

Abstract
Glomerular IgM and C3 deposits frequently accompany idiopathic FSGS and secondary glomerulosclerosis, but it is unknown whether IgM activates complement, possibly contributing to the pathogenesis of these diseases. We hypothesized that IgM natural antibody binds to neoepitopes exposed in the glomerulus after nonimmune insults, triggering activation of the complement system and further injury. We examined the effects of depleting B cells, using three different strategies, on adriamycin-induced glomerulosclerosis. First, we treated wild-type mice with an anti-murine CD20 antibody, which depletes B cells, before disease induction. Second, we evaluated adriamycin-induced glomerulosclerosis in Jh mice, a strain that lacks mature B cells. Third, we locally depleted peritoneal B cells via hypotonic shock before disease induction. All three strategies reduced deposition of IgM in the glomerulus after administration of adriamycin and attenuated the development of albuminuria. Furthermore, we found that glomerular IgM and C3 were detectable in a subset of patients with FSGS; C3 was present as an activation fragment and colocalized with glomerular IgM, suggesting that glomerular IgM may have bound a cognate ligand. Taken together, these results suggest that IgM activates the complement system within the glomerulus in an animal model of glomerulosclerosis. Strategies that reduce IgM natural antibody or that prevent complement activation may slow the progression of glomerulosclerosis.
Glomerular IgM deposition is observed in a wide range of primary and secondary renal diseases, although the significance of these deposits has remained elusive. Prominent mesangial deposits have been described in patients with idiopathic nephrotic syndrome, including patients with minimal change disease and idiopathic FSGS.1,2 In some patients, IgM is the primary immune factor detected in the kidney, and the term IgM nephropathy was coined to describe kidneys with minimal changes by light microscopy but with abundant mesangial deposition of IgM.3 Patients with mesangial expansion or mesangial proliferation by light microscopy frequently have abundant glomerular IgM deposition.4 Glomerular IgM is also observed in many secondary forms of glomerulosclerosis, including diabetic nephropathy5 and hypertensive nephropathy.6 Importantly, C3 and C4 are frequently detected in the glomeruli of patients who have IgM deposits.4,7,8
In spite of its promiscuous presence in glomerular lesions, a pathogenic role for glomerular IgM has not been demonstrated. Some series, however, have shown that the presence of IgM is of prognostic significance.9,10 Furthermore, recent reports suggest that rituximab, a mAb to CD20 that depletes mature B cells, may be beneficial in some patients with the nephrotic syndrome.11–16 However, the mechanisms by which rituximab alleviates the nephrotic syndrome are not yet known. Recent work has demonstrated that “natural antibody” contributes to tissue injury in a number of different diseases. Natural antibody refers to Ig that reacts to certain conserved epitopes, even without prior exposure to that epitope.17 In mice, natural antibody is primarily produced by CD5+CD11b+IgMhighB220low B-1a B cells, which are predominantly located in the peritoneum.18–20 Studies have demonstrated that natural antibody IgM binds to endogenous neoepitopes that are exposed after injury of the heart,21 intestine,22,23 skeletal muscle,24 and kidney.25 Once bound, IgM activates the complement system and can cause further tissue inflammation and injury. Strategies that deplete peritoneal B-1a cells or that block binding of specific natural antibody clones within injured tissues have proven successful for attenuating injury in a number of different models.23,26
We hypothesized that natural antibody IgM binds to glomerular epitopes that are exposed after adriamycin-induced injury of glomerular cells, and that bound IgM activates the complement system. To test this hypothesis, we utilized three strategies to deplete B cells in mice with adriamycin nephropathy. We also analyzed biopsy tissue from patients with idiopathic FSGS to determine whether the pattern of C3 deposition suggests complement activation by deposited IgM.
Results
Treatment of Mice with a mAb to CD20 Depletes Peritoneal B-1a Cells
Balb/c mice were injected intravenously with a murine mAb to mouse CD20 (clone 5D2, murine IgG2a) or with vehicle control. Treatment with this antibody reduced splenic total B cells as well as B-1a B cells (Table 1). As previously reported,20 anti-CD20 was not as effective at reducing peritoneal B cells. Two weeks after treating mice with the anti-CD20 antibody, serum levels of IgM were significantly reduced compared with vehicle-treated controls (Table 1).
Table 1.
Reduction in B cell subsets and Ig after intravenous injection with PBS or anti-CD20 mAb
| Compartment | Cells | Before Adriamycin (wk 0) | After Adriamycin (wk 4) | ||||
|---|---|---|---|---|---|---|---|
| PBS | Anti-CD20 | % Reduction | PBS | Anti-CD20 | % Reduction | ||
| Spleen | IgM+B220+ | 22.61 | 0.76 | 96.64 | 20.7 | 0.09 | 99.57 |
| IgMHighB220+CD5+ | 0.32 | 0.09 | 73.02 | 0.44 | 0.02 | 95.45 | |
| Peripheral blood | IgM+B220+ | 25.30 | 1.14 | 95.51 | 5.75 | 0 | 100 |
| IgMHighB220+CD5+ | 0.49 | 0.62 | −26.53 | 0.45 | 0 | 100 | |
| Peritoneum | IgM+B220+ | 29.80 | 19.97 | 32.98 | 17.28 | 1.43 | 91.72 |
| IgMHighB220+CD5+ | 0.41 | 0.23 | 44.44 | 0.605 | 0.4 | 33.88 | |
| Serum | IgM (mg/ml) | 7.80±1.10 | 2.60±0.30a | 66.7 | |||
Treatment was started 2 weeks before injection with adriamycin. Data are reported as the percentage of total cells.
P<0.001 versus PBS.
Treatment with a mAb to CD20 Reduces Glomerular IgM Deposition and Reduces Complement Activation in the Glomeruli of Mice with Adriamycin Nephropathy
Adriamycin nephropathy was induced in BALB/c mice, and the abundance of glomerular IgM was examined (Figure 1, A and B). Injection of the mice with adriamycin caused a significant increase in the abundance of glomerular IgM. Pretreatment of the mice with anti-CD20 prevented an increase in glomerular IgM after injection with adriamycin, and levels of glomerular IgM in mice that received both anti-CD20 and adriamycin were similar to those seen in healthy controls. To determine whether glomerular IgM deposition is an early event after treatment of the mice with adriamycin, a group of mice was harvested 1 week after injection of the adriamycin, a time point at which they have significant proteinuria but have not yet developed glomerulosclerosis.27,28 We found that glomerular IgM levels were significantly higher 1 week after injection with adriamycin than in control mice (Figure 1C).
