

Abstract
The endogenous protein antizyme inhibitor (AZI) is a potential oncogene which promotes cell growth by both inhibiting antizyme (AZ) activity and releasing ornithine decarboxylase (ODC) from AZ-mediated degradation. High levels of ODC and polyamines are associated with numerous types of neoplastic transformation, and the genomic region including AZI is frequently amplified in tumors of the ovary and prostate. To determine whether AZI functionally promotes prostate tumor growth, we made PC3M-LN4 (human) and AT6.1 (rat) cancer cell lines stably expressing shRNA to knockdown antizyme inhibitor 1 (AZI). AZI knockdown was confirmed by western blot, quantitative real-time PCR, and immunofluorescence. To examine the ability of these cells to form tumors in vivo, 1×106 cells were injected subcutaneously into nude mice either with (PC3M-LN4) or without (AT6.1) Matrigel. Tumor growth was measured two times per week by caliper. We found that cells in which AZI levels had been knocked down by shRNA formed significantly smaller tumors in vivo in both human and rat prostate cancer cell lines. These results suggest that not only does AZI promote tumor growth, but also that AZI may be a valid therapeutic target for cancer treatment.
Keywords: Antizyme inhibitor, cell proliferation, prostate cancer, tumor growth
Introduction
Antizyme inhibitor (AZI) is an endogenous protein which has been studied primarily due to its critical role in regulating polyamine biosynthesis. Polyamines are multivalent, organic cations which are essential for a variety of processes including normal cell growth, gene regulation, differentiation, and development (Gerner and Meyskens 2004; Basuroy and Gerner 2006; Pegg 2006; Kahana et al. 2005; Agostinelli et al. 2010). Polyamines are produced in a rate-limiting reaction catalyzed by the enzyme ornithine decarboxylase (ODC) (Pegg 2006), and ODC activity is primarily controlled by the balance between antizyme (AZ) and antizyme inhibitor (AZI) within the cell (Coffino 2001a). Antizyme, a tumor suppressor gene, acts as a negative regulator of the polyamine biosynthetic pathway, and production of antizyme protein is induced by high polyamine levels within the cell (Matsufuji et al. 1995). Antizyme inhibits polyamine synthesis by not only binding to ODC and preventing polyamine uptake, but also by targeting ODC for degradation (Mangold 2006; Keren-Paz et al. 2007; Coffino 2001a; Murakami et al. 1992; Li and Coffino 1992; Murakami et al. 1996; Coffino 2001b). AZI, a potential oncogene, prevents AZ from inducing ODC degradation, resulting in elevated cellular polyamine levels.
Increased ODC activity and elevated polyamine levels have been associated with numerous types of neoplastic transformation including intraepithelial neoplasias, the non-invasive precursors of epithelial cancers (Nishioka et al. 1995). ODC overexpression is also sufficient to transform mouse NIH-3T3 cells, and promote tumor growth in vivo (Auvinen et al. 1992), and these findings have been validated in several additional mouse models. Transgenic K6/ODC or K5/ODC mice overexpressing ODC from the keratin 6 or keratin 5 promoter, respectively, develop spontaneous squamous neoplasms, and treatment of these tumors with DFMO (difluoromethylornithine), an ODC inhibitor, leads to rapid tumor regression (Peralta Soler et al. 1998; Megosh et al. 1995; Smith et al. 1998; Lan et al. 2000). Based on its critical role in regulating cell growth and transformation, there has been long-term interest in targeting ODC as a tumor treatment, and several ODC inhibitors are now in clinical trials (Levin et al. 2003; Bachrach 2004; Bailey et al. 2010; Thompson et al. 2010; Zell et al. 2009; Meyskens et al. 2008).
Although elevated ODC activity and high polyamine levels are frequently associated with neoplastic transformation (Gerner and Meyskens 2004; Shantz and Levin 2007), very few studies to date have focused directly on the role of AZI in this process. Recent data has shown that AZI overexpression is sufficient to transform NIH-3T3 cells, resulting in both increased cell proliferation and increased ODC activity (Keren-Paz et al. 2006). Furthermore, NIH-3T3 cells overexpressing AZI formed tumors in vivo after subcutaneous injection into nude mice, while control cells did not (Keren-Paz Bercovich 2006). AZI levels have also been directly linked to rates of cell proliferation through knockdown experiments. Knockdown of AZI in A549 cells with siRNA resulted in decreased cell growth and decreased ODC activity in vitro, although the ability of these cells to form tumors in vivo was not determined (Choi et al. 2005). These preliminary studies suggest that modulating AZI expression may have a significant effect on tumor growth in vivo.
The genomic region including AZI has been found to be amplified in several tumor types including those of the ovary and prostate (Schaner et al. 2003; Schaner et al. 2005; van Duin et al. 2005), and AZI overexpression has been demonstrated in gastric tumors (Jung et al. 2000). Current data from Oncomine also suggests that AZI is upregulated in cancerous tissue from the breast, liver, skin, ovary, and cervix. Furthermore, AZI has been shown to be able to prevent AZ-mediated degradation of cyclin D1 (Newman et al. 2004), providing a second direct link between AZI levels and rates of cell proliferation. In addition, numerous studies have analyzed the effect of polyamine analogues in prostate cancer cell lines. Treatment with the polyamine analogues DENSPM, BE-4-4-4-4, or CGP-48664 led to inhibition of cell growth in a variety of androgen-independent prostate cancer cell lines, including AT6.1 and PC3 cells (Zagaja et al. 1998; Mi et al. 1998). In addition, animals with PC3 xenograft tumors exhibited decreased tumor growth following treatment with BE-4-4-4-4 (Mi et al. 1998). Together, these data suggest that increased AZI may be an important determinant in cell transformation and subsequent tumorigenesis.
To further explore the role of AZI in promoting tumor growth in vivo, we made a series of prostate cancer cell lines stably expressing shRNA (short hairpin RNA) to knockdown antizyme inhibitor 1 (AZI) or control shRNA. Our results demonstrate that suppression of AZI levels in prostate cancer cell lines results in repression of cell proliferation in vitro, and repression of tumor growth in vivo. These results suggest that in vivo suppression of AZI may be a worthwhile strategy for anti-cancer therapy.
