

Abstract
meso-Tetra(N-methyl-4-pyridyl) porphine tetra tosylate (TMP) is a photosensitizer that can be used in photodynamic therapy (PDT) to induce cell death through generation of reactive oxygen species in targeted tumor cells. However, TMP is highly hydrophilic, and therefore, its ability to accumulate intracellularly is limited. In this study, a strategy to improve TMP uptake into cells has been investigated by encapsulating the compound in a hydrogel-based chitosan/alginate nanoparticle formulation. Nanoparticles of 560 nm in diameter entrapping 9.1 μg of TMP per mg of formulation were produced and examined in cell-based assays. These particles were endocytosed into human colorectal carcinoma HCT116 cells and elicited a more potent photocytotoxic effect than free drug. Antibodies targeting death receptor 5 (DR5), a cell surface apoptosis-inducing receptor up-regulated in various types of cancer and found on HCT116 cells, were then conjugated onto the particles. The conjugated antibodies further enhanced uptake and cytotoxic potency of the nanoparticle. Taken together, these results show that antibody-conjugated chitosan/alginate nanoparticles significantly enhanced the therapeutic effectiveness of entrapped TMP. This novel approach provides a strategy for providing targeted site-specific delivery of TMP and other photosensitizer drugs to treat colorectal tumors using PDT.
Introduction
According to World Health Organization (WHO) statistics, colorectal cancer is the fourth most common cancer worldwide,1 and the combination of surgical resection and chemotherapeutics remain the frontline treatments for this disease. However, many advanced colorectal patients (stage IV) have tumors that cannot be resected and frequently develop resistance to current chemotherapies, while in earlier stage disease (stage II and, in particular, stage III) many patients relapse following surgery and adjuvant chemotherapy treatment. Therefore, novel therapeutic approaches are urgently needed. Photodynamic therapy (PDT) has been suggested as a novel approach for the treatment of such tumors, offering potential for increased efficacy and reduced off-target cytotoxic effects.2
PDT is a clinical treatment that has been used in the management of a range of different pathological conditions including cancer,3 infectious diseases,4 and autoimmune disorders.5 This technique is based on the administration of a photoreactive drug (a photosensitizer) to the patient followed by irradiation of the diseased tissue with a high intensity light, often of a specific wavelength, exciting the photosensitizer and producing reactive oxygen species, resulting in cell necrosis and apoptosis.6 A targeting effect can be achieved through focusing light only on the diseased tissue, thus, leaving other adjacent tissue relatively free of treatment and potential side effects.7
Photosensitizers currently used to treat cancer can suffer from poor selectivity8 and other drawbacks including prolonged skin photosensitization, scarring of healthy tissue following irradiation, interpatient fluctuations in response, and intralesion heterogeneity.9,10 These limitations are due to the difficulties in predicting the response to the drug dose and to the lack of specificity for the target tissue, which is frequently a consequence of the hydrophobic nature of many photosensitizer compounds.11,12 Furthermore, hydrophobic photosensitizers are characterized by poor solubility and aggregation in physiological environments.13 Therefore, the use of hydrophilic water-soluble compounds may have reduced side effects. However, suitable targeting and intracellular accumulation strategies are needed to deliver therapeutically useful concentrations of such compounds,14 which could be achieved using antibodies and nanoparticles.15,16 Nanoparticle formulations of these drugs have the potential to address biodistribution limitations by targeting the encapsulated drug to the tumor site by active and passive mechanisms. It has previously been suggested that nanoparticle delivery of photosensitizers could enhance their activity in comparison to free drug, a consequence of nanoparticle endocytosis leading to accumulation of the drug in the targeted cell.17 Indeed, previous studies have examined biocompatible nanoparticles including poly(lactide-co-glycolide) acid (PLGA)15,18 and polyacrylamide19 for the formulation of photosensitizers, yielding significantly improved efficacies.
meso-Tetra(N-methyl-4-pyridyl) porphine tetra tosylate (TMP) is a hydrophilic photosensitizer that has been previously demonstrated to possess antitumor activity in murine adenocarcinomas.20 TMP binds through electrostatic interactions with DNA, which, upon light activation, produces reactive oxygen species to induce cytotoxic DNA damage.21 It is a photosensitizer that absorbs two relatively low-energy photons, which means that activation of TMP is possible at greater tissue depth in patients exposed to activating light beams. This is due to the reduced scattering of the longer lower energy wavelengths passing through targeted tissue when compared to shorter wavelengths.22
The hydrophilic nature of TMP limits its cellular uptake and therefore to overcome these limitations we have evaluated a hydrogel chitosan/alginate nanoparticle formulation. We demonstrate the enhanced activity of these particles over free drug and show further improvement of their delivery through conjugation to active cell-targeting antibodies toward death receptor 5 (DR5). Moreover, we show that DR5-targeted antibody-conjugated nanoparticles are also able to activate caspase 8, further potentiating the antitumor effects of this formulation.
Experimental Section
Materials and Methods
Chitosan low molecular weight (75–84% deacylated), sodium alginate (molecular weight, 120–190 kDa) 87–89% hydrolyzed, sodium dodecyl sulfate (SDS), phosphate buffered saline (PBS), thiazolyl blue tetrazolium bromide (MTT), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), and rhodamine 6G were all obtained from Sigma Aldrich, U.K. meso-Tetra(N-methyl-4-pyridyl) porphine tetra tosylate (TMP-1363) was a kind gift from Frontier Scientific Europe and anti-DR5 antibody (AMG655/Conatumumab) was obtained from Amgen, U.S.A. Human colorectal cancer cell lines HCT116 were obtained from ATCC, U.S.A.
