

Abstract
Protein kinase C (PKC) isoforms have emerged as important regulators of cardiac contraction, hypertrophy, and signaling pathways that influence ischemic/reperfusion injury. This review focuses on newer concepts regarding PKC isoform-specific activation mechanisms and actions that have implications for the development of PKC-targeted therapeutics.
Protein kinase C (PKC) exists as a family of serine/threonine kinases that regulate a host of cellular effector responses. In the heart, PKC activation leads to rapid changes in contractile performance and more long-term effects on ventricular remodeling. Studies to date have interrogated the cellular actions of individual PKCs using pharmacologic inhibitors, peptide inhibitors of translocation, adenoviral vectors that drive expression of wild-type or mutant forms of PKC in cardiac cultures, or genetic models of PKC isoform overexpression or knockout in mice. This review will summarize and interpret these previous findings in the context of newer concepts regarding mechanisms that control PKC isoform maturation, localization, and catalytic activity in cardiomyocytes.
PKC isoform structure and mechanisms of activation
PKC isoforms share a similar overall structure consisting of an N-terminal regulatory domain that is joined through a flexible linker to a conserved C-terminal catalytic domain that binds ATP and substrates (Fig 1). PKC regulatory domains contain a pseudosubstrate domain that maintains the enzyme in an inactive conformation and membrane targeting modules that control the subcellular localization of the enzyme. PKC isoforms are subclassified based on these membrane targeting modules. Conventional PKC isoforms (cPKCs; α, the alternatively spliced βI and βII isoforms, and γ) contain tandem C1A/C1B motifs that bind diacylglycerol (DAG) or phorbol esters (such as PMA) and a C2 domain that binds anionic phospholipids in a calcium-dependent manner. Novel PKCs (nPKCs; δ, ε, θ, and η) also contain twin C1A/C1B domains and a C2 domain. However, the positions of the C1A/C1B and C2 domains in nPKCs are switched along the linear sequence of the protein (relative to cPKCs) and nPKC-C2 domains do not bind calcium (since they lack critically positioned calcium-coordinating acidic residues); nPKCs are maximally activated by lipid cofactors (DAG or PMA), without a calcium requirement. Atypical PKCs (aPKCs; ζ and ι/λ) contain an atypical C1 domain (that binds PIP3 and ceramide, but not DAG or PMA) and a protein-protein interaction PB1 (Phox and Bem 1) domain that binds PB-containing scaffolds; aPKC isoform activation is attributable to protein-protein interactions and activation loop phosphorylation by phosphoinositide-dependent kinase-1 (PDK-1).
Figure 1. Domain structure of PKC family enzymes.
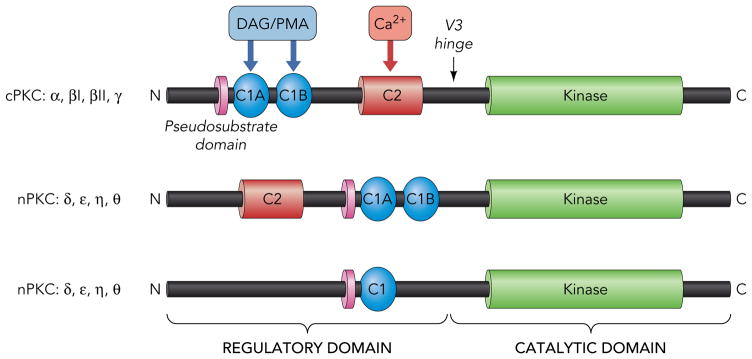
The conserved pseudosubstrate motif (shown in lavender) N-terminal to the C1 domain (shown in blue), the C2 domain (red), and the kinase domain (green) and the more variable regions (shown in black) are depicted. For further details see text.
The canonical model for PKC isoform activation derives from early studies of PKCα which localizes in a closed (inactive) conformation to the cytosolic fraction of resting cells (Fig 2). PKCα is activated by agonists that promote phosphoinositide hydrolysis and generate DAG and the calcium-mobilizing second messenger molecule IP3. Activation is via a two step process involving an initial low affinity electrostatic interaction between the calcium bound C2 domain and membranes, followed by a C1 domain interaction with DAG (the membrane-restricted product of phosphoinositide hydrolysis). Membrane-anchored PKCα then undergoes a conformational change that expels the autoinhibitory pseudosubstrate domain from the substrate-binding pocket. This traditional model of PKC activation views PKC as a generic kinase that achieves specificity exclusively through translocation events that co-localize the enzyme with target substrates in specific membrane compartments. This model predicts that PKC responses will reflect the ensemble actions of individual PKC isoforms coexpressed in any particular cell type; it also predicts that PKC substrates will be restricted to DAG-enriched membranes. However, recent studies indicate that the cellular actions of several PKC isoforms also are controlled by alternative lipid-independent mechanisms involving proteolytic cleavage and/or phosphorylation. These non-canonical mechanisms for PKC activation can lead to profound changes in the subcellular compartmentation and/or catalytic activity of the enzyme, in some cases generating forms of PKC that display high levels of activity throughout the cell (not just at DAG-containing membranes). Several landmark studies exposing new paradigms for PKC isoform activation were performed in cardiomyocyte models and are discussed in this review which focuses on emerging concepts of PKC isoform-specific actions in the heart.
