

Abstract
Fibrates are used as lipid-lowering drugs to prevent cardiovascular pathology. Fibrates are ligands of peroxisome proliferator-activated receptor α (PPARα). Besides altering lipid metabolism, PPARα ligands exert anti-inflammatory effects on various cell types. In this study, we hypothesized that PPARα agonists exert beneficial effects on osteoarthritis (OA) and rheumatoid arthritis (RA) by their local anti-inflammatory effects, but also by their systemic influences.
A systematic literature search of Medline and EMBASE databases was performed up to August 2011. The main search items were osteoarthritis, rheumatoid arthritis, peroxisome proliferator-activated receptor alpha and fibrates. Inclusion criteria were in vivo or in vitro studies regarding humans or animals in which the effects of PPARα ligands were studied.
Six in vivo human studies, four in vivo animal studies and seven in vitro studies were included. The in vivo human studies showed all beneficial clinical effects of PPARα ligands, but studies were small and only four were randomized. Ligands for PPARα significantly reduced pain, swelling of the joints and decreased systemic inflammatory markers. In vitro and in vivo animal studies indicate that PPARα agonists inhibit bone resorption, and reduce inflammatory and destructive responses in cartilage and synovium.
PPARα agonists such as fibrates should be considered as potential therapeutic strategy for RA. There is no clinical evidence for their use in OA, although in vitro studies indicate that PPARα agonists demonstrate different joint-protective effects locally, and systemic effects on inflammation, serum lipid levels and vascular pathology. Animal studies should be performed and after confirmation of the protective effects of PPARα, large randomized controlled trials could investigate fibrates in OA and RA.
Keywords: fibrates, osteoarthritis, peroxisome proliferator activated receptor α, rheumatoid arthritis
Introduction
Osteoarthritis (OA) is primarily described as a disease of cartilage damage, although there is growing evidence that synovium (hyperplasia and infiltration of monocytes), subchondral bone (remodeling) and adipose tissue (secretion of chemokines, cytokines and growth factors) are also involved [Aspden, 2008; Hunter, 2009; Clockaerts et al. 2010]. Recently, OA has been linked with atheromatous vascular disease, systemic inflammation caused by obesity, and dyslipidemia [Conaghan et al. 2005; Wang et al. 2008, 2009; Davies-Tuck et al. 2009]. Atheromatous vascular disease in the subchondral bone might enhance the OA disease process by decreasing cartilage nutrition or through direct ischemic effects on the subchondral bone [Conaghan et al. 2005]. In addition, visceral adipose tissue secretes cytokines and adipokines that are able to stimulate inflammatory and catabolic processes in the cartilage. Fatty acids have been shown to influence cartilage metabolism [Henrotin et al. 2003]. Based on these studies, OA might partly be considered as a systemic and metabolic disease [Katz et al. 2010].
Rheumatoid arthritis (RA) is a systemic immune disease, in which activated monocytes/macrophages produce proinflammatory cytokines and chemokines, and stimulate CD4+ T-cell-mediated immune reactions which induce inflammatory and destructive processes in synovium, cartilage, bone and bone marrow [Jenkins et al. 2002; Pratt et al. 2009; Schett and Firestein, 2010]. The destructive processes in cartilage might be stimulated by lipoproteins, such as low-density lipoprotein (LDL). Lipoproteins are involved in cartilage degradation in patients with RA by enhancing production of matrix metalloproteinase (MMP)-3 and reactive oxygen species (ROS) [Henrotin et al. 2003; Kakinuma et al. 2004]. Similar to OA, RA is associated with cardiovascular disease, type II diabetes mellitus and dyslipidemia, indicating that metabolic diseases are a part of the RA disease process [Shahin et al. 2010].
The search for disease-modifying drugs for OA has mainly focused on cartilage. However, the disease process in both OA and RA is probably influenced by systemic and metabolic disturbances and inflammatory processes. In this regard, peroxisome proliferator-activated receptor α (PPARα) ligands such as fibrates might be of interest. PPARα agonists reduce serum triglycerides, increase serum high-density lipoproteins and have a moderate lowering effect on serum LDLs. PPARα ligands exert anti-inflammatory effects on various cell types, including macrophages, smooth muscle cells, endothelial cells, lymphocytes and other cell types. The activation of PPARα inhibits the nuclear translocation of nuclear factor κB (NFκB) by the upregulation of inhibitor κB (IκB) [Chung et al. 2008; Lalloyer and Staels, 2010]. Other inflammatory pathways such as mitogen-activated protein kinase phosphorylation may also be influenced by fibrates [Crisafulli and Cuzzocrea, 2009]. As shown in Figure 1, fibrates may have a pleiotropic effect on OA and RA through their effect on several pathways. By exerting anti-inflammatory effects, they can decrease cartilage and bone destruction and have less synovial and systemic inflammation. In addition, fibrates decrease the influence of the dislipidemiae and vascular pathology [Findlay, 2007; Wang et al. 2008; Alagona, 2010; Clockaerts et al. 2010; Rubenfire et al. 2010; Krysiak et al. 2011].
Figure 1.

Pleiotropic effect of fibrates on osteoarthritis and rheumatoid arthritis.
We performed a systematic search for studies investigating the effects of fibrates or other PPARα ligands in in vitro and in vivo studies on OA and RA. In this study, we hypothesized that PPARα agonists exert beneficial effects on OA and RA by their anti-inflammatory effects, but also by their systemic influences.
Methods
Data sources and searches
A systematic literature search of Medline and EMBASE databases was performed for studies in English, Dutch and German up to August 2011. The main search items were osteoarthritis, rheumatoid arthritis, fibrates and PPARα. The following keywords were used: (arthritis OR osteoarthrit*) OR (cartilage OR synovium OR chondrocytes OR synoviocytes OR subchondral bone OR osteoclasts OR osteoblasts) AND ((‘peroxisome proliferator activated’ NEAR/2 receptor*) OR PPAR* OR (fibric NEAR/2 acid*) OR clofibr* OR fibrate* OR atromid OR beclobrate OR beclobrinic acid OR bezafibrate OR binifibrate OR ciprofibrate OR clinofibrate OR dulofibrate OR eniclobrate OR etofibrate OR etofylline clofibrate OR fenofibrate OR fenofibric acid OR halofenate OR metaglidasen OR methylclofenapate OR nicofibrate OR simfibrate OR tazasubrate).
