

Abstract
Basic fibroblast growth factor (bFGF) offers some measure of protection against excitotoxic neuronal injuries by upregulating the expression of the calcium-binding protein calbindin-D28k (Calb). The newly synthesized small molecule 4-({4-[[(4-amino-2,3,5,6-tetramethylanilino)acetyl](methyl)amino]-1-piperidinyl}methyl)benzamide (SUN11602) mimics the neuroprotective effects of bFGF, and thus, we examined how SUN11602 exerts its actions on neurons in toxic conditions of glutamate. In primary cultures of rat cerebrocortical neurons, SUN11602 and bFGF prevented glutamate-induced neuronal death. This neuroprotection, which occurred in association with the augmented phosphorylation of the bFGF receptor-1 (FGFR-1) and the extracellular signal-regulated kinase-1/2 (ERK-1/2), was abolished by pretreatment with PD166866 (a FGFR-1 tyrosine kinase-specific inhibitor) and PD98059 (a mitogen-activated protein kinase [MAPK]/[ERK-1/2] kinase [MEK] inhibitor). In addition, SUN11602 and bFGF increased the levels of CALB1 gene expression in cerebrocortical neurons. Whether this neuroprotection was linked to Calb was investigated with primary cultures of cerebrocortical neurons from homozygous knockout (Calb–/–) and wild-type (WT) mice. In WT mice, SUN11602 and bFGF increased the levels of newly synthesized Calb in cerebrocortical neurons and suppressed the glutamate-induced rise in intracellular Ca2+. This Ca2+-capturing ability of Calb allowed the neurons to survive severe toxic conditions of glutamate. In contrast, Calb levels remained unchanged in Calb–/– mice after exposure to SUN11602 or bFGF, and due to a loss of function of the gene, these neurons were no longer resistant to toxic conditions of glutamate. These findings indicated that SUN11602 activated a number of cellular molecules (FGFR-1, MEK/ERK intermediates, and Calb) and consequently contributed to intracellular Ca2+ homeostasis as observed in the case of bFGF.
Keywords: SUN11602, basic fibroblast growth factor, neuroprotection, PD166866, calbindin-D28k
Basic fibroblast growth factor (bFGF), which is an 18 kDa protein, exhibits potent trophic support for the survival and neurite outgrowth of a variety of brain neurons.1−4 According to these studies, this neurotrophic activity appears to be very specific and characteristic to bFGF. The trophic activity of a closely related peptide, acidic FGF, was not as effective.4
The neuroprotective activity of bFGF has been investigated with different types of in vitro and in vivo experimental models. A research group conducted a series of in vitro studies and demonstrated protection of cultured neurons from cell death by suppressing abnormal increases in intracellular Ca2+ levels in a hypoglycemic or β-amyloid protein-mediated toxic model that was pretreated for 24 h with bFGF.5,6 These studies indicated that the neuroprotective mechanisms of bFGF were closely related to the mechanisms underlying intracellular Ca2+ metabolism. Another in vitro study showed that both glutamate-induced abnormalities in intracellular Ca2+ concentrations and subsequent neuronal death were abolished by maintaining the cultures in the presence of bFGF and that the neuroprotective activities of bFGF required new protein synthesis to antagonize the glutamate-induced increase in intracellular Ca2+ levels.7 The subsequent study from this research group indicated that the calcium-binding protein, calbindin-D28k (Calb), played an important role in the intracellular Ca2+ regulation of cultured neurons that were exposed to toxic levels of glutamate.8
In vivo experimental studies have been conducted on different animal models with a combination of different administration protocols. In a rat model of unilateral middle cerebral artery occlusion (MCAO), the neuroprotective activity of exogenous bFGF, which was infused continuously and intraventricularly for 3 days prior to the MCAO, was assessed through the reductions of infarct volume in the impaired cerebral hemisphere.9 After a 3 h intravenous (iv) infusion of exogenous bFGF starting 30 min after unilateral MCAO, the cerebral infarct volume in the bFGF-administered rats was reduced in size to about half that observed in control animals.10 Using the same type of rat model, comparative studies of therapeutic treatments have revealed that the early administration of exogenous bFGF reduces cerebral infarct volume, whereas late administration does not affect infarct volume but does improve the recovery of the motor functions of the impaired limbs.11,12 The neuroprotective activity of exogenous bFGF in these rat models has been confirmed in a MCAO cat model, in which the continuous iv infusion of exogenous bFGF was started 60 min after the MCAO and continued until the end of the 4 h experiment.13 According to in vivo studies, it is likely that early treatment with exogenous bFGF after brain insults can minimize the severity of tissue damage, and even delayed treatment (later than 24 h) can exert significant effects on the recovery of the motor and cognitive functions of the animals.11,12,14
One clinical trial of bFGF (Fiblast [trafermin]) for the treatment of acute stroke patients has been conducted, but it failed to demonstrate a notable efficacy that would support its therapeutic use.15 Although this clinical trial was suspended because of an interim analysis of the efficacy data, the investigators suggested that Fiblast (trafermin), when used according to the proper treatment regimen, might be given safely to stroke patients to result in some advantages in the course of their long-term recovery. Such a slight advantage over placebo in human cases may partly be supported by preclinical behavioral studies in MCAO rats, in which delayed treatment with exogenous bFGF (i.e., starting 24 h postsurgery with intracisternal infusion) improved several behavioral scores without reducing the infarct volume in the impaired hemispheres.16,17 According to a number of in vivo studies that included human cases, it is likely that the sooner animals/humans can receive treatment with exogenous bFGF after brain insults, the better their neurological and behavioral outcomes. The delayed treatment of brain insults with exogenous bFGF (even later than 24 h) can also exert significant effects on the recovery of the motor and cognitive functions of the animals/humans.11,12,14−17
In addition, the activities of endogenous bFGF in various brain structures have been reported following MCAO-induced infarctions.18 The levels of bFGF mRNA expression have been shown to be increased in astrocytes in the penumbra regions of focal brain infarcts, and the intensity of the immunoreactivity for endogenous bFGF became increasingly conspicuous as the postoperative days proceeded. These findings suggest that increased endogenous bFGF levels that are derived from astrocytes play an important role in damage-healing mechanisms, such as neuronal survival, glial proliferation, and the reconstruction of vasculature. Thus, the molecular functions of either exogenous or endogenous bFGF might have essential clues of the neuroprotective mechanisms and/or functional recoveries that are induced following various brain insults (e.g., ischemia, infarction, hemorrhage, and trauma). Thus, bFGF and its functional analogues deserve continued attention in order to develop effective medications and presumed therapeutic practices.
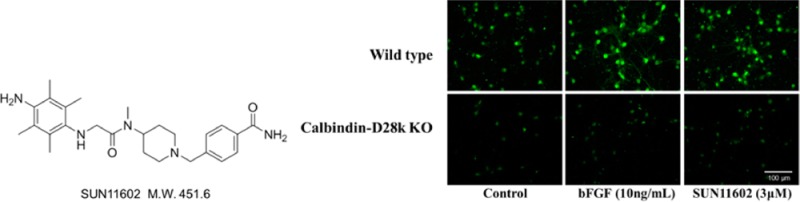
The present study was conducted on the effects on primary cultures of rat cerebrocortical neurons of a novel small synthetic compound, 4-({4-[[(4-amino-2,3,5,6-tetramethylanilino)acetyl](methyl)amino]-1-piperidinyl}methyl)benzamide (SUN11602; Figure 1 and Supporting Information), which stimulates several cellular events of neuroprotection that are induced by bFGF. It is worth noting that SUN11602 activated key molecules in the FGF receptor-1-mitogen-activated protein kinase/extracellular signal-regulated kinase-1/2 kinase (FGFR-1-MEK/ERK) signaling pathway for neuroprotection, and these molecules have been shown to be involved in neuroprotection in a number of previous studies that examined the natural peptide bFGF, which is physiologically active. In addition, the neuroprotective effectiveness of SUN11602 was quite comparable to that of exogenous bFGF.
Figure 1.

Chemical structure of 4-({4-[[(4-amino-2,3,5,6-tetramethylanilino)acetyl](methyl)amino]-1-piperidinyl}methyl)benzamide (SUN11602).
Results
Neuroprotective Cellular Events Induced by SUN11602 and bFGF
To assess the effectiveness of SUN11602 (Figure 1) and bFGF in rescuing neurons from excitotoxic cell death, we utilized 10-day cultures of rat cerebrocortical neurons that were exposed to 150 μM of glutamate. In control cultures, glutamate exposure resulted in cell death of greater than 50% in a (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H tetrazolium bromide (MTT) assay, but both SUN11602 (0.1, 0.3, and 1 μM) and bFGF (5 and 10 ng/mL), which were applied 1 day prior to the toxic injury, significantly reduced the rate of excitotoxic cell death in a concentration-dependent manner (Figure 2; Dunnett’s two-tailed test).