Figure 2. Growth factor-dependent mechanisms that allosterically activate cPKCs.

G protein coupled receptors (or receptor tyrosine kinases) that activate phospholipase C (PLC) generate DAG and the calcium mobilizing second messenger IP3; these signaling molecules stabilize the active conformation of cPKCs to membranes.
PKCα
Cardiomyocytes co-express multiple PKC isoforms, but PKCα has been viewed as a particularly attractive therapeutic target since it is the predominant isoform in most cardiomyocyte preparations and its expression and/or activity increases further in many models of cardiac injury, hypertrophy, or failure (5; 64; 65). Early studies implicated PKCα as a mediator of hypertrophic responses in cultured neonatal cardiomyocytes (7). However, subsequent studies in genetic models of PKCα overexpression or knockout in mice failed to expose any significant in vivo growth regulatory functions for PKCα. Rather, these studies identified a role for PKCα in the regulation of cardiac contraction, showing that PKCα−/− hearts manifest enhanced contractility and relaxation at baseline and that cardiac contractility is depressed in PKCα transgenic mice (8). These changes in cardiac contraction are intrinsic to the heart; they are retained ex vivo in working heart preparations. They also reflect the direct and specific actions of PKCα. Adenoviral-mediated overexpression of wild-type or dominant-negative PKCα leads to similar antithetical effects on cardiac contractility, whereas calcium transients and twitches are not altered by PKCβ and/or PKCγ gene ablation (8; 44). PKCα-dependent changes in calcium cycling and contraction have been attributed to inhibitor-1 phosphorylation at S67. This modification increases protein phosphatase 1 (PP1) activity and leads to the dephosphorylation of phospholamban, decreased sarcoplasmic reticulum (SR) Ca2+ ATPase (SERCA-2) activity, reduced SR Ca2+ loading, and decreased contractility (8). However, other PKCα substrates that also may contribute to changes in cardiac contractility. These include [1] various sarcomeric proteins that influence myofilament calcium sensitivity and myocardial stiffness, including cardiac troponin I (cTnI), cardiac troponin T, myosin binding protein C, and titin, (26; 39; 70; 71), [2] G protein-coupled receptor kinase 2 (GRK2), a Ser/Thr kinase that phosphorylates the agonist-occupied β-adrenergic receptor (βAR) and is a target for PKCα-dependent phosphorylation at S29 (a modification that increases GRK2 activity, enhances βAR desensitization, and decreasesβAR responsiveness (47)), and [3] the α1C subunit of the L-type calcium channel, which is phosphorylated by PKCα and several other PKCs (PKCε and PKCζ) at Ser1928 - a site traditionally viewed as a target for protein kinase A (PKA) (81). While α1C subunit phosphorylation by PKA increases channel activity, PKCα-dependent changes in L-type calcium channel activity are less straightforward, since PKCα also phosphorylates residues in other regions of the α1C subunit. Current models hold that activation of PKCα, PKA, other PKC isoforms, or other Ser/Thr kinases (such as Ca2+/calmodulin-dependent kinase or protein kinase G) leads to distinct ensemble phosphorylation patterns that result in distinctive changes in calcium channel activity (80).