Additionally, reference lists of the selected papers were screened for further publications.
Study selection
Articles were screened by their title and abstract by one observer (ICMvE) and checked by a second observer (SC). After inclusion, full text articles were read for further screening. Inclusion criteria were in vitro studies using human or animal material, in vivo animal studies, randomized controlled trials (RCTs) or clinical trials in which the effects of PPARα ligands on OA or RA were studied. Case reports, editorials, reviews and letters were excluded.
Assessment of quality and data extraction
Methodological quality of the clinical studies was assessed by one observer (ICMvE) and checked by a second observer (SC) using the quality scoring table of the Cochrane reviews with a maximum score of five points and a minimum score of 0 points representing sequence generation, allocation concealment, blinding of participants, personnel and outcome assessors, data outcome and selective outcome reporting. Quality items of in vivo animal studies were analyzed with a list of quality criteria compiled by Sniekers and colleagues [Sniekers et al. 2008]. For in vitro studies no quality items were scored. The scores of publications were noted and data were pooled independently of outcome quality.
Data of the in vivo human and animal studies were extracted by one observer (ICMvE) and in vitro data were summarized by another observer (SC). All general features, such as author, journal, year of publication, aim, type of drug used and patient information were extracted. The specific variables for in vivo human studies were level of pain [visual analog score (VAS) of the patient], scores for clinical assessment based on morning stiffness, grip strength and articular index [Ritchie et al. 1969], inflammatory markers [C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR)], and blood pressure as a marker for cardiovascular disease.
The variables extracted from the animal studies consisted of joint swelling, assessment of histology (synovial hyperplasia and infiltration of immune cells) serum parameters (such as CRP, ESR), radiographic changes and bone volume.
Data synthesis and analysis
Clinical data were standardized by calculating effect sizes [with mean ± standard deviation (SD); difference in outcomes for both groups divided by the SD of the outcome]. If the data were not expressed as mean ± SD, we entered an increase (↑) or reduction (↓) of the symptoms, or no effect (o) depending on the statistical significance of the results (p < 0.05).
The results were summarized separately for clinical effects (clinical severity and occurrence of RA or OA) and mechanistic effects (systemic inflammatory parameters, inflammation and destruction in cartilage, synovial immune cell infiltration, bone changes, improvement in systemic and local lipid profile and prevention of vascular pathology).
Results
A total of 19 publications met all the inclusion criteria: six in vivo human studies, four in vivo animal studies and nine in vitro studies (Figure 2). The six in vivo studies were scored for methodological quality resulting in a maximum score of 5. As shown in Table 1, all in vivo human studies were of poor methodological quality, resulting in a high risk of bias. The study of Goto scored best with 3 (missing clear allocation concealment and free of other bias) and the noncontrolled trial of Forster and colleagues scored worst with 0 (Table 1) [Forster and McConkey, 1986; Goto, 2010]. The animal studies were screened based on the list of quality compiled by Sniekers and colleagues [Sniekers et al. 2008]. The most prevalent shortcomings were the mentioning of dropouts, sample size number, age and body weight of the animals.
Figure 2.
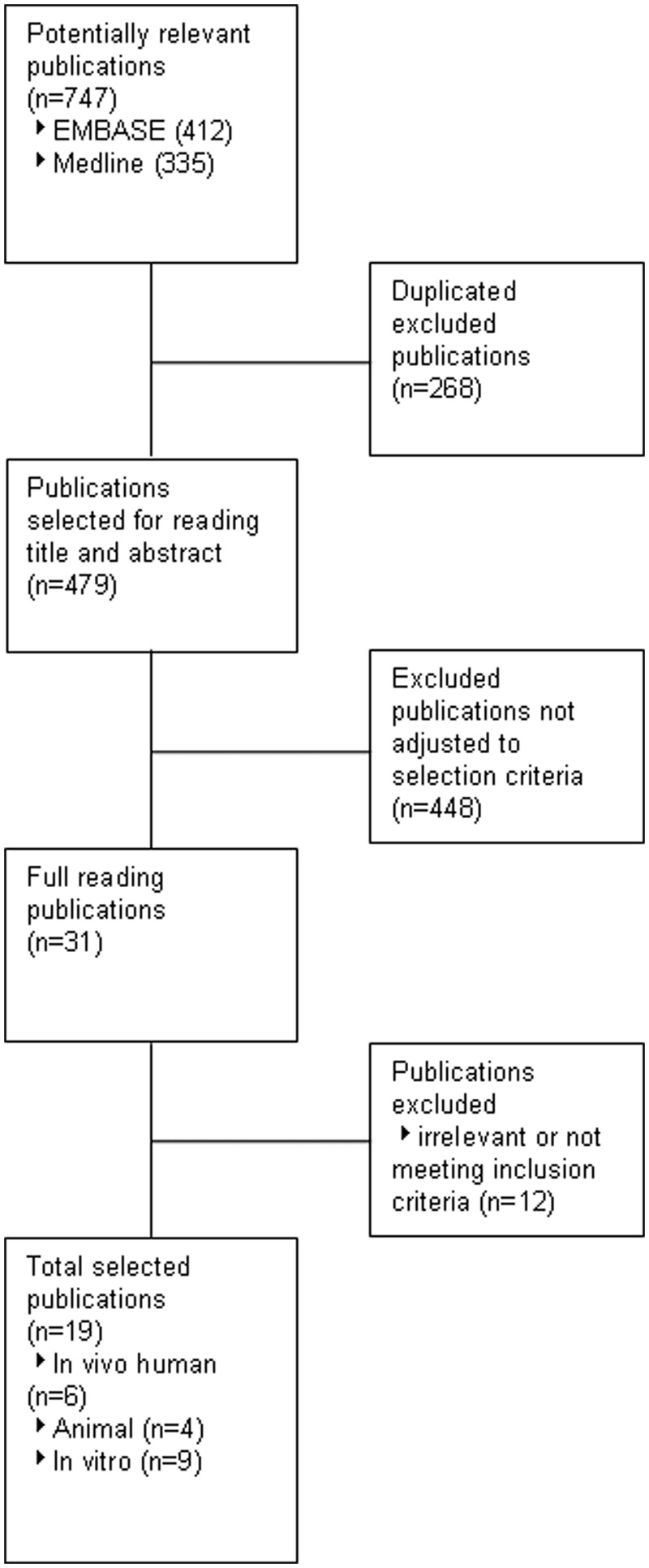
Flow chart.