Figure 1.
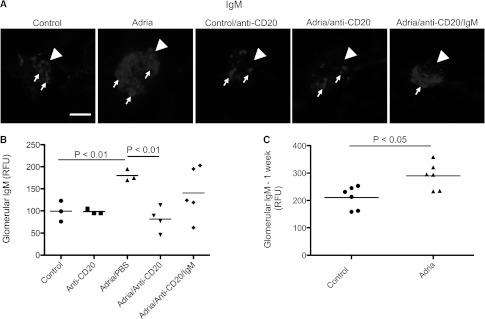
B cell depletion with an anti-CD20 antibody reduces glomerular IgM deposition in mice with adriamycin nephropathy. Mice are injected with anti-CD20 mAb to deplete their B cells before the induction of adriamycin nephropathy. (A) Immunofluorescence microscopy is performed on kidneys 4 weeks after induction of adriamycin nephropathy to assess the abundance of glomerular IgM. Kidneys from three to five mice per group are examined. Thirty glomeruli per section are visualized, and the average for each mouse is determined. Mice injected with adriamycin demonstrate a substantial increase in glomerular IgM deposition compared with control animals. Mice treated with anti-CD20 demonstrate less glomerular IgM. Mice with adriamycin nephropathy that had been injected with the anti-CD20 but were also reconstituted with purified IgM eluted from the kidneys of mice with adriamycin nephropathy (Adria/anti-CD20/IgM) demonstrate an apparent increase in glomerular IgM. Glomeruli are indicated with arrowheads and areas of IgM deposition within the glomeruli are indicated with small arrows. After converting the images to grayscale, the brightness and contrast of the shown images are adjusted. All images are adjusted equally. (B) Quantitative analysis of the glomerular IgM in the different treatment groups confirms that glomerular IgM deposition is increased after injection with adriamycin, but that this increase is attenuated in mice injected with anti-CD20 therapy. (C) Analysis of kidneys taken from mice 1 week after injection with adriamycin demonstrates that IgM deposits develop within the first week of disease. Original magnification, ×200. Scale bar, 100 μM.
We previously found that polyclonal IgM purified from mouse serum does not bind within the glomeruli when injected systemically.25 To determine whether IgM eluted specifically from kidneys with adriamycin nephropathy would bind to glomerular epitopes when re-injected into mice, we purified IgM from the kidneys of adriamycin-treated mice. We then injected the purified IgM into mice with adriamycin nephropathy that had previously undergone B cell depletion with the anti-CD20. This was repeated every week during the course of the study. A trend toward greater glomerular IgM was seen in the reconstituted mice (Figure 1, A and B). Although the injection of anti-CD20 mice with purified IgM did not fully restore injury to the level seen in BALB/c mice that did not receive anti-CD20, it is possible that isolation of the IgM from kidney lysates diminishes the binding of the IgM, and it loses affinity for its target ligands.
In mice with adriamycin nephropathy, low levels of glomerular C4d deposition were detected in control mice but significantly increased in mice treated with adriamycin (Figure 2A). Treatment of mice with anti-CD20 reduced glomerular C4d, confirming that the glomerular C4d deposits are caused by Ig-mediated complement activation. Immunostaining of kidneys for C3 deposits showed a similar pattern. C3 deposition increases in mice after treatment with adriamycin, and treatment of the mice with anti-CD20 prevents this increase (Figure 2B). Kidney sections from mice with adriamycin nephropathy were dual-stained for IgM and C3 (Figure 2C). IgM and C3 colocalized within the glomerulus (appearing yellow in the overlay), supporting the hypothesis that the glomerular IgM activates complement. Tubulointerstitial C3 did not colocalize with IgM, consistent with our previous work demonstrating that complement activation in the tubulointerstitium occurs via the alternative pathway.28 C3d, the final cleavage product of activated C3 was detected it the glomeruli of adriamycin-treated mice (Figure 2D). This indicates that the C3 deposits are due to complement activation, not simply to passive trapping of circulating C3.
Figure 2.

Treatment with anti-CD20 reduces glomerular complement activation in mice with adriamycin nephropathy. Mice are injected with anti-CD20 mAb to deplete B cells before the induction of adriamycin nephropathy. Complement activation in the glomeruli is examined by immunofluorescence microscopy 4 weeks after injection with adriamycin. (A) Staining for C4 demonstrates that glomerular C4 deposition increases after injection with adriamycin, but that treatment of the mice with anti-CD20 prevents this increase. (B) Staining for C3 demonstrates that glomerular C3 deposition increases after injection with adriamycin. Treatment of the mice with anti-CD20 prevents this increase in C3 deposition. Glomeruli are indicated with arrowheads and areas of C3 and C4 staining within the glomeruli are indicated with small arrows. Kidneys from three to five mice per group are examined. Thirty glomeruli per section are visualized, and the average for each mouse is determined. After converting the images to grayscale, the brightness and contrast of the shown images are adjusted. All images are adjusted equally. (C) Dual staining of kidneys from mice with adriamycin nephropathy for IgM and C3 demonstrates colocalization of IgM and C3 within glomeruli (appearing yellow in the overlay). Glomeruli are indicated with arrowheads and areas of colocalization are indicated with small arrows. (D) Staining of kidneys from mice with adriamycin nephropathy for C3d confirms that the glomerular deposits contain activated complement proteins. Original magnification, ×200 in A–C; ×400 in D. Scale bar, 100 μM.
Treatment with a mAb to CD20 Reduces Proteinuria and Attenuates Histologic Injury in Mice with Adriamycin Nephropathy
Urine albumin/creatinine ratios were measured as a marker of glomerular injury. Mice with adriamycin nephropathy were grossly albuminuric (Figure 3A), and treatment of the mice with anti-CD20 reduced the degree of albuminuria in mice with adriamycin nephropathy. When anti-CD20–treated mice were treated with adriamycin and then re-injected with purified glomerular IgM, however, the degree of albuminuria was comparable with that seen in mice that received adriamycin alone. This demonstrates that IgM is an important mediator of injury in this model.
Figure 3.

