombrita et al., 2009; Bosco et al., 2010; Freibaum et al., 2010; Liu-Yesucevitz et al., 2010; Dewey et al., 2011). Neither is required for stress granule formation, but TDP-43 regulates stress granule dynamics, both through direct recruitment of T-cell intracellular antigen 1 (TIA-1) to stress granules and through regulating the expression of another key stress granule protein, GTPase-activating protein binding protein 1 (G3BP) (McDonald et al., 2011).
Given the significant functional overlaps between TDP-43 and FUS, it is perhaps not surprising that their direct or indirect interaction might be important in disease pathogenicity (Ling et al., 2010; Kryndushkin et al., 2011; Lanson et al., 2011). In cells stably transfected with normal human or mutant TDP-43, FUS was identified by SILAC coupled Mass Spectroscopy and immunoprecipitation studies as a TDP-43 interacting protein (Ling et al., 2010). Interestingly, this interaction was enhanced for TDP-43 mutants (Ling et al., 2010). In yeast, FUS and TDP-43 have a high propensity of co-aggregation that is not seen with other aggregate-prone proteins (Kryndushkin et al., 2011). In Drosophila, FUS expression drives a neurodegenerative eye phenotype that is enhanced by co-expression of normal human or mutant TDP-43 (Lanson et al., 2011). Moreover, FUS and TDP-43 form a functional complex at endogenous expression levels in mammalian cells (Kim et al., 2010). Knockdown of either TDP-43 or FUS leads to decreased expression of histone deacetylase 6 (HDAC6) mRNA and protein (Fiesel et al., 2010; Kim et al., 2010). In the case of TDP-43, maintenance of HDAC6 mRNA expression requires an intact RNA binding domain which is partially lost in mutant forms of TDP-43 (Fiesel et al., 2010). Thus, there is a direct convergence of FUS and TDP-43 in common biochemical pathways, suggesting specific activities of these proteins that may be important in disease pathogenesis.
Given the multiplicity of important cellular processes in which TDP-43 and FUS are involved, it has been somewhat difficult to identify a specific pathway that is critical for ALS disease pathogenesis. An important step towards this end was recently achieved via identification of TDP-43 RNA targets and altered transcriptional and splicing patterns in disease and in the absence of TDP-43 (Polymenidou et al., 2011; Sephton et al., 2011; Tollervey et al., 2011). TDP-43 interacts with a remarkably large fraction of the mouse transcriptome (upwards of 30%), predominately through interactions with UG rich sequences in long intronic regions as well as the 3′ UTR regions of mRNAs, especially in the cytoplasm. TDP-43 also associates with a large number of non-coding RNAs and these showed the greatest expression alterations in diseased brain tissue samples, although the overall differences in bound messages were relatively small (Tollervey et al., 2011). These studies also identified approximately 900 misspliced genes in the setting of TDP-43 knockdown and another set of genes whose expression were decreased in this same setting. Although a number of interesting candidate genes emerged from these studies, they provided perhaps more questions than answers at this stage. Importantly, similar experiments to identify RNA targets of FUS are ongoing. Once these are available, identifying areas of significant overlap between the FUS and TDP-43 in terms of transcriptome alterations, splicing changes and RNA targets may allow for a more precise definition of the critical pathogenic cascades shared by these two proteinopathies.
Overall, the findings to date support a model whereby redistribution of TDP-43 and/or FUS from the nucleus to the cytoplasm, either into stress granules or inclusions, leads to both a nuclear loss of function as well as a gain of function phenotype associated with excess cytoplasmic accumulation, including the maintenance and propagation of stress granules or altered cytoplasmic translational regulation (see Fig. 2). The initial trigger for this redistribution remains unclear, but it is tempting to speculate that some chronic cellular insult could trigger ongoing activation of the stress granule response which would favor TDP-43 redistribution. With the identification of specific TDP-43 RNA targets and effects that can be defined as predominantly nuclear or cytoplasmic, it is now becoming possible to more effectively test this hypothesis and identify which, if either, of these two components is critical to disease pathogenesis in TDP-43 associated disorders.
Fig. 2.
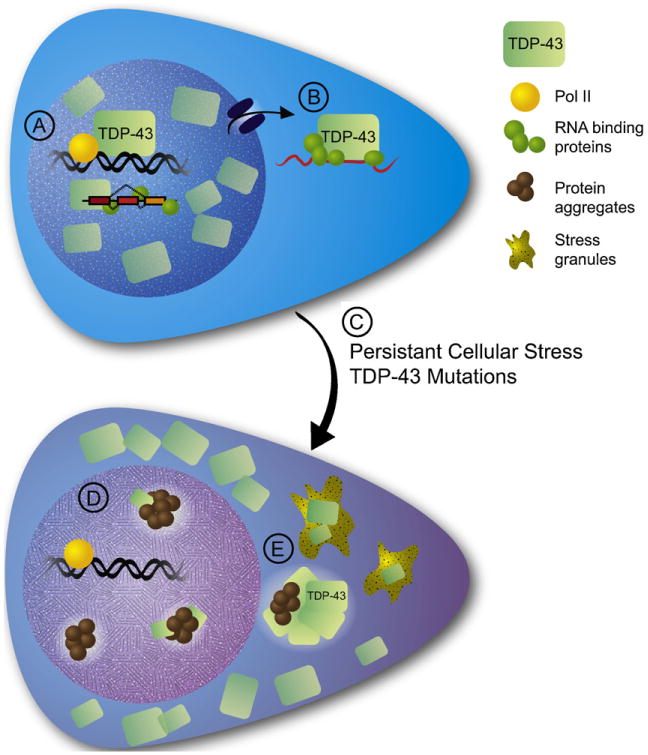
TDP-43 alterations in ALS and other neurodegenerative disorders. (A) TDP-43 is a nuclear protein which associates with the RNA polymerase Pol II on target DNA sequences, and participates in mRNA splicing. (B) Some TDP-43 is also exported from the nucleus associated with RNA-binding proteins on a subset of mRNAs. (C) Chronic neuronal stressors or mutations in TDP-43 lead to the translocation of the majority of TDP-43 to the cytoplasm. (D) This results in a loss of TDP-43s normal nuclear functions related to splicing and transcriptional regulation. (E) The increase in cytoplasmic TDP-43 results in a toxic gain-of-function via generation of TDP-43 protein aggregates as well as the accumulation of TDP-43 in cytoplasmic stress granules.
5. MicroRNAs in neurodegeneration
MicroRNAs (miRNAs) are short non-coding RNAs which regulate mRNA stability and translation (Eacker et al., 2009). In addition to direct RNA toxicity and the primary roles of TDP-43 and FUS in neurodegenerative disorders, numerous lines of research now implicate miRNAs and altered miRNA processing in neuro-degeneration (Hebert and De Strooper, 2009; Delay and Hebert, 2011; Enciu et al., 2011). Generally, these studies have taken one of two approaches: studying the impact of global miRNA synthesis on neuronal function and neurodegeneration, or identifying alterations in the expression of particular miRNAs in models of neurodegenerative diseases. However,a few recent studies suggest a model by which mutated disease proteins interfere with complicated transcription factor/miRNA regulatory networks as a central component of their pathogenic cascades (Packer et al., 2008; Gehrke et al., 2010; Schonrock et al., 2010; Wang et al., 2010).
A number of groups have looked at the effects of suppressing global miRNA processing by knocking down or eliminating critical components of the miRNA machinery, such as Dicer (Bilen et al., 2006; Schaefer et al., 2007; Davis et al., 2008; Lee et al., 2008; Konopka et al., 2010; Tao et al., 2011). Based on these studies, it appears that miRNAs are required for normal neuronal development and function, and their loss eventually leads to neuronal degeneration (Davis et al., 2008; Konopka et al., 2010). In cerebellar purkinje neurons, selective knockout of Dicer postnatally leads to a slowly progressive selective degeneration of these cells starting about 8 weeks post-gene inactivation (Schaefer et al., 2007). Interestingly, these dying neurons recapitulate many common features associated with neurodegenerative disorders, including cytoplasmic inclusion formation, the appearance of autophagic vacuoles, and an increase in apoptotic markers. When Dicer is knocked out in forebrain neurons during development, these animals demonstrate abnormalities in the maintenance of dendrite branching and spine morphology in the hippocampus (Davis et al., 2008). Intriguingly, these deleterious effects of impairing the miRNA machinery are time-dependent (Konopka et al., 2010). If Dicer is knocked out postnatally in these same forebrain neurons, during the first 8 weeks post inactivation, these animals actually display enhanced learning and memory and enhanced hippocampal long term potentiation (Konopka et al., 2010). However, as time progresses, neurodegeneration ensues, suggesting a dichotomous function for miRNA translational control throughout the life of neurons (Schaefer et al., 2007; Konopka et al., 2010). Dicer expression is also critical for the normal function of astroglial populations. As in forebrain neuron knockout experiments, mice lacking Dicer expression in astrocytes develop normally; but display early neurodegeneration consisting of motor abnormalities, seizures, and death by 10 weeks of age (Tao et al., 2011). This finding indicates that miRNA astrocytic function is necessary for neuronal survival (Tao et al., 2011). Broad miRNA alterations also exacerbate neurodegenerative phenotypes in disease models. For example, reducing Dicer function in Drosophila increases polyglutamine-mediated cell toxicity (Bilen et al., 2006). Additionally, down-regulation of specific miRNAs which normally target the ataxin1 transcript leads to increased cell death in human cell models of spinocerebellar ataxia type 1, suggesting a normal role for miRNAs in translational regulation of potentially toxic proteins (Lee et al., 2008).