Isothermal Titration Calorimetry
The interaction of TMP and alginate was studied by isothermal titration calorimetry (ITC) using a MicroCalorimeter iTC200 (GE Healthcare) essentially following a previously described protocol.23 The concentration of alginate was calculated based on the monomer concentration and all solutions were prepared in deionized water. The sample cell was filled with alginate (15 mM) and 25 injections of 1 μL TMP (1 mM) were titrated into the cell over 2 s with 2 min spacing between injections. All titrations were carried out at 25 °C with stirring at 1000 rpm. Heats of dilution of TMP into water were subtracted from the heats of binding of TMP into alginate to generate the final figure. The data was analyzed by MicroCal Origin software and the thermodynamic parameters were calculated by fitting the data using the one set of sites model.
Chitosan/Alginate Nanoparticle Preparation
As schematically presented in Figure 1, the chitosan/alginate nanoparticle formulation was performed using ionotropic gelation. Briefly, 5 mL of alginate solution at 3 mg/mL (pH 5) containing 0.75 mg TMP were prepared in water. Chitosan solution (1.2 mL) at variable concentrations from 0.5 to 3 mg/mL at pH 5 was added dropwise under pulsary sonication to optimize the formulation. The particles were left stirring for 30 min, centrifuged at 16700 g for 20 min and washed twice with water. All following nanoparticle formulations were carried out using chitosan at an optimal concentration of 1.5 mg/mL. Rhodamine 6G-loaded chitosan/alginate nanoparticles were prepared by adding 100 μg of the fluorescent dye into the sodium alginate solution.
Figure 1.

Nanoparticle formulation scheme. Illustration of the nanoparticle preparation process through first the formation of a polyelectrolyte complex and subsequent antibody conjugation through free carboxyl and amino moieties on both antibody and polymers.
Nanoparticle Characterization
Particle size and zeta potential were measured in water by dynamic light scattering (DLS) using the 3000 HS system from Malvern instruments. Measurements were carried out at room temperature with each size determination done in triplicate and with the average particle size expressed as the mean diameter (Zavg). Routine size measurements were confirmed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The nanoparticle solutions at a concentration of 1 mg/mL were dried on aluminum stubs, coated in gold, and visualized by SEM (Jeol 6500 field emission gun). Sample preparation for TEM was performed by adding a more dilute nanoparticle solution onto holey carbon grids (Agar Scientific) with subsequent imaging by a field emission gun TEM (Philips TECNAI F20).
Stability of TMP-Loaded Chitosan/Alginate Nanoparticles
The stability of the chitosan/alginate nanoparticles at 2 mg/mL was studied in 10% fetal bovine serum (FBS, PAA) containing medium at 37 °C. At each time point, an aliquot was diluted in water and size was analyzed by DLS.
Entrapment and Drug Release Monitoring
TMP entrapment was determined by measuring the nonencapsulated drug remaining in the supernatant postformulation. The solution was diluted in PBS, pH 7.4, containing 2% SDS and analyzed by comparison to a calibration curve using a fluorimeter at wavelengths of 426/654 nm for λex/λem. The drug release study was carried out in PBS, pH 7.4, at 37 °C under shaking. At each time point the nanoparticles were centrifuged and drug released into the supernatant analyzed as described above. Similarly, the release of rhodamine 6G over a 2 h incubation period was carried out under the same conditions and compared to a calibration curve measured at 480/520 nm for λex/λem.
Conjugation of Anti-DR5 Antibody to Chitosan/Alginate Nanoparticles
For the conjugation of the anti-DR5 antibody to the surface of the nanoparticles, 150 μg of the humanized antibody was added to the colloidal suspension in MES buffer (pH 5) in the presence of 2 mg EDC. The suspension was left stirring for 4 h followed by a centrifugation step to separate the nanoparticles from unbound antibody and EDC as schematically shown in Figure 1. Quantification of protein attached to the particles was measured using the BCA assay according to manufacturer’s instructions compared to a calibration curve (Pierce, U.S.A.).
Cytotoxicity Assay
In vitro analysis was carried out on the human colorectal cell line HCT116, which was cultured in McCoy medium (Invitrogen) supplemented with 10% FBS and 1% penicillin/streptomycin (Invitrogen). Cells were seeded into 96-well plates at 5000 cells per well and incubated overnight to allow adherence. A series of free TMP, TMP-loaded, or blank chitosan/alginate nanoparticles as a control were added to the wells at the indicated concentrations and incubated for 16 h. The medium was then exchanged and a red light beam (100 J/cm2) was applied by placing a Paterson light system (BL1000A, 630 ± 15 nm filters; Photo Therapeutics Ltd., Altricham, U.K.) at a distance of 1.8 cm from the 96-well plate for 5 min. A control plate was kept in the dark. After 0, 2, and 12 h, cell viability was assessed using the MTT assay. Briefly, MTT solution (20 μL of 5 mg/mL in water) was added and cells were incubated for 2 h at 37 °C. Formazan crystals were then dissolved in 100 μL of dimethylsulfoxide and absorbance measured at 570 nm. Viability was expressed as a percentage in comparison to untreated control cells.