Materials and methods
Cell culture and growth curves
PC3M-LN4 prostate cancer cells were maintained in RPMI media (Invitrogen) supplemented with 10% FBS (fetal bovine serum) and 1% GPS (glutathione/penicillin/streptomycin). PC3M-LN4 is a human, androgen-independent, highly-metastatic cell line derived by serial inoculation of cells into the prostate followed by isolation of lymph node metastases. AT6.1 prostate cancer cells were maintained in RPMI media with 10% FBS, 1% GPS, and 250nM dexamethasone (Sigma-Aldrich) as previously described. AT6.1 is an androgen-independent highly-metastatic variant of Dunning rat R3327 cells, and was isolated by serial transplantation in Copenhagen rats. All cells were grown at 37°C in a humidified atmosphere with 5% CO2.
shRNA plasmids to knockdown human and mouse AZI were obtained from Origene. Each shRNA set contained empty vector pRS plasmid, non-effective shRNA to knockdown GFP, or 4 individual shRNA sequences to knockdown AZI. shRNA plasmids were tested for knockdown efficacy in each cell line, and the two shRNA sequences which gave the greatest degree of knockdown were chosen for further experiments. For human PC3M-LN4 cells, shAZI H81 and shAZI H84 which both target human AZI were selected. For rat AT6.1 cells shAZI H83 and shAZI M35 were chosen since they target regions of human and mouse AZI, respectively, that are conserved in the rat gene. A summary of the shRNA sequences used is shown in Table 1.
Table 1.
shRNA sequences used to knockdown AZI and GFP.
| Target Gene | shRNA | Sequence (5' - 3') |
|---|---|---|
| Human AZI | shAZIH81 | GAACTGTAGGCATCTCTTGGAATGTGCTA |
| shAZI H83 | AGGAAGATGAGCCTCTGTTTACAAGCAGC | |
| shAZI H84 | TGCAAGATGCTGGAATTACTTCAGACTCA | |
| Mouse AZI | shAZI M35 | TGTGCCAAGGAACTTGATGTCCAAATAAT |
| GFP | shGFP | TGACCACCCTGACCTACGGCGTGCAGTGC |
To make stable cell lines, cells were plated in 6 well plates (4×105 cells/well for PC3M-LN4, and 2.5×105 cells/well for AT6.1) and transfected the following day with pRS, shGFP, and shAZI plasmids using Lipofectamine 2000 (Invitrogen), according to manufacturer's protocol (1μg shRNA/well and 5μl Lipofectamine). The following day selection media containing either 2μg/ml puromycin (PC3M-LN4 cells) or 26μg/ml puromycin (AT6.1 cells) was added. Following two weeks in selection media, stable non-clonal pools were established.
To compare rates of cell growth in vitro, PC3M-LN4 and AT6.1 cells stably expressing pRS control vector, shGFP control vector, or shAZI plasmids were plated at 1×104 cells/well in 6 well plates. Each day, duplicate wells of each cell line were collected and cells were counted using a TC10 automated cell counter (Bio-Rad). Cells plated in remaining wells were given new media every 2–3 days for the duration of the experiment.
Western Blot Analysis
Protein lysates were made using MPER (Mammalian Protein Extraction Reagent, Pierce ThermoScientific) containing complete mini protease inhibitor cocktail (Roche), according to manufacturer's protocols. Protein concentrations were measured using the BCA Assay (Pierce, ThermoScientific). Equal protein volumes were loaded onto polyacrylamide gels, and transferred to 0.2μm nitrocellulose using the Semi-Dry transfer system (Bio-Rad). The following antibodies were used for western analysis: mouse monoclonal to AZI (1:1000, clone HI-12, CosmoBio Co., LTD), rabbit polyclonal to ODC (1:500, 16061, Progen Biotechnik), rabbit polyclonal to AZ (1:250, Mitchell) and mouse monoclonal to Actin (1:10,000, Clone AC-15, Sigma). HRP-conjugated secondary antibodies were obtained from GE Healthcare (1:10,000). All western blots were developed with ECL Plus reagent (GE Healthcare), following manufacturer's directions.
qRT-PCR
RNA samples from various cell lines were harvested using RNeasy Mini kits (Qiagen), and 0.5μg of each RNA sample was used for cDNA synthesis with the Superscript III First-Strand Synthesis System (Invitrogen). Real time quantitative PCR reactions were then performed using 1μl cDNA and iQ SYBR Green Supermix (Bio-Rad) on an Opticon 2 instrument (Bio-Rad). The following cycling conditions were used: 95°C 3 min followed by 45 cycles of 95°C 15 sec, 60°C 15 sec, 72°C 30 sec, 77°C 1 sec plate read, followed by 72°C 5 min, melt curve from 65 to 98°C read every 0.2°C, 72°C 5 min, 10°C 5 min. Data points were collected using the MJ Opticon Monitor 3.1 program (Bio-Rad) and analyzed using the ΔCt method (Pfaffl 2001). Expression of beta-2-microglobulin was used as a control. Primer sequences used for qRT-PCR include the following: AZI Forward 5'- ATGTGTGTTTGACATGGCTGGAG -3'; AZI Reverse 5' – GAGGCTCATCTTCCTTGTATTTCTTG -3'; B2M Forward 5' – CAATCCAAATGCGGCATCTTCAAAC -3'; B2M Reverse 5' – GAATGGAGAGAGAATTGAAAAAGTGGAGCA -3'.