Treatment with anti-CD20 reduces glomerular injury in mice with adriamycin nephropathy. Mice are injected with anti-CD20 mAb to deplete B cells before the induction of adriamycin nephropathy. (A) Urine albumin/creatinine levels are measured. Treatment of mice with adriamycin causes high-level albuminuria, but this is significantly attenuated by treatment with anti-CD20. Mice treated with anti-CD20 that are re-injected with IgM purified from diseased kidneys have levels of albuminuria similar to those seen in adriamycin-treated mice. (B) Glomerulosclerosis in the different treatment groups is assessed on sections stained with periodic acid–Schiff. Glomeruli are indicated with arrowheads and areas of sclerosis are indicated with small arrows. (C) The percentage of each glomerulus occupied by matrix is determined as an index of glomerulosclerosis. The reduced glomerulosclerosis is observed in mice treated with adriamycin and anti-CD20 compared with adriamycin-treated mice, and mice that are reconstituted with IgM demonstrate an increase in glomerulosclerosis. (D) Staining of kidneys for collagen IV demonstrates that injection of mice with adriamycin causes an increase in glomerular collagen IV deposition, and this is not significantly affected by treatment with anti-CD20. Kidneys from three mice per group are examined. Thirty glomeruli per section are visualized, and the average for each mouse is determined. Representative glomeruli from mice in each group are shown. Glomeruli are indicated with arrowheads and areas of collagen IV deposition are indicated with small arrows. (E) Staining of kidneys for synaptopodin demonstrates that injection of mice with adriamycin disrupts synaptopodin expression in the glomerulus. Some expression is detected in mice treated with adriamycin and anti-CD20, but the expression is lower than in control mice. Glomeruli are indicated with arrowheads and areas of synaptopodin staining are indicated with small arrows. (F) Electron microscopy demonstrates severe foot process effacement in mice treated with adriamycin. Mice treated with adriamycin and anti-CD20 demonstrate patchy foot process effacement. Small arrows indicate areas of effacement, and the arrowhead indicates an area of preserved foot processes. Original magnification, ×400 in B, D, and E. Scale bar, 100 μM.
Glomerular matrix deposition was measured as an index of glomerulosclerosis. Treatment with the anti-CD20 antibody reduced glomerulosclerosis, and injection of the mice with IgM restored glomerulosclerosis (Figure 3, B and C). Mice injected with adriamycin demonstrated greater glomerular collagen IV deposition than control mice, but treatment with anti-CD20 did not significantly reduce glomerular collagen IV deposition (Figure 3D). Synaptopodin is an actin-associated protein expressed in podocytes. Staining of the kidneys for synaptopodin (Figure 3E) demonstrated that synaptopodin expression was lost in mice with adriamycin nephropathy, indicating podocyte injury. The pattern of synaptopodin staining was preserved in mice treated with anti-CD20. Electron microscopy also demonstrated widespread foot process effacement in adriamycin-treated mice (Figure 3F). Although there were areas of effacement in the anti-CD20 treated mice, the effacement was not as severe as in mice with adriamycin nephropathy that did not receive the anti-CD20.
B Cell–Deficient Mice Are Protected from Adriamycin Nephropathy
There is evidence that anti-CD20 can reduce proteinuria through direct effects on podocytes.29 To confirm that B cells are involved with the pathogenesis of this disease, we induced adriamycin nephropathy in BALB/c mice, Rag1−/− mice (a strain of mice that lacks T cells and B cells), and Jh mice (a strain of mice deficient in mature B cells). There was a trend toward lower albuminuria in the Rag1−/− mice compared with wild-type control mice, but albuminuria was significantly lower in Jh mice than in wild-type controls (Figure 4A). Electron microscopy demonstrated patchy foot process effacement (Figure 4B), but the effacement was not as widespread as in the adriamycin-treated BALB/c mice (Figure 3F). In wild-type mice, IgM and C3 colocalize within the glomeruli (see Figure 2C). As expected, no IgM was seen in the glomeruli of the Jh mice (Figure 4C). Glomerular C3 deposition was also lower in Jh mice, indicating that glomerular IgM activates the complement system in wild-type mice. Tubulointerstitial C3 deposition was still evident, however, consistent with our previous observation that complement activation in this compartment proceeds exclusively via the alternative pathway.28
Figure 4.

B cell–deficient mice are protected from adriamycin nephropathy. Balb/c, Rag-1 deficient mice (which lack T cells and B cells) on a Balb/c background, and Jh mice (which lack mature B cells) on a Balb/c background are injected with adriamycin. (A) There is significantly less albuminuria in Jh mice than in Balb/c mice 4 weeks after injection with adriamycin. (B) Electron microscopy demonstrates areas of preserved foot processes in the Jh mice. (C) No IgM is detected in the Jh mice. C3 is detected in the tubulointerstitium and along Bowman’s capsule, but is not seen within the glomerular tuft. Glomeruli are indicated with arrowheads and areas of C3 staining are indicated with small arrows.
Depletion of Peritoneal B cells by Hypotonic Shock Prevents Glomerular IgM Deposition in Adriamycin Nephropathy
Natural antibody IgM is predominantly produced by peritoneal B-1a cells. As described above, treatment of mice with anti-CD20 also depletes peritoneal B cells, although less effectively than it depletes splenic B cells (Table 1). To effectively reduce the peritoneal B cells without affecting splenic B cell function, we lysed peritoneal cells by hypotonic shock. This treatment causes lysis of all peritoneal cells, including macrophages, but it does not affect splenic B cells (Table 2). We did see some reduction in circulating B cells using this strategy, suggesting that the peritoneal compartment is the source of some circulating B cells. Although all peritoneal cells are reduced by this method, the treatment does not reduce the number of B-1a cells as a percentage of total peritoneal cells. Consequently, the effects of this treatment may not be specific to depletion of B-1a cells. The level of total IgM in the serum was unaffected by this treatment (Table 2).
Table 2.
Reduction in B cell subsets and Ig after intraperitoneal hypotonic shock was performed every 3 days for a total of 14 days
| Compartment | Cells | PBS | H2O | % Reduction |
|---|---|---|---|---|
| Spleen | IgM+B220+ | 26.34 | 24.45 | 7.19 |
| IgMHighB220+CD5+ | 0.15 | 0.16 | −3.33 | |
| Peripheral blood | IgM+B220+ | 25.165 | 11.72 | 53.45 |
| IgMHighB220+CD5+ | 0.355 | 0.09 | 76.06 | |
| Peritoneum | IgM+B220+ | 26.44 | 7.86 | 70.29 |
| IgMHighB220+CD5+ | 0.21 | 0.19 | 11.90 | |
| Serum | IgM (mg/ml) | 13.76±3.01 | 16.67±2.54 | — |
Data are reported as the percentage of total cells.
