

Abstract
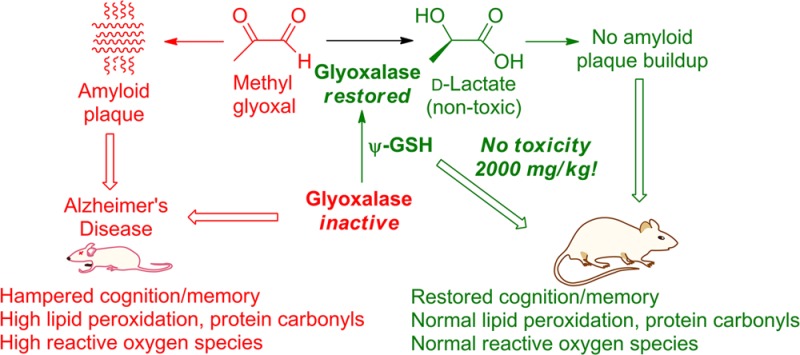
Pathologically high brain levels of reactive dicarbonyls such as methylglyoxal or glyoxal initiate processes that lead ultimately to neurodegeneration, presented clinically as Alzheimer’s disease and other cognitive or motor impairment disorders. Methylglyoxal and glyoxal result from glycolysis and normal metabolic pathways. Their reaction products with proteins (advanced glycation end products), and their primary chemical toxicities are both linked unequivocally to the primary pathologies of Alzheimer’s disease, namely, amyloid plaques and neurofibrillary tangles. Generation of dicarbonyls is countered through the reduction of dicarbonyls by the glutathione-dependent glyoxalase enzyme system. Although glyoxalase-I is overexpressed in early and middle stages of Alzheimer’s disease, glutathione depletion in the Alzheimer’s afflicted brain cripples its efficacy. Due to the lack of a suitable pharmacological tool, the restoration of glyoxalase enzyme activity in pre-Alzheimer’s or manifest Alzheimer’s remains yet unvalidated as a means for anti-Alzheimer’s therapy development. Disclosed herein are the results of a preclinical study into the therapeutic efficacy of ψ-GSH, a synthetic cofactor of glyoxalase, in mitigating Alzheimer’s indicators in a transgenic mouse model (APP/PS1) that is predisposed to Alzheimer’s disease. ψ-GSH administration completely averts the development of spatial mnemonic and long-term cognitive/cued-recall impairment. Amyloid β deposition and oxidative stress indicators are drastically reduced in the ψ-GSH-treated APP/PS1 mouse. ψ-GSH lacks discernible toxicity at strikingly high doses of 2000 mg/kg. The hypothesis that restoring brain glyoxalase activity would ameliorate neurogeneration stands validated, thus presenting a much needed new target for design of anti-Alzheimer’s therapeutics. Consequently, ψ-GSH is established as a candidate for drug-development.
Keywords: Alzheimer’s disease, glyoxalase, ψ-GSH, oxidative stress, methylglyoxal, β-amyloid peptide, advanced glycation end products
The canonical case of Alzheimer’s disease (AD) is an individual of age whose brain presents an environment progressively deficient in restorative biochemical mechanisms that normally counteract the effects of oxidative stress on neuronal tissue. Reactive dicarbonyls such as methylglyoxal (MG) are formed in large quantities through normal metabolic pathways and without detoxification are potent originators of pathological cascades underlying neurodegenerative disorders.1 The activity of the glyoxalase (GLO) enzyme system is vital to mitigation of the dicarbonyl component of oxidative stress.2 Dicarbonyls are neurotoxic at almost every biomolecular level. MG forms advanced glycation end products (AGEs) with proteins, leading to protein misfolding.3 Specifically, it accelerates amyloid β (Aβ) peptide aggregation leading to plaque deposition, one of the hallmarks of AD.4 MG also induces τ hyperphosphorylation and causes memory impairment in rats through AGE formation.3 Brain GLO activity depletion has been unequivocally shown to cause neurodegeneration and the appearance of AD pathologies.5 Depletion of glyoxalase activity during AD development is not attributable to low levels of the constituent enzymes, because glyoxalase-I (GLO-1) is upregulated in early and middle stages of AD.6 Rather, the depletion can be linked to progressively decreasing levels of the essential cofactor glutathione (GSH) during such pathological conditions.7 GSH deficiency may affect the GLO system also through indirect mechanisms. Aside from the involvement of GSH as a cofactor in the GLO mechanism, cellular redox potential, expressed as the balance between oxidized and reduced glutatione, also affects the efficiency of GLO-mediated MG detoxification. Oxidized glutathione (GSSG) was found recently to actively inactivate GLO-1 through covalent modification.8 Therefore, restoration of glyoxalase activity is a plausible modality toward rectification of the pathological tilt toward oxidative processes in the AD-afflicted brain. Given that early and middle staged AD-afflicted brain is deficient in the cofactor GSH and not in GLO, provision of the cofactor would be a logical solution. Indeed, supplementation with GSH or its precursors has been very thoroughly explored. Unfortunately, the development of a clinically efficacious anti-Alzheimer’s therapeutic that acts through restoring glyoxalase activity remains unrealized, perhaps due to the particularly involved biosynthetic, metabolic, and catabolic maze of GSH that leaves many facets of its catabolism unaddressed. (+)-Cysteine is the limiting precursor for the biosynthesis of GSH with only micromolar amounts being available from sequestered GSH in neuronal tissue, while millimolar concentrations of l-(−)-glutamate and glycine are available in the CSF.9N-Acetyl-l-cysteine (NAC) is a clinically utilized, blood–brain barrier (BBB) permeative cysteine precursor that protects neuronal cells against oxidative stress and delays cognitive impairment in senescence-accelerated mice (SAMP8).10 Augmentation of l-cysteine levels in this manner with an aim to increase or restore GSH levels does not address glutamylcysteine ligase (GCL) deficiency in the AD-afflicted brain.11,12 Administration of γ-glutamylcysteine as its BBB-permeative diethyl ester may address GCL deficiency but not the efficacy-limiting γ-glutamyltranspeptidase (γ-GGT) mediated catabolism. Administration of GSH itself does not cause brain accumulation due to the its rapid metabolism by intestinal and hepatic γ-GGTs.13 Thus, although the link between insufficient glyoxalase activity in the brain during pre-AD and AD has been well-defined and traced to a probable cause (GSH deficiency), there is no extant tool that can evaluate the hypothesis that restoration of glyoxalase activity in the brain can mitigate the ravages of AD.