Immunofluorescence
Cells were plated at 2×104 cells/well onto autoclaved glass coverslips previously coated with 10μg/ml fibronectin solution (BD Biosciences). Cells were fixed with cold methanol at −20° for 10 minutes, and permeabilized with a solution of 0.25% TritonX-100 in PBS for 10 minutes. After blocking for 1 hr at room temperature in 1% normal goat serum (Invitrogen)/PBS, cells were incubated in primary antibody. Antibodies used include mouse monoclonal to AZI (1:500, Cosmo Bio Co., LTD) and rabbit polyclonal to gamma-tubulin (1:500, T5192, Sigma). Cells were stained with primary antibody for 1 hour at room temperature, washed with PBS, and stained with secondary antibodies for 1 hour at room temperature. AlexaFluor-488 and -568 conjugated secondary antibodies were obtained from Invitrogen Molecular Probes, and used at a dilution of 1:200. Cells were stained with a solution of 150nM DAPI for 2 minutes prior to mounting on microscope slides. Mounting media was made containing 20mM Tris (pH 8.0), 0.5% N-propylgallate (Sigma-Aldrich), and 90% glycerol. Immunofluorescence images were obtained with a Nikon Fluorescence microscope, a Nikon Plan Apo VC 100× oil objective, and a CCD camera. Focal Check slides (Invitrogen) were used to correct microscope registration errors and for calibration.
Animal Experiments
For xenograft studies, stable PC3M-LN4 and AT6.1 cells expressing shGFP, or two individual shRNAs to knockdown AZI were injected into the hind flank of 8-week old male athymic (nu/nu) nude mice (Massachusetts General Hospital). A total of 10 mice were used for each cell line. All cells were injected at 1×106 cells/mouse either with (PC3M-LN4) or without (AT6.1) Matrigel (BD Biosciences). Following injection, tumor growth was measured twice per week by caliper. Tumor volume was calculated using the equation 1/2 × (length × width2). At the end of the experiment, all animals were humanely euthanized, tumors were excised, and tumor weights were measured. All animals were kept in specific pathogen-free housing and given abundant food and water under guidelines approved by the Children's Hospital Boston Animal Care and Use Committee.
SAGE and Oncomine analysis
Expression of AZI transcripts in normal vs. cancer tissue was analyzed using the SAGE (Serial Analysis of Gene Expression) Digital Northern from the National Cancer Institute's Cancer Genome Anatomy Project (CGAP) at the website http://cgap.nci.nih.gov/SAGE. For AZI, the SAGE Genie program assigned the following unique identifying sequence: TCTTTCACCC from the 3' UTR. The SAGE library database was then analyzed for the number of AZI-specific hits for every 200,000 hits in the library. AZI expression was directly compared between normal prostate epithelial tissue and prostate carcinoma (PC3).
AZI expression in tumor tissue was also analyzed using the Oncomine database (http://www.oncomine.org) and the filter “Cancer vs. Normal Analysis” to detect changes in DNA copy number.
Statistical Analysis
Statistical analysis was performed using one-way analysis of variance (ANOVA) or Kruskal –Wallis one way analysis of variance tests in SPSS. Where appropriate, results were analyzed using 2-tailed student's t-test in Microsoft Excel 2003 at the 95% confidence level. If initial results showed that a statistically significant difference was detected between various groups, post-hoc tests were used to identify which groups gave significantly different results. In each case, the Tukeys HSD (Honestly Significant Difference) statistic was determined, and the corresponding p value was reported. In all cases, p <.05 was considered statistically significant.
Results
Expression of AZI in Prostate Cancer
Our laboratory has previously shown that manipulating the levels of AZI in numerous prostate cancer cell lines has a direct effect on rates of cell proliferation (Newman et al. 2004; Kim et al. 2006), and cells possessing a higher AZI:AZ ratio have increased rates of cell growth. To explore whether AZI levels are elevated in prostate cancer, we analyzed SAGE (Serial Analysis of Gene Expression) data and data from Oncomine. As shown in Fig. 1a, AZI levels were significantly elevated (p=.011) in samples from prostate carcinoma as compared to normal prostate epithelium. We also used Oncomine to determine if AZI copy number was altered in various cancers. This analysis showed that AZI copy number is elevated in cancers of the breast and bone marrow, and may also be slightly elevated in prostate cancer (Fig. 1b). Based on our laboratory's long-term interest in prostate cancer, we chose to examine the role of AZI in two aggressive prostate cancer cell lines: human PC3M-LN4 cells and rat AT6.1 cells
Fig. 1.
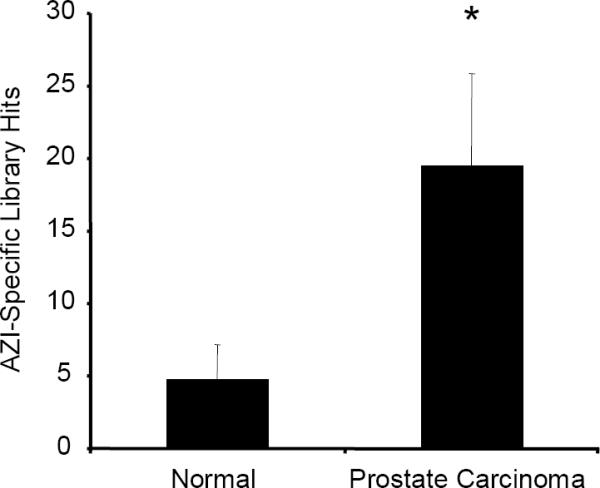
SAGE and Oncomine Analysis of AZI Expression in Normal Tissue versus Prostate Carcinoma
Using the unique AZI identifying tag TCTTTCACCC, AZI expression in normal prostate epithelium was compared to levels in prostate carcinoma using SAGE (Serial Analysis of Gene Expression). As shown above, AZI levels were significantly elevated in samples from prostate carcinoma (*, p=.011).
In vitro characterization of AZI Knockdown Cell Lines
PC3M-LN4 cells were transfected with control shRNA plasmids (empty vector pRS, shGFP), or two different shRNAs to knockdown AZI (shAZI H81, shAZI H84), and stable pools were generated. As shown by western blot, expression of either AZI shRNA led to a substantial decrease in protein levels of both AZI and ODC (Fig. 2a), although the resulting effect on cellular polyamine levels was not determined. These results were confirmed at the mRNA level by quantitative real-time PCR (qRT-PCR), as shown in Fig. 2b. To determine if AZI knockdown led to a change in proliferation rates, we measured cell growth over a period of 7 days (Fig 2c). Knockdown of AZI with shAZI H81 led to a 23.6% reduction in cell growth, and knockdown with shAZI H84 resulted in a 31.4% reduction in vitro as compared to control cells expressing shGFP.