Screening for differences in miRNA expression using either microarray platforms or next generation sequencing technology has facilitated numerous studies into alterations of miRNA levels in neurodegenerative diseases. Altered expression of several miRNAs has been found in both mouse models and human Huntington's disease (HD) patients (Marti et al., 2010; Lee et al., 2011). Among these altered in HD is mir9, which suppresses translation of the RE1-silencing transcription factor (REST) (Packer et al., 2008). REST is normally sequestered in the cytoplasm through its interaction with the huntingtin protein (Zuccato et al., 2003). Polyglutamine expansion in the huntingtin protein abrogates the interaction between huntingtin and REST, allowing REST to translocate to the nucleus and inhibit the transcription of a large group of genes normally suppressed in mature neurons (Zuccato et al., 2007). miR9 itself is downstream of an RE1 repressor sequence, leading to lower miR9 expression in HD patient tissues. This dysregulated feedback loop results in further increased expression of REST, which triggers wide-scale effects on gene transcription in HD (Packer et al., 2008).
A second potential alteration in miRNA pathways in HD involves Drosha, the RNAse that cleaves nuclear pri-miRNAs into pre-miRNAs prior to nuclear export. Drosha expression is decreased in HD patient brains (Lee et al., 2011). Further, there are alterations in the final 3′ end sequences in a number of miRNAs in HD, which might alter their stability and regulatory capabilities (Marti et al., 2010). Thus, miRNA-mediated translational regulation in HD could be altered by changes in miRNA processing, rather than just miRNA expression (Marti et al., 2010). However, the mechanism by which this occurs is not currently known.
A number of studies have identified miRNA abnormalities in human samples and animal and cellular models of Alzheimer's disease (AD) (Hebert et al., 2008; Lukiw et al., 2008; Niwa et al., 2008; Patel et al., 2008; W.X. Wang et al., 2008; Boissonneault et al., 2009; Hebert and De Strooper, 2009; Hebert et al., 2009; Lau and de Strooper, 2010; Schonrock et al., 2010; Wang et al., 2010; Yao et al., 2010). Interestingly, direct application of Aβ42, the presumed toxic peptide in AD, to neuronal cultures triggers alterations in the expression of numerous miRNAs (Schonrock et al., 2010; Wang et al., 2010). It is not clear how Aβ42 elicits this effect, although it is known that Aβ42 application activates a number of neuronal receptors and leads to altered synaptic physiology (Freir et al., 2001; Lee et al., 2009; Min et al., 2011). Some evidence also suggests that miRNA alterations in AD can lead to abnormal expression of certain cytoskeletal proteins. Specifically, one study found the actin-binding protein cofilin to be up-regulated in AD, possibly due to reduced levels of miR107 and miR103 (Yao et al., 2010). However, it remains unclear whether miRNA dysregulation is a proximal event in AD pathogenesis or is a downstream consequence of ongoing neurodegeneration that is not specific to AD. One approach to this question is analysis of how miRNA pathways may directly influence Aβ42 production. Along these lines, in vitro experiments demonstrate a number of specific miRNAs that target APP translation, though these interactions have not yet been demonstrated in human disease tissue (Patel et al., 2008; Hebert and De Strooper, 2009).
A number of recent studies implicate microRNA pathways in the pathogenesis of Parkinson's disease (PD) (Kim et al., 2007; Asikainen et al., 2010; Gehrke et al., 2010; Le et al., 2011). miRNAs are broadly required for dopaminergic neuronal function and survival, as the absence of a critical component of the miRNA machinery, Dicer, leads to apoptosis of dopamine neurons in mice (Kim et al., 2007). Mutations in the leucine rich kinase protein LRRK2 are the most common cause of familial Parkinson's disease (Paisan-Ruiz et al., 2004; Zimprichetal., 2004).Arecent studyfinds that LRRK2 directly effects miRNA processing by binding to Argonaute, a component of the RISC complex. This interaction alters global neuronal miRNA activity and influences neurodegeneration in a Drosophila model of PD (Gehrke et al., 2010). A second study implicates miR133, which is reduced in human PD patients, as a particularly important miRNA in Parkinson's disease (Kim et al., 2007). One target for miR133 is the transcription factor Pitx3, which is required for the differentiation and longevity of dopaminergic neurons. Intriguingly, single nucleotide polymorphisms in and near Pitx3 were found to be associated with Parkinson's disease in genome wide association studies (Kim et al., 2007; Le et al., 2011). Pitx3 itself regulates transcription of miR133, creating a feedback loop for Pitx3 expression that may prove pivotal to dopaminergic neurodegeneration in PD (Kim et al., 2007).
6. The new RNAs on the block: a future role in neurodegeneration?
While miRNAs have been widely studied to date, a number of other important classes of non-coding RNAs have not yet been extensively examined for roles in neurodegeneration. Long non-coding RNAs likely play critical roles in neuronal function and thus might be expected to contribute to neurodegenerative processes (Mercer et al., 2008; Qureshi et al., 2010). Long intergenic non-coding RNAs (lincRNAs) are evolutionarily conserved sequences that are transcribed, spliced and polyadenylated in a fashion resembling mRNAs (Mercer et al., 2009; Ponting et al., 2009; Guttman et al., 2010). LincRNAs are purported to mediate chromatin modifications and transcriptional regulation, although the full spectrum of their potential functions remains unknown (Khalil et al., 2009; Huarte et al., 2010; Tsai et al., 2010; Gong and Maquat, 2011; X. Wang et al., 2011). Specific lincRNAs such as the metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) and nuclear enriched abundant transcript 1 (NEAT1) are critical not only for correct splicing of other transcripts, but also for proper synapse formation (Bernard et al., 2010; Tripathi et al., 2010). Intriguingly, these two lncRNAs are specifically altered in the setting of TDP-43 pathology in FTD, suggesting an active role in neurodegenerative processes (Tollervey et al., 2011).
To date, there are few other published examples of dysregulated lncRNAs in neurodegenerative disease. One recent study identified the non-coding RNA 17A, which participates in alternative splicing of target mRNAs, as up-regulated in AD patient tissues (Massone et al., 2011). Interestingly, 17A expression leads to an increased secretion of toxic Aβ peptide, and is capable of facilitating plaque-forming protein aggregates (Massone et al., 2011). Further, in a recent study identifying a novel mechanism of RNA control in AD, Faghihi and colleagues describe a non-coding antisense transcript of BACE, the enzyme responsible for cleaving APP to generate the toxic Aβ42 peptides (Faghihi et al., 2008). This antisense transcript is up-regulated by Aβ42, and stabilizes BACE sense transcripts, thereby conferring a feed-forward mechanism to increase Aβ42 production (Faghihi et al., 2008). In HD, there is decreased expression of the lncRNA human accelerated region 1 (HAR1), which plays roles in mRNA splicing (Gupta et al., 2010). HAR1 is under the transcriptional regulation of REST – the transcriptional inhibitor which dissociates from mutant huntingtin to translocate into the nucleus in disease states (Johnson et al., 2010). In addition to the dysregulated miRNA pathways due to aberrant REST control, HAR1 may mediate splicing abnormalities in Huntington's disease. Given the potentially broad roles of lincRNAs in diseases like cancer (Gupta et al., 2010) and drug addiction (Michelhaugh et al., 2011), it seems likely that these early examples represent the tip of the iceberg regarding this class of molecules in neuronal death and neurodegenerative disease.