In additional to the canonical growth factor receptor-dependent mechanism for PKCα activation (involving PIP2-derived second messengers), PKCα also is activated via a receptor-independent mechanisms (that alter the enzyme’s subcellular compartmentation and pharmacologic profile) in cardiomyocytes subjected to hypoxia, ischemia, or oxidative stress (19; 31; 73). One such mechanism involves cleavage by calpains, calcium-dependent cysteine proteases that are activated in the calcium-overloaded ischemic heart (Fig 3). Calpain cleaves PKCα at sites in the V3 region, generating a C-terminal catalytic fragment (termed PKMα) that partitions to the nucleus and displays a high level of constitutive activity. The observation that overexpression of even relatively modest levels of PKMα leads to massive biventricular dilatation and contractile dysfunction (a phenotype that is considerably more severe than the phenotype induced by full-length PKCα) suggests that the unregulated/mislocalized PKCα catalytic fragment is an important mediator of pathologic cardiac remodeling (31). Studies of mechanism indicate that PKMα acts as a ‘rogue’ kinase; it phosphorylates substrates that are not (or are only weakly) phosphorylated by full-length PKCα (31; 82). For example, G protein-coupled receptors neutralizes the antihypertrophic actions of HDAC5 (a signal-responsive repressor of MEF-dependent pathologic gene programs and pathological cardiac remodeling) by activating PKD; PKCs (generally novel PKC isoforms, rather than PKCα) contribute to this pathway indirectly as activators of PKD. However, PKMα directly phosphorylates HDAC5, leading to the nuclear egress of HDAC5 and de-repressesion of MEF-dependent gene expression (82). Of note, while this proteolytic activation mechanism is not specific for PKCα, (since calpain cleaves other PKC isoforms (35; 78)), the cardiac actions of PKMα might predominate since PKCα is the most abundant PKC isoform in cardiomyocytes.
Figure 3. Proteolytic mechanisms that activate PKCα and PKCδ.

see text.
PKCβ
PKCβ expression and activity are increased in end stage human heart failure (5). PKCβ activity also increases in the setting of uncontrolled diabetes as a result of glucose-induced de novo synthesis of DAG from glycolytic intermediates and/or hyperglycemia-induced generation of reactive oxygen species (30; 53). The hyperglycemia-induced metabolic derangements that activate PKCβ do not activate PKCα.
The cardiac actions of PKCβ have been examined in transgenic mouse models. Studies in a binary transgenic system that allows for cardiac-specific and conditional PKCβ overexpression link PKCβ activation to sudden death associated with increased L-type calcium channel activity and calcium cycling abnormalities in neonatal cardiomyocytes (1; 6). In contrast, conditional low levels of PKCβ overexpression in adult cardiomyocytes leads to mild/progressive ventricular hypertrophy and impaired diastolic relaxation, but no gross histologic pathology (6). Cardiomyocytes isolated from PKCβ transgenic hearts show enhanced contractility in association with an increase in the calcium transient peak amplitude; this increase in calcium delivery to the myofilaments functionally overrides the more minor effect of PKCβ to decrease maximal tension generation by myofilaments (28). In contrast, transgenic mice that overexpress high levels of PKCβ from birth through adult life develop a more severe cardiomyopathic phenotype characterized by histologic evidence of multifocal fibrosis and dystrophic calcification and functional evidence of depressed cardiomyocyte contractility (associated with increased cTnI phosphorylation, decreased myofilament calcium responsiveness, and no gross changes in calcium transients (72; 76)). These results emphasize that the cellular actions of PKCβ can vary considerably depending upon the timing and/or intensity of enzyme activation. Finally, while these studies indicate that PKCβ activation is sufficient to drive pathologic remodeling of the adult heart, PKCβ is not required for the induction of cardiac hypertrophy; hypertrophic responses to aortic banding or phenylephrine infusion are preserved in PKCβ null mice (59). Rather, some studies link PKCβ activation to the induction of profibrotic cytokines and the development of tissue fibrosis, diastolic stiffness, and contractile dysfunction (14; 77). Finally, there is evidence that chronic hyperglycemia-induced PKC activation can contribute to cardiovascular risk by blunting cellular insulin responsiveness (15). Specifically, while PKC isoforms mediate certain insulin responses - chronic PKC activation in diabetes leads to a loss of insulin responsiveness due to inhibitory Ser/Thr phosphorylations on the insulin receptor itself or its downstream signaling partners such as insulin receptor substrate proteins or the regulatory subunit phosphatidylinositol 3-kinase (which links receptor tyrosine kinases to the activation of AKT (41; 46; 50; 75)).