Confocal Microscopy
HCT116 cells were seeded at 30000 cells/1.7 cm2 well on a LAB-TEK chamber slide (Thermo Scientific Nunc, U.K.). The growth medium was then changed to serum-free medium supplemented with rhodamine 6G-loaded chitosan/alginate nanoparticles (100 μg/mL) and incubated for 60 min. Immediately after incubation, the cells were washed using ice cold PBS (6 × 1 min) and fixed using 250 μL ice cold 4% paraformaldehyde for 20 min at room temperature (in darkness) followed by further washing steps (6 × 1 min). The slides were sealed with a coverslip and Prolong Gold Antifade reagent with DAPI (Invitrogen, U.S.A.). Slides were viewed on a Leica SP5 Confocal Microscope and fluorescent images were captured with a 63× lens zoomed 1–4× with a 1024 × 1024 frame and 400 Hz scanning speed. Images were analyzed using Leica LAS AF software. The images presented were captured using standardized setting and exposure times. Quantitative analysis of nanoparticle uptake was determined by comparison of relative intensity values of intracellular rhodamine 6G. Bright field images were used to draw regions of interest (ROI) over an area of imaged cells. Mean intensity of the pixels within the same ROI was then assessed in corresponding single channel fluorescent images. Data analysis was performed using ImageJ software.
DR5 Silencing
DR5 expression in HCT116 was silenced using a DR5 specific siRNA sequence and scramble control sequence as published before.24,25 Subconfluent cells were incubated with siRNA (10 μM) in Opti-MEM along with oligofectamine (Life Technologies) for 4 h and then incubated for 48 h in supplemented medium. DR5 surface expression was analyzed by flow cytometry using PE-labeled antibodies for human DR5 and the IgG1 control (Biolegend). A total of 10000 cells per well were seeded into 96-well plates. After adherence, the cells were treated with nanoparticle solutions for 16 h at indicated concentrations, medium was exchanged and cell viability was analyzed after 12 h by the MTT assay as described above.
Caspase 8 Activity Assay
For the analysis of active Caspase 8, HCT116 cells were seeded into a white 96-well plate (6000 cells per well) and incubated overnight to allow adherence. The cells were then treated with different concentrations of anti-DR5 antibody conjugated nanoparticles, free antibody or PBS for 15 h. Caspase 8 activity within the cells was analyzed by the caspase-Glo 8 substrate (Promega, U.S.A.) following the manufacturer’s instructions.
Statistical Analysis
Statistical analysis was performed using the Student’s t test (Graph-Pad Prism, U.S.A.). Rejection of the null hypothesis was considered when the p value was <0.05.
Results
Optimization of the TMP-Loaded Nanoparticle Formulation
Various polymeric systems have been evaluated for encapsulating drugs into nanoparticles. TMP is hydrophilic and therefore an alginate-based hydrogel system was selected to increase the likelihood of drug entrapment. Furthermore, TMP is cationic and therefore should interact with the alginate to facilitate effective drug loading. To investigate this possible interaction, ITC was employed as shown in Figure 2. This titration thermogram reveals an exothermic interaction between TMP and alginate, for which the thermodynamic parameters are presented in the inset table of Figure 2. The results show the binding affinity (K) is in the order of 105 M–1, which indicates strong binding between the two substances. A negative enthalpy change (ΔH) and positive entropy change (ΔS) is indicative of an electrostatic interaction between the drug and polymer. This highlights the potential of an alginate-based nanoparticle to facilitate loading of TMP through its ability to interact with the polymer.
Figure 2.

Isothermal titration calorimetry analysis of TMP and alginate binding. Thermogram of the titration of TMP (1 mM) into an alginate solution (15 mM) indicating an exothermic reaction showing the thermodynamic parameters of the reaction in the inset table. Data representative of three individual experiments.
The ability to form TMP-loaded nanoparticles was then investigated. We decided to use chitosan to stabilize the formulated alginate nanoparticle,26,27 as described in the Materials and Methods and shown schematically in Figure 1. Our results show that increasing the amount of chitosan led to an increase in drug loading (Table 1) but also led to increased particle size and heterogeneity. Therefore, a concentration of 1.5 mg/mL chitosan in the formulations was selected for further experiments, producing particles of 563 ± 93 nm in diameter entrapping 9.1 μg TMP per mg particle formulation (Table 1 and Figure 3A,B). The polydispersity (PI) of 0.45 is indicative of a broad size distribution, and this was further confirmed by SEM and TEM analysis; the latter highlighting an uneven surface, characteristic of alginate-based particles (Figure 3B, panel ii). The release profile of the drug from the nanoparticles was then examined, demonstrating a typical biphasic release with over 60% released within two days, reaching over 80% release after 8 days (Figure 3C). Finally, we examined the stability of these particles in 10% FBS over 48 h, and found that although an upward trend in PI was observed, there was no significant alteration in mean diameters (Figure 3D).
Table 1. Effect of Chitosan Concentration on TMP-Loaded Chitosan/Alginate Nanoparticle Formulationsa.
| chitosan concentration (mg/mL) | particle size (nm) | PI | zeta potential (mV) | loading (μg/mg formulationb) |
|---|---|---|---|---|
| 0.5 | 434 ± 39 | 0.31 ± 0.21 | –32 ± 2 | 2.4 ± 0.3 |
| 1.5 | 563 ± 93 | 0.45 ± 0.15 | –37 ± 6 | 9.1 ± 0.2 |
| 3 | 970 ± 65 | 0.71 ± 0.13 | –31 ± 7 | 18.8 ± 1.2 |
Results are expressed as mean ± standard deviation (SD; n = 3).
Formulation weight includes the total of chitosan and alginate weight.
Figure 3.