Fig. 2.

In vitro Characterization of PC3M-LN4 shAZI Stable Cell Lines
a Western blot analysis of human PC3M-LN4 cells expressing empty vector control pRS, control shRNA to knockdown GFP, or two shRNAs to knockdown AZI (shAZI H81 and shAZI H84). AZI levels were substantially reduced in both shAZI cell lines compared to control vectors. Knockdown of AZI was also associated with a decrease in levels of ODC protein. Levels of AZ appeared to be slightly elevated in shAZI H84 cells compared to controls. b Quantitative real-time PCR (qRT-PCR) analysis shows decreased levels of AZI mRNA in both shAZI knockdown cell lines compared to control cells. Significance was determined by one-way ANOVA (p=.047). c Comparison of cell proliferation rates between control and AZI knockdown cells after 7 days in culture. shAZI H81 cells had approximately a 24% reduced rate of cell growth compared to control cell lines, while a 31% reduction was observed in shAZI H84 cells. Representative results are shown from three independent experiments. (ANOVA, *, p<.05). d Immunofluorescence analysis of AZI levels in control pRS and shAZI H84 cells. Cells were stained with antibodies to AZI and gamma-tubulin, and counterstained with DAPI. shAZI H84 cells had decreased cytoplasmic AZI staining, but still retained a portion of AZI protein at the centrosome, as shown by staining for the centrosome marker gamma-tubulin. Bar = 10μm
We further examined AZI levels in our shRNA cell lines by immunofluorescence microscopy (Fig. 2d). Cytoplasmic AZI staining was reduced in both cell lines expressing shAZI, as compared to control shRNA, and representative images from pRS control cells and shAZI H84 knockdown cells are included. As we and others have previously reported, a portion of endogenous AZI localizes to the centrosome in several different cell lines (Mangold et al. 2008; Murakami et al. 2009). We observed that a portion of the endogenous AZI protein remained localized to the centrosome in all of our PC3M-LN4 stable cell lines, as seen by colocalization with the centrosome marker gamma-tubulin.
To extend these results to a second cell line, we made rat prostate AT6.1 cells stably expressing shRNA control plasmids (empty vector pRS, shGFP) or shAZI plasmids which target regions conserved in rat AZI protein (shAZI H83, shAZI M35). As shown by western blot in Fig. 3a, AZI knockdown with plasmid H83 led to decreased AZI protein levels, and a slight decrease was also observed with shAZI M35 as compared to control cells. Although only a slight decrease in AZI protein was observed with shAZI M35, qRT-PCR analysis confirmed that both shRNA sequences were effective at decreasing AZI mRNA levels (Fig. 3b). When growth rates of AT6.1 control and shAZI cells were compared, shAZI H83 cells had a 24.4% decrease in cell proliferation and shAZI M35 cells showed a 17.0% decrease compared to shGFP cells after 6 days in culture.
Fig. 3.

In vitro Characterization of AT6.1 shAZI Stable Cell Lines
a Rat AT6.1 cells stably expressing control (pRS, shGFP) or AZI shRNA (shAZI H83, shAZI M35) were analyzed by western blot for AZI levels. shAZI H83 cells had the greatest degree of knockdown, and slight knockdown was also observed with shAZI M35. No substantial changes were observed in AZ levels. b qRT-PCR analysis confirms decreased AZI levels in both shAZI cell lines compared to shGFP control cells (ANOVA, p=.044). c Following 6 days of growth in culture, AT6.1 shAZI H83 cells had approximately a 24% reduced rate of cell growth compared to control cell lines, while a 17% decrease was observed in shAZI M35 cells. (ANOVA, p=.042)
Knockdown of AZI Decreases Tumor Growth in vivo
To determine whether cellular AZI levels are correlated with the ability of cells to promote tumor growth in vivo, we performed xenograft studies. shGFP control cells and two shAZI cell lines were implanted subcutaneously into nude mice. Results from the PC3M-LN4 shAZI cells are summarized in Fig. 4. AZI knockdown with either shRNA led to decreased tumor growth compared to control shGFP cells (Fig. 4a). shAZI H81 cells showed a 56.1% decrease in tumor growth in vivo, while shAZI H84 cells had a 43.5% decrease. This decrease in tumor size in shAZI cells also correlated with a significant decrease in end-tumor weights (shAZI H81, p=.031) (Fig. 4b). Similar results were observed with AT6.1 cells stably expressing shAZI. AT6.1 cells expressing shAZI H83 had a 54.9% reduction in tumor growth compared to shGFP cells, and tumor growth in shAZI M35 cells was decreased by 35.8% (Fig. 5a). As before, the decrease in tumor growth correlated with a decrease in tumor weight at the end of the experiment (shAZI H83, p=.011) (Fig. 5b). We are currently analyzing tissue samples from shAZI and shGFP tumors to determine if AZI knockdown results in significant changes in cellular polyamine content. We are also currently analyzing whether AZI knockdown affects either microvessel density or cell invasiveness.
Fig. 4.
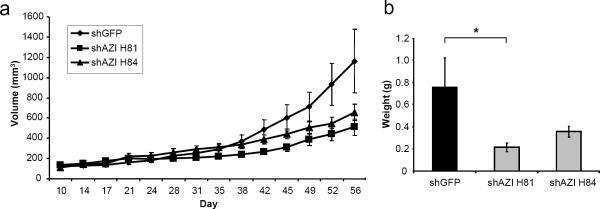
Knockdown of AZI in PC3M-LN4 Cells Decreases Tumor Growth in vivo
a Human prostate cancer cells stably expressing either control shGFP or shAZI were injected subcutaneously into nude mice. 1×106 cells were injected in a 1:1 mix with Matrigel, and ten animals were used for each cell line. Calipers were used to measure tumor size twice per week for two months, and tumor volume was calculated as previously described. As shown above, knockdown of AZI with shAZI H81 led to a 56% decrease in tumor growth in vivo, and knockdown with shAZI H84 resulted in a 44% decrease in tumor growth compared to control cells. b At the end of the experiment, tumors from each group were excised and weighed. Decreased growth of shAZI tumors was also detected as a decrease in end-tumor weight. shAZI H81 tumors had significantly decreased tumor weights (*, p=.031) compared to shGFP control cells
Fig. 5.