Our laboratories have developed a synthetic cofactor of glyoxalase-I, ψ-GSH (Figure 1), whose ureide isostere linkage confers upon it stability toward γ-GGT, while its skeletal framework confers upon it the ability to permeate the blood–brain barrier. ψ-GSH is a validated GLO-1 substrate. Additionally, we had previously shown that ψ-GSH protects cells against direct chemical oxidative insult (peroxide) and against Aβ peptide.14 Secondary to restoration of GLO activity are other biochemical effects of ψ-GSH depicted in Figure 1, namely, inhibition of γ-GGT and direct counteraction of free-radicals by the thiol function, both of which can be expected to assist in neutralizing brain oxidative stress.
Figure 1.

The multipronged activity of ψ-GSH (a); chemical basis for resistance of ψ-GSH to γ-GGT (b).
The working hypothesis of the current study, the latter serving also as a preclinical evaluation of ψ-GSH as an anti-AD drug entity, is that the administration of ψ-GSH would ward off or reduce indicators of AD pathology. Specific indicators monitored here are behaviorial (cognitive decline), microscopic (Aβ plaque deposition), and biochemical (Aβ load, lipid peroxidation, protein carbonyl content, and reactive oxygen species (ROS) load). The test subject is a transgenic mouse model (APP/PS1) that is predisposed to AD.
Results and Discussion
Short-Term Toxicity Study
Short-term toxicity of ψ-GSH was accessed by administration of ψ-GSH ip three times per week for 2 weeks with 2000 mg/kg being the highest dose. Body weights (Supporting Information; Figure S1) and complete blood count (CBC) were insignificantly affected. Unaffected alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), blood urea nitrogen (BUN), and creatinine levels (Supporting Information; Figure S2) suggest unaltered hepatic or renal function. Histopathology of lung, liver, and kidney tissue reflected no significant difference between the ψ-GSH-treated and saline-treated mice (data not shown).
In Vivo Pharmacokinetics of ψ-GSH
A. Indirect, Utilizing Total Brain Thiol as a Surrogate
The ability of ψ-GSH to elevate brain thiol would reflect the former’s efficacy in affecting overall brain redox potential regardless of its distribution across compartments. It is therefore worthwhile to measure total brain thiol levels during ψ-GSH administration (Figure 2). Although the toxicity study suggested tolerance for far higher doses, we chose to administer 500 mg/kg ip since it was the minimum dose and route that caused reproducible and statistically significant changes in plasma and brain thiol levels. A single ip administration of ψ-GSH (500 mg/kg) elevated total plasma thiol 46-fold, from 0.29 ± 0.05 to 13.55 ± 0.73 nmol/μL within 30 min (Figure 2b). This decreased to 15-fold over basal concentration within 1 h and dropped to basal levels at 4 h. Brain thiol levels, however, elevated 5-fold from 0.78 ± 0.06 to 4.16 ± 0.16 nmol/mg of tissue over 2 h. This level was sustained until the discontinuation of the experiment at 4 h (Figure 2a). Oral administration of ψ-GSH (500 mg/kg) elevated plasma thiol levels from 0.21 ± 0.05 to 3.68 ± 0.480 nmol/μL over 30 min, reducing to basal levels over 2 h (Figure 2d). Corresponding brain thiol levels plateaued at 2.1-fold higher (1.10 ± 0.04 nmol/mg of tissue) than basal levels (0.51 ± 0.05 nmol/mg of tissue) at the 4 h time point (Figure 2c). Clearly, elevation of brain thiol levels caused by either ip or oral administration of ψ-GSH is sustained for a duration far longer than that in plasma. We have previously also examined the ability of ψ-GSH to permeate the blood–brain barrier through utilization of the active-transport mechanism for GSH in in vitro cell-monolayer experiments.14 The present finding that ψ-GSH administration causes sustained elevation of brain thiol (Figure 2) suggests that the aforementioned transport mechanism is relevant in the intact animal.
Figure 2.

Temporal dependence of brain and plasma thiol concentrations of C57BL/6 mice following ip (a, b) and oral (c, d) administration of 500 mg/kg ψ-GSH. Data is expressed as the mean ± SEM; ∗ indicates p < 0.0001, n = 4. Plasma thiols reached maxima within 30 min of ψ-GSH injection, while brain concentrations plateaued within 2–4 h.