Table 1.
Overview of in vivo human trials.
| Study | Type | Score | Patients (n) |
Drug |
Follow up | Outcome |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control | Intervention | Type | Dosage | Painmarker |
BPD |
Function |
||||||||||
| VAS | CRP | ESR | Sys | Dias | Grip strength | Morning stiffness | Articular index | |||||||||
| Goto$ [2010] | RCT | RA | 3 | 21 | 23 | Fenofibrate | 200 mg/day | 6 months | 0.67* | 0.1 | –0.03 | 0.06 | –0.07 | - | - | – |
| Bird et al.$ [1983] | RCT | RA | 2 | 31‡ | 25 | Clobuzarit | 100 and 300 mg/day | 24 weeks | ↓* | ↓* | ↓*/↓* | – | – | ↑*/↑* | o/↓* | o/↑* |
| Dixon et al.$ [1983] | RCT | RA | 2 | 60‡ | 30 | Clobuzarit | 100 and 300 mg/day | 24 weeks | – | 0.10/0.85* | – | – | – | 0.60/0.53 | 0.82/0.86* | 0.70/0.75* |
| Reynolds et al.$ [1981] | RCT | RA | 1 | 20 | 20 | Clobuzarit | 50 mg/day | 6 months | – | ↓ | ↓ | – | – | ↑ | ↓ | ↑ |
| McConkey et al.$ [1980] | T | RA | 1 | – | 34 | Clobuzarit | 50–200 mg/day § | 1 year | – | ↓* | ↓* | – | – | – | - | – |
| Forster and McConkey [1986] | T | RA | 0 | - | 3 | Clobuzarit | 200 mg/day | 2 years | – | ↓ | – | – | – | – | – | – |
Outcomes given as effect size (calculated with difference in outcomes for both groups divided by the standard deviation of the outcome).
p < 0.05; –, missing value; ↑, increase, effect size cannot be determined; ↓, reduction, effect size cannot be determined.
Compares with baseline only.
Four groups taken together into patients given clofibrate (100 mg and 300 mg a day) and not given clofibrate.
Started with 50 mg/day and increased in stages to 200 mg/day.
BPD, blood pressure; co, crossover; CRP, C-reactive protein; DAS, disease activity score; Dias, diastolic; ESR, erythrocyte sedimentation rate; RA, rheumatoid arthritis; RCT, randomized controlled trial, SJC, swollen joint count; Sys, systolic; T, trial one group only; TJC, tender joint count; VAS, visual analog score.
Clinical effects
Human studies
Four randomized controlled trials and two noncontrolled trials have been reported for the use of fibrates in RA (Table 1). Goto and Bird and colleagues reported a significant reduction in VAS score in patients with RA and dislipidemia after treatment with fibrates [Bird et al. 1983, Goto, 2010]. Reynolds and colleagues mentioned a nonsignificant reduction in pain [Reynolds et al. 1981]. McConkey and colleagues demonstrated a significant reduction in clinical score (subjective clinical state score) for RA based on questionnaires in patients feeling better or worse after treatment with fibrates than before treatment [McConkey et al. 1980]. Dixon and colleagues and Bird and colleagues reported a significant improvement in the articular index [Ritchie et al. 1969] and early morning stiffness after treatment with 300 mg clobuzarit. This reduction was not seen with 100 mg clobuzarit [Bird et al. 1983; Dixon et al. 1983]. Bird and colleagues also showed a significant improvement in grip strength when 100 mg or 300 mg clobuzarit was given, which was not seen in the study by Dixon and colleagues [Bird et al. 1983; Dixon et al. 1983]. Reynolds and colleagues found no improvement in all assessments of function after the use of 50 mg clobuzarit [Reynolds et al. 1981]. No clinical studies for OA have been performed.
Animal studies
Three publications reported the clinical effect on RA through collagen-induced arthritis (CIA) or adjuvant-induced arthritis (AIA) (Table 2). Bloxham and colleagues investigated swelling of the joints in rats and found that 100 mg/kg clobuzarit significantly reduced swelling in AIA and 60 mg/kg clobuzarit significantly reduced joint swelling in CIA [Bloxham et al. 1990]. Okamoto and colleagues showed a significant reduction in swelling of joints after treatment with fenofibrate [Okamoto et al. 2005]. This was confirmed by Castillero and colleagues, who found a significant reduction in swelling and erythema in rats treated with 300 mg/kg fenofibrate [Castillero et al. 2011].
Table 2.