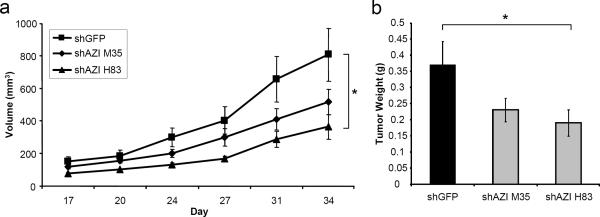
Knockdown of AZI in AT6.1 Cells Decreases Tumor Growth in vivo
a Rat AT6.1 cells stably expressing either shGFP control of shAZI (shAZI H83, shAZI M35) were injected subcutaneously in nude mice for xenograft studies. For each cell line, 1×106 cells resuspended in Hank's balanced salt solution (HBSS) were injected, and a total of ten mice were used in each experimental group. Tumor size was measured by caliper, and tumor volume was calculated. AT6.1 cells expressing shAZI H83 had a 55% reduction in tumor growth in vivo, and shAZI M35 cells had a 36% reduction in tumor growth, as compared to control cells (ANOVA, p=.032). b Knockdown of AZI in AT6.1 cells also led to significantly decreased end-tumor weights (*, p=.011)
Discussion
Despite being cloned and sequenced over ten years ago (Murakami et al. 1996), very little is known about the functions of AZI outside of its role in polyamine biosynthesis. Like ODC, high levels of AZI promote increased polyamine production and increased cell growth, and a high intracellular ratio of AZI:AZ within the cell favors cell proliferation. Numerous lines of evidence indicate that AZI levels are elevated in various cancers, particularly prostate cancer, and our objective was to better understand how AZI contributes to neoplastic transformation. To date, only a few studies have addressed the role of AZI in vivo, and many of these reports focus on expression of AZI during normal development (Murakami et al. 2010). Attempts to make AZI knockout mice established that homozygous AZI deletion results in neonatal lethality (Tang et al. 2009), and AZI−/− embryos are currently being characterized to better understand normal AZI function (Wan et al. 2010).
The goal of this study was to explore the contribution of AZI to neoplastic transformation by determining whether AZI has a functional role in promoting prostate tumor growth in vivo. The experimental approach we chose was to make cells stably expressing AZI shRNA, and then characterize these cells with regard to cell proliferation rates in vitro, and subcutaneous tumor growth rates in vivo.
We found that expression of AZI shRNA in PC3M-LN4 cells significantly decreased both AZI protein and mRNA levels. As expected, depletion of intracellular AZI resulted in decreased rates of cell proliferation in vitro. When PC3M-LN4 shAZI cells were used for xenograft experiments, both cell lines expressing AZI shRNA had substantially reduced tumor growth in vivo. Intriguingly, knockdown of AZI had a much greater effect on tumor growth in vivo, than on in vitro cell proliferation. In PC3M-LN4 cells, tumor growth in shAZI cell lines was reduced by an additional 1.39–2.38 fold compared to the in vitro data. This suggests that conditions in the tumor microenvironment may better support the growth of cells expressing high levels of AZI. One common way in which the microenvironment can influence tumor growth is through the balance between angiogenesis and hypoxic stress, and hypoxic conditions have recently been shown to directly influence polyamine levels. Culturing HeLa cells under hypoxic conditions in vitro enhanced polyamine uptake, increased ODC expression, and helped to protect cells against hypoxic stress (Svensson et al. 2008). Interestingly, this effect was dependent on expression of AZI, since cells treated with AZI siRNA no longer had increased polyamine levels after hypoxic treatment. It is possible that high levels of AZI in a developing tumor allow it to better overcome hypoxic stress, resulting in increased tumor growth in vivo. Our current studies analyzing microvessel density in shGFP compared to shAZI tumors will help us to determine how AZI knockdown affects angiogenesis in this model.
To validate these results, we repeated these experiments in a second prostate cancer cell line, rat AT6.1 cells. Although AZI expression was not inhibited to the same degree as in the PC3M-LN4 cells, AT6.1 shAZI cells did exhibit both decreased cell proliferation in vitro, and significantly decreased subcutaneous tumor growth in vivo. Tumor growth in AT6.1 shAZI cell lines was reduced by an additional 2.11–2.25 fold compared to the in vitro data.
Historically, much of the focus on inhibiting the polyamine pathway in cancer has been centered on preventing ODC activity, (Shantz and Levin 2007; Tsuji et al. 1998; Meyskens and Gerner 1999; Basuroy and Gerner 2006). We believe, however, that targeting AZI may prove to be a more useful therapeutic approach for tumor treatment. The reasons for this are two-fold. First, due to such tight control of polyamine homeostasis, AZI is in a position to regulate flux through the entire polyamine pathway. Classically, drug targets are generally enzymes, but even though AZI has no enzymatic activity our results and previously published data have clearly shown that modulating AZI levels has a direct effect on both cell proliferation and polyamine levels. Second, AZI can promote cell proliferation not only through the polyamine pathway, but also by preventing AZ-mediated degradation of cyclin D1 (Newman et al. 2004), as well as other proteins implicated in cell cycle regulation (Gruendler et al. 2001; Lim and Gopalan 2007). Therefore, any strategy designed to inhibit AZI would likely result in affecting cell proliferation through both the polyamine pathway and through normal cell cyclin levels. Our preliminary data suggests that levels of cyclin D1 are slightly decreased in PC3M-LN4 shAZI cells compared to shGFP controls, and this may contribute to the decrease in cell proliferation we observed.
One of the limitations associated with using DFMO to inhibit ODC, is that although DFMO can prevent cells from synthesizing their own intracellular polyamines, it does not prevent cells from taking polyamines up from the microenvironment. In contrast, inhibiting AZI may actually release AZ that could inhibit polyamine uptake. In the future, we plan to directly analyze polyamine uptake rates in our shGFP cells compared to the shAZI knockdown cell lines. The pathway involved in polyamine uptake in mammalian cells has not yet been fully characterized, and identifying the proteins involved could have a significant impact on the design of current and future therapeutics (Palmer and Wallace 2010).