In conjunction with lincRNAs, another intriguing family of non-coding genomic sequences is long interspersed nuclear elements (LINE1 or L1). L1 elements modulate expression of coding genes by retrotransposition. This effect has been seen to alter neural precursors in rat hippocampal stem cells, and affect cell fate choices (Muotri et al., 2005). Furthermore, active retrotransposition may happen in adult animals and lead to heterogeneous and mosaic gene expression profiles (Muotri et al., 2005; Coufal et al., 2009). In addition to altering neurodevelopment, data demonstrate LINEs may also be regulated by activity; mice given access to voluntary exercise were seen to have more new LINE1 insertions in subgranual layer neural stem cells than in controls (Muotri et al., 2009). Whether these transposition events serve a meaningful purpose or are perhaps harmful remains to be elucidated, but it is tempting to speculate a role for L1 elements in maintaining genomic flexibility in adult animals. LINE retrotransposition has also been evaluated in the context of neurological dysfunction; mutations in methyl-CpG-binding protein 2 (MeCP2) which cause the neurodevelopmental disorder Rett's Syndrome, lead to an increased incidence of LINE1 retrotransposition (Muotri et al., 2010). However, what impact LINE retrotransposition has in the context of neurodegenerative disease is unknown. As transcriptional and translational control of toxic RNAs and aberrant proteins becomes increasingly paramount to pathology in neurodegeneration, investigating lincRNAs and LINEs may prove to be fruitful future avenues of research.
7. Concluding remarks
This review has addressed but a few of the myriad of ways in which RNA and RNA processing might contribute to the pathogenesis of neurodegenerative disease. mRNAs that contain nucleotide repeat sequences can directly elicit neuronal dysfunction by binding to and sequestering critical proteins, preventing them from their normal functions. RNA binding proteins themselves, such as TDP-43 and FUS/TLS, can be mutated or inappropriately localized within neurons and thus contribute to neurodegeneration. Moreover, alterations in the expression and activity of microRNAs and other noncoding RNAs are a common finding in neurodegenerative disease, where they contribute to the pathogenic process in important, though still largely undefined ways.
Yet these lead findings likely represent only the tip of a larger iceberg. Emerging evidence suggests a convergence of pathogenic mechanisms in otherwise disparate neurodegenerative diseases. For example, mutations in TDP-43 have recently been associated with isolated Parkinson's disease phenotypes, with the same mutation leading to ALS in other family members (Quadri et al., 2011). Similarly, “gray zone” CGG repeat expansions (between 40 and 55 CGGs) in the FMR1 gene are associated with an increased risk of parkinsonism (Hall et al., 2009, 2010, 2011; Loesch et al., 2009, 2011), and non-penetrant CAG repeat expansions in Ataxin-2 (the gene associated with the polyglutamine disorder SCA2) have been identified as a risk factor for ALS and Progressive Supra-nuclear Palsy (Elden et al., 2010; Ross et al., 2011). Perhaps most provocatively, a very recent study identifies a dominantly inherited intronic GGGGCC hexanucleotide repeat expansion in the C9ORF72 gene as a most common inherited cause of both ALS and FTLD (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Although the mechanisms by which these genetic variations contribute to different disease phenotypes are areas of active study, it seems likely that altered RNA networks and RNA processing will play a role.
As next generation sequencing technologies promise ever greater insights into how coding and noncoding RNA networks function, we anticipate significant advances in our understanding of RNA's roles in neuronal function and neurodegenerative disease in the coming years. It is hoped that such advances in our knowledge base will translate to improved therapeutic options for patients. Indeed, preclinical trials of such therapies are beginning to emerge. In myotonic dystrophy, where a CUG mRNA repeat drives disease pathogenesis, morpholino and antisense oligonucleotide based therapies have shown promise in animal models (Mulders et al., 2009; Wheeler et al., 2009). Small molecules designed to block the interactions of the CUG RNA with associated RNA binding proteins are also able to suppress some molecular aspects of the disease phenotype in mice (Pushechnikov et al., 2009; Warf et al., 2009). Similarly, a small peptide screen recently identified a compound capable of interfering with CUG RNA hairpin formation, and its expression or delivery suppressed toxicity in flies and mice (Khateb et al., 2007; Ofer et al., 2009; Pushechnikov et al., 2009). The application of similar approaches to other nucleotide repeat disorders has been proposed and some are already underway (Khateb et al., 2007; Ofer et al., 2009; Pushechnikov et al., 2009). In ALS, the identification of TDP-43 and FUS/TLS as critical proteins in disease pathogenesis have not yet led to targeted compounds in clinical or pre-clinical trials. However, the emergence of new animal models based on mutated forms of these proteins promise to serve as valuable tools for biomarker identification and drug development (Zinman and Cudkowicz, 2011). Lastly, there are emerging efforts to therapeutically target alterations in miRNAs and other noncoding RNAs in neurodegeneration (Eacker et al., 2009; Lau and de Strooper, 2010). While these efforts are largely in their infancy, as our understanding of these RNA networks improves, so should our potential to effectively intervene and correct abnormalities. Thus, although significant work lies ahead, there is great hope that manipulating the activity and expression of noncoding RNAs will provide a powerful toolset with which to treat neurodegenerative disease.
Acknowledgments
This work was supported by NIH K08NS069809 and the Bucky and Patti Harris Professorship to PK Todd and the Systems and Integrative Biology Training Grant to AJ Renoux.
Abbreviations
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- APP
amyloid precursor protein
- Aβ42
amyloid β-42 amino acids
- CUGBP1
CUG binding protein 1
- DM1
myotonic dystrophy type 1
- DM2
myotonic dystrophy type 2
- FTD
frontotemporal dementia
- FTLD
frontotemporal lobar dementia
- FUS/TLS
Fused in Sarcoma/ Translocated in Liposarcoma protein
- FXS
Fragile X Syndrome
- FXTAS
Fragile X-associated Tremor/Ataxia Syndrome
- HAR1
human accelerated region 1
- HD
Huntington's disease
- HDL2
Huntington disease-like 2
- hnRNP
heterogeneous nuclear ribonucleoprotein particles
- HSP70
heat shock protein 70
- lincRNA
large intergenic non-coding RNA
- LINE
long interspersed nuclear element
- lncRNA
long non-coding RNA
- MALAT1
metastasis-associated lung adenocarcinoma transcript 1
- MBNL
muscleblind-like
- MeCP2
methyl-CpG-binding protein 2
- miRNA
microRNA
- mRNA
messenger RNA
- NEAT1
nuclear enriched abundant transcript 1
- NLS
nuclear localization signal
- PD
Parkinson's disease
- RAN
repeat-associated non-ATG
- REST
RE1-silencing transcription factor
- SCA10
spinocerebellar ataxia type 10
- SCA8
spinocerebellar ataxia type 8
- SFRS
serine-rich splicing factors
- TDP-43
Tar DNA binding protein of 43 kD
- UTR
untranslated region
References
- Amador-Ortiz C, Lin WL, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61(5):435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Mackenzie IR, et al. Phosphorylated TDP-43 in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117(2):125–136. doi: 10.1007/s00401-008-0480-1. [DOI] [PubMed] [Google Scholar]
- Arocena DG, Iwahashi CK, et al. Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum Mol Genet. 2005;14(23):3661–3671. doi: 10.1093/hmg/ddi394. [DOI] [PubMed] [Google Scholar]
- Ash PE, Zhang YJ, et al. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet. 2010;19(16):3206–3218. doi: 10.1093/hmg/ddq230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asikainen S, Rudgalvyte M, et al. Global microRNA expression profiling of Caenorhabditis elegans Parkinson's disease models. J Mol Neurosci. 2010;41(1):210–218. doi: 10.1007/s12031-009-9325-1. [DOI] [PubMed] [Google Scholar]
- Barmada SJ, Skibinski G, et al. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. 2010;30(2):639–649. doi: 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basuta K, Narcisa V, et al. Clinical phenotypes of a juvenile sibling pair carrying the fragile X premutation. Am J Med Genet A. 2011;155A(3):519–525. doi: 10.1002/ajmg.a.33446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell MV, Hirst MC, et al. Physical mapping across the fragile X: hypermethylation and clinical expression of the fragile X syndrome. Cell. 1991;64(4):861–866. doi: 10.1016/0092-8674(91)90514-y. [DOI] [PubMed] [Google Scholar]
- Belly A, Moreau-Gachelin F, et al. Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci Lett. 2005;379(3):152–157. doi: 10.1016/j.neulet.2004.12.071. [DOI] [PubMed] [Google Scholar]
- Berman RF, Willemsen R. Mouse models of fragile x-associated tremor ataxia. J Investig Med. 2009;57(8):837–841. doi: 10.231/JIM.0b013e3181af59d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard D, Prasanth KV, et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010;29(18):3082–3093. doi: 10.1038/emboj.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilen J, Liu N, et al. A new role for microRNA pathways: modulation of degeneration induced by pathogenic human disease proteins. Cell Cycle. 2006;5(24):2835–2838. doi: 10.4161/cc.5.24.3579. [DOI] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissonneault V, Plante I, et al. MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J Biol Chem. 2009;284(4):1971–1981. doi: 10.1074/jbc.M807530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontekoe CJ, Bakker CE, et al. Instability of a (CGG)98 repeat in the Fmr1 promoter. Hum Mol Genet. 2001;10(16):1693–1699. doi: 10.1093/hmg/10.16.1693. [DOI] [PubMed] [Google Scholar]
- Bosco DA, Lemay N, et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19(21):4160–4175. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braida C, Stefanatos RK, et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum Mol Genet. 2010;19(8):1399–1412. doi: 10.1093/hmg/ddq015. [DOI] [PubMed] [Google Scholar]