PKCδ
PKCδ is activated or up-regulated in many models of cardiac ischemia and hypertrophy. PKCδ’s cardiac actions were initially characterized in cultured neonatal cardiomyocytes. Here, PKCδ overexpression leads to down-regulation of SERCA2, activation of JNK and p38-MAPK, changes in the phosphorylation/compartmentation of p66Shc (an adapter protein that amplifies mitochondrial ROS generation and enhances susceptibility to oxidative stress-induced apoptosis), and induction of apoptosis (21; 23; 25; 57). These results provided the first hints that PKCδ might be a mediator of pathological cardiac remodeling. Subsequent studies used translocation inhibitor peptides to implicate PKCδ in the injury response resulting from reperfusion of ischemic myocardium. This approach (developed by Mochly-Rosen and colleagues) is based upon the assumption that all PKC responses require a docking interaction between the enzyme and its cognate Receptor for Activated C Kinase (RACK), a family of scaffolding proteins that localize activated forms of individual PKCs to specific membrane microdomains in close proximity with their unique target substrates (12; 34). PKCδ responses were defined in studies with Tat-δV1-1, a PKCδ translocation inhibitor peptide consisting of the putative RACK-binding sequence in PKCδ (8SFNSYELGSL17) conjugated to TAT (a carrier peptide that delivers fusion peptides into cells). These studies showed that restoration of blood flow to a region of ischemic myocardium leads to a series of PKCδ-dependent events in mitochondria that result in a decline in intracellular pH and ATP production, increased mitochondrial ROS accumulation, inhibition of the cytoprotective AKT-BAD phosphorylation pathway, release of cytochrome c, activation of caspase 3, and induction of apoptosis (13; 29; 51). Subsequent studies showed that Tat-δV1-1 reduces ischemia/reperfusion injury in animal models of acute myocardial infarction and stroke, providing a rationale to develop Tat-δV1-1 for clinical indications in humans (4; 10; 29). The initial dose-escalation study with Tat-δV1-1 (which was marketed for clinical use as KAI-9803) was encouraging, showing that catheter infusion of KAI-9803 into jeopardized myocardium is well tolerated and leads to non-significant reductions in some biomarkers of myocardial necrosis (4). However, this initial study was not powered to establish efficacy. Enthusiasm for KAI-9803 largely evaporated (and efforts by the pharmaceutical industry to develop KAI-9803 for acute cardiac indications were largely abandoned) when a subsequent Phase 2b efficacy trial failed to show any therapeutic effect of KAI-9803 to decrease myocardial injury or improve clinical outcome in 1,176 patients undergoing percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. While there are many possible reasons for drug failure (related to dose, timing, or route of drug delivery), it is worth noting that the momentum to develop TAT-δV1-1 as a therapeutic compound in humans was never accompanied by an equally vigorous and unbiased analysis of PKCδ’s cardiac actions and the molecular underpinnings of KAI-9803-dependent responses. Several issues deserve comment:
First, PKCδ−/− mice have been available since 2001 - and have been used to implicate PKCδ in the regulation of ROS production and stress-induced apoptosis in the vasculature (43). However, this model has not been used to validate the prevailing concept that PKCδ functions exclusively in pro-apoptotic pathways that mediate ischemia/reperfusion injury. Alternative roles for PKCδ in receptor-dependent pathways that activate PKD (that influence transcriptional programs controlled by HDAC5 and/or survival pathways regulated by CREB) or other pathways that might mitigate ischemic injury are not generally considered (22; 24; 54). In this context, there is evidence that PKCδ knockout leads to a decrease in infarct size – but a paradoxical defect in ischemic preconditioning-induced cardioprotection (49). Since acute myocardial infarction is preceded by preinfarction angina (a syndrome that mimics ischemic preconditioning) in ~30% of patients, a PKCδ inhibitor that prevents ischemic preconditioning might have deleterious consequences in some patient populations. As a separate issue, PKCδ−/− mice also have not been used to validate the conclusion that the cardiac actions of KAI-9803 are attributable exclusively to a mechanism involving PKCδ - and are lost in PKCδ−/− mice. PKCδ-independent “off-target” mechanisms for KAI-9803 are possible and have not been considered.
Second, studies that define the cardiac actions of PKCδ with a translocation inhibitor peptide are based upon the assumption that all PKCδ actions require docking interactions with RACK proteins. This assumption is problematic for several reasons: [1] A PKCδ-specific RACK, that anchors PKCδ to membranes, has not been identified unambiguously. While p32/gC1qBP was characterized as a protein that selectively associates with the allosterically-activated form of PKCδ, it does not fit the traditional criteria of a PKCδ-RACK since it also constitutively interacts with PKCθ (58). [2] PKCδ localization patterns are influenced by interactions with cytoskeletal proteins that are not RACKs and would not necessarily be inhibited by KAI-9803 (33). [3] The inherent assumption that PKCδ activation is a uniform process - and that a single translocation inhibitor peptide will act in a uniform manner to inhibit PKCδ translocation to signaling microdomains in multiple subcellular compartments (including the surface membrane, mitochondria, nucleus, sarcomere, and cytoskeleton) - also seems tenuous, since the kinetics and regulatory controls for PKCδ localization to various signaling compartments are quite different. The notion that KAI-9803 might inhibit PKCδ translocation to only selected compartments (or it might drive PKCδ to an aberrant location leading to the phosphorylation of non-physiologic substrates) has never been considered. [4] The use of KAI-9803 as an inhibitor of RACK-driven compartmentation of PKCδ is based upon the assumption that all PKCδ responses require translocation to membranes. This ignores a substantial literature showing that catalytically active forms of the constitutively-active PKMδ fragment or full-length PKCδ accumulate in the soluble fraction during apoptosis or oxidative stress (18; 63). There is no a priori reason to expect that a translocation inhibitor peptide would block the actions of a catalytically active form of PKCδ in the soluble fraction.