Characterization of TMP-loaded alginate/chitosan nanoparticles. (A) Size distribution of TMP-loaded nanoparticles using dynamic light scattering. (B) SEM (left-hand panel) and TEM pictures (right-hand panel) of nanoparticles. (C) Controlled drug release of TMP in PBS at 37 °C under shaking, quantified by comparison to a calibration curve of TMP at fluorescence of 426/654 nm. (D) Stability of nanoparticles in 10% FBS supplemented media over time at 37 °C.
Evaluation of the Photocytotoxicity of TMP toward HCT116 Colorectal Carcinoma Cells
To examine the effectiveness of these TMP-loaded chitosan/alginate nanoparticles, we first examined the sensitivity of a human colorectal carcinoma cell line HCT116 toward TMP. In order to find an effective concentration of TMP that can elicit a photodynamic effect, a range of free nonencapsulated drug concentrations were studied in the dark and after exposure to high intensity red-light activated conditions (100 J/cm2). Residual cell viability was assessed using the MTT assay. The results showed that although the drug possesses cytotoxicity in dark conditions, its effectiveness was significantly increased by treatment with light. Figure 4 illustrates the two dose–response curves of HCT116 cells treated with TMP showing clear dose-dependent effects. Analysis of the potency (EC50) revealed a 10-fold drop from 181 to 17 μg/mL in the light-treated cells. This clearly indicates the necessity of light for drug activation and demonstrates that this cell line was sensitive to the drug; both key factors pertinent to the application of nanoparticle formulations in subsequent experiments.
Figure 4.
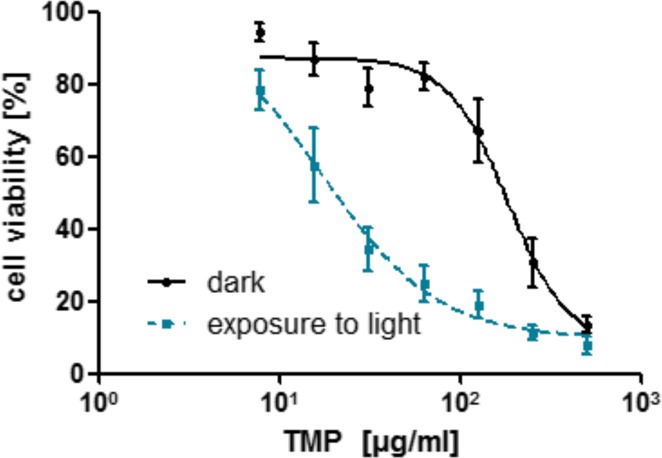
Photocytotoxicity of TMP under light and dark conditions toward HCT116. Incubation of various amounts of TMP for 16 h, exposure to red light (100 J/cm2), and incubation for 12 h in fresh medium prior to analysis of cell viability by MTT assay, dose-response curve (EC50) fit performed using Prism Graph Pad.
Nanoencapsulation of TMP in Chitosan/Alginate Nanoparticles Improves Its Cytotoxic Effect
The potential photocytotoxic effects of the TMP-loaded chitosan/alginate nanoparticles were then assessed toward the HCT116 cells. The cells were incubated with the drug formulations for 16 h, prior to irradiation (designated t = 0). In cells treated with light, a clear cytotoxic effect was apparent within 2 h and became more pronounced and significant at 12 h (Figure 5A). This analysis clearly revealed that treatment of the cells with identical concentrations of the drug in its encapsulated form elicited a further significant enhancement of its activity. A control of TMP-loaded nanoparticles kept in the dark could not induce such cytotoxic effects at t = 12 h (Figure 5B). A further control of blank nanoparticles for the full incubation period produced no significant effect on cell viability demonstrating the potential compatibility of this formulation at this concentration range (Figure 5C).
Figure 5.

Time-dependent photocytotoxicity of TMP-loaded chitosan/alginate nanoparticles toward HCT116 in comparison to free drug and controls. Preincubation of cells with TMP, TMP-loaded, or blank nanoparticles for 16 h, followed by various incubation times prior to analysis of cell viability via MTT assay. (A) Light exposure (100 J/cm2) followed by incubation for 0, 2, 12 h. (B) Control plate in the dark with incubation for 12 h. (C) Incubation of blank nanoparticles at the appropriate range of polymer concentration with 12 h incubation. **P < 0.05, ***P < 0.005.
Conjugation of Anti-DR5 Antibody to the Surface of the Chitosan/Alginate Nanoparticles
Based on the findings that entrapment of TMP in chitosan/alginate nanoparticles enhanced its activity, it was assessed whether active targeting of the nanoparticles to cell-surface receptors could further enhance this effect. Previously, we have demonstrated that polymeric nanoparticles coated with an antibody to DR5 enhances their uptake into HCT116 cells.28 Therefore, methodologies to covalently attach the anti-DR5 antibody to chitosan/alginate nanoparticles were examined. To chemically conjugate the nanoparticles with the DR5 specific antibody, carbodiimide chemistry was used to directly link the antibody via available amino and carboxyl groups to exposed reciprocal carboxyl and amino groups on the alginate and chitosan polymers, respectively, as illustrated in Figure 1. As shown in Table 2, the presence of the EDC cross-linker significantly increased the amount of protein associated with the nanoparticle, indicating the successful covalent attachment of the antibody to the nanoparticle. There were no significant changes in nanoparticle size, PI, or zeta potential upon addition of antibody, probably due to the relatively small amounts of antibody successfully conjugated.