- Brannan CI, Dees EC, et al. The product of the H19 gene may function as an RNA. Mol Cell Biol. 1990;10(1):28–36. doi: 10.1128/mcb.10.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook JD, McCurrach ME, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;69(2):385. doi: 10.1016/0092-8674(92)90418-c. [DOI] [PubMed] [Google Scholar]
- Brouwer JR, Huizer K, et al. CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome. J Neurochem. 2008;107(6):1671–1682. doi: 10.1111/j.1471-4159.2008.05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CJ, Ballabio A, et al. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature. 1991;349(6304):38–44. doi: 10.1038/349038a0. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276(39):36337–36343. doi: 10.1074/jbc.M104236200. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010;7(4):420–429. doi: 10.4161/rna.7.4.12205. [DOI] [PubMed] [Google Scholar]
- Buratti E, Brindisi A, et al. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280(45):37572–37584. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- Buratti E, Brindisi A, et al. Nuclear factor TDP-43 binds to the polymorphic TG repeats in CFTR intron 8and causes skipping of exon 9: a functional link with disease penetrance. Am J Hum Genet. 2004;74(6):1322–1325. doi: 10.1086/420978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Dork T, et al. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20(7):1774–1784. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet. 2006;7(12):940–952. doi: 10.1038/nrg1906. [DOI] [PubMed] [Google Scholar]
- Casafont I, Bengoechea R, et al. TDP-43 localizes in mRNA transcription and processing sites in mammalian neurons. J Struct Biol. 2009;167(3):235–241. doi: 10.1016/j.jsb.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Chanson JB, Echaniz-Laguna A, et al. TDP43-positive intraneuronal inclusions in a patient with motor neuron disease and Parkinson's disease. Neurodegener Dis. 2010;7(4):260–264. doi: 10.1159/000273591. [DOI] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Lee VM, et al. TAR DNA-binding protein 43 in neurode-generative disease. Nat Rev Neurol. 2010;6(4):211–220. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Tassone F, et al. Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum Mol Genet. 2010;19(1):196–208. doi: 10.1093/hmg/ddp479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Tassone F, et al. Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum Mol Genet. 2009;19(1):196–208. doi: 10.1093/hmg/ddp479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yang M, et al. Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein Cell. 2011;2(6):477–486. doi: 10.1007/s13238-011-1065-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho DH, Thienes CP, et al. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol Cell. 2005;20(3):483–489. doi: 10.1016/j.molcel.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Clark MB, Amaral PP, et al. The reality of pervasive transcription. PLoS Biol. 2011;9(7):e1000625. doi: 10.1371/journal.pbio.1000625. discussion e1001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombrita C, Zennaro E, et al. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111(4):1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- Cooper TA, Wan L, et al. RNA and disease. Cell. 2009;136(4):777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cost GJ, Boeke JD. Targeting of human retrotransposon integration is directed by the specificity of the L1 endonuclease for regions of unusual NA structure. Biochemistry. 1998;37(51):18081–18093. doi: 10.1021/bi981858s. [DOI] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, et al. L1 retrotransposition in human neural progenitor cells. Nature. 2009;460(7259):1127–1131. doi: 10.1038/nature08248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CL, Martinez Cerdeno V, et al. Premutation CGG-repeat expansion of the Fmr1 gene impairs mouse neocortical development. Hum Mol Genet. 2010;20(1):64–79. doi: 10.1093/hmg/ddq432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daughters RS, Tuttle DL, et al. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 2009;5(8):e1000600. doi: 10.1371/journal.pgen.1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TH, Cuellar TL, et al. Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J Neurosci. 2008;28(17):4322–4330. doi: 10.1523/JNEUROSCI.4815-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mezer M, Wojciechowska M, et al. Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res. 2011;39(9):3852–3863. doi: 10.1093/nar/gkq1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GYR, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011 doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delay C, Hebert SS. MicroRNAs and Alzheimer's disease mouse models: current insights and future research avenues. Int J Alzheimer's Dis. 2011;2011:894938. doi: 10.4061/2011/894938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey CM, Cenik B, et al. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol. 2011;31(5):1098–1108. doi: 10.1128/MCB.01279-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaenens CM, Schraen-Maschke S, et al. Overexpression of MBNL1 fetal isoforms and modified splicing of Tau in the DM1 brain: two individual consequences of CUG trinucleotide repeats. Exp Neurol. 2008;210(2):467–478. doi: 10.1016/j.expneurol.2007.11.020. [DOI] [PubMed] [Google Scholar]
- Dhaenens CM, Tran H, et al. Missplicing of Tau exon 10 in myotonic dystrophy type 1 is reproduced by overexpression of CELF2 but not by MBNL1 silencing. Biochim Biophys Acta. 2011;1812(7):732–742. doi: 10.1016/j.bbadis.2011.03.010. [DOI] [PubMed] [Google Scholar]
- Doi H, Koyano S, et al. The RNA binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci Res. 2010;66(1):131–133. doi: 10.1016/j.neures.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Dorsey ER, Constantinescu R, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68(5):384–386. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- Du H, Cline MS, et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol. 2010;17(2):187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eacker SM, Dawson TM, et al. Understanding microRNAs in neurode-generation. Nat Rev Neurosci. 2009;10(12):837–841. doi: 10.1038/nrn2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim HJ, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466(7310):1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvira G, Wasiak S, et al. Characterization of an RNA granule from developing brain. Mol Cell Proteomics. 2006;5(4):635–651. doi: 10.1074/mcp.M500255-MCP200. [DOI] [PubMed] [Google Scholar]
- Enciu AM, Popescu BO, et al. MicroRNAs in brain development and degeneration. Mol Biol Rep. 2011 doi: 10.1007/s11033-011-0973-1. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Entezam A, Biacsi R, et al. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene. 2007;395(1–2):125–134. doi: 10.1016/j.gene.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes PS, Boehringer A, et al. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum Mol Genet. 2011;20(12):2308–2321. doi: 10.1093/hmg/ddr124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faghihi MA, Modarresi F, et al. Expression of a noncoding RNA is elevated in Alzheimer's disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008;14(7):723–730. doi: 10.1038/nm1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Moran JV, et al. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87(5):905–916. doi: 10.1016/s0092-8674(00)81997-2. [DOI] [PubMed] [Google Scholar]
- Feng Y, Zhang F, et al. Translational suppression by trinucleotide repeat expansion at FMR1. Science. 1995;268(5211):731–734. doi: 10.1126/science.7732383. [DOI] [PubMed] [Google Scholar]
- Fiesel FC, Voigt A, et al. Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J. 2010;29(1):209–221. doi: 10.1038/emboj.2009.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippova GN, Thienes CP, et al. CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat Genet. 2001;28(4):335–343. doi: 10.1038/ng570. [DOI] [PubMed] [Google Scholar]
- Finch N, Baker M, et al. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 2009;132(Pt 3):583–591. doi: 10.1093/brain/awn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Chitta RK, et al. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res. 2010;9(2):1104–1120. doi: 10.1021/pr901076y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freir DB, Holscher C, et al. Blockade of long-term potentiation by beta-amyloid peptides in the CA1 region of the rat hippocampus in vivo. J Neurophysiol. 2001;85(2):708–713. doi: 10.1152/jn.2001.85.2.708. [DOI] [PubMed] [Google Scholar]
- Fu YH, Kuhl DP, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67(6):1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- Fu YH, Pizzuti A, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255(5049):1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- Fujii R, Okabe S, et al. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol. 2005;15(6):587–593. doi: 10.1016/j.cub.2005.01.058. [DOI] [PubMed] [Google Scholar]
- Fushimi K, Long C, et al. Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy. Protein Cell. 2011;2(2):141–149. doi: 10.1007/s13238-011-1014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal J, Zhang J, et al. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging. 2010;32(12):2323, e27–40. doi: 10.1016/j.neurobiolaging.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo JM, Jin P, et al. The role of RNA and RNA processing in neurode-generation. J Neurosci. 2005;25(45):10372–10375. doi: 10.1523/JNEUROSCI.3453-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Arocena D, Yang JE, et al. Fibroblast phenotype in male carriers of FMR1 premutation alleles. Hum Mol Genet. 2010;19(2):299–312. doi: 10.1093/hmg/ddp497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrke S, Imai Y, et al. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466(7306):637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Josephs KA, et al. Review: transactive response DNA-binding protein 43 (TDP-43): mechanisms of neurodegeneration. Neuropathol Appl Neurobiol. 2010;36(2):97–112. doi: 10.1111/j.1365-2990.2010.01060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem D, Tran H, et al. Altered splicing of Tau in DM1 is different from the foetal splicing process. FEBS Lett. 2009;583(4):675–679. doi: 10.1016/j.febslet.2008.12.065. [DOI] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428(6982):493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Giordana MT, Piccinini M, et al. TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol. 2010;20(2):351–360. doi: 10.1111/j.1750-3639.2009.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitcho MA, Baloh RH, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63(4):535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godena VK, Romano G, et al. TDP-43 regulates Drosophila neuromuscular junctions growth by modulating Futsch/MAP1B levels and synaptic microtubules organization. PLoS One. 2011;6(3):e17808. doi: 10.1371/journal.pone.0017808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature. 2011;470(7333):284–288. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco CM, Hagerman RJ, et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125(Pt 8):1760–1771. doi: 10.1093/brain/awf184. [DOI] [PubMed] [Google Scholar]