Overview of animal studies.
| Study | Animal (strain) | Treatment | Administration | Dosage | Frequency | Age | Number | Follow up | Outcome |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Swollen joints | Infiltration immune cells | Bone | Effect | ||||||||||
| Okamoto et al. [2005] | Rats (Lewis) | AIA | Fenofibrate | Oral | 10 mg/kg 100 mg/kg |
Daily | 7 weeks | – | 18 days | ↓* | ↓* | – | – |
| Castillero et al. [2011] | Rats (Wistar) | AIA | Fenofibrate | Oral | 300 mg/kg | Daily | N = 16/16/16 | 15 days | ↓* | – | – | ||
| Bloxham et al. [1990] | Rats (AHH/R) Rats (AP1/R2) |
AIA CIA |
Clobuzarit | Oral | 10 mg/kg 30 mg/kg 100 mg/kg 60 mg/kg |
Daily |
N = 5/5/5/5/5/5/5 N = 8/8/8/8/8/8/8 |
5 days 15 days |
↓ ↓ ↓* ↓* |
– | Radiographic assessment | 0.97* | |
| Still et al. [2008] | Rats (Wistar) |
- | Bezafibrate | Subcutaneous injection | 10 mg/kg | Daily | 3 months | N = 15/15/15 | 12 weeks | – | – | Trabecular bone Cortical bone Bone mass |
0.55*
0.96* 0.13 |
Outcomes given as effect size (calculated with difference in outcomes for both groups divided by the standard deviation of the outcome).
p < 0.05; ↓, reduction, effect size cannot be determined.
AIA, adjuvant-induced arthritis; CIA, collagen-induced arthritis.
Mechanisms
We reviewed possible etiological factors of OA and RA, such as systemic inflammation, destruction of cartilage, synovitis, bone changes, vascular pathology and altered lipid profiles, which are potentially altered by fibrates.
Reduction of systemic inflammatory parameters
McConkey and colleagues reported a significant reduction in CRP and ESR serum levels with fibrate treatment in patients with RA, while in the study by Goto, fibrates had no effect [McConkey et al. 1980; Goto, 2010]. Bird and colleagues demonstrated a significant reduction in CRP and ESR after treating patients with RA with 300 mg clofibrate, which was also shown by Dixon and colleagues for CRP [Bird et al. 1983; Dixon et al. 1983]. Reynolds and colleagues reported a nonsignificant reduction in CRP and ESR levels [Reynolds et al. 1981].
Reduction of inflammation and destruction in cartilage
None of the human trials investigated the effects of fibrates on cartilage. In addition, cartilage was never evaluated in the studies examining the effects of fibrates using animal models for OA or RA. However, seven in vitro studies investigated the effect of fibrates or other PPARα ligands on cartilage.
In in vitro studies, PPARα agonists were able to counteract the interleukin (IL)-1β-induced increase in MMP1, -3 and -13 production in cartilage [Francois et al. 2006; Clockaerts et al. 2011]. Similar effects were also observed for production of nitric oxide, prostaglandin E2 and release of glycosaminoglycans, although this was contradicted by Bordji and colleagues and Fahmi and colleagues, who reported that the addition of PPARα ligands did not counteract the effects of IL1β on the production of nitric oxide proteoglycan synthesis and MMP-13 by rat and human chondrocytes [Bordji et al. 2000; Fahmi et al. 2001; Clockaerts et al. 2011]. PPARα ligands inhibited the transforming growth factor (TGF)-β stimulated production of proteoglycans and collagen in chondrocytes [Poleni et al. 2007, 2010]. PPARα ligands also inhibited the stimulatory effect of TGF-β1 on tissue inhibitor of matrix metalloproteinase 1 gene expression and protein synthesis [Poleni et al. 2010].
Reduction in synovial immune cell infiltration
No human clinical studies have examined the effect of fibrates on synovium.
Okamoto and colleagues significantly suppressed the progression of arthritis by adding fenofibrate in an adjuvant-induced arthritis model in female Lewis rats. Histological assessment demonstrated a reduction in infiltration of immune cells in synovial tissue and less pannus formation [Okamoto et al. 2005].
In vitro data demonstrate that production of IL-6, IL-8 and granulocyte monocyte colony-stimulating factor were inhibited by PPARα ligands in IL-1β-stimulated synovial fibroblasts from patients with RA [Okamoto et al. 2005]. However, Cheng and colleagues showed no effect of Wy14643 (PPARα ligand) on IL-1-induced prostaglandin E2 production in synovial fibroblasts [Cheng et al. 2004].
Inhibition of bone changes in arthritic joints
No clinical studies have been published on the effect of fibrates on subchondral bone.
Using rats with collagen-induced arthritis, Bloxham and colleagues showed a significant reduction of erosive bone changes and new periosteal bone formation in rats treated with fibrates versus nontreated rats [Bloxham et al. 1990]. Still and colleagues demonstrated an elevated serum concentration of osteocalcin that is indicative for an increase in bone formation in rats and a significantly higher metaphyseal bone mineral density in animals treated with fibrates, while there was no significant difference between their body weights [Still et al. 2008]. Still and colleagues also showed a significantly greater trabecular bone volume and trabecular number, and total cross-sectional areas at the tibia–fibula junction were higher due to increases in cortical bone area [Still et al. 2008].
The in vitro differentiation of peripheral blood monocytes to osteoclasts is inhibited by PPARα ligands as well as the resorptive activity of mature osteoclasts [Okamoto et al. 2005; Chan et al. 2007; Still et al. 2008]. PPARα ligands are also able to increase the number of osteoblastic colonies and induce osteoblastic maturation [Jackson and Demer, 2000; Still et al. 2008].
Improvement of systemic and local lipid profile, prevention of vascular pathology
No reports have been published concerning the indirect effect of lipid lowering and prevention of vascular pathology on the OA or RA disease process. No studies have investigated the effect of fibrate therapy on synovial fluid lipid concentrations.
Discussion
This study indicates that fibrates or other PPARα agonists might lead to clinical improvement in RA. For OA, we found no clinical studies concerning these products. This overview of in vitro, animal studies and human clinical studies shows that PPARα ligands target many tissues, including tissues of the joint and its environment. This multiple targeting results in diminished systemic parameters of inflammation in patients with RA, but may also reduce pain, as indicated by decreased VAS scores, improved clinical scores and reduced swelling of the joints. PPARα ligands might also inhibit bone resorption. An effective dose of fibrates which could benefit patients with RA cannot be determined and no dose–response relationship was presented in any study.