Table 2. Conjugation of Humanized Anti-DR5 Antibody to the Surface of Chitosan/Alginate Nanoparticlesa.
| formulation | particle size (nm) | PI | zeta potential (mV) | conjugation efficiency (μg/mg formulationb) |
|---|---|---|---|---|
| conjugation in presence of EDC | 512 ± 42 | 0.28 ± 0.12 | –31 ± 7 | 6.3 ± 1.5c |
| conjugation in absence of EDC | 486 ± 78 | 0.38 ± 0.09 | –32 ± 12 | 0.4 ± 0.1 |
Results are expressed as mean ± standard deviation (SD; n = 3).
Formulation weight includes the total of chitosan and alginate weight.
p < 0.05.
Cellular Uptake of DR5-Targeted and Nude Rhodamine 6G-Loaded Chitosan/Alginate Nanoparticles
The potential of the DR5-targeted nanoparticles to preferentially target HCT116 cells was first assessed using rhodamine 6G-loaded chitosan/alginate nanoparticles. Confocal imaging and quantitative analysis of the punctate staining, indicative of nanoparticle localization in these cells, revealed that antibody conjugated nanoparticles showed higher levels of internalization after 1 h incubation, suggesting that the anti-DR5 antibody was actively targeting the nanoparticles to the DR5 expressing cells (Figure 6A,B). The intensity of fluorescence in the areas of interest revealed a significant difference between the two nanoparticle formulations, as shown in Figure 6C. To ensure that these effects were due to rhodamine 6G entrapped in the nanoparticles and not dye leached from the structures, a release study over 2 h was undertaken to investigate the release from nontargeted and targeted nanoparticles (Supporting Information, Figure 1), demonstrating no significant release of the dye in the time periods used for this microscopy analysis. These results indicate that DR5 targeting has successfully resulted in enhanced cellular internalization. Therefore, it was next determined whether increased nanoparticle uptake could further enhance the efficacy of TMP that we had observed toward these tumor cells.
Figure 6.

Enhanced cellular uptake of chitosan/alginate nanoparticles by DR5 targeting to HCT116. Incubation of DR5-targeted (A) or nude (B) nanoparticle (NP) suspensions (100 μg/mL formulation) for 60 min before fixation, cell nucleus demonstrated in blue and rhodamine 6G labeled nanoparticles in red. (C) Quantification of cellular rhodamine 6G content by fluorescence intensities in regions of interest (ROI) performed on cells in three fields of view using ImageJ software. ***P < 0.005.
Antibody Targeting of the TMP-Loaded Nanoparticles Further Enhances Their Photocytotoxicity
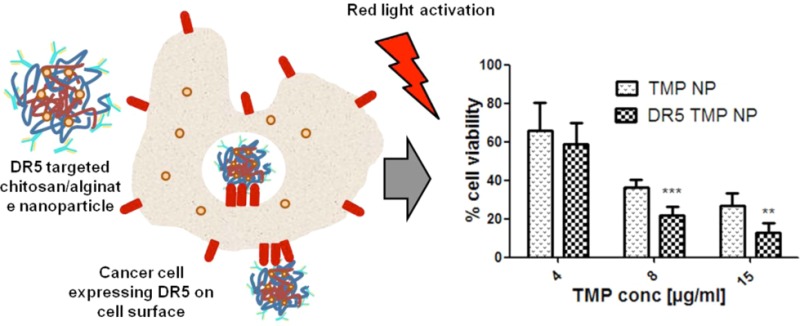
To evaluate whether active targeting of the chitosan/alginate nanoparticles with the anti-DR5 antibody enhanced photocytotoxicity, HCT116 cells were incubated with both nontargeted and DR5-targeted TMP-loaded chitosan/alginate particles for 16 h prior to irradiation, with cell viability assessed after a further 12 h incubation. The results showed that DR5 targeting significantly enhanced cytotoxic effects over nontargeted nanoparticles (Figure 7).
Figure 7.

Enhancement of photocytotoxicity by DR5 targeting of TMP-loaded nanoparticles to HCT116. Standard incubation of TMP-loaded nanoparticles (TMP-NP) and DR5-targeted TMP-loaded nanoparticles (DR5 TMP NP) for 16 h prior to exposure to red light (100 J/cm2), followed by 12 h incubation and analysis of cell viability by MTT assay. **p < 0.05, ***p < 0.005.
DR5 Conjugated Chitosan/Alginate Nanoparticles Produce Cytotoxic Effects Independent of TMP
On the basis of the enhanced cytotoxic effects observed, it was then investigated if the display of anti-DR5 antibody on the nanoparticles was inducing a cytotoxic effect, independent of TMP, through binding and activation of DR5. To examine this in more detail, the ability of free antibody and antibody conjugated to the nanoparticles to induce caspase 8 activation was assessed as a direct readout of receptor engagement. The results in Figure 8A clearly show that increasing the amounts of DR5-targeted nanoparticles induced activation of caspase 8, whereas a comparable amount of free DR5 antibody was unable to activate the receptor.
Figure 8.

Assessment of cytotoxicity of DR5-targeted chitosan/alginate nanoparticles by DR5 silencing and caspase 8 activation toward HCT116. (A) Treatment with comparable ratios of DR5-targeted nanoparticles and free anti-DR5 antibody for 15 h, followed by analysis of caspase 8 activity using caspase Glo 8 substrate (Promega). (B) FACS analysis of DR5 silencing using PE labeled anti-DR5 (clear peak) and IgG isotype control antibodies (filled peak). (C) Incubation of blank DR5-targeted nanoparticles for 16 h with DR5 silenced and control HCT116 cells prior to a further 12 h incubation and assessment of cell viability by MTT assay. ***p < 0.005.