Figure 2.

Neuroprotective effects of SUN11602 and basic fibroblast growth factor (bFGF) against glutamate-induced toxicity (150 μM) in primary cultures of rat cerebrocortical neurons. The open bars reveal that the effectiveness of SUN11602 and bFGF is ascribed to unimpaired protein synthesis, by which they can activate their neuroprotective mechanisms. Their neuroprotective effects were abolished by treatment with either actinomycin D (1 μg/mL, black bar) or cycloheximide (1 μg/mL, gray bar), which can interfere with transcription or translation, respectively. Actinomycin D or cycloheximide was first added to the cultures, and, 2 h later, SUN11602 or bFGF was added. After a 24 h incubation, neurons in the cultures were exposed to 150 μM glutamate for another 24 h, and cell viability was determined by a (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H tetrazolium bromide (MTT) assay. ***p < 0.001 compared to glutamate alone (means ± SEM, n = 6, Dunnett’s multiple comparison test).
Because the neuroprotective mechanisms of bFGF have been assumed to be closely related to the mechanisms underlying intracellular Ca2+ metabolism and Ca2+-binding proteins,6−8 the neuroprotective effects of SUN11602 and bFGF that required de novo protein synthesis were investigated in the presence of actinomycin D (a transcription inhibitor) and cycloheximide (a protein synthesis inhibitor). The neuroprotective effects of SUN11602 and bFGF were abolished by pretreating the cultures of cortical neurons with the inhibitors (Figure 2). Thus, these results implied that the neuroprotective actions of SUN11602 involved newly synthesized protein(s), as was observed with bFGF, and that the nuclear signaling pathway involved in SUN11602 stimulation was indispensable for the neuroprotective mechanisms (i.e., transcription and translation).
Neuroprotection and Phosphorylation of the FGF Receptor by SUN11602 and bFGF
In order to investigate whether SUN11602 acts through the FGF signaling cascade, we examined the neuroprotective effects of SUN11602 in the presence of PD166866, which specifically inhibits the FGFR-1 tyrosine kinase. The neuroprotective effects of SUN11602 and bFGF were completely antagonized when PD166866 (0.3 μM) was added to the cultures of cortical neurons 30 min before the application of SUN11602 and bFGF (Figure 3A). PD166866, which is a specific inhibitor of the FGFR-1 tyrosine kinase (IC50 of approximately 52 nM),19 is regarded as being selective for but less effective on the tyrosine kinases of the platelet-derived growth factor receptor (PDGFR), the epidermal growth factor receptor (EGFR), the insulin-like growth factor-1 receptor (IGF1R), and others.20 It is, therefore, important to know the molecules that are directly affected in their intracellular signaling by PD166866.
Figure 3.

Different effects of PD166866 on the neuroprotective effects of SUN11602 and several growth factors against glutamate-induced toxicity in primary cultures of rat cerebrocortical neurons. (A) Neuroprotective effects of SUN11602 and bFGF were abolished by pretreating the cultures with PD166866 (0.3 μM). This indicates that SUN11602 and bFGF activate their neuroprotective mechanisms through the phosphorylation of the FGF receptor-1 (FGFR-1), although it has not yet been demonstrated whether the receptor is directly phosphorylated by SUN11602. (B) Other growth factors (nerve growth factor [NGF], brain-derived neurotrophic factor [BDNF], insulin-like growth factor-1 [IGF-1], neurotrophin-3 [NT-3], vascular endothelial growth factor-A [VEGF-A], heparin binding-EGF [HB-EGF], and platelet-derived growth factor [PDGF]) were investigated in order to discriminate the important factors in the intracellular signaling of SUN11602 and bFGF. Unlike bFGF, the bioactive effects of the other growth factors were not affected by PD166866. The neuroprotective mechanisms of SUN11602 and bFGF appear to differ from those of the other growth factors. The toxic concentrations of glutamate in the figures (150 or 75 μM) were employed compared to those that were suitable for the growth factors in order to show their neuroprotective effects clearly. *p < 0.05 and ***p < 0.001 compared to glutamate alone (means ± SEM, n = 6, Dunnett’s multiple comparison test).
The responses of Trk,21 IGF1R,22,23 EGFR,24 PDGFR,25 and vascular endothelial growth factor receptor (VEGFR)26 were investigated in order to clarify whether PD166866 inhibited neuroprotection as a result of the phosphorylation of the tyrosine kinase domains of these receptors. Because the binding of natural ligands to the corresponding receptors has been established to induce actions that protect vulnerable neurons, the inhibitory effects of PD166866 on these receptors were examined in the cultured cortical neurons by MTT assay (Figure 3B). When the natural ligands of these receptors were applied 1 day prior to the onset of glutamate toxicity, cultured cortical neurons were able to survive in the adverse conditions. However, unlike bFGF, the neuroprotective effects of the other growth factors were not affected in the presence of PD166866 (Figure 3B). As for VEGF and VEGFR, the neuroprotective effects were decreased slightly by PD166866 (Figure 3B). These results indicated that the inhibitory actions of PD166866 were not involved in the neuroprotection of these growth factors, implying that there are discrete intracellular signaling pathways for neuroprotection acting through these receptors, which differs from the pathway involving FGFR-1.
Because the phosphorylation of FGFR-1 by SUN11602 or bFGF is linked to the neuroprotection of endangered neurons in vitro, the phosphorylation of the cytosolic tyrosine kinase domain of the receptor must be indispensable for the initiation of the neuroprotective processes involved in the intracellular signaling of SUN11602 and bFGF. Furthermore, SUN11062 did not competitively disturb the binding of 125I-labeled bFGF to the extracellular domain of FGFR in in vitro binding assays of the cultures of brain neurons (hippocampus) and baby hamster kidney cells (BHK-21) that endogenously express FGFR (unpublished data). Thus, SUN11602 can either directly or indirectly trigger the phosphorylation of the cytosolic domain of the FGFR without binding to the extracellular domain of the FGFR-1, although how such phosphorylation occurs remains to be determined.
Neuroprotective Effects of SUN11602 and bFGF against Glutamate Toxicity through Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Phosphorylation
As for the neuroprotection described above, it is crucial to know what types of intracellular signaling pathways are directly associated with the survival of cultured cortical neurons in toxic environments. A survey of intracellular signaling molecules elucidated that extracellular signal-regulated kinase 1/2 (ERK1/2) was phosphorylated in neurons following stimulation with either SUN11602 (10 or 100 μM) or bFGF (10 ng/mL) for 20 min. Western blot analyses showed that the phosphorylation of ERK1/2 occurred without alterations in the protein levels of ERK1/2 (Figure 4A). Moreover, the involvement of MEK/ERK signaling cascades in the neuroprotection of SUN11602 and bFGF was established by inhibiting MEK’s activity with PD98059 (a MEK inhibitor). The activation of MEK/ERK was eliminated in a concentration-dependent manner by pretreating cultured cerebrocortical neurons with the inhibitor (Figure 4B).
Figure 4.