OA has primarily been described as a ‘wear and tear’ disease with loss of cartilage structure. However, inflammation is considered an important aspect of the OA pathogenesis and therefore some etiopathogenic mechanisms might be similar for OA and RA. Levels of cytokines in the synovial fluid of patients with OA are in some cases as high as in patients with RA, and inflammatory and destructive responses have been described in OA and RA cartilage [Bondeson et al. 2010; Simopoulou et al. 2010; Sverdrup et al. 2010]. Furthermore, OA and RA are both characterized by infiltration of immune cells in the synovium. Subchondral bone alterations occur in both diseases and both diseases are not limited to the joint, but may have systemic consequences or systemic causes. Low-grade systemic inflammation has been described in both OA and RA [Hulejova et al. 2007]. In general, OA and RA have a multifactorial etiopathogenesis. Therefore, it might be possible to expect some of the observed effects of fibrates in patients with RA, and in patients with OA.
In the available studies we found no evidence for metabolic mechanisms of actions of PPARα agonists in OA and RA, although there is growing evidence that reinforces the hypothesis that PPARα agonists can prevent OA and RA by their systemic effects. First, cytokines and adipokines secreted by intra-articular adipose tissue may enhance inflammatory and destructive processes in cartilage [Clockaerts et al. 2010]. We found no studies reporting the effects of PPARα agonists on adipose tissue, but studies show that PPARα agonists lower CRP serum levels and inhibit monocyte release of IL-6, IL-1β, monocyte chemoattractant protein-1 and tumor necrosis factor α [Krysiak et al. 2011]. A recent meta-analysis concluded that short-term treatment of fenofibrate decreases CRP levels in a heterogeneous group. Although not all PPARα ligands have exactly the same pharmacokinetics, the main mechanisms of action are thought to be comparable [Miller and Spence, 1998; Ye et al. 2011]. In people with obesity, it is known that inflammation is partly caused by visceral and subcutaneous adipose tissue, and even more specific, by adipose tissue-related macrophages [Clockaerts et al. 2010]. Second, studies have reported an association between serum lipid levels and the development of OA and RA [Nurmohamed, 2007; Wang et al. 2008, 2009; Davies-Tuck et al. 2009; Nurmohamed and Dijkmans, 2009]. The concentration of cholesterol and fatty acids in serum might be correlated with synovial fluid concentrations, and these lipids might be capable of altering cartilage metabolism [Findlay, 2007; Wang et al. 2008]. Therefore, it is likely that PPARα agonists may have a preventive effect on the development of RA and OA through their lipid-lowering modalities. Finally, the link between vascular pathology and OA is becoming more and more established, and vascular pathology is also an important consequence of RA. A recent study by Hoeven and colleagues showed an association between the intima media thickness (or carotic plaque) and the prevalence of OA in the knee, distal interphalangeal and metacarpophalangeal OA in women [Hoeven et al. 2012].
Since PPARα agonists such as fibrates are known to prevent atherosclerosis by altering lipid composition and by their anti-inflammatory effects, they have the potential to prevent OA by counteracting this underlying risk factor [Alagona, 2010; Ji et al. 2010; Rubenfire et al. 2010].
Other lipid-lowering drugs, such as statins, are also shown to exert pleiotropic effects [Ridker et al. 2009]. Several studies already described that statins have potential beneficial effects on RA and OA [Paraskevas, 2008; Lazzerini et al. 2010; Baker et al. 2011] and this was also observed by Clockaerts and colleagues in the Rotterdam Study, a large open population study, where there was a 50% decrease in incidence and progression of knee OA in statin users [Clockaerts et al. 2012]. Statins and fibrates might exhibit similar potential beneficial effects on the joint and on systemic metabolic dysregulations. However, fibrates are drugs with a higher bioavailability compared with statins. For example, fenofibrate is rapidly converted into its active metabolite, fenofibric acid, which is protein bound (primarily to albumin) for 99%. Plasma levels of fenofibric acid peak after 6–8 h after oral administration and it has a half life of 20 h and may therefore reach the joint in higher concentrations and over a longer period [Tziomalos and Athyros, 2006]. Statins, however, are largely taken up by the liver and undergo first-pass metabolism [Lazzerini et al. 2010]. The overall tolerability of fibrates compared with statins or the combination shows no difference, and there is no difference in adverse advents [Farnier et al. 2012].
Despite the availability of RCTs investigating the effect of fibrates on RA, we could not perform a meta-analysis due to the differences in outcome parameters used. We found no clinical studies investigating the effect of PPARα ligands on OA. Other limitations of our study include the use of data of different kinds of species. The effects of PPARα ligands are species dependent, which can influence the results. This is however a well-known problem in in vitro research.
Well-designed RCTs are needed to reinforce the potential inhibitory effect of fibrates on OA and RA development and progression, but only after examination of potential underlying mechanisms, such as the role of systemic inflammation caused by obesity, vascular pathology and dyslipidemia. Spontaneous OA and RA animal models with metabolic alterations such as vascular pathology and disturbed lipid metabolism should be performed to investigate the effect of fibrates on OA and RA development and progression. These studies will help to determine the inclusion criteria for a well-designed human randomized clinical trial to investigate the effect of fibrates on OA and RA development.
Conclusion
OA and RA have a complex and multifactorial etiology. PPARα agonists such as fibrates may prevent disease progression by their ability to modify cartilage, synovium and bone responses in both diseases. For RA, fibrates seem to improve pain and function. For OA, however, there is no clinical evidence.
The effect of PPARα agonists on OA or RA development through the lowering of serum or intra-articular lipid levels, the prevention of vascular pathology or the inhibition of cytokine or adipokine production remains to be elucidated. Animal models for OA and RA, with metabolic alterations, should be performed. After confirmation of a protective effect on joints, well-designed RCTs, using well-defined inclusion criteria, can be started to investigate the symptom improvement and disease-modifying properties of fibrates in OA and RA.