- Griggs RC, Wood DS. Criteria for establishing the validity of genetic recombination in myotonic dystrophy. Neurology. 1989;39(3):420–421. doi: 10.1212/wnl.39.3.420. [DOI] [PubMed] [Google Scholar]
- Guinto JB, Ritson GP, et al. Valosin-containing protein and the pathogenesis of frontotemporal dementia associated with inclusion body myopathy. Acta Neuropathol. 2007;114(1):55–61. doi: 10.1007/s00401-007-0224-7. [DOI] [PubMed] [Google Scholar]
- Gupta RA, Shah N, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458(7235):223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Garber M, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multiexonic structure of lincRNAs. Nat Biotechnol. 2010;28(5):503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Leehey M, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57(1):127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- Hall DA, Berry-Kravis E, et al. FMR1 gray-zone alleles: association with Parkinson's disease in women? Mov Disord. 2011 doi: 10.1002/mds.23755. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DA, Howard K, et al. Parkinsonism in FMR1 premutation carriers may be indistinguishable from Parkinson disease. Parkinsonism Relat Disord. 2009;15(2):156–159. doi: 10.1016/j.parkreldis.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DA, Jennings D, et al. FMR1 gene expansion and scans without evidence of dopaminergic deficits in parkinsonism patients. Parkinsonism Relat Disord. 2010;16(9):608–611. doi: 10.1016/j.parkreldis.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa V, Goldwater D, et al. Long CGG-repeat tracts are toxic to human cells: implications for carriers of Fragile X premutation alleles. FEBS Lett. 2005;579(12):2702–2708. doi: 10.1016/j.febslet.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Hanson KA, Kim SH, et al. Ubiquilin modifies TDP-43 toxicity in a Drosophila model of amyotrophic lateral sclerosis (ALS) J Biol Chem. 2010;285(15):11068–11072. doi: 10.1074/jbc.C109.078527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi T, Terada S, et al. Coexistence of TDP-43 and tau pathology in neurodegeneration with brain iron accumulation type 1 (NBIA-1, formerly Hallervorden–Spatz syndrome) Neuropathology. 2011;31(5):531–539. doi: 10.1111/j.1440-1789.2010.01186.x. [DOI] [PubMed] [Google Scholar]
- Hashem V, Galloway JN, et al. Ectopic expression ofCGG containing mRNA is neurotoxic in mammals. Hum Mol Genet. 2009;18(13):2443–2451. doi: 10.1093/hmg/ddp182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert SS, De Strooper B. Alterations of the microRNA network cause neurodegenerative disease. Trends Neurosci. 2009;32(4):199–206. doi: 10.1016/j.tins.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Hebert SS, Horre K, et al. MicroRNA regulation of Alzheimer's amyloid precursor protein expression. Neurobiol Dis. 2009;33(3):422–428. doi: 10.1016/j.nbd.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Hebert SS, Horre K, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105(17):6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessl D, Wang JM, et al. Decreased fragile X mental retardation protein expression underlies amygdala dysfunction in carriers of the fragile X premutation. Biol Psychiatry. 2011;70(9):859–865. doi: 10.1016/j.biopsych.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks GG, Singh N, et al. Fus deficiency in mice results in defective Blymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet. 2000;24(2):175–179. doi: 10.1038/72842. [DOI] [PubMed] [Google Scholar]
- Higashi S, Iseki E, et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain Res. 2007;1184:284–294. doi: 10.1016/j.brainres.2007.09.048. [DOI] [PubMed] [Google Scholar]
- Hou L, Antion MD, et al. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51(4):441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Houseley JM, Wang Z, et al. Myotonic dystrophy associated expanded CUG repeat muscleblind positive ribonuclear foci are not toxic to Drosophila. Hum Mol Genet. 2005;14(6):873–883. doi: 10.1093/hmg/ddi080. [DOI] [PubMed] [Google Scholar]
- Hsu RJ, Hsiao KM, et al. Long tract of untranslated CAG repeats is deleterious in transgenic mice. PLoS One. 2011;6(1):e16417. doi: 10.1371/journal.pone.0016417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WT, Josephs KA, et al. Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol. 2008;116(2):215–220. doi: 10.1007/s00401-008-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Zhou H, et al. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011;7(3):e1002011. doi: 10.1371/journal.pgen.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huarte M, Guttman M, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142(3):409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker MR, Wenzel HJ, et al. Progressive spatial processing deficits in a mouse model of the fragile X premutation. Behav Neurosci. 2009;123(6):1315–1324. doi: 10.1037/a0017616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest. 2011;121(2):726–738. doi: 10.1172/JCI44867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Seki M, et al. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol. 2011;69(1):152–162. doi: 10.1002/ana.22246. [DOI] [PubMed] [Google Scholar]
- Itoh K, Mitani M, et al. Neuropathology does not correlate with regional differences in the extent of expansion of CTG repeats in the brain with myotonic dystrophy type 1. Acta Histochem Cytochem. 2010;43(6):149–156. doi: 10.1267/ahc.10019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahashi CK, Yasui DH, et al. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129(Pt 1):256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- Jansen G, Groenen PJ, et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat Genet. 1996;13(3):316–324. doi: 10.1038/ng0796-316. [DOI] [PubMed] [Google Scholar]
- Jaspert A, Fahsold R, et al. Myotonic dystrophy: correlation of clinical symptoms with the size of the CTG trinucleotide repeat. J Neurol. 1995;242(2):99–104. doi: 10.1007/BF00887824. [DOI] [PubMed] [Google Scholar]
- Jiang H, Mankodi A, et al. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13(24):3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- Jin P, Duan R, et al. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007;55(4):556–564. doi: 10.1016/j.neuron.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Zarnescu DC, et al. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39(5):739–747. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- Johnson R, Richter N, et al. The Human Accelerated Region 1 noncoding RNA is repressed by REST in Huntington's disease. Physiol Genomics. 2010;41(3):269–274. doi: 10.1152/physiolgenomics.00019.2010. [DOI] [PubMed] [Google Scholar]
- Ju S, Tardiff DF, et al. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 2011;9(4):e1001052. doi: 10.1371/journal.pbio.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kanadia RN, Johnstone KA, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302(5652):1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- Kanadia RN, Shin J, et al. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci U S A. 2006;103(31):11748–11753. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama S, Tomaru Y, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309(5740):1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- Kenneson A, Zhang F, et al. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet. 2001;10(14):1449–1454. doi: 10.1093/hmg/10.14.1449. [DOI] [PubMed] [Google Scholar]
- Ketting RF, Fischer SE, et al. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001;15(20):2654–2659. doi: 10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Faghihi MA, et al. A novel RNA transcript with antiapoptotic function is silenced in fragile X syndrome. PLoS One. 2008;3(1):e1486. doi: 10.1371/journal.pone.0001486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106(28):11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khateb S, Weisman-Shomer P, et al. The tetraplex (CGG)n destabilizing proteins hnRNP A2 and CBF-A enhance the in vivo translation of fragile X premutation mRNA. Nucleic Acids Res. 2007;35(17):5775–5788. doi: 10.1093/nar/gkm636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Inoue K, et al. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317(5842):1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Shanware NP, et al. Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J Biol Chem. 2010;285(44):34097–34105. doi: 10.1074/jbc.M110.154831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim VN, Han J, et al. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10(2):126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- Klesert TR, Cho DH, et al. Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nat Genet. 2000;25(1):105–109. doi: 10.1038/75490. [DOI] [PubMed] [Google Scholar]
- Konopka W, Kiryk A, et al. MicroRNA loss enhances learning and memory in mice. J Neurosci. 2010;30(44):14835–14842. doi: 10.1523/JNEUROSCI.3030-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob MD, Moseley ML, et al. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8) Nat Genet. 1999;21(4):379–384. doi: 10.1038/7710. [DOI] [PubMed] [Google Scholar]