Finally, a conventional allosteric model of PKCδ activation by lipid cofactors - which focuses on translocation events that deliver the enzyme in an active conformation to target substrates in membranes - assumes that PKCδ activity is an inherent/immutable property of the enzyme that is not altered by the activation process. However, there is recent evidence that PKCδ accumulates as pools of enzyme with distinct phosphorylation patterns and catalytic activities in different subcellular compartments (48; 61). The phosphorylation reactions that regulate the cellular actions of PKCδ are summarized in the sections that follow.
PKCδ activity is regulated by autophosphorylation at a highly conserved Thr residue in the activation loop (T505) (60; 63). This mode of regulation is unique to PKCδ; for other PKCs, activation loop phosphorylation is a stable priming event that is completed during de novo synthesis of the enzyme, mediated by PDK-1, and required to structure the catalytic pocket in a favorable conformation for catalysis (52; 68). In contrast, PKCδ is catalytically active without activation loop phosphorylation. While newly synthesized PKCδ can be phosphorylated at the activation loop by PDK-1, native PKCδ is recovered with little-to-no activation loop phosphorylation in resting cardiomyocytes and several other differentiated cell types (presumably because this site is exposed and phosphorylation is reversed by cellular phosphatases). PKCδ autophosphorylates at T505 during treatment with PMA or various growth factor receptor agonists; this dynamically regulated modification plays a key role to regulate activity toward selected target substrates (see below (11; 60; 63; 69; 71)). Additional autophosphorylation sites at other strategic positions in PKCδ have recently been identified; their regulatory functions are the focus of ongoing studies (62).
PKCδ also is phosphorylated by Src at Y311 and Y332 (Tyr residues in the V3 hinge region numbering based upon rodent sequence) in cells exposed to oxidative stress or other proapoptotic stimuli (37; 38; 67; 68); Tyr phosphorylation plays little-to-no role in the regulation of other PKCs. The Tyr-phosphorylated form of PKCδ accumulates in the cytosol of H2O2-treated cardiomyocytes as a lipid-independent enzyme (37; 63). This observation resolves a longstanding dilemma regarding PKCδ’s cardiac actions. The conventional model of PKCδ activation focuses exclusively on PKCδ’s membrane-delimited actions; it does not account for PKC-dependent regulation of cardiac contraction by phosphorylating myofibrillar proteins such as cTnI (the “inhibitory” subunit of the troponin complex). The Tyr-phosphorylated form of PKCδ is poised to phosphorylate substrates throughout the cell (not just on lipid membranes)
Recent studies also implicate phosphorylation as a mechanism that alter PKCδ’s enzymology (Fig 4). Specifically, PKCδ phosphorylates cTnI at S23/S24 when it is allosterically activated by PMA; studies in detergent-extracted cardiomyocytes link this modification to reduced force development at sub-maximal calcium (with no change in force at maximal calcium, as would be expected for cTnI-S23/S24 phosphorylation). However, the dually T505/Y311-phosphorylated form of PKCδ (that accumulates during oxidative stress) phosphorylates cTnI at both S23/S24 and T144; S23/S24-T144-phosphorylated cTnI decreases tension and cross-bridge kinetics at maximal calcium (i.e., the S23/S24 phosphorylation-dependent change in force development at sub-maximal calcium is prevented by the additional phosphorylation at T144). Finally, mutagenesis studies show that single residue substitutions in PKCδ at Y311F or T505A prevent the Src-dependent acquisition of cTnI-T144 kinase activity. Collectively, these studies describe distinct signalling modes for PKCδ as both an allosterically-activated enzyme (during growth factor stimulation) and as a Tyr-phosphorylated enzyme (during oxidative stress). The dynamic changes in PKCδ’s substrate specificity - due to changes in T505 autophosphorylation and Y311 phosphorylation by Src - represent a novel form of cardiac adaption which could be particularly important in the context of cardiac hypertrophy or failure, where coordinate increases in PKC isoform expression and cTnI phosphorylation are linked to reduced actin-myosin interactions and depressed contractile function 11–13.