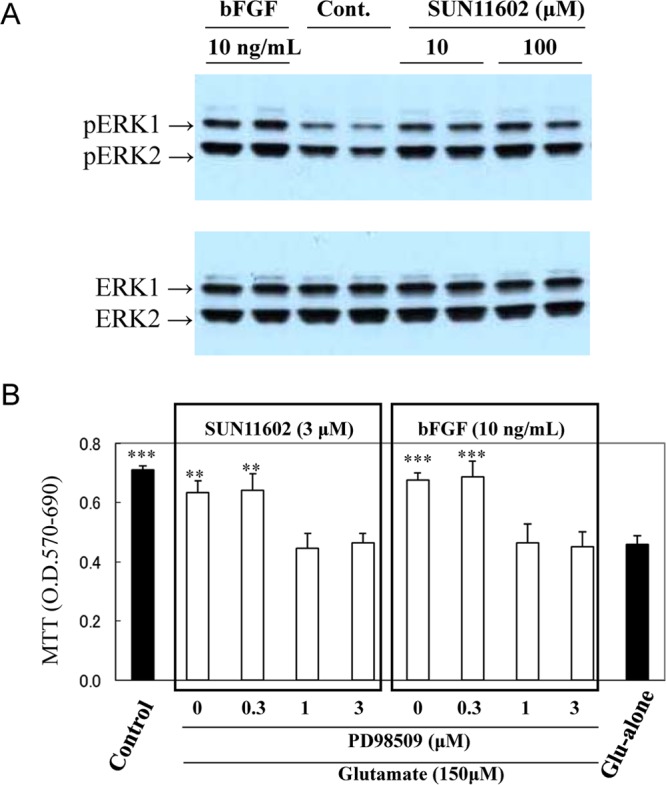
Phosphorylation of extracellular signal-regulated kinase-1/2 (ERK-1/2), which is necessary for the neuroprotection by SUN11602 and bFGF against glutamate-induced toxicity in primary cultures of rat cerebrocortical neurons. (A) Cultured cortical neurons express ERK1/2 proteins at a constant level in normal conditions (lower panel: ERK1, ERK2), whereas, after treatment with 10 or 100 μM SUN11602 and 10 ng/mL bFGF for 20 min, both ERK1/2 proteins of the neurons were phosphorylated substantially (upper panel: p-ERK1, p-ERK2) in comparison to the control lysates. The whole-cell lysates of the cultured neurons were subjected to 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and immunodetected with an antiphosphorylated-ERK1/2 antibody. (B) Inhibition by PD98059 on the neuroprotective effects of SUN11602 and bFGF. Impairment of the ERK kinase/ERK pathway resulted in eliminating the effects of SUN11602 and bFGF, implying that signaling through the pathway plays a pivotal role in activating the neuroprotective mechanisms. The cultures pretreated with PD98059 for 30 min were incubated with SUN11602 or bFGF for 24 h. Thereafter, neurons in the cultures were exposed to 150 μM glutamate for another 24 h, and cell viability was determined by the MTT assay. **p < 0.01, ***p < 0.001 compared to the glutamate group (means ± SEM, n = 6, Dunnett’s multiple comparison test).
Calbindin-D28k (Calb) Is a Requisite for Neuroprotection by SUN11602 and bFGF
In order to characterize the FGFR-1/Calb-mediated neuroprotection by SUN11602 and bFGF, the relationships of FGFR-1 and Calb were investigated using primary cultures of cerebral cortices from knockout (Calb–/–) and wild-type (WT) mice. The immunocytochemistry of the mouse brains clearly showed the gene deletion of the knockout mice (Figure 5A). The expression of the FGFR1 was confirmed immunocytochemically in cultured cortical neurons from Calb–/– and WT mice (Figure 5A). However, endogenous Calb was not detected immunocytochemically in either the cultured cortical neurons or brain tissues from Calb–/– mice (Figure 5A, B), whereas those from WT mice expressed Calb.
Figure 5.
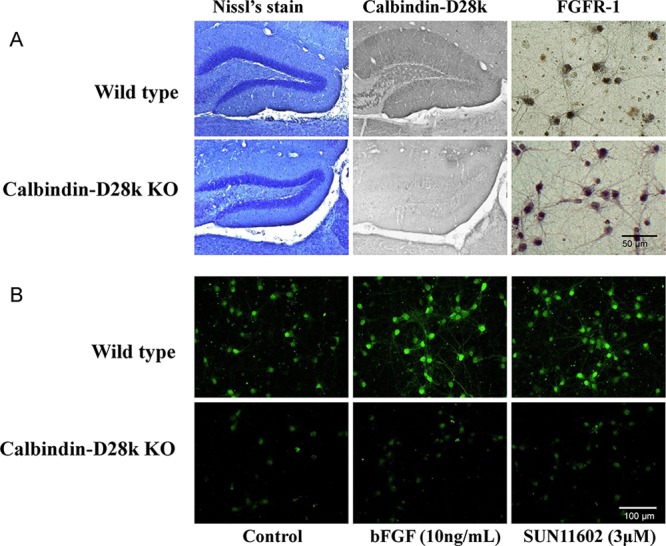
Immunocytochemical characterization of brain neurons from wild-type (WT) and homozygous calbindin-knockout (Calb–/–) mice. (A) Immunoreactivity to calbindin-D28k and fibroblast growth factor receptor-1 (FGFR-1) in neurons from WT and Calb–/– mice. In order to show the gene deletion of calbindin-D28k, typical brain tissue sections were employed for confirmation. Primary cultures from the brains of WT and Calb–/– mice displayed that they contained neurons with immunoreactive FGFR-1. (B) Increased levels of expression of calbindin-D28k by bFGF and SUN11602 in primary cultures of cerebrocortical neurons from WT and Calb–/– mice. Culture neurons that were grown on coverslips in the presence of 10 ng/mL bFGF or 3 μM SUN11602 for 48 h were fixed and immunostained with a calbindin-D28k antibody. Calbindin D28k-immunoreactivity was mostly eliminated from the cultures of cerebrocortical neurons from Calb–/– mice.
Most cortical neurons from Calb–/– mice were destined to cell death following stimulation with 100 μM glutamate. Because the toxicity of 100 μM glutamate appeared to be too severe for the cultured cortical neurons from Calb–/– mice, the strength of the toxic stimulation to these neurons was lowered to 50 μM glutamate, and the excitotoxic effects were then evaluated based on the rate of neuronal survival. The typical neuroprotective effects of SUN11602 and bFGF were abolished completely in cultured cortical neurons from Calb–/– mice in toxic environments of 50 μM glutamate (Figure 6). In contrast to the Calb–/– mice, the cultured cortical neurons from WT mice were more resistant to the toxic concentration of 100 μM of glutamate (Figure 6). SUN11602 and bFGF exerted protective actions on cultured cortical neurons from WT mice in a concentration-dependent manner, even when the neurons were subjected to the toxic conditions of 100 μM glutamate. The cultured cortical neurons exhibited increased levels of Calb, which regulates the concentration of intracellular free Ca2+.
Figure 6.

Neuroprotective effects of SUN11602 and bFGF against glutamate-induced toxicity in primary cultures of cerebrocortical neurons from WT and Calb–/– mice. These data indicate that calbindin-D28k in WT mice may play important roles in the neuroprotective mechanisms by SUN11602 and bFGF. In Calb–/– mice, the molecular function of calbindin-D28k was mostly diminished, although there was a slight improvement in neuroprotection with higher concentrations of SUN11602. Following the 24 h incubation with SUN11602 and bFGF, the cultures were exposed to either 100 or 50 μM glutamate for another 24 h, and cell viability was then determined by MTT assay. The toxic concentrations of glutamate (100 or 50 μM) were employed for which those concentrations were suitable for the cultures in order to react sensitively and appropriately. *p < 0.05, **p < 0.01, ***p < 0.001 compared to the glutamate group (means ± SEM, n = 6, Dunnett’s multiple comparison test).
In addition, it is worth noting that higher concentrations (1 and 3 μM) of SUN11602 were neuroprotective for cultured cortical neurons from Calb–/– mice in toxic conditions of glutamate, but bFGF was not (Figure 6). The intensity of the neuroprotection was reduced but effective compared to the corresponding results in neurons from WT mice (Figure 6). This may account for the possibility that SUN11602 can activate a given set of intracellular molecules other than those involved with Calb.
In experiments of intracellular free Ca2+ imaging, pretreatment with SUN11602 and bFGF suppressed the increase in the fluorescence levels of the free Ca2+ indicator (fluo 3-AM) in cultured cortical neurons from WT mice following stimulation with 100 μM glutamate for 5 min (Figure 7). In contrast, in cultured cortical neurons from Calb–/– mice, neither SUN11602 nor bFGF suppressed the glutamate-induced increase in intracellular Ca2+ levels. These data indicated that SUN11602 and bFGF were able to protect fragile neurons that were affected by glutamate toxicity from dying by maintaining intracellular Ca2+ homeostasis with the aid of increased intracellular Calb.
Figure 7.
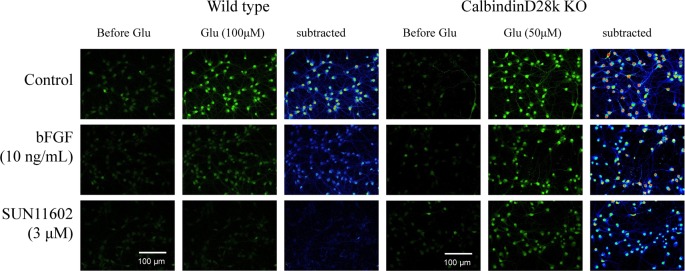
Intracellular Ca2+ overload in primary cultures of cerebrocortical neurons from WT and Calb–/– mice following a 5 min stimulation with glutamate (100 μM for WT, 50 μM for Calb–/–). Basic FGF and SUN11602 were added to the cultures 1 day before glutamate stimulation. The fluctuations in the intracellular Ca2+ levels in the cultures were photographed with a laser confocal microscope with fluorescence indicators of free Ca2+ (fluo 3-AM). These data suggest, along with Figure 5, that the physiological actions of bFGF and SUN11602 are closely associated with the regulators of intracellular Ca2+, especially calbindin-D28k. All micrographs were taken under the same conditions and at the same magnification for comparison.