B. Direct Determination of ψ-GSH Pharmacokinetics and Comparison with GSH
In vivo fates of ψ-GSH and GSH were compared by administration to wild-type C57BL/6 mice followed by plasma sampling and DTNB derivatization of samples before HPLC analysis. Following the peak in plasma levels upon administration, plasma concentrations of both declined in a logarithmically linear fashion. The plasma concentration–time curve fitted a one-compartment model with first-order absorption and elimination (Figure 3a). The Cmax of ψ-GSH (109.7 μg/mL) was 2.5-fold higher than that of GSH (43.94 μg/mL; Figure 3b), while its corresponding Tmax value (0.566 h) was 3-fold higher than that of GSH (0.336 h). ψ-GSH was eliminated considerably more slowly (elimination half-life, t1/2 = 1.227 h) than GSH (t1/2 = 0.495 h). Combined with a lower t1/2, the higher Cmax for ψ-GSH resulted in a 5.3-fold higher area under the curve (AUC) (267.4 h·μg/mL) compared with GSH (50.22 h·μg/mL) (Table 1). This substantiates the in vivo significance of the γ-GGT-resistant ureide isostere contained in ψ-GSH from comparison of its pharmacokinetic profile with GSH.
Figure 3.

(a) Nonlinear pharmacokinetic estimation of ψ-GSH (n = 4 per time point). The observed (○) and predicted (−) plasma concentrations are plotted as a function of time for ψ-GSH. Data were best fit by the initial one-compartment open model with first-order absorption and elimination. (b) Concentrations of ψ-GSH (●) and GSH (○) in plasma after a single ip dose of 500 mg/kg. Each time point represents the mean of four animals.
Table 1. Estimated Pharmacokinetic Parameters for ψ-GSH and GSH.
| PK Parameter | ψ-GSH | GSH |
|---|---|---|
| Cmax (μg/mL) | 109.7 | 43.94 |
| Tmax (h) | 0.566 | 0.336 |
| AUC (h·μg/mL) | 267.4 | 50.22 |
| K01 absorption half-life (h) | 0.171 | 0.127 |
| K10 elimination half-life (h) | 1.227 | 0.495 |
| CL_F clearance (mL/(h·kg)) | 1869 | 9955 |
Effects of ψ-GSH on Cognitive Function
Mild cognitive impairment (MCI) that precedes early AD (eAD) reflects synapse loss in the dentate gyrus of the hippocampal formation, with cognitive descent (expressed as lower spatial mnemonic and cued recall abilities) becoming tractable.15,16 The “Morris water maze” tool is a widely accepted modality for isolation of cognitive decline of hippocampal origin from unrelated behavioral impairment.17,18 In the hidden platform version of the Morris water maze, increasing ability of a mouse to locate this platform and to thereby “escape” the water during the training period indicates spatial memory acquisition. Specifically, lowering in the “escape latency”, that is, the time required for a mouse to locate the platform, reflects the degree of place-cell development in the hippocampus and the firing of neurons of the postsubiculum to afford momentary directional heading.19 These regions of the hippocampal formation are known to be among the primary foci of AD, and thus, it is expected that hippocampal lesion would increase escape latency.20 The transgenic mouse model (APP/PS1) employed in this study expresses human APPs and PS1-ΔE9 and develops elevated Aβ1–42 levels at the age of 4 months with manifest plaque deposition a month later.21 ψ-GSH (500 mg/kg ip) was administered to 3-month old APP/PS female mice three times per week for 12 consecutive weeks. Nontransgenic WT C57BL/6 mice were employed as negative controls. At 11 weeks of ψ-GSH treatment, all groups were evaluated in the “Morris water maze” learning evaluation technique (Figure 4). Mice treated with ψ-GSH showed significant learned behavior inculcation during the first week of testing, while untreated transgenic mice did not (Figure 4a). On days 3 and 4 of the training, escape latencies for mice treated with ψ-GSH (day 3, 17.91 ± 4.65 s; day 4, 14.64 ± 2.61 s) were similar to those for WT saline-treated mice (day 3, 17.29 ± 2.13 s; day 4, 12.42 ± 2.18 s). The escape latency values for ψ-GSH-treated APP/PS1 mice and those for saline-treated APP/PS1 mice were also significantly different (one-way ANOVA, p < 0.001). Path lengths traversed by the ψ-GSH-treated APP/PS1 mice correlated well with corresponding escape latencies (Figure 4b); thus on the last day of training, shorter path lengths were traversed by APP/PS1 mice treated with ψ-GSH (2.69 ± 0.47 m) than saline-treated APP/PS1 mice (5.76 ± 0.63 m). In fact, ψ-GSH-treated APP/PS1 mice traversed paths of lengths similar to saline-treated WT mice. The possibility of motor function improvement contributing to these improved abilities is excludable because swim speeds were similar across the groups (Supporting Information; Figure S3).
Figure 4.

Effect of ψ-GSH administration on spatial reference memory in a Morris water maze. (a, b) Performance during acquisition is expressed as the mean (±SEM) latency and path length required to locate a submerged platform by APP/PS1 saline, APP/PS1 ψ-GSH ip, and nontransgenic wild-type (WT) saline groups during four consecutive days of training (four trials per day). (c, d) Results of visible platform trials executed at the end of hidden platform training and the probe trial are represented as escape latency and path length as well. Retention of memory tested in the probe trial (e) 24 h after the hidden platform training showed a greater bias of ψ-GSH-treated mice to spend time in the area previously containing the platform (∗ p < 0.05, ∗∗ p < 0.001 compared with APP/PS1 saline group).