Figure 4. Phosphorylation-dependent changes in the catalytic activity of PKCδ.
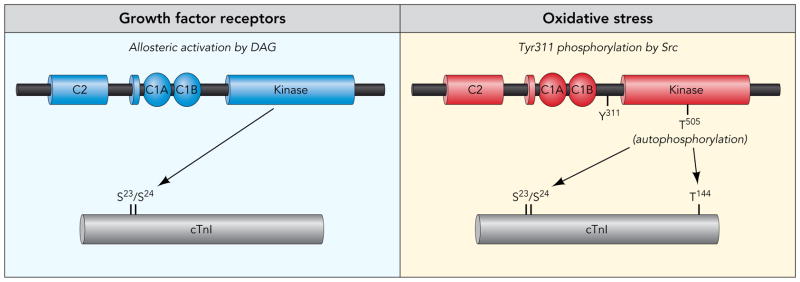
PKCδ phosphorylates cardiac troponin I (cTnI) when it is allosterically activated by lipid cofactors. PKCδ phosphorylates cTnI at both S23/S24 and T144 when it is tyrosine phosphorylated by Src. The Src-dependent acquisition of cTnI-T144 kinase activity is abrogated by single residue substitutions in PKCδ at Y311 or T505.
While our studies identify an important role for Y311 phosphorylation to ‘fine tune’ PKCδ’s substrate specificity, the functional consequences of PKCδ-Y332 phosphorylation remain less obvious. Leitges et al. reported that the Y332-phosphorylated hinge region of PKCδ functions as a docking site for the SH2 domain of Shc (providing a mechanism to nucleate signaling pathways that activate the MAPK cascade (42)), but recent studies indicate that PKCδ interacts with Shc proteins via a phospho-Y332-independent mechanism in H2O2-treated neonatal cardiomyocytes (21). These studies also failed to link Y332 phosphorylation to any gross changes in full-length PKCδ catalytic activity (71). However, a role for Y332 phosphorylation to indirectly regulate PKCδ activity remains possible, since Y332 phosphorylation is reported to facilitate caspase 3-dependent cleavage of the V3 hinge region of PKCδ at 324DIPD327 (a cleavage event that liberates a proapoptotic constitutively active PKMδ catalytic domain fragment; Fig 3 (45)).
PKCε
PKCε is activated by various hypertrophic stimuli, but PKCε’s role in cardiac growth responses remains uncertain, since transgenic cardiac-specific overexpression of a constitutively active PKCε mutant leads to only a modest hypertrophic phenotype (without any gross derangement in contractile function (74)), PKCε knock-out mice lack a baseline cardiac phenotype, and PKCε knock-out mice develop hypertrophy (with preserved systolic contractile function) in response to pressure overload (32; 36). More recent studies have focused on PKCε’s role in ischemic preconditioning (IPC), a mechanism whereby transient ischemic episodes protect against subsequent severe ischemia/reperfusion injury (17; 20). There is evidence that PKCε translocates to mitochondria during IPC (56) and that peptide activators of PKCε (that deliver PKCε to the mitochondrial compartment) confer resistance to ischemia/reperfusion injury, suggesting that cardioprotection is due to PKCε-regulated events in mitochondria (17). Several components of the mitochondrial IPC signaling machinery have been characterized as PKCε targets, including the mitochondrial permeability transition pore (which regulates the mitochondrial membrane potential) and mitochondrial aldehyde dehydrogenase 2 (an enzyme that detoxifies reactive aldehydes and plays an important role in oxidative stress responses (9)).
Studies in PKCε knock-out mice also suggest a role for PKCε in IPC (17; 20). However, the interpretation of studies in PKCε−/− mice remains somewhat ambiguous, since total body PKCε-KO from embryonic life onward leads to a compensatory increase in the expression and phosphorylation (i.e., activity) of PKCδ (20; 36). In fact, Klein et al. attributed the myocardial fibrosis and diastolic dysfunction that develops during chronic pressure overload hypertrophy in PKCε-KO (but not wild-type) mice to activation of PKCδ and its effector p38MAPK (36). Moreover, while studies in PKCε-KO mice suggest that PKCε functions as a tonic inhibitor PKCδ, a completely different form of nPKC isoform cross-regulation is detected in cultured cardiomyocytes. Here, PKCε overexpression leads to an increase in PKCδ phosphorylation (60). These results highlight the limitations of current knowledge regarding the molecular machinery that governs nPKC isoform cross-regulation. As a result, extrapolations from data obtained in genetic and molecular models of altered PKCε expression may be premature.