To deplete the peritoneal B cells in mice with adriamycin nephropathy, the peritoneal cells were lysed by hypotonic shock every 3 days for 2 weeks before injection with adriamycin in order to give sufficient time for preformed IgM to turn over.25 Depletion of the cells was maintained by intraperitoneal injections of distilled water every three days during the course of the study. Although this treatment is very effective at immediately reducing the peritoneal B cell numbers,25 the cells do re-accumulate and the depletion at day 14 was not complete (Table 2). As controls, another group of mice received peritoneal injections with PBS according to the same schedule. When reported as a percentage of total peritoneal cells, the B-1a cells were not reduced by this treatment (Table 2) because the treatment reduces all peritoneal cells equally.
In mice that underwent peritoneal B cell depletion, the accumulation of glomerular IgM was significantly attenuated after injection of the mice with adriamycin (Figure 5A). Depletion of the peritoneal cells showed a trend toward reduction of glomerular C4 deposition, and glomerular C3 deposition was reduced by this treatment (Figures 5, B and C). Tubulointerstitial C3 deposition was unaffected by this treatment.
Figure 5.

Depletion of peritoneal B cells reduces the glomerular deposition of IgM, C3, and C4 in mice with adriamycin nephropathy. Peritoneal cells are depleted with hypotonic shock, starting 2 weeks before inducing adriamycin nephropathy. (A) Immunofluorescence microscopy demonstrates that depletion of the peritoneal cells attenuates the glomerular deposition of IgM. (B) Immunofluorescence microscopy for C4 demonstrates that depletion of the peritoneal cells also reduces C4 deposition within the glomeruli, although this does not reach statistical significance. (C) Immunofluorescence microscopy for C3 demonstrates that depletion of the peritoneal cells prevents complement C3 activation within the glomerulus. Kidneys from three to four mice per group are examined. Thirty glomeruli per section are visualized, and the average for each mouse is determined. Glomeruli are indicated with arrowheads, and areas of positive staining within the glomeruli are indicated with small arrows. Original magnification, ×200. Scale bar, 100 μM.
These results demonstrate that peritoneal B cells generate a significant proportion of the IgM deposited in the glomeruli in this model. Levels of circulating IgM were not affected by the peritoneal cell depletion even though glomerular deposition was reduced, suggesting that the glomerular IgM deposits are composed of IgM that binds to specific glomerular antigens. As with treatment of mice with anti-CD20, depletion of the peritoneal B cells reduced glomerular complement activation.
Depletion of Peritoneal B-1 Cells by Hypotonic Shock Reduces Proteinuria and Histologic Injury in Mice with Adriamycin Nephropathy
Urine albumin/creatinine ratios were measured in samples collected 1 or 4 weeks after injection with adriamycin (Figure 6A). The level of albuminuria was significantly lower in mice that had undergone peritoneal cell depletion at both the early and the later time points. The reduction in albuminuria at the 1-week time point indicates that glomerular IgM contributes to injury at an early phase of disease in this model. Depletion of the peritoneal B cells attenuated the overall degree of glomerulosclerosis (Figure 6, B and C), but it did not prevent collagen IV accumulation in adriamycin-treated mice (Figure 6D).
Figure 6.

Depletion of peritoneal B cells reduces albuminuria in mice with adriamycin nephropathy. Peritoneal cells are depleted with hypotonic shock, starting 2 weeks before the induction of adriamycin nephropathy. Urine albumin/creatinine levels are measured. (A) Peritoneal depletion of B cells significantly attenuates the level of albuminuria 1 week and 4 weeks after injection of the adriamycin. (B) Representative glomeruli from mice in each group are shown. The kidneys are stained with periodic acid–Schiff, and glomeruli are indicated with arrowheads. (C) The percentage of each glomerulus occupied by matrix is determined as an index of glomerulosclerosis. Peritoneal cell depletion reduces the degree of glomerulosclerosis compared with PBS-treated mice with adriamycin nephropathy. (D) Staining of kidneys for collagen IV demonstrates that injection of mice with adriamycin causes an increase in glomerular collagen IV deposition, and this is not significantly affected by depletion of peritoneal B cells. Thirty glomeruli per section are visualized, and the average for each mouse is determined. Representative glomeruli from mice in each group are shown. Original magnification, ×400. Scale bar, 100 μM.
Glomerular IgM and C3d Colocalize in Human FSGS
Previous series have reported that glomerular IgM and/or C3 may be detected in up to 90% of patients with FSGS.1 We reviewed the biopsy reports of 174 cases of FSGS evaluated over the past 8 years by one of the authors of this study (D.L.). Of those biopsies, approximately 23% demonstrated glomerular IgM without C3, approximately 2% demonstrated glomerular C3 without IgM, and 7% of the biopsies demonstrated glomerular IgM and C3 (Figure 7A). The percentage of biopsies demonstrating IgM and/or C3 in our patients is lower than that reported in other series, and indicates that natural IgM may only contribute to a small subset of FSGS patients. It is possible, however, that a focused examination of fresh tissue specifically for IgM or C3 would have identified these proteins in a greater percentage of the patients. We performed dual staining for IgM and for C3d on tissue in which both factors were detectable (Figure 7B). C3d and IgM showed a similar pattern of distribution throughout the glomerulus. Dual staining for IgM and C4 was also performed (Figure 7C). This staining demonstrated similar colocalization of these factors within the glomerulus.
Figure 7.

IgM and C3d colocalize in glomeruli of patients with FSGS. (A) The biopsy reports of 174 patients with FSGS are reviewed. Of these, 23% are noted to have IgM deposition, 7% have both IgM and C3, and 2% have detectable C3 in the absence of IgM. Available tissue from 19 patients with FSGS is dual-stained for IgM and C3d (B) or for IgM and C4d (C). In biopsies that contain IgM and complement deposits, the two immune factors colocalize within the glomeruli. Nonspecific binding of the FITC-conjugated secondary antibody is examined (control) in order to confirm specificity of the staining. The brightness and contrast of the shown images are adjusted to improve localization of the factors in the overlay. Original magnification, ×400. Scale bar, 100 μM.
Discussion
Glomerulosclerosis is a common histologic finding in both primary and secondary glomerular diseases. Glomerular IgM and complement proteins are frequently detected within glomeruli that bear segmental scars. The significance of these deposits has been uncertain, and some investigators have proposed that these large molecules are nonspecifically trapped in areas of glomerular injury.30 In this study, we utilized three strategies to deplete B-1a B cells, the primary source of natural antibody IgM. Our first approach was to treat mice with a murine anti-murine CD20 antibody. Treatment of the mice with anti-CD20 reduced glomerular IgM deposition and glomerular complement activation in mice with adriamycin nephropathy. Treatment with the anti-CD20 reduced the degree of proteinuria that developed after injection with adriamycin, demonstrating that B cells contribute to glomerular injury in this model. Treatment with anti-CD20 preserved glomerular synaptopodin expression and reduced the degree of foot process effacement, indicating that this treatment protected the podocytes from injury in this model. Mice that received the same dose of anti-CD20 but that were reconstituted with IgM purified from kidneys with adriamycin nephropathy showed a trend toward greater proteinuria than mice that were not reconstituted with the IgM, supporting the hypothesis that IgM is an important mediator of glomerular injury in this model. Injection of the mice with purified IgM did not restore injury to the level seen in mice that did not receive anti-CD20, however, possibly because the purification process altered the binding of the IgM with glomerular epitopes.