Learned tasks retention and acquired memory were evaluated by a single probe trial 24 h after conclusion of training (Figure 4e). APP/PS1 mice spent only 16.39% ± 3.39% of the time in the target quadrant, while the APP/PS1 (ip ψ-GSH) and nontransgenic vehicle controls retained significantly greater memory, spending 38.43% ± 3.73% and 36.67% ± 5.30%, respectively, in the target quadrant. The APP/PS1 mice treated with ψ-GSH also entered the target quadrant with significantly higher frequency than did the untreated APP/PS1 mice (data not shown). When the platform was rendered visible again (Figure 4c), the escape latencies of all the treatement groups were similar at the end of four trials (APP/PS1/saline, 6.64 ± 1.58 s; APP/PS1/ψ-GSH, 6.75 ± 1.49 s; WT/saline, 6.53 ± 0.75 s). Path lengths (Figure 4d) and swimming speeds (Supporting Information; Figure S3) across various groups also were similar, indicating no impact of the transgene with regard to reference memory.
Effect of ψ-GSH Therapy on Microscopic and Biochemical Indicators of Neurodegeneration
Excised brain tissue was examined for AD pathology indicators. Aβ plaque deposits were estimated by immunostaining brain sections with Aβ-antibody (Figure 5). Brain tissue sections of ψ-GSH-treated APP/PS1 mice had significantly lower Aβ load than the corresponding untreated mice. The number and sizes of Aβ plaques were drastically reduced in the ψ-GSH-treated group. Aβ1–42-specific ELISA revealed PBS and guanidine-soluble Aβ levels in the brain homogenates (Figure 5d,e). Brain tissue of APP/PS1 mice treated ip with ψ-GSH showed robust decrease in insoluble Aβ1–42 (45.4% ± 6.01% of vehicle-treated APP/PS1 mice; p < 0.0001; Figure 5d). Insoluble Aβ plaques are the ultimate result of preponderant β-sheet conformation in Aβ populations, presence of Aβ-derived AGEs, and their interactions with other glyco- and lipoproteins in the cytoplasm, CSF, or IST. ψ-GSH thus seems to have countered AGE-initiated or derived pathological processes. In contrast to Aβ plaques, PBS-soluble Aβ loads over all the groups of mice were not significantly different (Figure 5e). Soluble unaggregated Aβ1–42 is not toxic of its own accord, and higher levels of soluble Aβ1–42 are expected in the APP/PS1 mouse due to the transgene. Aβ1–42 aggregation orders are indeterminate through the ELISA utilized in this study. Behavioral results, however, allude to reduction of Aβ1–42 aggregation to toxic soluble oligomers.22 ψ-GSH is not designed to affect the formation of soluble Aβ; rather, it is designed to inhibit the misfolding and aggregation of Aβ. Unaffected soluble Aβ levels are thus expected, particularly in a transgenic mouse.
Figure 5.

Immunohistochemical detection of Aβ1–42 in ψ-GSH-treated mouse brain, magnification 10×: (a) wild-type (WT) saline control; (b) APP/PS1 saline; (c) APP/PS1 ψ-GSH ip. Amyloid plaques are observed as intensely yellow stained regions in sagital sections of brain. Mice treated with ψ-GSH showed dramatic reduction in size and number of plaques. (d, e) Quantitation of the effect of ψ-GSH treatment on amyloid load in APP/PS1 mice. Aβ1–42 levels in the brain homogenate of mice treated with saline or ψ-GSH for 12 weeks were measured by ELISA as described in Methods. (d) PBS-soluble Aβ1–42 levels were not different within treatments groups. (e) Significant reduction in PBS-insoluble (guanidine-soluble) Aβ1–42 was observed in ψ-GSH-treated APP/PS1 brain (∗∗∗, p < 0.0001 compared with saline-treated controls).
Levels of protein carbonyls, products of lipid peroxidation, and raw ROS herald incipient Aβ pathogenesis. Figure 6a,b shows the protein carbonyl levels and the extent of lipid peroxidation in ψ-GSH-treated and untreated APP/PS1 mice brains. The same were also measured in age-matched saline-treated WT mice as controls and were regarded as 100%. Protein carbonyls (detected as 2,4-dinitrophenylhydrazones) were elevated significantly in untreated APP/PS1 mice (154% ± 8.49%; p < 0.0001) relative to WT mice. Intraperitoneal ψ-GSH-treated APP/PS1 mice, however, had levels comparable to WT mice (97.8% ± 6.30%; p > 0.05; Figure 6a), lending credence to the premise that ψ-GSH acts upstream in the oxidative damage process by culling early originators of free-radical formation. Estimation of thiobarbituric acid reactive substance (TBARS) provides an empirical measure of the extent of lipid peroxidation, particularly that of the production of the peroxidation end product, malondialdehyde (Figure 6b). Lipid peroxidation is particularly concerning because it induces τ hyperphosphorylation, furthering tauopathy, the second etiologic component of AD.23 APP/PS1 mice treated with saline had significantly elevated (137% ± 9.58%; p < 0.001) levels of TBARS compared with WT controls. Treatment with ψ-GSH ip reduced TBARS levels in APP/PS1 mice to extents comparable to WT mice (95.3% ± 5.51%; p > 0.05). Consistent with protein carbonylation and lipid oxidation levels, total brain ROS levels were also found to be elevated in APP/PS1 mice (266% ± 11.78%; p < 0.0001) compared with WT mice (Figure 6c). The ip ψ-GSH treatment lowered these levels to a statistically insignificant 6% (p > 0.05) increase. Levels of MG in brain homogenates of the WT, APP/PS1, and ψ-GSH-treated APP/PS1 mouse were quantified to gauge the extent of its accumulation. As seen in Figure S4 (Supporting Information), the brains of ψ-GSH-treated mice were similar to the WT mice as far as their MG load status is concerned, while the untreated APP/PS1 mice had significantly higher MG loads. As discussed earlier, the GLO system is upregulated, albiet fruitlessly, in the Alzheimer’s afflicted brain. It must be understood that the GLO system in the intact animal cannot be studied without altering it. Obtaining affirmative proof of the underlying hypothesis of this study, that is, that “restoration of GLO activity corrects RCS accumulation and AD progression”, would require experimental correction of the GLO activity deficiency, essentially changing the system in question. The lowering of MG levels by ψ-GSH treatment is therefore the closest surrogate indicator of GLO activity restoration.