Key messages
PPARα agonists should be considered as a potential therapeutic strategy for OA and RA.
PPARα agonists seem to demonstrate different joint-protective effects locally and systematically.
Animal models for OA and RA with metabolic alterations should be performed.
Footnotes
Funding: This study/work was performed (partly) within the framework of the Dutch Top Institute Pharma project # T1-213. Stefan Clockaerts received a scholarship of the University of Antwerp and the Anna Foundation.
Conflict of interest statement: None of the authors have any financial or personal relationships with other people or organizations that could inappropriately influence this work to disclose.
Contributor Information
Inge C.M. van Eekeren, Department of Orthopaedics, Erasmus MC, University Medical Center, Rotterdam, The Netherlands
Stefan Clockaerts, Erasmus MC, University Medical Center Rotterdam, Room GK 1053, PO Box 2040, 3000 CA Rotterdam, The Netherlands Monica Orthopedic Research (MoRe) Foundation and Monica Hospital, Antwerp, Belgium.
Yvonne M. Bastiaansen-Jenniskens, Department of Orthopaedics, Erasmus MC, University Medical Center, Rotterdam, The Netherlands
Eric Lubberts, Departments of Rheumatology and Immunology, Erasmus MC, University Medical Center, Rotterdam, The Netherlands.
Jan A.N. Verhaar, Department of Orthopaedics, Erasmus MC, University Medical Center, Rotterdam, The Netherlands
Gerjo J.V.M. van Osch, Departments of Orthopaedics and Otorhinolaryngology, Erasmus MC, University Medical Center, Rotterdam, The Netherlands
Sita M. Bierma-Zeinstra, Departments of Orthopaedics and General Practice, Erasmus MC, University Medical Center, Rotterdam, The Netherlands
References
- Alagona P., Jr (2010) Fenofibric acid: a new fibrate approved for use in combination with statin for the treatment of mixed dyslipidemia. Vasc Health Risk Manag 6: 351–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspden R. (2008) Osteoarthritis: a problem of growth not decay? Rheumatology (Oxford) 47: 1452–1460 [DOI] [PubMed] [Google Scholar]
- Baker J., Walsh P., Mulhall K. (2011) Statins: a potential role in the management of osteoarthritis? Joint Bone Spine 78: 31–34 [DOI] [PubMed] [Google Scholar]
- Bird H., Dixon J., Finney P., Leatham P., Lowe J., Pickup M., et al. (1983) A Clinical and biochemical evaluation of clozic, a novel disease modifying drug in rheumatoid arthritis. Clin Exp Rheumatol 1: 93–99 [PubMed] [Google Scholar]
- Bloxham D., Bradshaw D., Cashin C., Dodge B., Lewis E., Westmacott D., et al. (1990) Biologic properties of romazarit (Ro 31-3948), a potential disease-modifying antirheumatic drug. J Pharmacol Exp Ther 252: 1331–1340 [PubMed] [Google Scholar]
- Bondeson J., Blom A., Wainwright S., Hughes C., Caterson B., Van Den Berg W. (2010) The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum 62: 647–657 [DOI] [PubMed] [Google Scholar]
- Bordji K., Grillasca J., Gouze J., Magdalou J., Schohn H., Keller J., et al. (2000) Evidence for the presence of peroxisome proliferator-activated receptor (PPAR) alpha and gamma and retinoid Z receptor in cartilage. PPARgamma activation modulates the effects of interleukin-1beta on rat chondrocytes. J Biol Chem 275: 12243–12250 [DOI] [PubMed] [Google Scholar]
- Castillero E., Nieto-Bona M., Fernandez-Galaz C., Martin A., Lopez-Menduina M., Granado M., et al. (2011) Fenofibrate, a PPAR{alpha} agonist, decreases atrogenes and myostatin expression and improves arthritis-induced skeletal muscle atrophy. Am J Physiol Endocrinol Metab 300: E790–E799 [DOI] [PubMed] [Google Scholar]
- Chan B., Gartland A., Wilson P., Buckley K., Dillon J., Fraser W., et al. (2007) PPAR agonists modulate human osteoclast formation and activity in vitro. Bone 40: 149–159 [DOI] [PubMed] [Google Scholar]
- Cheng S., Afif H., Martel-Pelletier J., Pelletier J., Li X., Farrajota K., et al. (2004) Activation of peroxisome proliferator-activated receptor gamma inhibits interleukin-1beta-induced membrane-associated prostaglandin E2 synthase-1 expression in human synovial fibroblasts by interfering with EGR-1. J Biol Chem 279: 22057–22065 [DOI] [PubMed] [Google Scholar]
- Chung J., Seo A., Chung S., Kim M., Leeuwenburgh C., Yu B., et al. (2008) Molecular mechanism of PPAR in the regulation of age-related inflammation. Ageing Res Rev 7: 126–136 [DOI] [PubMed] [Google Scholar]
- Clockaerts S., Bastiaansen-Jenniskens Y., Feijt C., Verhaar J., Somville J., De Clerck L., et al. (2011) Peroxisome proliferator activated receptor alpha activation decreases inflammatory and destructive responses in osteoarthritic cartilage. Osteoarthritis Cartilage 19: 895–902 [DOI] [PubMed] [Google Scholar]
- Clockaerts S., Bastiaansen-Jenniskens Y., Runhaar J., Van Osch G., Van Offel J., Verhaar J., et al. (2010) The infrapatellar fat pad should be considered as an active osteoarthritic joint tissue: a narrative review. Osteoarthritis Cartilage 18: 876–882 [DOI] [PubMed] [Google Scholar]
- Clockaerts S., Osch G., Bastiaansen-Jenniskens Y., Verhaar J., Glabbeek F., Meurs J., et al. (2012) Statin use is associated with reduced incidence and progression of knee osteoarthritis in the Rotterdam Study. Ann Rheum Dis 71: 642–647 [DOI] [PubMed] [Google Scholar]