Finally, to confirm that this effect was dependent on DR5 expression on the cells, HCT116 cells were transfected with a previously optimized siRNA to DR5 and a scrambled control for 48 h, with receptor reduction confirmed by FACS analysis (Figure 8B). The viability of these cell populations in the presence of blank DR5-targeted nanoparticles was assessed by MTT assay and it was found that the DR5-targeted particles significantly reduced cellular viability, whereas the cell viability of DR5 silenced HCT116 was not affected (Figure 8C).
Discussion
In this current study, we have shown that the photosensitizer TMP can be successfully formulated into chitosan/alginate-based nanoparticles. These particles elicit improved photodynamic cytotoxic effects over free drug toward human colorectal tumor cells, which is further enhanced by antibody targeting, exploiting the DR5 receptor that is frequently overexpressed in colorectal carcinomas.29
In contrast to most porphyrins, which are hydrophobic, TMP is relatively hydrophilic (log P = −2.64) and therefore is poorly absorbed by cells. One approach to improve internalization of this photosensitizer that has been evaluated is using direct conjugation to an antibody targeting epidermal growth factor receptors.16 This antibody targeting of the conjugated drug facilitated its uptake via receptor-mediated endocytosis. However, these studies showed that a high antibody to TMP ratio was needed to reach an efficient and preferential targeting of the drug to elicit useful biological effects. This is a common issue with antibody drug conjugates and, therefore, approaches that can maximize delivery of multiple payload molecules per antibody, such as antibody-targeted nanoparticles, may further enhance this targeting strategy.17,30
The results shown here reveal that TMP is amenable to encapsulation in alginate-based nanoparticle systems. Alginate-calcium suspensions have been shown to form nanoparticles on their own, but the nanoparticles are weak and disrupt easily in vivo due to the presence of monovalent ions that compete with calcium.31,32 Therefore, the addition of an alternative polyelectrolyte such as chitosan can increase the strength of the ionotropic gel complex.33 Although a number of studies have included calcium in chitosan/alginate nanoparticle formulations,34,35 other groups have shown that stable chitosan/alginate particles can be formed in its absence;36−38 in agreement with the results shown here. Moreover, TMP is a tetravalent cationic molecule that has been shown to interact with the anionic phosphate groups of DNA,39 and such electrostatic binding with alginate was consistent with the results found here using ITC. When this approach to formulation was used, TMP-entrapped nanoparticles were successfully generated, which were capable of producing a controlled release of the drug for up to 8 days.
In cell-based assays, the nanoparticles elicited an improved cytotoxic effect over free drug. This suggests that more drug was internalized by cells in its encapsulated form in comparison to the free compound and, indeed, images in Figure 6 clearly show that the nanoparticles can be taken up by the tumor cells. The internalization of polymeric nanoparticles such as chitosan/alginate into mammalian cancer cells has been well documented,36,40 where particles of 200 nm or less appear to enter through a clathrin-dependent pathway and larger nanoparticles of the size range reported here also internalize readily but through non-clathrin dependent mechanisms.41 Although nanoparticles can be internalized into cells through endocytosis, further enhancement, targeting, and control of this uptake can be achieved through the conjugation of antibodies on their surface. The use of targeting antibodies on nanoparticles has been shown to increase the therapeutic effect of loaded drugs.42,43 We have previously shown that PLGA nanoparticles can be targeted to cells using antibodies.44,45 In this current study, this targeting has been extended to chitosan/alginate nanoparticles using a similar carbodiimide conjugation approach conjugating free carboxyl groups to amino functions. This nonspecific conjugation strategy has the potential to facilitate high conjugation efficiency due to the presence of the high density of free functional groups in the polymers46 and avoidance of organic solvents which could denature the antibody. However, its efficiency is limited by potential cross-linking of the carboxyl groups in alginate to the amino functions of chitosan in the particles instead of conjugation to antibodies. Nonetheless, the results clearly showed that antibody was successfully conjugated to the particles, with retention of their binding activities. Future work will aim to produce methodologies allowing for site-specific modification of both the targeting antibodies and polymers to maximize the efficiencies of the reaction and activity of the resultant nanoconjugate.
The antibody chosen for targeting in these studies targeted DR5. DR5 is a member of the tumor necrosis factor receptor (TNFR) gene superfamily. These includes the TNF-R1 (CD120a), Fas (CD95), death receptor 3 (DR3, TRAMP), DR4 (TRAIL-R1), DR5 (TRAIL-R2), and DR6 (TR-7) receptors.47 DR5 is up-regulated in various types of cancer, including colorectal carcinomas, and can be exploited for selective apoptotic killing of cancer cells through recruitment and activation of caspase 8 on its cytoplasmic domain.48 A recombinant version of its cognate ligand TRAIL has been shown to selectively activate DR5 in tumors inducing synergistic cytotoxicity with other chemotherapeutic agents.49 Conatumumab (AMG655) is a fully humanized anti-DR5 antibody that is a poor activator of the receptor in cell-based assays. However, when multimerized through complexing to protein G to increase its avidity, the DR5 antibodies can activate the receptor through effective clustering of receptor monomers.50 Previously, we showed that the conjugation of AMG655 to the surface of a PLGA nanoparticle also drove clustering of the receptor.28 Here, using a chitosan/alginate system, similar activation of caspase 8 has been observed, suggesting that these particles are able to act in a similar mechanism, further increasing the antitumor potency of the TMP-loaded nanoparticle. This demonstrates that the antibodies can not only be applied for the active targeting of the drug-loaded nanoparticle to the cell, but can also elicit a therapeutically relevant cytotoxic effect themselves through the activation of the receptor.