In the future, we plan to use conditional knockdown models to further explore the function of AZI in tumor growth. One of the best models for these experiments would be to examine tumor growth in a genetically engineered mouse with conditional AZI knockdown. Our results showing that AZI knockdown can decrease prostate tumor growth in vivo add to the growing data that the AZI/AZ pathway is a critical component involved in modulating cell proliferation and cell transformation. Furthermore, this data suggests that inhibiting AZI in neoplastic tissues may be beneficial for treatment of prostate cancer.
Acknowledgements
The authors wish to thank Dr. Isaiah Fidler, University of Texas MD Anderson Cancer Center, for providing PC3M-LN4 cells, and Dr. John Isaacs, John Hopkins University, for providing AT6.1 cells. Funding for these studies was provided by grant CA037393 from the National Institutes of Health.
Abbreviations
- ANOVA
one-way analysis of variance
- AT6.1
Rat prostate cancer cells
- AZ
Antizyme
- AZI
Antizyme inhibitor
- BE-4-4-4-4
1,19-di-(ethylamino)-5,10,15-triazononadecane
- DAPI
4,6-diamidino-2-phenylindole
- DENSPM
N1,N11-di(ethyl)norspermine
- DFMO
Difluoromethylornithine
- GFP
Green Fluorescent Protein
- HBSS
Hank's Balanced Salt Solution
- ODC
Ornithine Decarboxylase
- PC3M-LN4
Human prostate cancer cells
- qRT-PCR
Quantitative Real-Time PCR
- SAGE
Serial Analysis of Gene Expression
- shRNA
short hairpin RNA
Footnotes
The authors declare that they have no conflict of interest.
References
- Agostinelli E, Marques MP, Calheiros R, Gil FP, Tempera G, Viceconte N, Battaglia V, Grancara S, Toninello A. Polyamines: fundamental characters in chemistry and biology. Amino Acids. 2010;38(2):393–403. doi: 10.1007/s00726-009-0396-7. doi:10.1007/s00726-009-0396-7. [DOI] [PubMed] [Google Scholar]
- Auvinen M, Paasinen A, Andersson LC, Holtta E. Ornithine decarboxylase activity is critical for cell transformation. Nature. 1992;360(6402):355–358. doi: 10.1038/360355a0. doi:10.1038/360355a0. [DOI] [PubMed] [Google Scholar]
- Bachrach U. Polyamines and cancer: minireview article. Amino Acids. 2004;26(4):307–309. doi: 10.1007/s00726-004-0076-6. doi:10.1007/s00726-004-0076-6. [DOI] [PubMed] [Google Scholar]
- Bailey HH, Kim K, Verma AK, Sielaff K, Larson PO, Snow S, Lenaghan T, Viner JL, Douglas J, Dreckschmidt NE, Hamielec M, Pomplun M, Sharata HH, Puchalsky D, Berg ER, Havighurst TC, Carbone PP. A randomized, double-blind, placebo-controlled phase 3 skin cancer prevention study of {alpha}-difluoromethylornithine in subjects with previous history of skin cancer. Cancer Prev Res (Phila) 2010;3(1):35–47. doi: 10.1158/1940-6207.CAPR-09-0096. doi:10.1158/1940-6207.capr-09-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basuroy UK, Gerner EW. Emerging concepts in targeting the polyamine metabolic pathway in epithelial cancer chemoprevention and chemotherapy. J Biochem. 2006;139(1):27–33. doi: 10.1093/jb/mvj022. doi:10.1093/jb/mvj022. [DOI] [PubMed] [Google Scholar]
- Choi KS, Suh YH, Kim WH, Lee TH, Jung MH. Stable siRNA-mediated silencing of antizyme inhibitor: regulation of ornithine decarboxylase activity. Biochem Biophys Res Commun. 2005;328(1):206–212. doi: 10.1016/j.bbrc.2004.11.172. doi:10.1016/j.bbrc.2004.11.172. [DOI] [PubMed] [Google Scholar]
- Coffino P. Antizyme, a mediator of ubiquitin-independent proteasomal degradation. Biochimie. 2001a;83(3–4):319–323. doi: 10.1016/s0300-9084(01)01252-4. [DOI] [PubMed] [Google Scholar]
- Coffino P. Regulation of cellular polyamines by antizyme. Nat Rev Mol Cell Biol. 2001b;2(3):188–194. doi: 10.1038/35056508. doi:10.1038/35056508. [DOI] [PubMed] [Google Scholar]
- Gerner EW, Meyskens FL., Jr. Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer. 2004;4(10):781–792. doi: 10.1038/nrc1454. doi:10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- Gruendler C, Lin Y, Farley J, Wang T. Proteasomal degradation of Smad1 induced by bone morphogenetic proteins. J Biol Chem. 2001;276(49):46533–46543. doi: 10.1074/jbc.M105500200. doi:10.1074/jbc.M105500200. [DOI] [PubMed] [Google Scholar]
- Jung MH, Kim SC, Jeon GA, Kim SH, Kim Y, Choi KS, Park SI, Joe MK, Kimm K. Identification of differentially expressed genes in normal and tumor human gastric tissue. Genomics. 2000;69(3):281–286. doi: 10.1006/geno.2000.6338. doi:10.1006/geno.2000.6338. [DOI] [PubMed] [Google Scholar]
- Kahana C, Asher G, Shaul Y. Mechanisms of protein degradation: an odyssey with ODC. Cell Cycle. 2005;4(11):1461–1464. doi: 10.4161/cc.4.11.2115. [DOI] [PubMed] [Google Scholar]
- Keren-Paz A, Bercovich Z, Kahana C. Antizyme inhibitor: a defective ornithine decarboxylase or a physiological regulator of polyamine biosynthesis and cellular proliferation. Biochem Soc Trans. 2007;35(Pt 2):311–313. doi: 10.1042/BST0350311. doi:10.1042/bst0350311. [DOI] [PubMed] [Google Scholar]