- Conaghan P., Vanharanta H., Dieppe P. (2005) Is progressive osteoarthritis an atheromatous vascular disease? Ann Rheum Dis 64: 1539–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisafulli C., Cuzzocrea S. (2009) The role of endogenous and exogenous ligands for the peroxisome proliferator-activated receptor alpha (PPAR-alpha) in the regulation of inflammation in macrophages. Shock 32: 62–73 [DOI] [PubMed] [Google Scholar]
- Davies-Tuck M., Hanna F., Davis S., Bell R., Davison S., Wluka A., et al. (2009) Total cholesterol and triglycerides are associated with the development of new bone marrow lesions in asymptomatic middle-aged women – a prospective cohort study. Arthritis Res Ther 11: R181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J., Sitton N., Surrall K., Martin M., Pickup M., Bird H. (1983) The effect of drugs on serum histidine levels in rheumatoid arthritis. Rheumatol Int 3: 145–149 [DOI] [PubMed] [Google Scholar]
- Fahmi H., Di Battista J., Pelletier J., Mineau F., Ranger P., Martel-Pelletier J. (2001) Peroxisome proliferator-activated receptor gamma activators inhibit interleukin-1beta-induced nitric oxide and matrix metalloproteinase 13 production in human chondrocytes. Arthritis Rheum 44: 595–607 [DOI] [PubMed] [Google Scholar]
- Farnier M., Marcereuil D., De Niet S., Ducobu J., Steinmetz A., Retterstol K., et al. (2012) Safety of a fixed-dose combination of fenofibrate/pravastatin 160 mg/40 mg in patients with mixed hyperlipidaemia: a pooled analysis from a database of clinical trials. Clin Drug Investig 32: 281–291 [DOI] [PubMed] [Google Scholar]
- Findlay D. (2007) Vascular pathology and osteoarthritis. Rheumatology (Oxford) 46: 1763–1768 [DOI] [PubMed] [Google Scholar]
- Forster P., McConkey B. (1986) The effect of antirheumatic drugs on circulating immune complexes in rheumatoid arthritis. Q J Med 58: 29–42 [PubMed] [Google Scholar]
- Francois M., Richette P., Tsagris L., Fitting C., Lemay C., Benallaoua M., et al. (2006) Activation of the peroxisome proliferator-activated receptor alpha pathway potentiates interleukin-1 receptor antagonist production in cytokine-treated chondrocytes. Arthritis Rheum 54: 1233–1245 [DOI] [PubMed] [Google Scholar]
- Goto M. (2010) A comparative study of anti-inflammatory and antidyslipidemic effects of fenofibrate and statins on rheumatoid arthritis. Mod Rheumatol 20: 238–243 [DOI] [PubMed] [Google Scholar]
- Henrotin Y., Bruckner P., Pujol J. (2003) The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthritis Cartilage 11: 747–755 [DOI] [PubMed] [Google Scholar]
- Hoeven T., Kavousi M., Clockaerts S., Kerkhof H., Van Meurs J., Franco O., et al. (2012) Association of atherosclerosis with presence and progression of osteoarthritis: the Rotterdam Study. Ann Rheum Dis 6 May (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- Hulejova H., Baresova V., Klezl Z., Polanska M., Adam M., Senolt L. (2007) Increased level of cytokines and matrix metalloproteinases in osteoarthritic subchondral bone. Cytokine 38: 151–156 [DOI] [PubMed] [Google Scholar]
- Hunter D. (2009) Osteoarthritis. Med Clin North Am 93: xv–xviii [DOI] [PubMed] [Google Scholar]
- Jackson S., Demer L. (2000) Peroxisome proliferator-activated receptor activators modulate the osteoblastic maturation of MC3T3-E1 preosteoblasts. FEBS Lett 471: 119–124 [DOI] [PubMed] [Google Scholar]
- Jenkins J., Hardy K., McMurray R. (2002) The pathogenesis of rheumatoid arthritis: a guide to therapy. Am J Med Sci 323: 171–180 [DOI] [PubMed] [Google Scholar]
- Ji Y., Wang Z., Li Z., Liu J. (2010) Modulation of LPS-mediated inflammation by fenofibrate via the TRIF-dependent TLR4 signaling pathway in vascular smooth muscle cells. Cell Physiol Biochem 25: 631–640 [DOI] [PubMed] [Google Scholar]
- Kakinuma T., Yasuda T., Nakagawa T., Hiramitsu T., Akiyoshi M., Akagi M., et al. (2004) Lectin-like oxidized low-density lipoprotein receptor 1 mediates matrix metalloproteinase 3 synthesis enhanced by oxidized low-density lipoprotein in rheumatoid arthritis cartilage. Arthritis Rheum 50: 3495–3503 [DOI] [PubMed] [Google Scholar]
- Katz J., Agrawal S., Velasquez M. (2010) Getting to the heart of the matter: osteoarthritis takes its place as part of the metabolic syndrome. Curr Opin Rheumatol 22: 512–519 [DOI] [PubMed] [Google Scholar]
- Krysiak R., Gdula-Dymek A., Okopien B. (2011) Monocyte-Suppressing Effect of Bezafibrate but Not Omega-3 Fatty Acids in Patients with Isolated Hypertriglyceridaemia. Basic Clin Pharmacol Toxicol 109: 23–29 [DOI] [PubMed] [Google Scholar]
- Lalloyer F., Staels B. (2010) Fibrates, glitazones, and peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol 30: 894–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzerini P., Capecchi P., Selvi E., Lorenzini S., Bisogno S., Baldari C., et al. (2010) Statins and the joint: multiple targets for a global protection? Semin Arthritis Rheum 40: 430–446 [DOI] [PubMed] [Google Scholar]