Next, we utilized mice with targeted genetic deletions that prevent the formation of mature B cells. We utilized Rag1−/− and Jh mice. The Jh mice developed milder albuminuria than BALB/c controls. As with the mice that received anti-CD20, there was less glomerular complement activation than BALB/C mice, indicating that IgM activates the complement system in the injured glomeruli. The foot processes in the kidneys of Jh mice were also relatively preserved, indicating that podocyte injury is reduced in B cell–deficient mice. Interestingly, the Rag1−/− mice were not protected to the same degree as the Jh mice, suggesting that the combined absence of T cells and B cells is not as protective as the absence of B cells. This difference suggests that some T cell populations, such as regulatory T cells, are protective in this model.31 This also raises the possibility that therapies that selectively target B cells will be more effective than those that target both B cells and T cells. It is worth noting, however, that B cells have additional immunologic functions beyond Ig production. For example, they affect T cell function through the production of cytokines and B cells also function as antigen presenting cells. These other B cell functions may contribute to their pathogenic role in this model.
Finally, we depleted peritoneal B cells by hypotonic shock. This treatment depletes all peritoneal cells (not just B cells), but it has only a minor effect on splenic B cells or serum levels of IgM. Depletion of the peritoneal B cells in mice reduced glomerular IgM deposition, glomerular complement activation, albuminuria, and glomerulosclerosis. Furthermore, this strategy reduced glomerular IgM despite the fact that circulating levels of IgM were unchanged. This indicates that the glomerular IgM deposits are composed of specific clones of IgM, and are not simply the result of deposition of total circulating IgM.
More than 70% of B-1a B cells reside in the peritoneum.18 The reduction in glomerular IgM in mice subjected to peritoneal hypotonic shock supports the concept that the nephritogenic IgM is natural antibody made primarily by this B cell subset. Our findings indicate that glomerular IgM deposition is due to the binding of specific clones of IgM to glomerular epitopes. Similarly, our results indicate that the deposition of C4 and C3 fragments in the glomeruli is caused by Ig deposition and complement activation because therapies that depleted B cells reduced the degree of glomerular complement activation. Because IgM activates the classic pathway when bound to a cognate ligand, complement activation by IgM within the glomeruli also indicates that the IgM has bound to specific glomerular epitopes.
As previously reported in several case series,1,2,32 we found that IgM is present in the glomeruli of a subset of patients diagnosed with primary FSGS. Dual staining of the tissue for IgM and C3d showed colocalization of these immune factors when they were both present. C3d is a cleavage fragment of C3 that is generated during complement activation, and IgM activates the complement system when in complex with a target ligand. Thus, the biopsy results indicate that IgM binds to glomerular epitopes in some patients with FSGS. Measurement of complement activation fragments in plasma from patients with FSGS further demonstrates that the complement system is activated in this disease (J.M. Thurman and H. Trachtman, unpublished data).33
Glomerular IgM and C3 are only detected in a subset of patients with primary and secondary forms of FSGS. FSGS is a very heterogeneous disease, however, both clinically and morphologically.34,35 Although B cell depletion and complement deficiency contribute to the early development of albuminuria in the adriamycin model,28,36 Ig- and complement-mediated injury may be secondary phenomena that occur after a primary renal insult. We have observed glomerular IgM deposition after ischemic and chemical injury of the kidneys,25 suggesting that diverse insults may generate glomerular neoepitopes. The degree to which the IgM-complement axis is engaged in a given patient, however, is likely influenced by the abundance of glomerular epitope and also by the degree to which local complement regulation remains intact. Nevertheless, the presence of these same immune factors in kidneys affected by systemic diseases such as hypertensive nephrosclerosis6 and diabetic nephropathy5 raises the possibility that recognition of glomerular epitopes by natural IgM is a common downstream mechanism for a wide range of renal insults.
As previously reported,25,37 we found that there are low levels of IgM and C4 in the glomeruli of normal mice. In unmanipulated mice, the IgM colocalized with C4, but only low levels of C3 were observed within the glomeruli (Figure 2). This may be because complement regulatory proteins expressed within the glomerulus effectively limit complement activation by the IgM.25 Studies of normal human tissue also demonstrate low levels of IgM and C4 deposition within the glomeruli,38,39 and mesangial C4d is so reliably present that it is often used as an internal staining control for C4d staining of transplant biopsies. Glomerular injury by adriamycin may impair local complement regulation, thus permitting greater complement activation by bound IgM. IgM is a classical pathway activator, but we and others previously demonstrated a role for alternative pathway amplification in this model.28,36 It is possible, therefore, that glomerular injury simultaneously increases classical pathway activation by natural IgM, which binds to injury associated epitopes, while also decreasing the ability of complement regulatory proteins within the glomerulus to control amplification of complement activation through the alternative pathway.
The primary treatments for idiopathic FSGS are immunosuppressive drugs, yet it has remained uncertain whether FSGS is an immune-mediated disease. Indeed, studies have indicated that the efficacy of some of these drugs may be due, in part, to nonimmune effects. Cyclosporine, for example, may reduce proteinuria by stabilizing the podocyte cytoskeleton.40 Even rituximab may confer benefit from direct (nonimmune) effects on the podocytes.29 We cannot exclude off-target effects of either of the treatments we have utilized. Taken together, however, the current studies demonstrate that B cells and IgM contribute to glomerular injury. Our previous study demonstrating that complement-deficient mice are also protected in this model further implicates the IgM-complement axis in the development of glomerulosclerosis.28
Conventional immunosuppressive treatments, such as corticosteroids and calcineurin inhibitors, have limited efficacy in reducing natural antibody formation and complement activation.41–43 Thus, even when this system is engaged, the standard immunomodulatory therapies used to treat FSGS may not be of benefit. Newer drugs that target B cells or the complement system may be more effective at reducing natural antibody deposition and complement activation within the glomeruli of patients with FSGS. Our findings raise the possibility that identification of glomerular IgM and C3 deposits may identify patients likely to benefit from treatment with rituximab or eculizumab (a mAb to the complement C5 protein).