Figure 6.

Increased oxidative stress in the brains of APP/PS1 mice was decimated by ψ-GSH treatment. Protein carbonyls (a), thiobarbituric acid reactive substances (TBARS, b), and ROS (c) were measured as markers of oxidative stress as described in Methods. Results are shown as a percentage increase over levels of each oxidative marker in the brains of the nontransgenic (wild-type) saline-treated controls. Statistical significance was assessed by one-way ANOVA (∗∗ p < 0.001, ∗∗∗ p < 0.0001 compared with APP/PS1 saline-treated mice).
Conclusions
To date, all of the approaches aimed at increasing brain thiol levels were either biochemically nonspecific “antioxidants,” consisted of chemical entities that were GSH surrogates labile to normal metabolic processing, particularly that by γ-GGT, or were dependent on GSH-biosynthetic machinery that is hampered during AD. Validation of brain GLO activity restoration as a means to combat AD remained therefore an elusive goal, and hence efforts at the clinical development of a drug aimed at this mechanism were stymied. ψ-GSH is the first entity designed rationally to bypass the metabolic maze of GSH and function independently of GSH-related machinery in acting as a cofactor for brain GLO. This permitted the first evaluation of brain glyoxalase activity restoration as a potential means of combating AD. The approach appears to be remarkably effective, showcased by the ability of ψ-GSH to preclude all of the studied indicators of AD development in the APP/PS1 mouse model. Consequently, ψ-GSH stands established as a candidate for drug development.
Methods
Drugs and Reagents
All the chemicals including glutathione used in this study were obtained from Sigma (St. Louis, MO), unless stated otherwise. ψ-GSH was synthesized in our laboratory using a previously published synthetic method.14 Purity of ψ-GSH was confirmed with elemental analysis upon combustion as well as through chromatographic separation on a HPLC, and it was found to be >96% pure. Amyloid-β antibody (Cat. no. 2454) used for immunohistochemistry was obtained from Cell Signaling Technology International (Beverly, MA). DyLight 594 conjugated donkey anti-rabbit IgG H&L (Cat no. ab96921, Abcam, Cambridge, MA) was used as a secondary antibody.
Animals
The C57BL/6 wild-type and APP/PS1 double transgenic Alzheimer’s mice (APPswe/PS1ΔE9) were used in the present study. The mice were acquired from the Jackson Laboratory (Bar Harbor, ME) at the age of 6–10 weeks. All experimental procedures and animal handling were executed in accordance with the national ethics guidelines, approved and complied with all protocol requirements at the University of Minnesota, Minneapolis, MN (IACUC). For all experiments, animals were housed three per cage in our facility, in a controlled environment (temperature 22 °C, humidity 50–60%, and light from 07:00–19:00); food and water were available ad libitum.
Detection of Nonprotein Thiols in Plasma and Brain
Wild-type C57BL/6 mice at the age of 6–8 weeks (four per group) were treated with ψ-GSH ip. Mice were euthanized by overdose of ketamine and xylazine at the desired time points from the administration of the drug (0, 0.5, 1, 2, and 4 h after drug administration). Brain and plasma samples were collected. Brains were perfused with ice-cold 0.9% NaCl and cut sagittally into left and right hemispheres. The right hemisphere was fixed in Zamboni’s fixative as described under Immunohistochemistry. The left hemispehere was immediately frozen in liquid nitrogen and utilized in further biochemical analysis. Brain tissue was homogenized in ice-cold buffer (50 mM phosphate, pH 7.0, containing 1 mM EDTA) containing 5% sulfosalicylic acid. After homogenization, samples were centrifuged at 8000 × g for 10 min at 4 °C. The clear supernatant was transferred to a new tube and used for analysis of free thiol concentration.
Blood was collected in heparin-coated tubes (Fisher, Pittsburgh, PA) and immediately centrifuged at 5000 × g for 10 min at 4 °C. The top plasma layer was transferred into a new tube and mixed with an equal volume of 5% sulfosalicylic acid in 50 mM potassium phosphate buffer containing 1 mM EDTA disodium salt (pH 7.5). This was followed by centrifugation of samples at 8000 × g for 10 min at 4 °C and analysis of thiol concentration.
For analysis of thiol, DTNB (5,5′-dithio-bis-2-nitrobenzoic acid, Ellman’s reagent) was used, which liberated a yellow colored 5-thio-2-nitrobenzoic acid (TNB) upon reaction with thiol. The rate of TNB production is directly proportional to the concentration of free thiols in the sample. Total thiol concentration was normalized to the weight of tissue or microliters of plasma employed. Assay conditions employed 150 μL of Tris·HCl, pH 8.9 (Trizma base, 0.2 M; EDTA 0.02 M), 20 μL of DTNB (1 mg/mL in methanol), and 50 μL of supernatant from sulfosalicylic acid precipitation. The changes in absorbance were measured at 412 nm after 10 min incubation in the dark at room temperature.