- Kosik KS. The neuronal microRNA system. Nat Rev Neurosci. 2006;7(12):911–920. doi: 10.1038/nrn2037. [DOI] [PubMed] [Google Scholar]
- Krol J, Fiszer A, et al. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol Cell. 2007;25(4):575–586. doi: 10.1016/j.molcel.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Kryndushkin D, Wickner RB, et al. FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast model of amyotrophic lateral sclerosis. Protein Cell. 2011;2(3):223–236. doi: 10.1007/s13238-011-1525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo PH, Doudeva LG, et al. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009;37(6):1799–1808. doi: 10.1093/nar/gkp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11(4):247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladd PD, Smith LE, et al. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum Mol Genet. 2007;16(24):3174–3187. doi: 10.1093/hmg/ddm293. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136(6):1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, et al. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19(R1):R46–R64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalmansingh AS, Urekar CJ, et al. TDP-43 is a transcriptional repressor: the testis-specific mouse acrv1 gene is a TDP-43 target in vivo. J Biol Chem. 2011;286(13):10970–10982. doi: 10.1074/jbc.M110.166587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lanson NA, Jr, Maltare A, et al. A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum Mol Genet. 2011;20(13):2510–2523. doi: 10.1093/hmg/ddr150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau P, de Strooper B. Dysregulated microRNAs in neurodegenerative disorders. Semin Cell Dev Biol. 2010;21(7):768–773. doi: 10.1016/j.semcdb.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Le W, Nguyen D, et al. Transcription factor PITX3 gene in Parkinson's disease. Neurobiol Aging. 2011;32(4):750–753. doi: 10.1016/j.neurobiolaging.2009.03.015. [DOI] [PubMed] [Google Scholar]
- Lee CC, Kuo YM, et al. Insulin rescues amyloid beta-induced impairment of hippocampal long-term potentiation. Neurobiol Aging. 2009;30(3):377–387. doi: 10.1016/j.neurobiolaging.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Lee ST, Chu K, et al. Altered microRNA regulation in Huntington's disease models. Exp Neurol. 2011;227(1):172–179. doi: 10.1016/j.expneurol.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Lee Y, Samaco RC, et al. miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat Neurosci. 2008;11(10):1137–1139. doi: 10.1038/nn.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy O, Wang J, et al. Brain-specific change in alternative splicing of Tau exon6 inmyotonic dystrophy type1. Biochim Biophys Acta. 2006;1762(4):460–467. doi: 10.1016/j.bbadis.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Li LB, Yu Z, et al. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature. 2008;453(7198):1107–1111. doi: 10.1038/nature06909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Ray P, et al. A Drosophila model for TDP-43 proteinopathy. Proc Natl Acad Sci U S A. 2010;107(7):3169–3174. doi: 10.1073/pnas.0913602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Pestova TV, et al. Translational control by a small RNA: dendritic BC1 RNA targets the eukaryotic initiation factor 4A helicase mechanism. Mol Cell Biol. 2008;28(9):3008–3019. doi: 10.1128/MCB.01800-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WL, Dickson DW. Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol. 2008;116(2):205–213. doi: 10.1007/s00401-008-0408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Ashizawa T. SCA10 and ATTCT repeat expansion: clinical features and molecular aspects. Cytogenet Genome Res. 2003;100(1–4):184–188. doi: 10.1159/000072853. [DOI] [PubMed] [Google Scholar]
- Ling SC, Albuquerque CP, et al. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci U S A. 2010;107(30):13318–13323. doi: 10.1073/pnas.1008227107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori CL, Ricker K, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293(5531):864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One. 2010;5(10):e13250. doi: 10.1371/journal.pone.0013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesch DZ, Godler DE, et al. Evidence for the toxicity of bidirectional transcripts and mitochondrial dysfunction in blood associated with small CGG expansions in the FMR1 gene in patients with parkinsonism. Genet Med. 2011;13(5):392–399. doi: 10.1097/GIM.0b013e3182064362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesch DZ, Khaniani MS, et al. Small CGG repeat expansion alleles of FMR1 gene are associated with parkinsonism. Clin Genet. 2009;76(5):471–476. doi: 10.1111/j.1399-0004.2009.01275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan DD, Korman MH, et al. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retro-transposition. Cell. 1993;72(4):595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- Ludwig AL, Hershey JW, et al. Initiation of translation of the FMR1 mRNA occurs predominantly through 5′-end-dependent ribosomal scanning. J Mol Biol. 2011;407(1):21–34. doi: 10.1016/j.jmb.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig AL, Raske C, et al. Translation of the FMR1 mRNA is not influenced by AGG interruptions. Nucleic Acids Res. 2009;37(20):6896–6904. doi: 10.1093/nar/gkp713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Zhao Y, et al. An NF-kappaB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J Biol Chem. 2008;283(46):31315–31322. doi: 10.1074/jbc.M805371200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Rademakers R, et al. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9(10):995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- Mahadevan M, Tsilfidis C, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255(5049):1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- Mahadevan MS, Yadava RS, et al. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat Genet. 2006;38(9):1066–1070. doi: 10.1038/ng1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankodi A, Logigian E, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289(5485):1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Takahashi MP, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10(1):35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Teng-Umnuay P, et al. Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann Neurol. 2003;54(6):760–768. doi: 10.1002/ana.10763. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Urbinati CR, et al. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet. 2001;10(19):2165–2170. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- Marti E, Pantano L, et al. A myriad of miRNA variants in control and Huntington's disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010;38(20):7219–7235. doi: 10.1093/nar/gkq575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SL. The ORF1 protein encoded by LINE-1: structure and function during L1 retrotransposition. J Biomed Biotechnol. 2006;2006(1):45621. doi: 10.1155/JBB/2006/45621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massone S, Vassallo I, et al. 17A, a novel non-coding RNA, regulates GABA B alternative splicing and signaling in response to inflammatory stimuli and in Alzheimer disease. Neurobiol Dis. 2011;41(2):308–317. doi: 10.1016/j.nbd.2010.09.019. [DOI] [PubMed] [Google Scholar]
- Matsuura T, Yamagata T, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26(2):191–194. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- McDonald KK, Aulas A, et al. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet. 2011;20(7):1400–1410. doi: 10.1093/hmg/ddr021. [DOI] [PubMed] [Google Scholar]
- McLaughlin BA, Spencer C, et al. CAG trinucleotide RNA repeats interact with RNA binding proteins. Am J Hum Genet. 1996;59(3):561–569. [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, et al. Noncoding RNAs in long-term memory formation. Neuroscientist. 2008;14(5):434–445. doi: 10.1177/1073858408319187. [DOI] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, et al. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10(3):155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- Michelhaugh SK, Lipovich L, et al. Mining Affymetrix microarray data for long non-coding RNAs: altered expression in the nucleus accumbens of heroin abusers. J Neurochem. 2011;116(3):459–466. doi: 10.1111/j.1471-4159.2010.07126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JW, Urbinati CR, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19(17):4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min SS, An J, et al. Neuregulin-1 prevents amyloid beta-induced impairment of long-term potentiation in hippocampal slices via ErbB4. Neurosci Lett. 2011;505(1):6–9. doi: 10.1016/j.neulet.2011.05.246. [DOI] [PubMed] [Google Scholar]
- Modoni A, Silvestri G, et al. Characterization of the pattern of cognitive impairment in myotonic dystrophy type 1. Arch Neurol. 2004;61(12):1943–1947. doi: 10.1001/archneur.61.12.1943. [DOI] [PubMed] [Google Scholar]
- Moseley ML, Zu T, et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38(7):758–769. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- Mulders SA, van den Broek WJ, et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc Natl Acad Sci U S A. 2009;106(33):13915–13920. doi: 10.1073/pnas.0905780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz DG, Neumann M, et al. FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009;118(5):617–627. doi: 10.1007/s00401-009-0598-9. [DOI] [PubMed] [Google Scholar]
- Muotri AR, Chu VT, et al. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435(7044):903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- Muotri AR, Marchetto MC, et al. L1 retrotransposition in neurons is modulated by MeCP2. Nature. 2010;468(7322):443–446. doi: 10.1038/nature09544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muotri AR, Zhao C, et al. Environmental influence on L1 retrotransposons in the adult hippocampus. Hippocampus. 2009;19(10):1002–1007. doi: 10.1002/hipo.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchison EP, Hannon GJ. miRNAs on the move: miRNA biogenesis and the RNAi machinery. Curr Opin Cell Biol. 2004;16(3):223–229. doi: 10.1016/j.ceb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Nakashima-Yasuda H, Uryu K, et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007;114(3):221–229. doi: 10.1007/s00401-007-0261-2. [DOI] [PubMed] [Google Scholar]