Discussion
The present study provids evidence for the neuroprotective actions of the novel aniline compound SUN11602 on cultured cerebrocortical neurons of rats. It is, in particular, worth noting that the physiological actions of SUN11602 mimic several phenomena of the neuroprotection that is induced by bFGF. These actions appeared to correspond to the cellular events that occur along the MEK/ERK signaling pathway acting through FGFR-1, which involves the following steps: (a) phosphorylation of FGFR-1, (b) subsequent phosphorylation of ERK1/2, and (c) buffering action of intracellular free Ca2+ by newly synthesized Calb.
a. Phosphorylation of FGFR-1
The actions of SUN11602 started with the phosphorylation of the cytosolic tyrosine kinase domain of FGFR-1, which is expressed in the cultured cortical neurons. The phosphorylation of FGFR-1 is an inevitable step in the neuroprotective mechanisms of SUN11602 because PD166866 has been shown to abolish the receptor-mediated effects.19 However, SUN11602 does not obstruct the binding of 125I-labeled bFGF to the extracellular domain of FGFR-1 (unpublished data). These results imply that SUN11602 can either directly or indirectly trigger the phosphorylation of the cytosolic domain of FGFR-1 without acting on the extracellular bFGF-binding site. Besides the bFGF-binding site, little information is available on the other extracellular functional sites that stimulate the autophosphorylation of FGFR-1, resulting in its physiological actions.27,28 As for the activation of FGFR-1, it still remains to be solved how the cytosolic tyrosine kinase domain of the receptor is phosphorylated.
There are a number of growth factors that more or less affect the survival of brain neurons in adverse environments, either in vitro or in vivo.29 Of their receptors, Trk, IGF1R, EGFR, PDGFR, and VEGFR were investigated in the present study in order to discriminate the distinctiveness of the intracellular signaling of SUN11602 because these growth factor receptors have a more than 40% homology with the amino acid sequence of the tyrosine kinase domain of FGFR-1.30 Even after pretreatment with PD166866, the natural ligands of these receptors, other than FGFR-1, exerted neuroprotective effects on cultured cortical neurons in the same manner as they did with no inhibitor. A minor decrease in the survival rate of the cultured neurons was observed for VEGFR-mediated neuroprotection in the presence of PD166866. This finding suggested that the higher-order structure of the FGFR-1 tyrosine kinase domain at V559 and V661 (the ATP-binding pocket), where PD166866 interacts, resembles that of the VEGF receptor.30 Accordingly, neuroprotection elicited by the activation of FGFR-1 proves to be specific and distinctive to SUN11602 as well as to bFGF. Thus, it is assumed that SUN11602 can activate the intracellular signaling molecules that are involved in the neuroprotective mechanisms following the interactions between bFGF and FGFR-1.
b. Phosphorylation of ERK1/2
ERKs are members of the MAPK superfamily, which is concerned with diverse physiological responses in various cells, and play pivotal roles in the MAPK signaling cascade.31,32 ERK1/2 was phosphorylated intensely after stimulation by SUN11602 and bFGF without notable alterations in its protein levels. This activation in the MAPK signaling cascade appeared to be inevitably important in the protection of the targeted neurons against glutamate toxicity because pretreating the neurons with the MEK kinase inhibitor PD98059 eliminated the protective activities of SUN11602 and bFGF and resulted in the cell death of the affected neurons. A similar cellular response was noted in an experiment designed to generate multipotential stem cells from microglia.29 Basic FGF and the FGFR were involved in the generation of the stem cells, and bFGF stimulation increased the amount of phosphorylated ERK1/2 without altering total ERK1/2 levels,29 as was observed with SUN11602 and bFGF in the present study.
An in vivo study of the nerve regeneration and the neuronal survival of frog retinal ganglion neurons (RGN) showed that bFGF application to the amputated nerve stump prolonged the increase in ERK activation, causing an ERK-dependent increase in antiapoptotic proteins (Bcl-2 and Bcl-xL) and preventing the apoptosis of the axotomized RGN.30 In a rat model of ischemia, the MEK/ERK signaling pathway was implicated in the neuroprotection against sublethal ischemia and reperfusion injury.33 The sublethal ischemia induced an initial robust activation and then a rapid inactivation of ERKs in the sequence of which activated-Raf-1 (phosphorylation at Ser259 after ischemia) was inhibited by the phosphorylation of Raf-1 (at Ser289/296/301 but not at Ser259) through a negative-feedback circuit, which caused the rapid inactivation of ERKs.33 This study suggests that the dual phase activation (ON and OFF) of ERKs plays a role in neuroprotection against lethal ischemia by blocking apoptotic cellular events. Taken together, these studies indicate that FGF receptors and ERKs are activated sequentially along the MAPK signaling cascade to elicit physiological responses in the targeted neurons following stimulation with SUN11602 as well as with bFGF.
With the use of the other growth factors (e.g., VEGF, NGF, IGF-1, and BDNF) and different experimental models, several experiments have been conducted to investigate the involvement of ERKs in cell survival and death.34−37 In an in vitro traumatic injury model, pretreatment with VEGF activated ERKs in the primary cultures of cortical neurons and decreased the amount of apoptotic neuronal death.34 Because the VEGF-induced activities of ERKs are suppressed with U0126 (a MEK inhibitor), it is assumed that VEGF is involved in neuroprotection against mechanical traumatic injury through the MEK/ERK signaling cascade. In an NGF-deprivation cell death model, PD98059 blocked the activation of ERKs, but the trophic effects of NGF on neuronal survival were not impaired in the primary cultures of sympathetic ganglion neurons.35 The trophic actions of NGF may not require the activation of ERKs in this model.35 Another study that used an NGF-deprivation model in pheochromocytoma cells (PC12 cells) has reported that p38 (MAP2), a member of the MAPK superfamily, works as the intermediate leading to cell death.36 Insulin-like growth factor-1 (IGF-1) and brain-derived neurotrophic factor (BDNF) have trophic effects with different potencies on dissociated hippocampal neurons, and their protective effects are mediated by the activation of the phosphatidylinositide 3 kinase (PI3K)/Akt kinase pathway but not the MAPK signaling cascade.37 Each growth factor above appears to be involved in trophic or cell-death mechanisms in targeted neurons, depending on the activation or inactivation of its own signaling pathway other than the MEK/ERK signaling cascade, which SUN11602 and bFGF use to transmit their signals for neuroprotection. Taken together, the neuroprotective mechanisms that act through the activation of ERKs appear to be specific to bFGF as well as to SUN11602.
Besides the trophic activities of ERKs, several studies have reported that ERKs are implicated in the apoptotic cellular events that occur in cell-culture models38,39 and in focal ischemia mouse models.40 The apoptotic neuronal death is antagonized by pretreatment with PD98059, so that the MEK/ERK signaling pathway is assumed to be involved in the mechanisms of cell death but not in neuroprotection in these in vitro and in vivo models. In hippocampal slice cultures, it has been reported that the sustained tyrosine phosphorylation of ERK1/2 resulted in neuronal death after direct treatment with okadaic acid (Ser/Thr protein phosphatase inhibitor).41 This neuronal death was markedly reduced after the inactivation of ERKs with K252a (a nonselective protein kinase inhibitor) and PD98059. Furthermore, an excessive amount of neurotransmitters is known to cause adverse conditions in the targeted neurons. High levels of extracellular dopamine, which acts through dopamine receptor 1, induces a sustained activation of ERKs (i.e., phosphorylated ERKs) through a cAMP/PKA and MAPK signaling pathway, instead of through c-jun N-terminal kinase (JNK) and p38, in primary cultures of rat striatal neurons.42 An increased amount of phosphorylated ERKs in the cytoplasm induces apoptotic cell death in striatal neurons.42 These studies may claim that the sustained activation of ERKs is not always associated with protective mechanisms on the affected or targeted neurons by extracellular signal molecules. It may be suggested that the neuroprotective mechanisms for the targeted neurons require ON and OFF negative-feedback control of the MEK/ERK signaling pathway.33 Furthermore, members of the MAPK superfamily may function as intermediates in order to transmit different extracellular signals in the right-signal-in-the-right-place manner in different cell types and in different conditions, as has been previously suggested.43
c. Intracellular Ca2+ Levels and Buffering Actions of Newly Synthesized Calb
The aberrant increase of intracellular Ca2+ concentrations is the most critical factor that leads affected cultured neurons to cell death, according to studies monitoring the actions of excitotoxic glutamate and the effects of the calcium ionophore, A23187.44−47 The subsequent in vitro experiments that used different means of toxic injuries, which included glutamate, replicated neuronal death in dishes by causing large increases in intracellular Ca2+ levels. Such high-Ca2+-induced neuronal death was mostly prevented by treating the neurons with bFGF.5−7 Unlike other growth factors (NGF, BDNF, EGF, PDGF, IGF, NT-3, and VEGF), bFGF suppressed the glutamate-induced increase in intracellular free Ca2+ and appeared to keep the ionic environments of the neurons at normal physiological levels. SUN11602, like bFGF, antagonized the glutamate-induced neuronal death by suppressing the abnormal increase in the intracellular free Ca2+ levels in the cultured cortical neurons of WT mice. The protective activities of SUN11602 and bFGF appeared to be mostly equivalent in intensity. However, in spite of the pretreatment with SUN11602 and bFGF prior to the toxic injuries, the glutamate-induced rise in intracellular Ca2+ levels occurred and resulted in the cell death of the cultured cortical neurons from Calb–/– mice.