- Keren-Paz A, Bercovich Z, Porat Z, Erez O, Brener O, Kahana C. Overexpression of antizyme-inhibitor in NIH3T3 fibroblasts provides growth advantage through neutralization of antizyme functions. Oncogene. 2006;25(37):5163–5172. doi: 10.1038/sj.onc.1209521. doi:10.1038/sj.onc.1209521. [DOI] [PubMed] [Google Scholar]
- Kim SW, Mangold U, Waghorne C, Mobascher A, Shantz L, Banyard J, Zetter BR. Regulation of cell proliferation by the antizyme inhibitor: evidence for an antizyme-independent mechanism. J Cell Sci. 2006;119(Pt 12):2583–2591. doi: 10.1242/jcs.02966. doi:10.1242/jcs.02966. [DOI] [PubMed] [Google Scholar]
- Lan L, Trempus C, Gilmour SK. Inhibition of ornithine decarboxylase (ODC) decreases tumor vascularization and reverses spontaneous tumors in ODC/Ras transgenic mice. Cancer Res. 2000;60(20):5696–5703. [PubMed] [Google Scholar]
- Levin VA, Hess KR, Choucair A, Flynn PJ, Jaeckle KA, Kyritsis AP, Yung WK, Prados MD, Bruner JM, Ictech S, Gleason MJ, Kim HW. Phase III randomized study of postradiotherapy chemotherapy with combination alpha-difluoromethylornithine-PCV versus PCV for anaplastic gliomas. Clin Cancer Res. 2003;9(3):981–990. [PubMed] [Google Scholar]
- Li X, Coffino P. Regulated degradation of ornithine decarboxylase requires interaction with the polyamine-inducible protein antizyme. Mol Cell Biol. 1992;12(8):3556–3562. doi: 10.1128/mcb.12.8.3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SK, Gopalan G. Antizyme1 mediates AURKAIP1-dependent degradation of Aurora-A. Oncogene. 2007;26(46):6593–6603. doi: 10.1038/sj.onc.1210482. doi:10.1038/sj.onc.1210482. [DOI] [PubMed] [Google Scholar]
- Mangold U. Antizyme inhibitor: mysterious modulator of cell proliferation. Cell Mol Life Sci. 2006;63(18):2095–2101. doi: 10.1007/s00018-005-5583-4. doi:10.1007/s00018-005-5583-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangold U, Hayakawa H, Coughlin M, Munger K, Zetter BR. Antizyme, a mediator of ubiquitin-independent proteasomal degradation and its inhibitor localize to centrosomes and modulate centriole amplification. Oncogene. 2008;27(5):604–613. doi: 10.1038/sj.onc.1210685. doi:10.1038/sj.onc.1210685. [DOI] [PubMed] [Google Scholar]
- Matsufuji S, Matsufuji T, Miyazaki Y, Murakami Y, Atkins JF, Gesteland RF, Hayashi S. Autoregulatory frameshifting in decoding mammalian ornithine decarboxylase antizyme. Cell. 1995;80(1):51–60. doi: 10.1016/0092-8674(95)90450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megosh L, Gilmour SK, Rosson D, Soler AP, Blessing M, Sawicki JA, O'Brien TG. Increased frequency of spontaneous skin tumors in transgenic mice which overexpress ornithine decarboxylase. Cancer Res. 1995;55(19):4205–4209. [PubMed] [Google Scholar]
- Meyskens FL, Jr., Gerner EW. Development of difluoromethylornithine (DFMO) as a chemoprevention agent. Clin Cancer Res. 1999;5(5):945–951. [PubMed] [Google Scholar]
- Meyskens FL, Jr., McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, Hawk E, Kelloff G, Lawson MJ, Kidao J, McCracken J, Albers CG, Ahnen DJ, Turgeon DK, Goldschmid S, Lance P, Hagedorn CH, Gillen DL, Gerner EW. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev Res (Phila) 2008;1(1):32–38. doi: 10.1158/1940-6207.CAPR-08-0042. doi:10.1158/1940-6207.capr-08-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi Z, Kramer DL, Miller JT, Bergeron RJ, Bernacki R, Porter CW. Human prostatic carcinoma cell lines display altered regulation of polyamine transport in response to polyamine analogs and inhibitors. Prostate. 1998;34(1):51–60. doi: 10.1002/(sici)1097-0045(19980101)34:1<51::aid-pros7>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Ichiba T, Matsufuji S, Hayashi S. Cloning of antizyme inhibitor, a highly homologous protein to ornithine decarboxylase. J Biol Chem. 1996;271(7):3340–3342. doi: 10.1074/jbc.271.7.3340. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Matsufuji S, Kameji T, Hayashi S, Igarashi K, Tamura T, Tanaka K, Ichihara A. Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature. 1992;360(6404):597–599. doi: 10.1038/360597a0. doi:10.1038/360597a0. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Suzuki J, Samejima K, Kikuchi K, Hascilowicz T, Murai N, Matsufuji S, Oka T. The change of antizyme inhibitor expression and its possible role during mammalian cell cycle. Exp Cell Res. 2009;315(13):2301–2311. doi: 10.1016/j.yexcr.2009.04.024. doi:10.1016/j.yexcr.2009.04.024. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Suzuki J, Samejima K, Oka T. Developmental alterations in expression and subcellular localization of antizyme and antizyme inhibitor and their functional importance in the murine mammary gland. Amino Acids. 2010;38(2):591–601. doi: 10.1007/s00726-009-0422-9. doi:10.1007/s00726-009-0422-9. [DOI] [PubMed] [Google Scholar]
- Newman RM, Mobascher A, Mangold U, Koike C, Diah S, Schmidt M, Finley D, Zetter BR. Antizyme targets cyclin D1 for degradation. A novel mechanism for cell growth repression. J Biol Chem. 2004;279(40):41504–41511. doi: 10.1074/jbc.M407349200. doi:10.1074/jbc.M407349200. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Melgarejo AB, Lyon RR, Mitchell MF. Polyamines as biomarkers of cervical intraepithelial neoplasia. J Cell Biochem Suppl. 1995;23:87–95. doi: 10.1002/jcb.240590912. [DOI] [PubMed] [Google Scholar]