- Neumann M, Mackenzie IR, et al. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007;66(2):152–157. doi: 10.1097/nen.0b013e31803020b9. [DOI] [PubMed] [Google Scholar]
- Neumann M, Roeber S, et al. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009;118(5):605–616. doi: 10.1007/s00401-009-0581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Niwa R, Zhou F, et al. The expression of the Alzheimer's amyloid precursor protein-like gene is regulated by developmental timing microRNAs and their targets in Caenorhabditis elegans. Dev Biol. 2008;315(2):418–425. doi: 10.1016/j.ydbio.2007.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke JR, Swanson MS. Mechanisms of RNA-mediated disease. J Biol Chem. 2009;284(12):7419–7423. doi: 10.1074/jbc.R800025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberle I, Rousseau F, et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252(5009):1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- Ofer N, Weisman-Shomer P, et al. The quadruplex r(CGG)n destabilizing cationic porphyrin TMPyP4 cooperates with hnRNPs to increase the translation efficiency of fragile X premutation mRNA. Nucleic Acids Res. 2009;37(8):2712–2722. doi: 10.1093/nar/gkp130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo JP, Chambon P, et al. Expanded CTG repeats within the DMPK 3′ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc Natl Acad Sci U S A. 2008;105(7):2646–2651. doi: 10.1073/pnas.0708519105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo JP, Ward AJ, et al. Alternative splicing dysregulation secondary to skeletal muscle regeneration. Ann Neurol. 2011;69(4):681–690. doi: 10.1002/ana.22278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne RJ, Lin X, et al. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum Mol Genet. 2009;18(8):1471–1481. doi: 10.1093/hmg/ddp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne RJ, Thornton CA. RNA-dominant diseases. Hum Mol Genet. 2006;15:R162–R169. doi: 10.1093/hmg/ddl181. Spec. No. 2. [DOI] [PubMed] [Google Scholar]
- Ou SH, Wu F, et al. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type1 TAR DNA sequence motifs. J Virol. 1995;69(6):3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer AN, Xing Y, et al. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington's disease. J Neurosci. 2008;28(53):14341–14346. doi: 10.1523/JNEUROSCI.2390-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44(4):595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Pandit S, Wang D, et al. Functional integration of transcriptional and RNA processing machineries. Curr Opin Cell Biol. 2008;20(3):260–265. doi: 10.1016/j.ceb.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao WC, Dickson DW, et al. Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer Dis Assoc Disord. 2011;25(4):364–368. doi: 10.1097/WAD.0b013e31820f8f50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N, Hoang D, et al. MicroRNAs can regulate human APP levels. Mol Neurodegener. 2008;3:10. doi: 10.1186/1750-1326-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson CE. Repeat associated non-ATG translation initiation: one DNA, two transcripts, seven reading frames, potentially nine toxic entities! PLoS Genet. 2011;7(3):e1002018. doi: 10.1371/journal.pgen.1002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peier AM, Nelson DL. Instability of a premutation-sized CGG repeat in FMR1 YAC transgenic mice. Genomics. 2002;80(4):423–432. doi: 10.1006/geno.2002.6849. [DOI] [PubMed] [Google Scholar]
- Philips AV, Timchenko LT, et al. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280(5364):737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- Pieretti M, Zhang FP, et al. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66(4):817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting CP, Oliver PL, et al. Evolution and functions of long noncoding RNAs. Cell. 2009;136(4):629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Poulos MG, Batra R, et al. Developments in RNA splicing and disease. Cold Spring Harb Perspect Biol. 2011;3(1):a000778. doi: 10.1101/cshperspect.a000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primerano B, Tassone F, et al. Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. RNA. 2002;8(12):1482–1488. [PMC free article] [PubMed] [Google Scholar]
- Pushechnikov A, Lee MM, et al. Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3. J Am Chem Soc. 2009;131(28):9767–9779. doi: 10.1021/ja9020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin M, Entezam A, et al. A mouse model of the fragile X premutation: effects on behavior, dendrite morphology, and regional rates of cerebral protein synthesis. Neurobiol Dis. 2011;42(1):85–98. doi: 10.1016/j.nbd.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri M, Cossu G, et al. Broadening the phenotype of TARDBP mutations: the TARDBP Ala382Thr mutation and Parkinson's disease in Sardinia. Neurogenetics. 2011;12(3):203–209. doi: 10.1007/s10048-011-0288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi IA, Mattick JS, et al. Long non-coding RNAs in nervous system function and disease. Brain Res. 2010;1338:20–35. doi: 10.1016/j.brainres.2010.03.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy S, Smith DB, et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat Genet. 1996;13(3):325–335. doi: 10.1038/ng0796-325. [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Raphael Gibbs J, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Hölttä-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chiò A, Restagno G, Borghero G, Sabatelli M, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard S. Reaching for the stars: linking RNA binding proteins to diseases. Adv Exp Med Biol. 2010;693:142–157. [PubMed] [Google Scholar]
- Ritson GP, Custer SK, et al. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci. 2010;30(22):7729–7739. doi: 10.1523/JNEUROSCI.5894-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Huber KM. Metabotropic glutamate receptors and fragile x mental retardation protein: partners in translational regulation at the synapse. Sci Signal. 2008;1(5):pe6. doi: 10.1126/stke.15pe6. [DOI] [PubMed] [Google Scholar]
- Ross OA, Rutherford NJ, et al. Ataxin-2 repeat-length variation and neurodegeneration. Hum Mol Genet. 2011;20(16):3207–3212. doi: 10.1093/hmg/ddr227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnicki DD, Holmes SE, et al. Huntington's disease-like 2 is associated with CUG repeat-containing RNA foci. Ann Neurol. 2007;61(3):272–282. doi: 10.1002/ana.21081. [DOI] [PubMed] [Google Scholar]
- Rudnicki DD, Pletnikova O, et al. A comparison of Huntington disease and Huntington disease-like 2 neuropathology. J Neuropathol Exp Neurol. 2008;67(4):366–374. doi: 10.1097/NEN.0b013e31816b4aee. [DOI] [PubMed] [Google Scholar]
- Sarkar PS, Appukuttan B, et al. Heterozygous loss of Six5 in mice is sufficient to cause ocular cataracts. Nat Genet. 2000;25(1):110–114. doi: 10.1038/75500. [DOI] [PubMed] [Google Scholar]
- Sato N, Amino T, et al. Spinocerebellar ataxia type 31 is associated with inserted penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009;85(5):544–557. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savkur RS, Philips AV, et al. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29(1):40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- Schaefer A, O'Carroll D, et al. Cerebellar neurodegeneration in the absence of microRNAs. J Exp Med. 2007;204(7):1553–1558. doi: 10.1084/jem.20070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonrock N, Ke YD, et al. Neuronal microRNA deregulation in response to Alzheimer's disease amyloid-beta. PLoS One. 2010;5(6):e11070. doi: 10.1371/journal.pone.0011070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C, Arai T, et al. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J Neuropathol Exp Neurol. 2008;67(12):1159–1165. doi: 10.1097/NEN.0b013e31818e8951. [DOI] [PubMed] [Google Scholar]
- Scott AF, Schmeckpeper BJ, et al. Origin of the human L1 elements: proposed progenitor genes deduced from aconsensus DNA sequence. Genomics. 1987;1(2):113–125. doi: 10.1016/0888-7543(87)90003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's Disease. Cold Spring Harb Perspect Biol. 2011;3(7):pii–a004457. doi: 10.1101/cshperspect.a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Rau F, et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010;29(7):1248–1261. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Cenik C, et al. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J Biol Chem. 2011;286(2):1204–1215. doi: 10.1074/jbc.M110.190884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant N, Sablonniere B, et al. Dysregulation of human brain microtubule-associated tau mRNA maturation inmyotonic dystrophy type 1. Hum Mol Genet. 2001;10(19):2143–2155. doi: 10.1093/hmg/10.19.2143. [DOI] [PubMed] [Google Scholar]
- Singer T, McConnell MJ, et al. LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends Neurosci. 2010;33(8):345–354. doi: 10.1016/j.tins.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Gaur P, et al. Fragile x mental retardation (Fmr-1) gene expression is down regulated in brain of mice during aging. Mol Biol Rep. 2007;34(3):173–181. doi: 10.1007/s11033-006-9032-8. [DOI] [PubMed] [Google Scholar]
- Sofola OA, Jin P, et al. Argonaute-2-dependent rescue of a Drosophila model of FXTAS by FRAXE premutation repeat. Hum Mol Genet. 2007a;16(19):2326–2332. doi: 10.1093/hmg/ddm186. [DOI] [PubMed] [Google Scholar]