The current treatment approach for patients with secondary glomerulosclerosis is treatment of the primary disease and nonspecific measures, such as BP control.44 CKD often progresses, however, even if the initiating process becomes quiescent. This suggests that common downstream mechanisms of disease progression may be engaged.45 The prevalence of glomerular IgM and C3 in diverse toxic, metabolic, hemodynamic, and autoimmune diseases of the kidney raises the possibility that IgM natural antibody binds epitopes exposed or generated by all of these insults. If so, strategies that block natural IgM in the kidney could be beneficial in a wide range of chronic, progressive renal diseases. Competitive antagonists have been used to block the binding of natural antibodies to tissue-specific epitopes.46
In conclusion, this study demonstrates that therapeutic strategies that deplete B cells ameliorate glomerular injury in adriamycin nephropathy. All three of the strategies we tested reduced the deposition of IgM in the glomeruli and prevented glomerular complement activation. These findings indicate that the deposition of IgM in this model is due to specific binding of IgM to glomerular epitopes, and therapies that deplete B-1a cells may be a useful therapy for glomerulosclerosis.
Concise Methods
Adriamycin Nephropathy Model
BALB/c and Rag1 deficient mice (C.129S7(B6)-Rag1tm1Mom/J; deficient in mature T cells and B cells) on a BALB/c background were obtained from the Jackson Laboratory (Bar Harbor, ME). Mice with targeted deletion of the Jh gene, which are deficient in mature B cells, were obtained from Taconic (Hudson, NY). All mice were housed and maintained at the University of Colorado Center for Laboratory Animal Care in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Adriamycin nephropathy was induced in Balb/c mice as previously described.27,28 Disease was induced in 8- to 10-week-old male mice with a single intravenous injection of 11 mg/kg of adriamycin, (doxorubicin; Bedford Laboratories, Bedford, OH). Mice were followed for 4 weeks after disease induction.
Urine Albumin Measurement
Urine albumin was measured by an ELISA, according to the manufacturer’s instructions (Bethyl Laboratories, Montgomery, TX). Urine creatinine was measured using an Alfa Wasserman ACE Chemistry Analyzer. To normalize urine albumin excretion, the values are reported as micrograms albumin per milligram creatinine.
Renal Morphology
After the kidneys were removed from the mice, sagittal sections were fixed, embedded in paraffin, and 4-μm sections were cut and stained with periodic acid–Schiff. To quantitate the degree of glomerular injury, glomeruli were examined using an Aperio digital microscope. The degree of fibrosis was quantified with the Scanscope digital program. Regions of interest were drawn around glomeruli, and the percentage of each glomerulus occupied by matrix was determined. An average percentage for each slide was calculated. Electron microscopy was performed at the University of Colorado Denver School of Medicine electron microscopy core facility.
Depletion of B Cells by CD20 Antibody
Mice were injected via the tail vein with 250 μg of Ab to CD20 (murine IgG2a [clone 5D2], generously provided by Genentech). This treatment was performed 2 weeks before disease initiation, and every 2 weeks for the duration of the study. Control mice received an equal volume of PBS as a vehicle control.
Depletion of Peritoneal B Cells by Hypotonic Shock
Peritoneal B cells were depleted, as previously described.25 Mice received intraperitoneal injections of distilled water (0.5 ml) starting 12 days before disease induction, and this treatment was continued every 3 days for the duration of the experiment. Control mice were injected with 0.5 ml of PBS according to the same schedule.
Flow Cytometry
Peritoneal cells were obtained by lavage, and were then washed and resuspended in 500 μl of PBS. To stain splenocytes, the spleen was mechanically dissociated and suspended in 10 ml of PBS, and the cell suspension was passed through a 70-μM nylon strainer. After washing in the cells in PBS, they were resuspended in 1 ml of 36% Percoll, gently applied to 72% Percoll, and centrifuged at 1000×g for 30 minutes at 6°C, and the interphase was collected. The cells were washed in PBS and resuspended in PBS containing 1% FCS. The cells were stained with antibodies to murine B220, CD5, and CD3 (all antibodies from Biolegend, San Diego, CA). Antibodies to murine CD19 (BD Pharmingen, San Diego, CA), IgM, and IgG (both from Novus Biologicals, Littleton, CO) were also used. Flow cytometry was performed using a FACS Calibur machine (BD Biosciences, San Jose, CA), and analyzed with CellQuest Pro software (BD Biosciences).
Immunofluorescence Microscopy
Sagittal sections of the kidneys were snap-frozen in OCT compound (Sakura Finetek, Torrance, CA). Four-micrometer sections were cut with a cryostat and stored at −70°C. At the time of staining, the slides were fixed with acetone and then incubated with the primary antibody. Sections were stained with FITC-conjugated anti-mouse C3 (MP Biomedicals, Solon, OH), an antibody to human C3d that cross-reacts with murine C3d (Dako, Carpinteria, CA),47 FITC-conjugated antibodies to mouse IgG and IgM (Jackson ImmunoResearch Laboratories, West Grove, PA), anti-synaptopodin (Sigma-Aldrich, St. Louis, MO), and anti-mouse C4 (Hycult Biotech, Uden, The Netherlands). Secondary antibodies were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). Sections were counterstained with hematoxylin (Vector Laboratories, Burlingame, CA). Staining was visualized using an Olympus BX51 microscope with a digital camera (Pixera, Santa Clara, CA) and Studio 3.0.1 software (Pixera). For quantitative analysis, high-powered images of 30 glomeruli were obtained per section at room temperature. The fluorescence intensity was determined using ImageJ software, and the mean value for each treatment group was determined. Image overlays were created using Adobe Photoshop CS3 software (San Jose, CA).
Serum IgM Measurement by ELISA
Briefly, ELISA plates (Corning Glass, Corning, NY) were coated with 100 ng of anti-mouse IgM (Southern Biotechnology Associates, Birmingham, AL). After the plates were washed and blocked with 1% BSA (Sigma-Aldrich) for 2 hours, serum samples were diluted 1:50 and added to the wells. Standard curves were generated using serial dilutions of purified IgM (7.8125–500 ng/ml). The plates were then washed, and IgM was detected with a horseradish peroxidase–conjugated anti-mouse IgM Ab (Southern Biotechnology Associates). The plates were then developed with ABTS (Sigma-Aldrich) and read on a Spectramax PLUS plate reader (Molecular Devices, Sunnyvale, CA).