- Palmer AJ, Wallace HM. The polyamine transport system as a target for anticancer drug development. Amino Acids. 2010;38(2):415–422. doi: 10.1007/s00726-009-0400-2. doi:10.1007/s00726-009-0400-2. [DOI] [PubMed] [Google Scholar]
- Pegg AE. Regulation of ornithine decarboxylase. J Biol Chem. 2006;281(21):14529–14532. doi: 10.1074/jbc.R500031200. doi:10.1074/jbc.R500031200. [DOI] [PubMed] [Google Scholar]
- Peralta Soler A, Gilliard G, Megosh L, George K, O'Brien TG. Polyamines regulate expression of the neoplastic phenotype in mouse skin. Cancer Res. 1998;58(8):1654–1659. [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaner ME, Davidson B, Skrede M, Reich R, Florenes VA, Risberg B, Berner A, Goldberg I, Givant-Horwitz V, Trope CG, Kristensen GB, Nesland JM, Borresen-Dale AL. Variation in gene expression patterns in effusions and primary tumors from serous ovarian cancer patients. Mol Cancer. 2005;4:26. doi: 10.1186/1476-4598-4-26. doi:10.1186/1476-4598-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaner ME, Ross DT, Ciaravino G, Sorlie T, Troyanskaya O, Diehn M, Wang YC, Duran GE, Sikic TL, Caldeira S, Skomedal H, Tu IP, Hernandez-Boussard T, Johnson SW, O'Dwyer PJ, Fero MJ, Kristensen GB, Borresen-Dale AL, Hastie T, Tibshirani R, van de Rijn M, Teng NN, Longacre TA, Botstein D, Brown PO, Sikic BI. Gene expression patterns in ovarian carcinomas. Mol Biol Cell. 2003;14(11):4376–4386. doi: 10.1091/mbc.E03-05-0279. doi:10.1091/mbc.E03-05-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shantz LM, Levin VA. Regulation of ornithine decarboxylase during oncogenic transformation: mechanisms and therapeutic potential. Amino Acids. 2007;33(2):213–223. doi: 10.1007/s00726-007-0531-2. doi:10.1007/s00726-007-0531-2. [DOI] [PubMed] [Google Scholar]
- Smith MK, Trempus CS, Gilmour SK. Co-operation between follicular ornithine decarboxylase and v-Ha-ras induces spontaneous papillomas and malignant conversion in transgenic skin. Carcinogenesis. 1998;19(8):1409–1415. doi: 10.1093/carcin/19.8.1409. [DOI] [PubMed] [Google Scholar]
- Svensson KJ, Welch JE, Kucharzewska P, Bengtson P, Bjurberg M, Pahlman S, Ten Dam GB, Persson L, Belting M. Hypoxia-mediated induction of the polyamine system provides opportunities for tumor growth inhibition by combined targeting of vascular endothelial growth factor and ornithine decarboxylase. Cancer Res. 2008;68(22):9291–9301. doi: 10.1158/0008-5472.CAN-08-2340. doi:10.1158/0008-5472.can-08-2340. [DOI] [PubMed] [Google Scholar]
- Tang H, Ariki K, Ohkido M, Murakami Y, Matsufuji S, Li Z, Yamamura K. Role of ornithine decarboxylase antizyme inhibitor in vivo. Genes Cells. 2009;14(1):79–87. doi: 10.1111/j.1365-2443.2008.01249.x. doi:10.1111/j.1365-2443.2008.01249.x. [DOI] [PubMed] [Google Scholar]
- Thompson PA, Wertheim BC, Zell JA, Chen WP, McLaren CE, LaFleur BJ, Meyskens FL, Gerner EW. Levels of rectal mucosal polyamines and prostaglandin E2 predict ability of DFMO and sulindac to prevent colorectal adenoma. Gastroenterology. 2010;139(3):797–805. 805, e791. doi: 10.1053/j.gastro.2010.06.005. doi:10.1053/j.gastro.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji T, Todd R, Meyer C, McBride J, Liao PH, Huang MF, Chou MY, Donoff RB, Wong DT. Reduction of ornithine decarboxylase antizyme (ODC-Az) level in the 7,12-dimethylbenz(a)anthracene-induced hamster buccal pouch carcinogenesis model. Oncogene. 1998;16(26):3379–3385. doi: 10.1038/sj.onc.1201887. doi:10.1038/sj.onc.1201887. [DOI] [PubMed] [Google Scholar]
- van Duin M, van Marion R, Vissers K, Watson JE, van Weerden WM, Schroder FH, Hop WC, van der Kwast TH, Collins C, van Dekken H. High-resolution array comparative genomic hybridization of chromosome arm 8q: evaluation of genetic progression markers for prostate cancer. Genes Chromosomes Cancer. 2005;44(4):438–449. doi: 10.1002/gcc.20259. doi:10.1002/gcc.20259. [DOI] [PubMed] [Google Scholar]
- Wan T, Hu Y, Zhang W, Huang A, Yamamura K, Tang H. Changes in liver gene expression of Azin1 knock-out mice. Z Naturforsch C. 2010;65(7–8):519–527. doi: 10.1515/znc-2010-7-816. [DOI] [PubMed] [Google Scholar]
- Zagaja GP, Shrivastav M, Fleig MJ, Marton LJ, Rinker-Schaeffer CW, Dolan ME. Effects of polyamine analogues on prostatic adenocarcinoma cells in vitro and in vivo. Cancer Chemother Pharmacol. 1998;41(6):505–512. doi: 10.1007/s002800050774. [DOI] [PubMed] [Google Scholar]
- Zell JA, Pelot D, Chen WP, McLaren CE, Gerner EW, Meyskens FL. Risk of cardiovascular events in a randomized placebo-controlled, double-blind trial of difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas. Cancer Prev Res (Phila) 2009;2(3):209–212. doi: 10.1158/1940-6207.CAPR-08-0203. doi:10.1158/1940-6207.capr-08-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]