- Sofola OA, Jin P, et al. RNA binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron. 2007b;55(4):565–571. doi: 10.1016/j.neuron.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopher BL, Ladd PD, et al. CTCF regulates ataxin-7 expression through promotion of a convergently transcribed, antisense noncoding RNA. Neuron. 2011;70(6):1071–1084. doi: 10.1016/j.neuron.2011.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Diaz Z, et al. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011;9(4):e1000614. doi: 10.1371/journal.pbio.1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja KL, McCurrach M, et al. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol. 1995;128(6):995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Wu H, et al. Deletion of astroglial dicer causes non-cell-autonomous neuronal dysfunction and degeneration. J Neurosci. 2011;31(22):8306–8319. doi: 10.1523/JNEUROSCI.0567-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Beilina A, et al. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA. 2007;13(4):555–562. doi: 10.1261/rna.280807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, et al. Transcription of the FMR1 gene in individuals with fragile X syndrome. Am J Med Genet. 2000a;97(3):195–203. doi: 10.1002/1096-8628(200023)97:3<195::AID-AJMG1037>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, et al. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet. 2000b;66(1):6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Iwahashi C, et al. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS) RNA Biol. 2004;1(2):103–105. doi: 10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]
- Tervonen TA, Louhivuori V, et al. Aberrant differentiation of glutamatergic cells inneocortex of mouse model for fragile X syndrome. Neurobiol Dis. 2009;33(2):250–259. doi: 10.1016/j.nbd.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Thies W, Bleiler L. 2011 Alzheimer's disease facts and figures. Alzheimers Dement. 2011;7(2):208–244. doi: 10.1016/j.jalz.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Thomas MG, Loschi M, et al. RNA granules: the good, the bad and the ugly. Cell Signal. 2011;23(2):324–334. doi: 10.1016/j.cellsig.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko NA, Cai ZJ, et al. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem. 2001;276(11):7820–7826. doi: 10.1074/jbc.M005960200. [DOI] [PubMed] [Google Scholar]
- Todd PK, Mack KJ, et al. The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc Natl Acad Sci U S A. 2003;100(24):14374–14378. doi: 10.1073/pnas.2336265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Oh SY, et al. Histone deacetylases suppress CGG repeat-induced neurodegeneration via transcriptional silencing in models of fragile X tremor ataxia syndrome. PLoS Genet. 2010;6(12):e1001240. doi: 10.1371/journal.pgen.1001240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Paulson HL. RNA-mediated neurodegeneration in repeat expansion disorders. Ann Neurol. 2010;67(3):291–300. doi: 10.1002/ana.21948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollervey JR, Curk T, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14(4):452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi V, Ellis JD, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39(6):925–938. doi: 10.1016/j.molcel.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Manor O, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329(5992):689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Nakashima-Yasuda H, et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol. 2008;67(6):555–564. doi: 10.1097/NEN.0b013e31817713b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam D, Errijgers V, et al. Cognitive decline, neuromotor and behavioural disturbances in a mouse model for fragile-X-associated tremor/ataxia syndrome (FXTAS) Behav Brain Res. 2005;162(2):233–239. doi: 10.1016/j.bbr.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Vance C, Rogelj B, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65(5):905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Vicente-Crespo M, Pascual M, et al. Drosophila muscleblind is involved in troponin T alternative splicing and apoptosis. PLoS One. 2008;3(2):e1613. doi: 10.1371/journal.pone.0001613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GS, Kearney DL, et al. Elevation of RNA binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J Clin Invest. 2007;117(10):2802–2811. doi: 10.1172/JCI32308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu J, et al. miR-106b aberrantly expressed in a double transgenic mouse model for Alzheimer's disease targets TGF-beta type II receptor. Brain Res. 2010;1357:166–174. doi: 10.1016/j.brainres.2010.08.023. [DOI] [PubMed] [Google Scholar]
- Wang IF, Wu LS, et al. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105(3):797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- Wang LC, Chen KY, et al. Muscleblind participates in RNA toxicity of expanded CAG and CUG repeats in Caenorhabditis elegans. Cell Mol Life Sci. 2011;68(7):1255–1267. doi: 10.1007/s00018-010-0522-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, Rajeev BW, et al. The expression of microRNA miR-107 decreases early in Alzheimer's disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28(5):1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Song X, et al. The long arm of long noncoding RNAs: roles as sensors regulating gene transcriptional programs. Cold Spring Harb Perspect Biol. 2011;3(1):a003756. doi: 10.1101/cshperspect.a003756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21(6):354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Ward AJ, Rimer M, et al. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum Mol Genet. 2010;19(18):3614–3622. doi: 10.1093/hmg/ddq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf MB, Nakamori M, et al. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc Natl Acad Sci U S A. 2009;106(44):18551–18556. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420(6915):520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Temiz P, et al. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2008;79(10):1186–1189. doi: 10.1136/jnnp.2007.131334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, et al. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A. 1997;94(10):5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TM, Lueck JD, et al. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. 2007;117(12):3952–3957. doi: 10.1172/JCI33355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TM, Sobczak K, et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009;325(5938):336–339. doi: 10.1126/science.1173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TM, Thornton CA. Myotonic dystrophy: RNA-mediated muscle disease. Curr Opin Neurol. 2007;20(5):572–576. doi: 10.1097/WCO.0b013e3282ef6064. [DOI] [PubMed] [Google Scholar]
- White MC, Gao R, et al. Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet. 2010;6(6):e1000984. doi: 10.1371/journal.pgen.1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilburn B, Rudnicki DD, et al. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-like 2 mice. Neuron. 2011;70(3):427–440. doi: 10.1016/j.neuron.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm BT, Marguerat S, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453(7199):1239–1243. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- Willemsen R, Hoogeveen-Westerveld M, et al. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum Mol Genet. 2003;12(9):949–959. doi: 10.1093/hmg/ddg114. [DOI] [PubMed] [Google Scholar]
- Williams AJ, Paulson HL. Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci. 2008;31(10):521–528. doi: 10.1016/j.tins.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wils H, Kleinberger G, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2010;107(8):3858–3863. doi: 10.1073/pnas.0912417107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton MJ, Igaz LM, et al. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283(19):13302–13309. doi: 10.1074/jbc.M800342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciechowska M, Krzyzosiak WJ. CAG repeats RNA as an auxiliary toxic agent in polyglutamine disorder. RNA Biol. 2011;8(4) doi: 10.4161/rna.8.4.15397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadava RS, Frenzel-McCardell CD, et al. RNA toxicity in myotonic muscular dystrophy induces NKX2-5 expression. Nat Genet. 2008;40(1):61–68. doi: 10.1038/ng.2007.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Hennessey T, et al. MicroRNA-related cofilin abnormality in Alzheimer's disease. PLoS One. 2010;5(12):e15546. doi: 10.1371/journal.pone.0015546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi R, Qin Y, et al. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17(24):3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoseki A, Shiga A, et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63(4):538–542. doi: 10.1002/ana.21392. [DOI] [PubMed] [Google Scholar]
- Yu S, Pritchard M, et al. Fragile X genotype characterized by an unstable region of DNA. Science. 1991;252(5010):1179–1181. doi: 10.1126/science.252.5009.1179. [DOI] [PubMed] [Google Scholar]
- Yu Z, Teng X, et al. Triplet repeat-derived siRNAs enhance RNA-mediated toxicity in a Drosophila model for myotonic dystrophy. PLoS Genet. 2011;7(3):e1001340. doi: 10.1371/journal.pgen.1001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalfa F, Giorgi M, et al. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell. 2003;112(3):317–327. doi: 10.1016/s0092-8674(03)00079-5. [DOI] [PubMed] [Google Scholar]
- Zaremba-Czogalla M, Dubinska-Magiera M, et al. Laminopathies: the molecular background of the disease and the prospects for its treatment. Cell Mol Biol Lett. 2011;16(1):114–148. doi: 10.2478/s11658-010-0038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Cullen BR. Recognition and cleavage of primary microRNA transcripts. Methods Mol Biol. 2006;342:49–56. doi: 10.1385/1-59745-123-1:49. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Yi R, et al. Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. EMBO J. 2005;24(1):138–148. doi: 10.1038/sj.emboj.7600491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, et al. Mutationsin LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Zinman L, Cudkowicz M. Emerging targets and treatments in amyotrophic lateral sclerosis. Lancet Neurol. 2011;10(5):481–490. doi: 10.1016/S1474-4422(11)70024-2. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Sok J, et al. TLS (FUS) binds RNA in vivo and engages in nucleocytoplasmic shuttling. J Cell Sci. 1997;110(Pt 15):1741–1750. doi: 10.1242/jcs.110.15.1741. [DOI] [PubMed] [Google Scholar]
- Zu T, Gibbens B, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A. 2011;108(1):260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Belyaev N, et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington's disease. J Neurosci. 2007;27(26):6972–6983. doi: 10.1523/JNEUROSCI.4278-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35(1):76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]