Conclusions
In conclusion, the preparation of a novel antibody-conjugated chitosan/alginate nanoparticle formulation has demonstrated therapeutic potential in a PDT strategy bringing together targeting, activation of extrinsic apoptosis and intracellular delivery of the TMP cargo. These nanoparticles may be able to provide passive targeting in vivo, as solid tumors have leaky malformed vasculature and poor lymphatic drainage, allowing nanoparticles to localize and accumulate at these sites simply through permeation of the nanoparticles through the gaps in the endothelial wall: the enhanced permeability and retention effect.51 Light fixation at the diseased tissues can provide further targeting, having been already demonstrated for detection and treatment of internal organ neoplasms such as the colon, stomach, and pancreas.52 Collectively, these mechanisms may ultimately improve the efficacy of TMP in PDT, providing improved chemotherapeutic effects at the tumor and minimizing off-target effects in normal tissues.
Acknowledgments
This work was funded in part by the Medical Research Council (Grant No. G1001805).
Supporting Information Available
Further supplementary data is available for this work. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): Chris Scott owns shares in the biotechnology company Fusion Antibodies Ltd. Fusion Antibodies has no financial interest in this current work. The project was supported by the MRC, as stipulated in the manuscript. No other authors have conflicts of interest to report.
Author Contributions
† Authors contributed equally to this study.
Supplementary Material
References
- Ferlay J.; Shin H. R.; Bray F.; Forman D.; Mathers C.; Parkin D. M.. GLOBOCAN 2008, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 10.; International Agency for Research on Cancer: Lyon, France, 2010; http://globocan.iarc.fr.
- Dougherty T. J. J. Clin. Laser Med. Surg. 2002, 20, 3–7. [DOI] [PubMed] [Google Scholar]
- Allison R. R.; Sheng C.; Cuenca R.; Bagnato V. S.; Austerlitz C.; Sibata C. H. Photodiagn. Photodyn. Ther. 2010, 7, 115–119. [DOI] [PubMed] [Google Scholar]
- Giomi B.; Pagnini F.; Cappuccini A.; Bianchi B.; Tiradritti L.; Zuccati G. Br. J. Dermatol. 2011, 164, 448–451. [DOI] [PubMed] [Google Scholar]
- Motta S.; Monti M. Photochem. Photobiol. Sci. 2007, 6, 1150–1151. [DOI] [PubMed] [Google Scholar]
- Bechet D.; Couleaud P.; Frochot C.; Viriot M. L.; Guillemin F.; Barberi-Heyob M. Trends Biotechnol. 2008, 26, 612–621. [DOI] [PubMed] [Google Scholar]
- Huang Z. Technol. Cancer Res. Treat. 2005, 4, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jori G. J. Photochem. Photobiol., B 1996, 36, 87–93. [DOI] [PubMed] [Google Scholar]
- Grosjean P.; Savary J. F.; Mizeret J.; Wagnieres G.; Woodtli A.; Theumann J. F.; Fontolliet C.; Van den Bergh H.; Monnier P. J. Clin. Laser Med. Surg. 1996, 14, 281–7. [DOI] [PubMed] [Google Scholar]
- Webber J.; Herman M.; Kessel D.; Fromm D. Ann. Surg. 1999, 230, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrejevic-Blant S.; Hadjur C.; Ballini J. P.; Wagnieres G.; Fontolliet C.; van den Bergh H.; Monnier P. Br. J. Cancer 1997, 76, 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borle F.; Radu A.; Fontolliet C.; van den Bergh H.; Monnier P.; Wagnieres G. Br. J. Cancer 2003, 89, 2320–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Pogue B. W.; Hasan T. Exp. Opin. Drug Delivery 2005, 2, 477–487. [DOI] [PubMed] [Google Scholar]
- Josefsen L. B.; Boyle R. W. Br. J. Pharmacol. 2008, 154, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konan Y. N.; Berton M.; Gurny R.; Allemann E. Eur. J. Pharm. Sci. 2003, 18, 241–249. [DOI] [PubMed] [Google Scholar]
- Vrouenraets M. B.; Visser G. W.; Loup C.; Meunier B.; Stigter M.; Oppelaar H.; Stewart F. A.; Snow G. B.; van Dongen G. A. Int. J. Cancer 2000, 88, 108–114. [DOI] [PubMed] [Google Scholar]
- Konan Y. N.; Gurny R.; Allemann E. J. Photochem. Photobiol., B 2002, 66, 89–106. [DOI] [PubMed] [Google Scholar]
- Konan Y. N.; Chevallier J.; Gurny R.; Allemann E. Photochem. Photobiol. 2003, 77, 638–644. [DOI] [PubMed] [Google Scholar]
- Kuruppuarachchi M.; Savoie H.; Lowry A.; Alonso C.; Boyle R. W. Mol. Pharm. 2011, 8, 920–931. [DOI] [PubMed] [Google Scholar]
- Colombo L. L.; Vanzulli S. I.; Villanueva A.; Canete M.; Juarranz A.; Stockert J. C. Int. J. Oncol. 2005, 27, 1053–1059. [PubMed] [Google Scholar]