Stabilizing intracellular Ca2+ concentrations at physiological levels is key in the prevention of cell death in cultured neurons and brain tissues. In neurons that are activated by prolonged afferent stimulation, chelating intracellular Ca2+ decreases the vulnerability to damage from activity-induced Ca2+ overload.48 This suggests that it is particularly important to identify the regulators of intracellular Ca2+ levels. Several calcium-capturing molecules, such as Calb, parvalbumin (Parv), and calretinin (Calr), have been recognized in the brain. In P-19 murine embryonic carcinoma cells that were transfected with the cDNA for Calb, Parv, and Calr and then transformed into neuron-like cells by retinoic acid, Calb and Calr transiently delayed the onset of cell death after excitotoxic stimulation.49 HEK293 cells that were transfected with human recombinant Calb exhibited intracellular Ca2+-buffering action.50 The dopaminergic cell line MN9D that was transfected with the cDNA for Calb exhibited a reduced rate of apoptotic cell death that was induced by 6-hydroxydopamine.51 Dissociated hippocampal neurons that possessed immunoreactivity for Calb were relatively resistant to excitotoxic insults by glutamate and the calcium ionophore A23187; that is, Calb-positive neurons were able to reduce intracellular Ca2+ levels more effectively in comparison to Calb-negative neurons.8 The present study demonstrated that Calb played a crucial role in intracellular Ca2+ homeostasis in Calb–/– mice and in the consequent neuronal survival in cell culture models.
Furthermore, the protective activities of SUN11602 and bFGF were exclusively achieved by newly synthesized protein(s) because their effects were completely abolished by pretreating targeted neurons with either a transcription or a translation inhibitor, as was also observed in a previous study.7 This suggested that the maximum protective effects were obtained in either targeted neurons or brain tissues when they were pretreated with exogenous bFGF.5−7,9,52 Thus, it may be concluded that the neuroprotective mechanisms of SUN11602, as well as of bFGF, were achieved through the gene expression of calbindin-D28k (CALB1) through intracellular signaling pathways (i.e., the activation of FGFR-1, the phosphorylation of ERK1/2, the transcription and translation of Calb) and the stabilization of intracellular Ca2+ metabolism in the affected neurons.
d. Neuroprotective Abilities
One of the most notable results of the present study was that SUN11602 rescued vulnerable neurons from cell death due to excitotoxic conditions of glutamate, which was similar to the observations of bFGF. The administration of SUN11602 and bFGF 1 day before the excitotoxic injury seemed to be long enough to completely elicit the neuroprotective effects. The protective effects of SUN11602 were mostly equivalent to those of bFGF in the primary cultured cortical neurons and were comparable with the results in previous in vitro5,6 and in vivo studies,9 in which bFGF was administered beforehand (more than 1 day). Accordingly, the promising protective actions of SUN11602 appeared to be supported by evidence that was derived from experiments with bFGF.
In addition, the time point of bFGF administration is the most intriguing issue when considering the therapeutic time window in various clinical cases of diseases. Different experiments that have used different administration protocols have been conducted in order to determine the protective effects of bFGF in cultured brain neurons and in animal models. Early treatment with bFGF after the onset of experimental injuries attenuated neuronal death as well as the disintegration of membranes and the cytoarchitecture.53−55 In animal models (rat and cat), early administration (less than 1 to 3 h, iv infusion) reduced the infarct volumes in the MCAO side of the cerebral hemisphere.10,13,56 On the other hand, delayed treatment with bFGF (started 1 day postinfarct, intracisternally) in the MCAO rat model did not ameliorate the cerebral infarct volumes but resulted in an enhancement of the recovery of the cerebral motor functions of the contralateral limbs in cases of prolonged survival.12,16,57−59 However, 2 day postinfarct treatment (chronic intraventricular infusion) in the MCAO rat model failed to retain normal conditions of the capillary bed density and a normal number of live neurons in the peri-infarct cortex compared to control animals.60 In a clinical trial,15 it has been reported that the administration of bFGF after cerebral infarctions suppresses the exacerbation of neurological symptoms, such as scores on the Barthel and Rankin scales, and that the therapeutic time window for clinical use may exceed 5 h.
Accordingly, when administered in advance or early enough so that bFGF can exert its full potential, bFGF treatment results in beneficial protective effects on vulnerable neurons in vitro and injured brain tissues in vivo. Studies of human cases have suggested that even the posthoc administration of bFGF may enhance the recovery of the cortical motor functions of the corresponding peripheral organs without a marked amelioration of brain tissue damage.15 It is, therefore, suggested that the use of bFGF may hold promise for enhancing the recovery mechanisms of motor cortical functions in prolonged survival, in addition to brain tissue protection in the acute phase after stroke. However, bFGF is a bioactive peptide with a short half-life in the blood and with poor blood-brain barrier permeability. Thus, the strategies for administration need to consider its physiological properties for practical use. Furthermore, it is alarming that the administration of bFGF may lead to leukocytosis and decreased blood pressure in human cases. Taken together, SUN11602, which mimics the major neuroprotective activities of bFGF, may be a promising candidate as a therapeutic drug for several neurological diseases of the brain and spinal cord.
In summary, SUN11602 is a small synthetic compound that mimics the neuroprotective activities of bFGF in experimental protocols for maintaining the structural and functional integrity of cultured cortical neurons. SUN11602 played a pivotal role in allowing primary cultured neurons to survive in adverse environments of glutamate toxicity and activating intracellular key molecules that were involved in the neuroprotective mechanisms. These actions were quite similar to those of bFGF. Such neuroprotective mechanisms were specific and distinctive to SUN11602 and bFGF and differed clearly from those of the other growth factors that were investigated. Furthermore, unlike bFGF, SUN11602 can either directly or indirectly trigger the phosphorylation of the cytosolic domain of the FGFR-1 without interacting with the extracellular bFGF-binding sites of the receptor. However, how SUN11602 interacts with FGFR-1 remains to be clarified at molecular levels.
Methods
Reagents
SUN11602, which was the small synthetic compound of interest in the present study, and PD166866 (pyrido [2, 3]pyrimidine), which is a specific phosphorylation inhibitor of the FGFR-1 cytosolic tyrosine kinase domain, were synthesized in the Biomedical Research Laboratories of Asubio Pharma Co., Ltd. (Hyogo, Japan). The synthetic schemes and supporting analytical data of SUN11602 are provided in the Supporting Information. The functional biomolecules (bFGF, NGF, BDNF, IGF-1, NT-3, VEGF-A, heparin binding-EGF [HB-EGF], and PDGF) and PD98059 (a phosphorylation inhibitor of ERK1/2) were purchased from EMD Millipore Corporation (Billerica, MA), fluo 3-AM was from DOJINDO LABORATORIES (Kumamoto, Japan), and all other reagents were from Nacalai Tesque, Inc. (Kyoto, Japan).