Concluding Remarks
This review has attempted to summarize recent advances toward defining the molecular controls and cardiac actions of individual PKC isoforms. It is interesting to note that several non-canonical activation mechanisms for PKCα and PKCδ involve phosphorylation or proteolytic cleavage at sites in the V3 hinge region of the enzyme; V3 domain phosphorylations that generate docking sites for 14-3-3 proteins and lead to lipid-independent activation of PKCε also have been identified (40). These results suggest that the V3 region may be an underappreciated and promising target for novel PKC-targeted pharmaceuticals.
Studies to date interrogating the cardiac actions of PKC isoforms have focused primarily on a single (presumably the main) protein product of individual PKC genes. This ignores alternative splicing mechanisms that lead to the generation of multiple RNA transcripts with distinct functional and/or regulatory properties, a mechanism that has been described for PKCβ and PKCδ. In the case of PKCβ, differential use of exons 17 and 18 results in the expression of splice variants with distinct C-terminal V5 domains that are expressed in a tissue-specific and developmentally regulated manner. PKCβI (which has a shorter C-terminus encoded by exon 18) and PKCβII (with the longer C-terminus encoded by exon 17) partition to distinct subcellular compartments (both at rest and following activation) and they activate distinct (and in some cases, opposing) functional responses in smooth muscle cells and several other non-cardiomyocytes models (16; 79). While studies to date suggest that PKCβI and PKCβII function similarly to regulate cardiac growth responses (66), differences in other aspects of PKCβ’s signaling repertoire in cardiomyocytes remain possible; these have not been excluded.
Splice variants of human and mouse PKCδ - that contain V3 domain inserts that disrupt the caspase-3 cleavage sites (i.e., prevent proteolytic PKCδ activation) - protect cells from proapoptotic stimuli, since the full-length caspase-resistant form of PKCδ and the freed PKCδ catalytic domain fragment exert diametrically opposite effects to prevent or induce cellular apoptosis, respectively. There is evidence that the vitamin A metabolite all-trans-retinoic acid and by insulin control alternative splicing of PKCδ in neuronal cells and that the resultant changes in the relative abundance of caspase-sensitive and caspase-resistant forms of PKCδ influence neuronal cell survival and cognitive function. (2; 3; 55). Alternative splicing mechanisms that control the cardiac actions of PKCδ have not been considered.
Finally, recent studies expose mechanisms that alter the pharmacologic profiles of PKCs. For example, C-terminal PKM catalytic fragments (generated during apoptosis) are inhibited by drugs that occupy the ATP binding pocket, but they are not inhibited by calphostin C (or other drugs that act at regulatory domain determinants). While this change in PKC’s pharmacologic profile is quite predictable, there is recent evidence that PKCβII becomes resistant to ATP-competitive inhibitors (such as BIS1 and staurosporin) when complexed with the scaffolding protein AKAP-79 (27). The notion that docking interactions with intracellular signaling partners and scaffolding proteins can create microdomains with persistent PKC activity in BIS I-treated cells could not be predicted from in vitro studies with purified enzymes. These issues provide further challenges for the development of clinically useful PKC-targeted compounds.
Acknowledgments
This work is supported in part by National Heart, Lung, and Blood Institute Grant HL-77860.