Purification IgM from Kidneys
IgM was purified from mouse kidneys using immobilized anti-IgM beads (rat anti-mouse IgM Sepharose 4B; Invitrogen, Grand Island, NY). Mouse kidneys were solubilized and IgM was dissociated from antigen in standard RIPA buffer. The supernatant was centrifuged at 20,000×g for 15 minutes and the soluble fraction was used for affinity purification. The IgM content was determined by ELISA for mouse IgM, and the purity was assessed by Coomassie staining and Western blot analysis. Approximately 3 μg of IgM was injected per mouse per week via the tail vein starting 1 week after disease initiation. The weekly injections were continued for the duration of the experiment. Natural IgM is generally polyreactive, and the specificity of the eluted IgM is not known. Therefore, control IgM was not used in these experiments.
Statistical Analyses
Data were analyzed using GraphPad Prism software (La Jolla, CA). Comparison between groups was performed with unpaired t testing and by ANOVA when multiple groups were compared. A P value <0.05 was considered significant.
Disclosures
V.M.H. and J.M.T. are paid consultants for Alexion Pharmaceuticals Inc.
Acknowledgments
Murine anti-CD20 antibody was generously provided by Dr. Andrew Chan at Genentech.
This work was supported in part by the National Institutes of Health (R01 DK076690) and the National Institute of Allergy and Infectious Disease (pilot project grant to J.M.T. and R01 AI311052 to V.M.H.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1.Gephardt GN, Tubbs RR, Popowniak KL, McMahon JT: Focal and segmental glomerulosclerosis. Immunohistologic study of 20 renal biopsy specimens. Arch Pathol Lab Med 110: 902–905, 1986 [PubMed] [Google Scholar]
- 2.Habib R, Girardin E, Gagnadoux MF, Hinglais N, Levy M, Broyer M: Immunopathological findings in idiopathic nephrosis: Clinical significance of glomerular “immune deposits”. Pediatr Nephrol 2: 402–408, 1988 [DOI] [PubMed] [Google Scholar]
- 3.Cohen AH, Border WA, Glassock RJ: Nehprotic syndrome with glomerular mesangial IgM deposits. Lab Invest 38: 610–619, 1978 [PubMed] [Google Scholar]
- 4.Lawler W, Williams G, Tarpey P, Mallick NP: IgM associated primary diffuse mesangial proliferative glomerulonephritis. J Clin Pathol 33: 1029–1038, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ainsworth SK, Hirsch HZ, Brackett NC, Jr, Brissie RM, Williams AV, Jr, Hennigar GR: Diabetic glomerulonephropathy: Histopathologic, immunofluorescent, and ultrastructural studies of 16 cases. Hum Pathol 13: 470–478, 1982 [DOI] [PubMed] [Google Scholar]
- 6.Mujais SK, Emmanouel DS, Kasinath BS, Spargo BH: Marked proteinuria in hypertensive nephrosclerosis. Am J Nephrol 5: 190–195, 1985 [DOI] [PubMed] [Google Scholar]
- 7.Cavallo T, Johnson MP: Immunopathologic study of minimal change glomerular disease with mesangial IgM deposits. Nephron 27: 281–284, 1981 [DOI] [PubMed] [Google Scholar]
- 8.Hsu HC, Chen WY, Lin GJ, Chen L, Kao SL, Huang CC, Lin CY: Clinical and immunopathologic study of mesangial IgM nephropathy: Report of 41 cases. Histopathology 8: 435–446, 1984 [DOI] [PubMed] [Google Scholar]
- 9.Zeis PM, Kavazarakis E, Nakopoulou L, Moustaki M, Messaritaki A, Zeis MP, Nicolaidou P: Glomerulopathy with mesangial IgM deposits: Long-term follow up of 64 children. Pediatr Int 43: 287–292, 2001 [DOI] [PubMed] [Google Scholar]
- 10.Alexopoulos E, Papagianni A, Stangou M, Pantzaki A, Papadimitriou M: Adult-onset idiopathic nephrotic syndrome associated with pure diffuse mesangial hypercellularity. Nephrol Dial Transplant 15: 981–987, 2000 [DOI] [PubMed] [Google Scholar]
- 11.Fernandez-Fresnedo G, Segarra A, González E, Alexandru S, Delgado R, Ramos N, Egido J, Praga M, Trabajo de Enfermedades Glomerulares de la Sociedad Española de Nefrología (GLOSEN) : Rituximab treatment of adult patients with steroid-resistant focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 4: 1317–1323, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gulati A, Sinha A, Jordan SC, Hari P, Dinda AK, Sharma S, Srivastava RN, Moudgil A, Bagga A: Efficacy and safety of treatment with rituximab for difficult steroid-resistant and -dependent nephrotic syndrome: Multicentric report. Clin J Am Soc Nephrol 5: 2207–2212, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pescovitz MD, Book BK, Sidner RA: Resolution of recurrent focal segmental glomerulosclerosis proteinuria after rituximab treatment. N Engl J Med 354: 1961–1963, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Ravani P, Magnasco A, Edefonti A, Murer L, Rossi R, Ghio L, Benetti E, Scozzola F, Pasini A, Dallera N, Sica F, Belingheri M, Scolari F, Ghiggeri GM: Short-term effects of rituximab in children with steroid- and calcineurin-dependent nephrotic syndrome: A randomized controlled trial. Clin J Am Soc Nephrol 6: 1308–1315, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sethna C, Benchimol C, Hotchkiss H, Frank R, Infante L, Vento S, Trachtman H: Treatment of recurrent focal segmental glomerulosclerosis in pediatric kidney transplant recipients: Effect of rituximab. J Transplant 2011: 389542, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suri M, Tran K, Sharma AP, Filler G, Grimmer J: Remission of steroid-resistant nephrotic syndrome due to focal and segmental glomerulosclerosis using rituximab. Int Urol Nephrol 40: 807–810, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Carroll MC, Holers VM: Innate autoimmunity. Adv Immunol 86: 137–157, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Baumgarth N: The double life of a B-1 cell: Self-reactivity selects for protective effector functions. Nat Rev Immunol 11: 34–46, 2011 [DOI] [PubMed] [Google Scholar]