Neuronal Cultures
Cerebrocortical neurons were prepared from embryonic brains (gestation/embryonic day 18 [E18]) of Wistar rats (Japan SLC, Inc., Shizuoka, Japan) and from embryonic brains of Calb–/– and WT mice (E15–16, C57BL/6J, The Jackson Laboratory, Sacramento, CA, USA and E15–16, C57BL/6J, CLEA Japan, Inc., Shizuoka, Japan, respectively). The cerebral cortices were dissociated with papain, suspended in neurobasal medium (Life Technologies Corporation, Grand Island, NY, USA) containing 5% NuSerum (Japan BD, Tokyo, Japan) and 2% B27 (Life Technologies Corporation), and then seeded onto either poly-d-lysine-coated 96-well microplates or polyethyleneimine-coated 35-mm glass dishes (Glass Base Dish, AGC Techno Glass Co., Chiba, Japan). The neuronal cultures were maintained in an atmosphere of 10% CO2 at 37 °C in a humidified incubator. Four days after seeding, the proliferation of non-neuronal cells was inhibited by the addition of 1 μM of AraC (Arabinofuranosyl Cytidine) at the final concentration. The subsequent experiments were performed in 7- to 10-day cultures. All animal experiments were approved by the Ethics Committee for Animal Experiments of Biomedical Research Laboratories, Asubio Pharma Co., Ltd., and performed in accordance with the Guideline for Animal Experiments of the laboratories.
Quantification of Neuronal Survival
The neuroprotective effects of the chemical compound SUN11602 and the growth factors (bFGF, NGF, BDNF, IGF-1, NT-3, VEGF-A, HB-EGF, and PDGF) were assessed with a survival indicator of live cells by means of a MTT (an assay of the cytotoxic environments of glutamate [75 or 150 μM at the final concentration]). Cerebrocortical neurons in poly-d-lysine-coated 96-well microplates were pretreated with vehicle (Hanks’ Balanced Salt Solution), SUN11602, bFGF, or the other growth factors for 24 h prior to the onset of glutamate toxicity. Subsequently, 10 μL of the MTT solution (5 mg/mL) was added to each well (200 μL of culture medium) of the microplates, which were further incubated for 15 min in order to allow for the reduction reaction. At the end of the incubation, the culture medium was discarded quickly in order to stop the reaction and to eliminate any uncertain factors that were derived from the prolonged reaction. Neurons in each well were then dried for 24 h, and 200 μL of dimethyl sulfoxide (DMSO) was poured into all of the wells in order to dissolve the reaction products thoroughly for the MTT assay. Each well of the microplate was measured on a microplate reader (Multiskan MS, Thermo BioAnalysis Corporation, Santa Fe, NM, USA) by the absorbance at the wavelength of 570 nm with reference to the wavelength of 650 nm.
Intracellular Signaling and the Inhibitors
In the course of the present study, SUN11602 and bFGF activated FGFR-1 and ERK1/2. In order to establish the involvement of the presumed intracellular signaling molecules, PD166866 and PD98059 were used to inhibit the activation of FGFR-1 and ERK1/2, respectively. PD166866 (0.3 μM at the final concentration) or PD98059 (0.3, 1, or 3 μM at the final concentration) were added to the neuronal cultures 30 min prior to the stimulation with SUN11602, bFGF, or each of the other growth factors. The inhibitory actions of each inhibitor against the neuroprotection of SUN11602 and the growth factors were assessed by the rates of survival of the neurons with the MTT assay in excitotoxic conditions of glutamate.
Protein Synthesis for Neuroprotection
Because the maximum neuroprotective effects of the vulnerable neurons were obtained with the prior administration of SUN11602 and bFGF, the requirement of the transcription and translation of the CALB1 gene for neuroprotection was examined in cytotoxic environments of glutamate. Actinomycin D and cycloheximide were separately employed in order to examine the inhibitory effects against neuroprotection by blocking transcription and translation, respectively. Each inhibitor (1 μg/mL at the final concentration) was added to the neuronal cultures 2 h prior to stimulation with SUN11602 or bFGF, and their inhibitory effects against neuroprotection were assessed by the rates of surviving neurons with the MTT assay.
Immunocytochemistry of Cultured Neurons
Cerebrocortical neurons that were cultured in polyethyleneimine-coated 35-mm glass dishes were prepared from the brains of mice, as described above. In brief, 7-day neuronal cultures were fixed in 2% paraformaldehyde and 4% sucrose in 0.1 M sodium phosphate buffer (pH 7.4) for 15 min. After treatment in 20% Block Ace solution (Dainippon Sumitomo Pharma Co., Ltd., Osaka, Japan) for 30 min at room temperature, neurons were incubated with primary antibodies against either Calb (1:1,000; EMD Millipore Corporation) or FGFR1 (1:1,000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 24 h at 4 °C. Then, the neurons were incubated with the biotinylated secondary antibody in the ABC Elite kit (Vector Laboratories, Inc., Burlingame, CA, USA), and the subsequent avidin–biotin complex formation was performed according to the kit protocol. Signals of the target molecules were finally visualized as insoluble colored precipitations using 3,3′-diaminobenzidine tetrahydrochloride (DAB) as the chromogenic substrate. For immunofluorescence staining, fluorescein isothiocyanate-conjugated goat antirabbit IgG was employed as the secondary antibody (1:1,000; MP Biomedicals LLC, Solon, OH, USA).
Immunocytochemistry of Brain Tissue
In order to ensure the phenotype of the Calb–/– mice, the brains were processed for immunocytochemistry. First, the mice were perfused intracardially with a small volume of saline, which was followed with 50 mL of ice-cold 4% paraformaldehyde and 1% sucrose in 0.1 M phosphate buffer (pH 7.4). Second, the brains were removed from the skulls and postfixed in the same fresh fixative at 4 °C overnight. Thereafter, the brains were rinsed in phosphate-buffered 20% (w/v) sucrose at 4 °C overnight. The brains were frozen in powdered dry ice, and serial coronal sections were cut at a thickness of 10 μm in a cryostat and thaw-mounted on slides (MAS-GP type A, Matsunami Glass Ind., Ltd., Osaka, Japan).
Several sections of mouse brain were stained with cresyl violet for the cytoarchitectural analyses of the brains of WT and Calb–/– mice. Immunocytochemistry for Calb was conducted in the same way as described above for the cultured neurons. In brief, brain sections were incubated with an anti-Calb antibody (1:1,000) for 2 days at 4 °C and then with the secondary antibody conjugated with horseradish peroxidase (1:500) for 12 h at 4 °C. Next, sections were presoaked in 0.02% DAB in 0.1 M phosphate buffer (pH 7.4) for 5 min, and the oxidative reaction for peroxidase was completed by adding 0.01% hydrogen peroxide at a final concentration for another 5 min.
Imaging Analyses of Intracellular Calcium Levels
Cerebrocortical neurons from mouse brains (WT and Calb–/– mice) in polyethyleneimine-coated 35-mm glass dishes were pretreated with vehicle, SUN11602, or bFGF for 24 h prior to the imaging analyses. The loading of 4 μM of fluo 3-AM into the neurons was conducted in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered saline (120 mM NaCl, 5.0 mM KCl, 0.62 mM MgSO4, 1.8 mM CaCl2, 10 mM HEPES, and 6.0 mM glucose; pH 7.4) containing 0.2% DMSO and 0.04% F-127 for 60 min at 37 °C. The loading of the Ca2+ indicator was stopped by replacing the loading buffer with fresh HEPES-buffered saline. Cytoplasmic free Ca2+ levels were measured by laser confocal microscopy with FluoView imaging software (Olympus Corporation, Tokyo, Japan). After a 5 min exposure to glutamate (WT, 100 μM; Calb–/– mice, 50 μM), increased levels of cytoplasmic free Ca2+ were analyzed.
Western Blot Analysis for ERK1/2 and p-ERK1/2
Cerebrocortical neurons from rat brains were grown in poly-d-lysine-coated 12-well microplates, scraped in the presence of SUN11602 (10 and 100 μM) or bFGF (10 ng/mL) for 20 min, and then lysed in Laemmli buffer. A total amount of 18.75 μg of protein was loaded into each well of 12% sodium dodecyl sulfate polyacrylamide gels, electrophoresed in order to separate the proteins by their molecular sizes, and then transferred to a nitrocellulose membrane using a Bio-Rad Mini Trans Blot electrophoretic transfer unit (Bio-Rad Laboratories, Inc., Hercules CA). Nonspecific staining of the membranes was blocked with 2% Block Ace (blocking reagent: AbD Serotec, Oxford, U.K.) in phosphate-buffered saline (PBS, pH 7.4). The target molecules (ERK1/2 and phosphorylated ERK1/2 [p-ERK1/2]) were probed on the membranes with their specific primary antibodies (1:1000, Cell Signaling Technology, Inc., Danvers, MA) either at room temperature for 1 h or at 48 °C overnight. Membranes were washed three times for 10 min each in PBS containing 0.05% Tween-20 at room temperature and then incubated with horseradish-peroxidase-conjugated secondary antibodies (1:1000). After washing in PBS, membranes were processed in order to complete the peroxidase activity and to yield signals of ERK1/2 and p-ERK1/2.