Dose Determination Study
ψ-GSH was injected intraperitoneally in wild-type C57BL/6 mice (6–8 week old, four per group) three times a week for 2 weeks. The doses of ψ-GSH employed ranged from 100 to 2000 mg/kg. Body weight (every other day), complete blood count (CBC) analysis, and liver, kidney, and lung histology was considered to be indicative of toxicity from the drug treatment. Histological sections of tissue were analyzed by a board-certified pathologist at the Masonic Cancer Center-Comparative Pathology Shared Resource, University of Minnesota.
Pharmacokinetic Evaluation of ψ-GSH
Eight week old C57BL/6 female mice (n = 4 animals per time point) were given 500 mg/kg of GSH or ψ-GSH in saline intraperitoneally. Blood samples were collected at 0, 5, 15, and 30 min and 1, 2, 4, 8, and 24 h after the drug administration through retro-orbital bleeding into heparin-coated tubes. Blood was immediately centrifuged at 5000 × g for 10 min at 4 °C. The top plasma layer was transferred into a new tube and mixed with an equal volume of 5% sulfosalicylic acid in 50 M potassium phosphate buffer containing 1 mM EDTA disodium salt (pH 7.2). This was followed by centrifugation of samples at 8000 × g for 10 min at 4 °C and analysis of thiol concentration. The derivatization reaction was realized utilizing 130 μL of the supernatant, 500 μL of Tris·HCl buffer 0.5 M, pH 8.9, and 350 μL of DTNB 10 mM in K2HPO4 (0.5 M, pH 8.0). After 5 min in an ice bath, the reaction mixture was acidified by addition of 100 μL of H3PO4 7 M and centrifuged at 3000 × g for 10 min. Then, derivatized GSH or ψ-GSH was filtered through a 0.22 μm membrane, and 20 μL was injected in HPLC at 330 nm. HPLC analysis was carried out on a Beckman Coulter Gold HPLC system, controlled by a system controller (32 Karat workstation with PC). The HPLC conditions employed a Varian Microsorb column (C18, 5 μm, 4.6 mm × 250 mm) using solvent A [water containing 0.9% v/v formic acid] and solvent B [acetonitrile] with 1 mL/min flow rate and detected at 330 nm. Solvent B was increased from 0 to 30% in 9 min, then to 80% in 7 min, and held at 80% for an additional 4 min. The retention time for derivatized ψ-GSH was 11.8 min.
Efficacy Study Design
To determine the effect of intraperitoneal ψ-GSH treatment, the study was divided in three groups (10 mice/group): (1) APP/PS1 mice–vehicle control; (2) APP/PS1 mice−ψ-GSH ip 500 mg/kg; (3) nontransgenic wild-type mice–vehicle control. All the animals in the above groups were treated with saline or ψ-GSH three times a week for 12 weeks, starting at the age of 3–4 months. Eleven weeks after the start of the treatment, the animals were tested in the Morris water maze, and 12 weeks after the start of the treatment, the animals were sacrificed for histopathological analysis.
Behavior Study
Eleven weeks after the beginning of the treatment, the animals were tested for 1 week in an open field water maze. The maze consists of a white circular tank (120 cm diameter) of water (23 ± 1 °C) rendered opaque with nontoxic crayola paint. The mice were placed in the water at the edge of the pool and allowed to swim in order to locate a hidden but fixed escape platform, using extra-maze cues.
On day 1, the mice were be placed in the pool and allowed to swim freely for 60 s. If an animal could not locate the hidden platform within that time, it was placed manually upon the platform and left there for 10 s. Each animal was tested in four trials per day. The intertrial interval was 15 min. Each start position was used equally in a pseudorandom order, and the animals were always placed in the water facing the wall. The platform was placed in the middle of the northeast quadrant of the pool (approximately 30 cm from the side of the pool). The mouse’s task throughout the experiment was to find and climb onto the platform, escaping the water. Once the mouse learned the task (day 4, trial 16), a probe trial was conducted 24 h after the last trial of acquisition on day 4.
In the probe trial (i.e., trial 17, day 5), the platform was removed from the pool and animals were allowed to swim for 60 s. Their ability to remember the location of the previously present escape platform was quantified in proportion to the residence time of the mouse in the quadrant previously containing the platform. Immediately following the probe trial, visible platform was reintroduced at a different location (i.e., southeast quadrant) along with a visible flag. Mice were trained to locate this cued platform in four trials conducted in the aforementioned manner.
Immunohistochemistry
Mice were anesthetized, and their brains were removed and cut sagittaly. Following fixation of right hemisphere in Zamboni’s fixative (4% paraformaldehyde and 15% picric acid in 0.25 M sodium phosphate, pH 7.5) for 48 h before being cryopreserved in a 30% (wt/vol) sucrose in phosphate-buffered saline solution, brains were then were embedded in O.C.T. Tissue Tek Compound (Miles Scientific) and were cryosectioned at 10 μm thickness for immunohistochemistry, mounted on Superfrost microscope slides (Erie Scientific, Portsmouth NH) and stored at −20 °C until their utilization.
The brain sections were stained with a primary antibody against β-amyloid peptide (Cell signaling, Cat. no. 2454) at a dilution of 1:200 in PBS. After an overnight incubation at 4 °C, the sections were rinsed three times for 10 min with PBS and then incubated for 2 h in donkey anti-rabbit IgG conjugated to Cy3 diluted 1:1000 in PBS. The sections were rinsed in PBS 3 times for 10 min and stained with a nuclear stain, DAPI, before being dehydrated in graded ethanol series and mounted in DPX prior to being coverslipped. Slides treated with the same technique but without incubation with the primary antibody were used as negative controls. Slides were observed under an Olympus microscope with confocal immunofluorescence.