- Tada-Oikawa S.; Oikawa S.; Hirayama J.; Hirakawa K.; Kawanishi S. Photochem. Photobiol. 2009, 85, 1391–1399. [DOI] [PubMed] [Google Scholar]
- Ishida S.; Nishizawa N. Biomed. Opt. Express. 2012, 3, 282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferstl M.; Strasser A.; Wittmann H. J.; Drechsler M.; Rischer M.; Engel J.; Goepferich A. Langmuir 2011, 27, 14450–14459. [DOI] [PubMed] [Google Scholar]
- Wang S.; El-Deiry W. S. Cancer Res. 2004, 64, 6666–6672. [DOI] [PubMed] [Google Scholar]
- Longley D. B.; Wilson T. R.; McEwan M.; Allen W. L.; McDermott U.; Galligan L.; Johnston P. G. Oncogene 2006, 25, 838–848. [DOI] [PubMed] [Google Scholar]
- Ping Li.; Dai Y.; Zhang J.; Wang A.; Wei Q. Int. J. Biomed. Sci. 2008, 4, 221–228. [PMC free article] [PubMed] [Google Scholar]
- Motwani S. K.; Chopra S.; Talegaonkar S.; Kohli K.; Ahmad F. J.; Khar R. K. Eur. J. Pharm. Biopharm. 2008, 68, 513–525. [DOI] [PubMed] [Google Scholar]
- Fay F.; McLaughlin K. M.; Small D. M.; Fennell D. A.; Johnston P. G.; Longley D. B.; Scott C. J. Biomaterials 2011, 32, 8645–8653. [DOI] [PubMed] [Google Scholar]
- Tang X.; Sun Y. J.; Half E.; Kuo M. T.; Sinicrope F. Cancer Res. 2002, 62, 4903–4908. [PubMed] [Google Scholar]
- Sharman W. M.; van Lier J. E.; Allen C. M. Adv. Drug Delivery Rev. 2004, 56, 53–76. [DOI] [PubMed] [Google Scholar]
- d’Ayala G. G.; Malinconico M.; Laurienzo P. Molecules 2008, 13, 2069–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.; Li W.; Siu R. K.; Whang J.; Zhang X.; Soo C.; Ting K.; Wu B. M. Biomaterials 2009, 30, 6094–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil J. S.; Kamalapur M. V.; Marapur S. C.; Kadam D. V. Dig. J. Nanomater. Biostruct. 2010, 5, 241–248. [Google Scholar]
- Schutz C. A.; Juillerat-Jeanneret L.; Kauper P.; Wandrey C. Biomacromolecules 2011, 12, 4153–4161. [DOI] [PubMed] [Google Scholar]
- Arora S.; Gupta S.; Narang R. K.; Budhiraja R. D. Sci. Pharm. 2011, 79, 673–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamman J. H. Mar. Drugs 2010, 8, 1305–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C. Y.; Yin B. C.; Zhang W.; Cheng S. X.; Zhang X. Z.; Zhuo R. X. Colloids Surf., B 2009, 68, 245–249. [DOI] [PubMed] [Google Scholar]
- Parveen S.; Mitra M.; Krishnakumarb S.; Sahoo S. K. Acta Biomater. 2010, 6, 3120–3131. [DOI] [PubMed] [Google Scholar]
- Strickland J. A.; Marzilli L. G.; Wilson W. D. Biopolymers 1990, 29, 1307–1323. [DOI] [PubMed] [Google Scholar]
- Yang S. J.; Lin F. H.; Tsai H. M.; Lin C.; F, Chin H. C.; Wong J. M.; Shieh M. J. Biomaterials 2011, 32, 2174–2182. [DOI] [PubMed] [Google Scholar]
- Rejman J.; Oberle V.; Zuhorn I. S.; Hoekstra D. Biochem. J. 2004, 377, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarron P. A.; Olwill S. A.; Marouf W. M.; Buick R. J.; Walker B.; Scott C. J. Mol. Interventions 2005, 5, 368–380. [DOI] [PubMed] [Google Scholar]
- Kocbek P.; Obermajer N.; Cegnar M.; Kos J.; Kristl J. J. Controlled Release 2007, 120, 18–26. [DOI] [PubMed] [Google Scholar]
- McCarron P. A.; Marouf W. M.; Quinn D. J.; Fay F.; Burden R. E.; Olwill S. A.; Scott C. J. Bioconjugate Chem. 2008, 19, 1561–1569. [DOI] [PubMed] [Google Scholar]
- Scott C. J.; Marouf W. M.; Quinn D. J.; Buick R. J.; Orr S. J.; Donnelly R. F.; McCarron P. A. Pharm. Res. 2008, 25, 135–46. [DOI] [PubMed] [Google Scholar]
- Kas H. S. J. Microencapsulation 1997, 14, 689–711. [DOI] [PubMed] [Google Scholar]
- Papenfuss K.; Cordier S. M.; Walczak H. J. Cell Mol. Med. 2008, 12, 2566–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver P. G.; LoBuglio A. F.; Zinn K. R.; Kim H.; Nan L.; Zhou T.; Wang W.; Buchsbaum D. J. Clin. Cancer Res. 2008, 14, 2180–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar S.; Srivastava R. K. Drug Resist. Updates 2004, 7, 139–56. [DOI] [PubMed] [Google Scholar]
- Kaplan-Lefko P. J.; Graves J. D.; Zoog S. J.; Pan Y.; Wall J.; Branstetter D. G.; Moriguchi J.; Coxon A.; Huard J. N.; Xu R.; Peach M. L.; Juan G.; Kaufman S.; Chen Q.; Bianchi A.; Kordich J. J.; Ma M.; Foltz I. N.; Gliniak B. C. Cancer Biol. Ther. 2010, 9, 618–631. [DOI] [PubMed] [Google Scholar]
- Gerlowski L. E.; Jain R. K. Microvasc. Res. 1986, 31, 288–305. [DOI] [PubMed] [Google Scholar]
- Wang J. B.; Liu L. X. Hepatogastroenterology 2007, 54, 718–24.17591048 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.