- 19.Hayakawa K, Hardy RR, Herzenberg LA, Herzenberg LA: Progenitors for Ly-1 B cells are distinct from progenitors for other B cells. J Exp Med 161: 1554–1568, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamaguchi Y, Uchida J, Cain DW, Venturi GM, Poe JC, Haas KM, Tedder TF: The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. J Immunol 174: 4389–4399, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Haas MS, Alicot EM, Schuerpf F, Chiu I, Li J, Moore FD, Carroll MC: Blockade of self-reactive IgM significantly reduces injury in a murine model of acute myocardial infarction. Cardiovasc Res 87: 618–627, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fleming SD, Shea-Donohue T, Guthridge JM, Kulik L, Waldschmidt TJ, Gipson MG, Tsokos GC, Holers VM: Mice deficient in complement receptors 1 and 2 lack a tissue injury-inducing subset of the natural antibody repertoire. J Immunol 169: 2126–2133, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Zhang M, Austen WG, Jr, Chiu I, Alicot EM, Hung R, Ma M, Verna N, Xu M, Hechtman HB, Moore FD, Jr, Carroll MC: Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proc Natl Acad Sci U S A 101: 3886–3891, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiser MR, Williams JP, Moore FD, Jr, Kobzik L, Ma M, Hechtman HB, Carroll MC: Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J Exp Med 183: 2343–2348, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Renner B, Strassheim D, Amura CR, Kulik L, Ljubanovic D, Glogowska MJ, Takahashi K, Carroll MC, Holers VM, Thurman JM: B cell subsets contribute to renal injury and renal protection after ischemia/reperfusion. J Immunol 185: 4393–4400, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peterson LK, Tsunoda I, Fujinami RS: Role of CD5+ B-1 cells in EAE pathogenesis. Autoimmunity 41: 353–362, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Wang YP, Tay YC, Harris DC: Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int 58: 1797–1804, 2000 [DOI] [PubMed] [Google Scholar]
- 28.Lenderink AM, Liegel K, Ljubanović D, Coleman KE, Gilkeson GS, Holers VM, Thurman JM: The alternative pathway of complement is activated in the glomeruli and tubulointerstitium of mice with adriamycin nephropathy. Am J Physiol Renal Physiol 293: F555–F564, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, Li J, Mattiazzi A, Ciancio G, Chen L, Zilleruelo G, Abitbol C, Chandar J, Seeherunvong W, Ricordi C, Ikehata M, Rastaldi MP, Reiser J, Burke GW, 3rd: Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med 3: 85ra46, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartz MM: Focal Segmental Glomerulosclerosis. In: Heptinstall's Pathology of the Kidney, edited by Jennette JC, Olson JL, Schwartz MM, Silva FG, 6th Ed., Philadelphia, Lippincott Williams & Wilkins, 2007, pp 155–204 [Google Scholar]
- 31.Wang Y, Wang Y, Feng X, Bao S, Yi S, Kairaitis L, Tay YC, Rangan GK, Harris DC: Depletion of CD4(+) T cells aggravates glomerular and interstitial injury in murine adriamycin nephropathy. Kidney Int 59: 975–984, 2001 [DOI] [PubMed] [Google Scholar]
- 32.Ji-Yun Y, Melvin T, Sibley R, Michael AF: No evidence for a specific role of IgM in mesangial proliferation of idiopathic nephrotic syndrome. Kidney Int 25: 100–106, 1984 [DOI] [PubMed] [Google Scholar]
- 33.Morita Y, Ikeguchi H, Nakamura J, Hotta N, Yuzawa Y, Matsuo S: Complement activation products in the urine from proteinuric patients. J Am Soc Nephrol 11: 700–707, 2000 [DOI] [PubMed] [Google Scholar]
- 34.D’Agati VD, Fogo AB, Bruijn JA, Jennette JC: Pathologic classification of focal segmental glomerulosclerosis: A working proposal. Am J Kidney Dis 43: 368–382, 2004 [DOI] [PubMed] [Google Scholar]
- 35.D’Agati V: The many masks of focal segmental glomerulosclerosis. Kidney Int 46: 1223–1241, 1994 [DOI] [PubMed] [Google Scholar]
- 36.Turnberg D, Lewis M, Moss J, Xu Y, Botto M, Cook HT: Complement activation contributes to both glomerular and tubulointerstitial damage in adriamycin nephropathy in mice. J Immunol 177: 4094–4102, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Park P, Haas M, Cunningham PN, Bao L, Alexander JJ, Quigg RJ: Injury in renal ischemia-reperfusion is independent from immunoglobulins and T lymphocytes. Am J Physiol Renal Physiol 282: F352–F357, 2002 [DOI] [PubMed] [Google Scholar]
- 38.Velosa J, Miller K, Michael AF: Immunopathology of the end-stage kidney. Immunoglobulin and complement component deposition in nonimmune disease. Am J Pathol 84: 149–162, 1976 [PMC free article] [PubMed] [Google Scholar]
- 39.Zwirner J, Felber E, Herzog V, Riethmüller G, Feucht HE: Classical pathway of complement activation in normal and diseased human glomeruli. Kidney Int 36: 1069–1077, 1989 [DOI] [PubMed] [Google Scholar]
- 40.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P: The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 14: 931–938, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Atkinson JP, Frank MM: Effect of cortisone therapy on serum complement components. J Immunol 111: 1061–1066, 1973 [PubMed] [Google Scholar]
- 42.Martin F, Chan AC: B cell immunobiology in disease: Evolving concepts from the clinic. Annu Rev Immunol 24: 467–496, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Lemercier C, Julen N, Coulpier M, Dauchel H, Ozanne D, Fontaine M, Ripoche J: Differential modulation by glucocorticoids of alternative complement protein secretion in cells of the monocyte/macrophage lineage. Eur J Immunol 22: 909–915, 1992 [DOI] [PubMed] [Google Scholar]
- 44.Jafar TH, Stark PC, Schmid CH, Landa M, Maschio G, de Jong PE, de Zeeuw D, Shahinfar S, Toto R, Levey AS, AIPRD Study Group : Progression of chronic kidney disease: The role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition: A patient-level meta-analysis. Ann Intern Med 139: 244–252, 2003 [DOI] [PubMed] [Google Scholar]
- 45.Kriz W, LeHir M: Pathways to nephron loss starting from glomerular diseases-insights from animal models. Kidney Int 67: 404–419, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Kulik L, Fleming SD, Moratz C, Reuter JW, Novikov A, Chen K, Andrews KA, Markaryan A, Quigg RJ, Silverman GJ, Tsokos GC, Holers VM: Pathogenic natural antibodies recognizing annexin IV are required to develop intestinal ischemia-reperfusion injury. J Immunol 182: 5363–5373, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sargsyan SA, Serkova NJ, Renner B, Hasebroock KM, Larsen B, Stoldt C, McFann K, Pickering MC, Thurman JM: Detection of glomerular complement C3 fragments by magnetic resonance imaging in murine lupus nephritis. Kidney Int 81: 152–159, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]