Amyloid-β1–42 ELISA
Total Aβ1–42 in the brain was quantified by sandwich ELISA (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Wild-type and transgenic Alzheimer’s mice brains were Dounce homogenized in phosphate-buffered saline (PBS), pH 7.2, containing a mixture of protease inhibitors. Brain homogenates were centrifuged, and supernatants were assayed for soluble Aβ1–42 per manufacturers’ protocols. The pellets were suspended in guanidine buffer (5.0 M guanidine·HCl/50 mM Tris·HCl, pH 8.0) to dissolve all intra- and extracellular aggregates, incubated for 3 h at room temperature, diluted with 1× Dulbecco’s phosphate-buffered saline containing 5% BSA and protease inhibitors to reduce the concentration of guanidine·HCl to 0.5 M, and centrifuged at 16 000 × g for 20 min at 4 °C. Supernatants were loaded into well-established commercially available anti-Aβ42 sandwich ELISA plates and processed according to the manufacturer’s instructions (Invitrogen). The concentration of both PBS soluble and insoluble (guanidine soluble) Aβ1–42 was calculated from a standard curve of human Aβ1–42 and was normalized to the weight of the tissue employed.
Protein Carbonyl Determination
Excised brain tissue was homogenized in the cold utilizing a buffer (50 mM phosphate, pH = 7.00 with 1 mM EDTA and supplemented with protease inhibitors), and spun at 10 000 × g at 4 °C for 15 min. The clear supernatant was diluted with 0.5 mL of 2,4-dinitrophenylhydrazine (DNPH) in 2 N HCl. Blanks consisted of 2 N HCl alone. Samples were incubated in the dark at ambient temperature for 1 h with intermittent agitation. Thereafter, 0.5 mL of TCA (20% aqueous) was added to precipitate all proteins, and the precipitate was separated through centrifugation for 10 min at 10 000 × g at 4 °C. The protein pellet was agitated with EtOH–EtOAc (1:1, 1 mL) for 1 min to remove unreacted DNPH, and the supernatant upon centrifugation of this mixture (10 000 × g, 10 min) was discarded. Washing in this manner was repeated two more times. The resultant pellet was then suspended in guanidine HCl (6 M aqueous). The mixtures were held at 37 °C for 30 min to promote protein dissolution. Centrifugation then removed any insoluble material. The clear supernatant was then evaluated for DNPH-derivatized protein at the λmax of a DNPH hydrazone (360 nm) and the ε of 22 000 M–1·cm–1. Total protein was determined through the BCA assay utilizing a kit (Pierce, Rockford, IL). Carbonyl content was proportioned to protein content and represented as percentage increase over the samples obtained from saline-treated APP/PS1 mice.
Lipid Peroxidation Assay
The extent of lipid peroxidation was correlated to the levels of thiobarbituric (TBA) acid-reactive entities (TBARS assay). Homogenized brains were exposed to TBA–trichloroacetic acid (TCA)–chloridic acid reagent (HCl; 0.37% TBA, 0.25 N HCl, and 15% TCA) (1:1:1, 2 mL) and incubated for 15 min at ambient temperature. Centrifugation of the incubate was followed by measurement of the absorption of the supernatant at 535 nm against a blank reference. Total protein was determined through the BCA protein assay. TBARS where then proportioned to total protein content and represented as percentage of the values in saline-treated nontransgenic control mice.
ROS Assay
The 2′,7′-dichlorofluorescein (DCFH-DA) assay was utilized for gauging ROS levels. To ensure the lack of interference of light-induced oxidation, this assay was conducted in the dark. Brain tissue, homogenized in phosphate buffer (50 mM with 1 mM EDTA, pH = 7.2), was centrifuged (10 000 × g) for 10 min. Supernatants were treated with DCFH-DA to a final concentration of 100 μM of DCFA, and the mixture was held at 37 °C for 20 min. The reaction was then halted by chilling it on ice, and the generation of the fluorescent quinone DCF was quantitated with a fluorescent plate reader (BioTek SynergyHT, VT, USA) using Xc = 488 nm and Ec = 525 nm. Background autofluorescence was accounted for by subtracting the values obtained immediately after addition of DCFH-DA. These values were proportioned to total protein concentration and finally expressed as percentages of the values obtained from similarly processed brain homogenates from saline-treated WT mice.
Acknowledgments
The authors profusely thank Linda Kotilinek of the N. Bud Grossman Center for Memory Research and Care, University of Minnesota, for guidance during the Morris water maze experiments and Professor Karen Ashe for providing the facility to conduct the experiments. Professor Gerard O’Sullivan of Masonic Cancer Center-Comparative Pathology Shared Resource, University of Minnesota, generously performed histopathological analysis of organ samples. Professor Marna Ericson, Center for Drug Design, University of Minnesota, is gratefully acknowledged for her guidance during immunohistochemistry and imaging. The authors thank Jaime Nugent, Center for Drug Design, University of Minnesota, for technical assistance during the in vivo experiments.
Supporting Information Available
Short-term toxicity study of ψ-GSH in WT mice describing weight changes, liver and kidney function analyses, effect of ψ-GSH treatment on speeds of mice in hidden and visible platform training, and effect of ψ-GSH treatment on MG concentration in mouse brain. This material is available free of charge via the Internet at http://pubs.acs.org.
This research was supported by funds from the Center for Drug Design at the University of Minnesota.
The authors declare no competing financial interest.