Statistical Analysis
All numerical data were expressed as the mean value ± standard error of the mean (SEM) and analyzed by Dunnett’s multiple comparison tests (EXSUS Ver. 7.6). A p value with statistical significance is indicated by asterisks (e.g., *p < 0.05, **p < 0.01, and ***p < 0.001).
Acknowledgments
The authors would like to thank Dr. Tsugio Kaneko and Dr. Yoshiaki Fukuda at Asubio Pharma and Prof. Thomas E. Finger at Colorado University School of Medicine for their encouragement and valuable suggestions for this study.
Glossary
Abbreviations
- NGF
nerve growth factor
- BDNF
brain-derived neurotrophic factor
- IGF-1
insulin-like growth factor-1
- NT-3
neurotrophin-3
- VEGF-A
vascular endothelial growth factor-A
- HB-EGF
heparin-binding EGF-like growth factor
- PDGF
platelet-derived growth factor
- ERK
extracellular signal-regulated kinase
Supporting Information Available
Synthesis and characterization of SUN11602. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
N.M., T.K., S.K., Y.M., and I.T. designed research; N.M., T.K., N.T., S.K., T.T., R.O., T.N., and T.O. performed research; S.U., M.K., and Y.S. analyzed data; N.M., Y.M. and I.T. wrote the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Morrison R. S.; Sharma A.; de Vellis J.; Bradshaw R. A. (1986) Basic fibroblast growth factor supports the survival of cerebral cortical neurons in primary culture. Proc. Natl. Acad. Sci. U.S.A. 83, 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walicke P.; Cowan W. M.; Ueno N.; Baird A.; Guillemin R. (1986) Fibroblast growth factor promotes survival of dissociated hippocampal neurons and enhances neurite extension. Proc. Natl. Acad. Sci. U.S.A. 83, 3012–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers J.; Hausmann B.; Unsicker K.; Berry M. (1987) Fibroblast growth factors promote the survival of adult rat retinal ganglion cells after transection of the optic nerve. Neurosci. Lett. 76, 157–162. [DOI] [PubMed] [Google Scholar]
- Walicke P. A. (1988) Basic and acidic fibroblast growth factors have trophic effects on neurons from multiple CNS regions. J. Neurosci. 8, 2618–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B.; Mattson M. P. (1992) Glucose deprivation elicits neurofibrillary tangle-like antigenic changes in hippocampal neurons: prevention by NGF and bFGF. Exp. Neurol. 117, 114–123. [DOI] [PubMed] [Google Scholar]
- Mattson M. P.; Tomaselli K. J.; Rydel R. E. (1993) Calcium-destabilizing and neurodegenerative effects of aggregated beta-amyloid peptide are attenuated by basic FGF. Brain Res. 621, 35–49. [DOI] [PubMed] [Google Scholar]
- Mattson M. P.; Murrain M.; Guthrie P. B.; Kater S. B. (1989) Fibroblast growth factor and glutamate: opposing roles in the generation and degeneration of hippocampal neuroarchitecture. J. Neurosci. 9, 3728–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P.; Rychlik B.; Chu C.; Christakos S. (1991) Evidence for calcium-reducing and excito-protective roles for the calcium-binding protein calbindin-D28k in cultured hippocampal neurons. Neuron 6, 41–51. [DOI] [PubMed] [Google Scholar]
- Koketsu N.; Berlove D. J.; Moskowitz M. A.; Kowall N. W.; Caday C. G.; Finklestein S. P. (1994) Pretreatment with intraventricular basic fibroblast growth factor decreases infarct size following focal cerebral ischemia in rats. Ann. Neurol. 35, 451–457. [DOI] [PubMed] [Google Scholar]
- Fisher M.; Meadows M. E.; Do T.; Weise J.; Trubetskoy V.; Charette M.; Finklestein S. P. (1995) Delayed treatment with intravenous basic fibroblast growth factor reduces infarct size following permanent focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 15, 953–959. [DOI] [PubMed] [Google Scholar]
- Kawamata T.; Speliotes E. K.; Finklestein S. P. (1997) The role of polypeptide growth factors in recovery from stroke. Adv. Neurol. 73, 377–382. [PubMed] [Google Scholar]
- Ay H.; Ay I.; Koroshetz W. J.; Finklestein S. P. (1999) Potential usefulness of basic fibroblast growth factor as a treatment for stroke. Cerebrovasc. Dis. 9, 131–135. [DOI] [PubMed] [Google Scholar]
- Bethel A.; Kirsch J. R.; Koehler R. C.; Finklestein S. P.; Traystman R. J. (1997) Intravenous basic fibroblast growth factor decreases brain injury resulting from focal ischemia in cats. Stroke 28, 609–615. [DOI] [PubMed] [Google Scholar]
- Sun D.; Bullock M. R.; McGinn M. J.; Zhou Z.; Altememi N.; Hagood S.; Hamm R.; Colello R. J. (2009) Basic fibroblast growth factor-enhanced neurogenesis contributes to cognitive recovery in rats following traumatic brain injury. Exp. Neurol. 216, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogousslavsky J., Victor S. J., Salinas E. O., Pallay A., Donnan G. A., Fieschi C., Kaste M., Orgogozo J. M., Chamorro A., Desmet A., and European-Australian Fiblast (Trafermin) in Acute Stroke Group. (2002) Fiblast (trafermin) in acute stroke: results of the European-Australian phase II/III safety and efficacy trial. Cerebrovasc. Dis. 14, 239–251. [DOI] [PubMed] [Google Scholar]
- Kawamata T.; Alexis N. E.; Dietrich W. D.; Finklestein S. P. (1996) Intracisternal basic bibroblast growth factor (bFGF) enhances behavioral recovery following focal cerebral infarction in the rat. J. Cereb. Blood Flow Metab. 16, 542–547. [DOI] [PubMed] [Google Scholar]
- Kawamata T.; Dietrich W. D.; Schallert T.; Gotts J. E.; Cocke R. R.; Benowitz L. I.; Finklestein S. P. (1997) Intracisternal basic bibroblast growth factor (bFGF) enhances functional recovery and up-regulates the expression of a molecular marker of neuronal sprouting following focal cerebral infarction. Proc. Natl. Acad. Sci. U.S.A. 94, 8179–8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speliotes E. K.; Caday C. G.; Do T.; Weise J.; Kowall N. W.; Finklestein S. P. (1996) Increased expression of basic fibroblast growth factor (bFGF) following focal cerebral infarction in the rat. Mol. Brain Res. 39, 31–42. [DOI] [PubMed] [Google Scholar]
- Hamby J. M.; Connolly C. J.; Schroeder M. C.; Winters R. T.; Showalter H. D.; Panek R. L.; Major T. C.; Olsewski B.; Ryan M. J.; Dahring T.; Lu G. H.; Keiser J.; Amar A.; Shen C.; Kraker A. J.; Slintak V.; Nelson J. M.; Fry D. W.; Bradford L.; Hallak H.; Doherty A. M. (1997) Structure-activity relationships for a novel series of pyrido[2,3-d]pyrimidine tyrosine kinase inhibitors. J. Med. Chem. 40, 2296–2303. [DOI] [PubMed] [Google Scholar]
- Panek R. L.; Lu G. H.; Dahring T. K.; Batley B. L.; Connolly C.; Hamby J. M.; Brown K. J. (1998) In vitro biological characterization and antiangiogenic effects of PD 166866, a selective inhibitor of the FGF-1 receptor tyrosine kinase. J. Pharmacol. Exp. Ther. 286, 569–577. [PubMed] [Google Scholar]
- Ablooglu A. J.; Frankel M.; Rusinova E.; Ross J. B.; Kohanski R. A. (2001) Multiple activation loop conformations and their regulatory properties in the insulin receptor’s kinase domain. J. Biol. Chem. 276, 46933–46940. [DOI] [PubMed] [Google Scholar]
- Hubbard S. R. (1997) Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 16, 5572–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till J. H.; Ablooglu A. J.; Frankel M.; Bishop S. M.; Kohanski R. A.; Hubbard S. R. (2001) Crystallographic and solution studies of an activation loop mutant of the insulin receptor tyrosine kinase: insights into kinase mechanism. J. Biol. Chem. 276, 10049–10055. [DOI] [PubMed] [Google Scholar]
- Stamos J.; Sliwkowski M. X.; Eigenbrot C. (2002) Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 277, 46265–46272. [DOI] [PubMed] [Google Scholar]