- McConkey B., Amos R., Billingham M., Constable T., Crockson R., Crockson A., et al. (1980) Rheumatoid arthritis: effects of a new agent (ICI 55 897) on serum acute phase proteins and the erythrocyte sedimentation rate. Ann Rheum Dis 39: 18–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D., Spence J. (1998) Clinical pharmacokinetics of fibric acid derivatives (fibrates). Clin Pharmacokinet 34: 155–162 [DOI] [PubMed] [Google Scholar]
- Nurmohamed M. (2007) Atherogenic lipid profiles and its management in patients with rheumatoid arthritis. Vasc Health Risk Manag 3: 845–852 [PMC free article] [PubMed] [Google Scholar]
- Nurmohamed M., Dijkmans B. (2009) Dyslipidaemia, statins and rheumatoid arthritis. Ann Rheum Dis 68: 453–455 [DOI] [PubMed] [Google Scholar]
- Okamoto H., Iwamoto T., Kotake S., Momohara S., Yamanaka H., Kamatani N. (2005) Inhibition of NF-kappab signaling by fenofibrate, a peroxisome proliferator-activated receptor-alpha ligand, presents a therapeutic strategy for rheumatoid arthritis. Clin Exp Rheumatol 23: 323–330 [PubMed] [Google Scholar]
- Paraskevas K. (2008) Statin treatment for rheumatoid arthritis: a promising novel indication. Clin Rheumatol 27: 281–287 [DOI] [PubMed] [Google Scholar]
- Poleni P., Bianchi A., Etienne S., Koufany M., Sebillaud S., Netter P., et al. (2007) Agonists of peroxisome proliferators-activated receptors (PPAR) alpha, beta/delta or gamma reduce transforming growth factor (TGF)-beta-induced proteoglycans’ production in chondrocytes. Osteoarthritis Cartilage 15: 493–505 [DOI] [PubMed] [Google Scholar]
- Poleni P., Etienne S., Velot E., Netter P., Bianchi A. (2010) Activation of PPARs α, β/δ, and γ impairs TGF-β1-induced collagens’ production and modulates the TIMP-1/MMPS balance in three-dimensional cultured chondrocytes. PPAR Res 2010: 635912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt A., Isaacs J., Mattey D. (2009) Current concepts in the pathogenesis of early rheumatoid arthritis. Best Pract Res Clin Rheumatol 23: 37–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds P., McLeod M., Dick W. (1981) Clozic (ICI 55,897) in rheumatoid arthritis – a controlled comparison with gold. Br J Clin Pract 35: 306–308 [PubMed] [Google Scholar]
- Ridker P., Danielson E., Fonseca F., Genest J., Gotto A., Jr, Kastelein J., et al. (2009) Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the Jupiter trial. Lancet 373: 1175–1182 [DOI] [PubMed] [Google Scholar]
- Ritchie D., Boyle J., McInnes J., Jasani M., Dalakos T., Grieveson P., et al. (1969) Evaluation of a simple articular index for joint tenderness in rheumatoid arthritis. Ann Rheum Dis 28: 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenfire M., Brook R., Rosenson R. (2010) Treating mixed hyperlipidemia and the atherogenic lipid phenotype for prevention of cardiovascular events. Am J Med 123: 892–898 [DOI] [PubMed] [Google Scholar]
- Schett G., Firestein G. (2010) Mr Outside and Mr Inside: classic and alternative views on the pathogenesis of rheumatoid arthritis. Ann Rheum Dis 69: 787–789 [DOI] [PubMed] [Google Scholar]
- Shahin D., Eltoraby E., Mesbah A., Houssen M. (2010) Insulin resistance in early untreated rheumatoid arthritis patients. Clin Biochem 43: 661–665 [DOI] [PubMed] [Google Scholar]
- Simopoulou T., Malizos K., Poultsides L., Tsezou A. (2010) Protective effect of atorvastatin in cultured osteoarthritic chondrocytes. J Orthop Res 28: 110–115 [DOI] [PubMed] [Google Scholar]
- Sniekers Y., Weinans H., Bierma-Zeinstra S., Van Leeuwen J., Van Osch G. (2008) Animal models for osteoarthritis: the effect of ovariectomy and estrogen treatment - a systematic approach. Osteoarthritis Cartilage 16: 533–541 [DOI] [PubMed] [Google Scholar]
- Still K., Grabowski P., Mackie I., Perry M., Bishop N. (2008) The peroxisome proliferator activator receptor alpha/delta agonists linoleic acid and bezafibrate upregulate osteoblast differentiation and induce periosteal bone formation in vivo. Calcif Tissue Int 83: 285–292 [DOI] [PubMed] [Google Scholar]
- Sverdrup F., Yates M., Vickery L., Klover J., Song L., Anglin C., et al. (2010) Protein geranylgeranylation controls collagenase expression in osteoarthritic cartilage. Osteoarthritis Cartilage 18: 948–955 [DOI] [PubMed] [Google Scholar]
- Tziomalos K., Athyros V. (2006) Fenofibrate: a novel formulation (triglide) in the treatment of lipid disorders: a review. Int J Nanomedicine 1: 129–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Davies-Tuck M., Wluka A., Forbes A., English D., Giles G., et al. (2009) Dietary fatty acid intake affects the risk of developing bone marrow lesions in healthy middle-aged adults without clinical knee osteoarthritis: a prospective cohort study. Arthritis Res Ther 11: R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Wluka A., Hodge A., English D., Giles G., O’Sullivan R., et al. (2008) Effect of fatty acids on bone marrow lesions and knee cartilage in healthy, middle-aged subjects without clinical knee osteoarthritis. Osteoarthritis Cartilage 16: 579–583 [DOI] [PubMed] [Google Scholar]
- Ye J., Kiage J., Arnett D., Bartolucci A., Kabagambe E. (2011) Short-term effect of fenofibrate on c-reactive protein: a meta-analysis of randomized controlled trials. Diabetol Metab Syndr 3: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]