Supplementary Material
References
- Chen K.; Maley J.; Yu P. H. (2006) Potential implications of endogenous aldehydes in beta-amyloid misfolding, oligomerization and fibrillogenesis. J. Neurochem. 99, 1413–1424. [DOI] [PubMed] [Google Scholar]
- Rabbani N.; Thornalley P. J. (2012) Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids 42, 1133–1142. [DOI] [PubMed] [Google Scholar]
- Li X. H., Xie J. Z., Lv B. L., Cheng X. S., Du L. L., Zhang J. Y., Wang J. Z., Zhou X. W. (2012) Methylglyoxal Induces Tau Hyperphosphorylation via Promoting AGEs Formation. Neuromolecular Med. Epub ahead of print, DOI: 10.1007/s12017-012-8191-0. [DOI] [PubMed] [Google Scholar]
- Gomes R.; Silva M. S.; Quintas A.; Cordeiro C.; Freire A.; Paulino P.; Martins A.; Monteiro E.; Barroso E.; Freire A. P. (2005) Argpyrimidine, a methylglyoxal-derived advanced glycation end-product in familial amyloidotic polyneuropathy. Biochem. J. 385, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhla B.; Luth H. J.; Haferburg D.; Weick M.; Reichenbach A.; Arendt T.; Munch G. (2006) Pathological effects of glyoxalase I inhibition in SH-SY5Y neuroblastoma cells. J. Neurosci. Res. 83, 1591–1600. [DOI] [PubMed] [Google Scholar]
- Kuhla B.; Boeck K.; Schmidt A.; Ogunlade V.; Arendt T.; Munch G.; Luth H. J. (2007) Age- and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer’s disease brains. Neurobiol. Aging. 28, 29–41. [DOI] [PubMed] [Google Scholar]
- Pocernich C. B.; Butterfield A. D. (2012) Age- and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer’s disease brains. Biochem. Biophys. Acta 1822, 625–630.22015471 [Google Scholar]
- Birkenmeier G.; Stegemann C.; Hoffmann R.; Günther R.; Huse K.; Birkemeyer C. (2010) Posttranslational Modification of Human Glyoxalase 1 Indicates Redox-Dependent Regulation. PLoS One 5, e10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A. J. L. (1997) Glutathione in the brain: Disorders of glutathione metabolism. In The Molecular and Genetic Basis of Neurological Disease. (Rosenberg R. N., Prusiner S. B., Rimauro S., Barchi R. L., Kunk L. M. eds) pp 1195–1230, Butterworth-Heinemann, Boston. einemann. [Google Scholar]
- Farr S. A.; Poon H. F.; Dogrukol-Ak D.; Drake J.; Banks W. A.; Eyerman E.; Butterfield D. A.; Morley J. E. (2003) The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J. Neuro-chem. 84, 1173–1183. [DOI] [PubMed] [Google Scholar]
- Liu H.; Wang H.; Shenvi S.; Hagen T. M.; Liu R. M. (2006) Glutathione metabolism during aging and in Alzheimer disease. Ann. N.Y. Acad. Sci. 1019, 346–349. [DOI] [PubMed] [Google Scholar]
- Chen C. N.; Brown-Borg H. M.; Rakoczy S. G.; Ferrington D. A.; Thompson L. V. (2010) Aging impairs the expression of the catalytic subunit of glutamate cysteine ligase in soleus muscle under stress. J. Gerontol. Ser. A 65, 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel A.; Cikryt P. (1980) The level and half-life of glutthione in human plasma. FEBS Lett. 120, 209–211. [DOI] [PubMed] [Google Scholar]
- More S. S.; Vince R. (2012) Potential of a γ-glutamyltranspeptidase-stable glutathione analogue against amyloid-β toxicity. ACS Chem. Neurosci. 3, 204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff S. W.; Price D. A.; Schmitt F. A.; Mufson E. J. (2006) Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging. 27, 1372–1384. [DOI] [PubMed] [Google Scholar]
- Laczó J.; Andel R.; Vyhnalek M.; Vlcek K.; Magerova H.; Varjassyova A.; Nedelska Z.; Gazova I.; Bojar M.; Sherdova K.; Hort J. (2012) From morris water maze to computer tests in prediction of Alzheimer’s disease. Neurodegener. Dis. 10, 153–157. [DOI] [PubMed] [Google Scholar]
- Vorhees C. V.; Williams M. T. (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nature protocols 1, 848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. G.; Garrud P.; Rawling J. N.; O’Keefe J. (1982) Place navigation imparied in rats with hippocampal lesions. Nature 297, 681–683. [DOI] [PubMed] [Google Scholar]
- Brown M. A.; Sharp P. E. (2004) Simulation of spatial learning in the morris water maze by a neural network model of the hippocampal formation and nucleus accumbens. Hippocampus 5, 171–188. [DOI] [PubMed] [Google Scholar]
- Taube J. S.; Muller R. U.; Ranck J. B. Jr. (1990) Head-direction cells recorded from the postsubiculum in freely moving rats. I. description and quantitative analysis. J. Neurosci. 10, 420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchese F.; Liu S.; Battaglia F.; Walter S.; Mathews P. M.; Arancio O. (2004) Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann. Neurol. 55, 801–814. [DOI] [PubMed] [Google Scholar]
- Tabaton M.; Gambetti P. (2006) Soluble amyloid-beta in the brain: the scarlet pimpernel. J. Alzheimers Dis. 9, 127–132. [DOI] [PubMed] [Google Scholar]
- Gómez-Ramos A.; Díaz-Nido I.; Smith M. A.; Perry G.; Avila J. (2003) Effect of lipid peroxidation product acrolein on tau phosphorylation in neural cells. J. Neurosci. Res. 15, 863–870. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.