- Oefner C.; D’Arcy A.; Winkler F. K.; Eggimann B.; Hosang M. (1992) Crystal structure of human platelet-derived growth factor BB. EMBO J. 11, 3921–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller Y. A.; Li B.; Christinger H. W.; Wells J. A.; Cunningham B. C.; de Vos A. M. (1997) Vascular endothelial growth factor: crystal structure and functional mapping of the kinase domain receptor binding site. Proc. Natl. Acad. Sci. U.S.A. 94, 7192–7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae J. H.; Boggon T. J.; Tomé F.; Mandiyan V.; Lax I.; Schlessinger J. (2010) Asymmetric receptor contact is required for tyrosine autophosphorylation of fibroblast growth factor receptor in living cells. Proc. Natl. Acad. Sci. U.S.A. 107, 2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourse M. B.; Rolle M. W.; Pabon L. M.; Murry C. E. (2007) Selective control of endothelial cell proliferation with a synthetic dimerizer of FGF receptor-1. Lab. Invest. 87, 828–835. [DOI] [PubMed] [Google Scholar]
- Mattson M. P. (2008) Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann. N.Y. Acad. Sci. 1144, 97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M.; Froum S.; Hamby J. M.; Schroeder M. C.; Panek R. L.; Lu G. H.; Eliseenkova A. V.; Green D.; Schlessinger J.; Hubbard S. R. (1998) Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. EMBO J. 17, 5896–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R. J. (1993) The mitogen-activated protein kinase signal transduction pathway. J. Biol. Chem. 268, 14553–14556. [PubMed] [Google Scholar]
- Seger R.; Krebs E. G. (1995) The MAPK signaling cascade. FASEB J. 9, 726–735. [PubMed] [Google Scholar]
- Cao Q.; Qian M.; Wang X. F.; Wang B.; Wu H. W.; Zhu X. J.; Wang Y. W.; Guo J. (2011) Negative feedback regulation of Raf/MEK/ERK cascade after sublethal cerebral ischemia in the rat hippocampus. Neurochem. Res. 36, 153–162. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Liu W.; Wang Y.; Chao X.; Qu Y.; Wang K.; Fei Z. (2011) VEGF protects rat cortical neurons from mechanical trauma injury induced apoptosis via the MEK/ERK pathway. Brain Res. Bull. 86, 441–446. [DOI] [PubMed] [Google Scholar]
- Creedon D. J.; Johnson E. M.; Lawrence J. C. (1996) Mitogen-activated protein kinase-independent pathways mediate the effects of nerve growth factor and cAMP on neuronal survival. J. Biol. Chem. 271, 20713–20718. [DOI] [PubMed] [Google Scholar]
- Xia Z.; Dickens M.; Raingeaud J.; Davis R. J.; Greenberg M. E. (1995) Opposing effects of ERK and JNK-p38MAP kinases on apoptosis. Science 270, 1326–1331. [DOI] [PubMed] [Google Scholar]
- Zheng W. H.; Quirion R. (2004) Comparative signaling pathways of insulin-like growth factor-1 and brain-derived neurotrophic factor in hippocampal neurons and the role of the PI3 kinase pathway in cell survival. J. Neurochem. 89, 844–852. [DOI] [PubMed] [Google Scholar]
- Murray B.; Alessandrini A.; Cole A. J.; Yee A. G.; Furshpan E. J. (1998) Inhibition of the p44/42 MAP kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc. Natl. Acad. Sci. U.S.A. 95, 11975–11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanciu M.; Wang Y.; Kentor R.; Burke N.; Watkins S.; Kress G.; Reynolds I.; Klann E.; Angiolieri M. R.; Johnson J. W.; DeFranco D. B. (2000) Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 275, 12200–12206. [DOI] [PubMed] [Google Scholar]
- Alessandrini A.; Namura S.; Moskowitz M. A.; Bonventre J. V. (1999) MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A. 96, 12866–12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundén E.; Seglen P. O.; Haug F. M.; Ottersen O. P.; Wieloch T.; Shamloo M.; Laake J. H. (1998) Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: evidence for a MAP kinase-dependent mechanism. J. Neurosci. 18, 7296–7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Rusnak M.; Lombroso P. J.; Sidhu A. (2009) Dopamine promotes striatal neuronal apoptotic death via ERK signaling cascades. Eur. J. Neurosci. 29, 287–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton T. G.; Nye S. H.; Robbins D. J.; Ip N. Y.; Radziejewska E.; Morgenbesser S. D.; DePinho R. A.; Panayotatos N.; Cobb M. H.; Yancopoulos G. D. (1991) ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 65, 663–675. [DOI] [PubMed] [Google Scholar]
- Choi D. W. (1987) Ionic dependence of glutamate neurotoxicity. J. Neurosci. 7, 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D. W. (1988) Glutamate neurotoxicity and diseases of the nervous system. Neuron 1, 623–634. [DOI] [PubMed] [Google Scholar]
- Mattson M. P.; Dou P.; Kater S. B. (1988) Outgrowth-regulating actions of glutamate in isolated hippocampal pyramidal neurons. J. Neurosci. 8, 2087–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P.; Guthrie P. B.; Kater S. B. (1988) Intracellular messengers in the generation and degeneration of hippocampal neuroarchitecture. J. Neurosci. Res. 21, 447–464. [DOI] [PubMed] [Google Scholar]
- Scharfman H. E.; Schwartzkroin P. A. (1989) Protection of dentate hilar cells from prolonged stimulation by intracellular calcium chelation. Science 246, 257–260. [DOI] [PubMed] [Google Scholar]
- D’Orlando C.; Fellay B.; Schwaller B.; Salicio V.; Bloc A.; Gotzos V.; Celio M. R. (2001) Calretinin and calbindin D-28k delay the onset of cell death after excitotoxic stimulation in transfected P19 cells. Brain Res. 909, 145–158. [DOI] [PubMed] [Google Scholar]
- Rintoul G. L.; Raymond L. A.; Baimbridge K. G. (2001) Calcium buffering and protection from excitotoxic cell death by exogenous calbindin-D28k in HEK 293 cells. Cell Calcium 29, 277–287. [DOI] [PubMed] [Google Scholar]
- Sun S.; Li F.; Gao X.; Zhu Y.; Chen J.; Zhu X.; Yuan H.; Gao D. (2011) Calbindin-D28K inhibits apoptosis in dopaminergic neurons by activation of the PI3-kinase-Akt signaling pathway. Neuroscience 199, 359–367. [DOI] [PubMed] [Google Scholar]
- Freese A.; Finklestein S. P.; DiFiglia M. (1992) Basic fibroblast growth factor protects striatal neurons in vitro from NMDA-receptor mediated excitotoxicity. Brain Res. 575, 351–355. [DOI] [PubMed] [Google Scholar]
- Himmelseher S.; Pfenninger E.; Georgieff M. (1996) The effect of basic fibroblast growth factor on glutamate-injured neuroarchitecture and arachidonic acid release in adult hippocampal neurons. Brain Res. 707, 54–63. [DOI] [PubMed] [Google Scholar]
- Himmelseher S.; Pfenninger E.; Georgieff M. (1997) Effects of basic fibroblast growth factor on hippocampal neurons after axonal injury. J. Trauma 42, 659–664. [DOI] [PubMed] [Google Scholar]
- Himmelseher S.; Pfenninger E.; Georgieff M. (1998) Basic fibroblast growth factor reduces lactic acid-induced neuronal injury in rat hippocampal neurons. Crit. Care Med. 26, 2029–2036. [DOI] [PubMed] [Google Scholar]
- Ren J. M.; Finklestein S. P. (1997) Time windlow of infarct reduction by intravenous basic fibroblast growth factor in focal cerebral ischemia. Eur. J. Pharmacol. 327, 11–16. [DOI] [PubMed] [Google Scholar]
- Berry D.; Ren J.; Kwan C. P.; Sietsma D. K.; Sasisekharan R.; Finklestein S. P. (2005) Dimeric fibroblast growth factor-2 enhances functional recovery after focal cerebral ischemia. Restor. Neurol. Neurosci. 23, 251–256. [PubMed] [Google Scholar]
- Ren J. M.; Finklestein S. P. (2005) Growth factor treatment of stroke. Curr. Drug Targets: CNS Neurol. Disord. 4, 121–125. [DOI] [PubMed] [Google Scholar]
- Kawamata T.; Ren J.; Cha J. H.; Finklestein S. P. (1999) Intracisternal antisense oligonucleotide to growth associated protein-43 blocks the recovery-promoting effects of basic fibroblast growth factor after focal stroke. Exp. Neurol. 158, 89–96. [DOI] [PubMed] [Google Scholar]
- Tenjin H.; Anderson R. E.; Meyer F. B. (1995) Treatment with basic fibroblastic growth factor following focal cerebral ischemia does not prevent neuronal injury. J. Neurol. Sci. 128, 66–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.