References
- 1.Alden KJ, Goldspink PH, Ruch SW, Buttrick PM, Garcia J. Enhancement of L-type Ca2+ current from neonatal mouse ventricular myocytes by constitutively active PKC-β II. Am J Physiol. 2002;282:C768–C774. doi: 10.1152/ajpcell.00494.2001. [DOI] [PubMed] [Google Scholar]
- 2.Apostolatos A, Song S, Acosta S, Peart M, Watson JE, Bickford P, Cooper DR, Patel NA. Insulin promotes neuronal survival via the alternatively spliced protein kinase C-δ II (PKCδII) isoform. J Biol Chem. 2012 doi: 10.1074/jbc.M111.313080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Apostolatos H, Apostolatos A, Vickers T, Watson JE, Song S, Vale F, Cooper DR, Sanchez-Ramos J, Patel NA. Vitamin A metabolite, all-trans-retinoic acid, mediates alternative splicing of protein kinase C-δVIII (PKCδVIII) isoform via splicing factor SC35. J Biol Chem. 2010;285:25987–25995. doi: 10.1074/jbc.M110.100735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bates E, Bode C, Costa M, Gibson CM, Granger C, Green C, Grimes K, Harrington R, Huber K, Kleiman N, Mochly-Rosen D, Roe M, Sadowski Z, Solomon S, Widimsky P. Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. Circulation. 2008;117:886–896. doi: 10.1161/CIRCULATIONAHA.107.759167. [DOI] [PubMed] [Google Scholar]
- 5.Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, Mintze K, Pickard T, Roden R, Bristow MR, Sabbah HN, Mizrahi JL, Gromo G, King GL, Vlahos CJ. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- 6.Bowman JC, Steinberg SF, Jiang T, Gennan D, Fishman GI, Buttrick PM. Expression of protein kinase C-β in the heart causes hypertrophy in adult mice and sudden death in neonates. J Clin Invest. 1997;100:2189–2195. doi: 10.1172/JCI119755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braz JC, Bueno OF, De Windt LJ, Molkentin JD. PKCα regulates the hypertrophic growth of cardiomyocytes through extracellular signal-regulated kinase1/2 (ERK1/2) J Cell Biol. 2002;156:905–919. doi: 10.1083/jcb.200108062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, Paoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. PKC-α regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10:248–254. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 9.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, Cavallaro G, Banci L, Guo Y, Bolli R, Dorn GW, Mochly-Rosen D. Opposing cardioprotective actions and parallel hypertrophic effects of δ PKC and ε PKC. Proc Natl Acad Sci U S A. 2001;98:11114–11119. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng N, He R, Tian J, Dinauer MC, Ye RD. A critical role of protein kinase Cδ activation loop phosphorylation in formyl-methionyl-leucyl-phenylalanine-induced phosphorylation of p47phox and rapid activation of nicotinamide adenine dinucleotide phosphate Oxidase. J Immunol. 2007;179:7720–7728. doi: 10.4049/jimmunol.179.11.7720. [DOI] [PubMed] [Google Scholar]
- 12.Churchill EN, Qvit N, Mochly-Rosen D. Rationally designed peptide regulators of protein kinase C. Trends Endocrinol Metab. 2009;20:25–33. doi: 10.1016/j.tem.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Churchill EN, Szweda LI. Translocation of δPKC to mitochondria during cardiac reperfusion enhances superoxide anion production and induces loss in mitochondrial function. Arch Biochem Biophys. 2005;439:194–199. doi: 10.1016/j.abb.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 14.Connelly KA, Kelly DJ, Zhang Y, Prior DL, Advani A, Cox AJ, Thai K, Krum H, Gilbert RE. Inhibition of protein kinase C-β by ruboxistaurin preserves cardiac function and reduces extracellular matrix production in diabetic cardiomyopathy. Circ Heart Fail. 2009;2:129–137. doi: 10.1161/CIRCHEARTFAILURE.108.765750. [DOI] [PubMed] [Google Scholar]
- 15.Davidoff AJ, Davidson MB, Carmody MW, Davis ME, Ren J. Diabetic cardiomyocyte dysfunction and myocyte insulin resistance: role of glucose-induced PKC activity. Mol Cell Biochem. 2004;262:155–163. doi: 10.1023/b:mcbi.0000038231.68078.4b. [DOI] [PubMed] [Google Scholar]
- 16.Disatnik MH, Buraggi G, Mochly-Rosen D. Localization of protein kinase C isozymes in cardiac myocytes. Experimental Cell Research. 1994;210:287–297. doi: 10.1006/excr.1994.1041. [DOI] [PubMed] [Google Scholar]
- 17.Dorn GW, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ, Csukai M, Wu G, Lorenz JN, Mochly-Rosen D. Sustained in vivo cardiac protection by a rationally designed peptide that causes ε protein kinase C translocation. Proc Natl Acad Sci U S A. 1999;96:12798–12803. doi: 10.1073/pnas.96.22.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y, Pandey P, Datta R, Huang Y, Kharbanda S, Allen H, Kamen R, Wong W, Kufe D. Proteolytic activation of protein kinase C δ by an ICE/CED 3-like protease induces characteristics of apoptosis. J Exp Med. 1996;184:2399–2404. doi: 10.1084/jem.184.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldberg M, Zhang HL, Steinberg SF. Hypoxia alters the subcellular distribution of protein kinase C isoforms in neonatal rat ventricular myocytes. J Clin Invest. 1997;99:55–61. doi: 10.1172/JCI119133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gray MO, Zhou HZ, Schafhalter-Zoppoth I, Zhu P, Mochly-Rosen D, Messing RO. Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase C-ε. J Biol Chem. 2004;279:3596–3604. doi: 10.1074/jbc.M311459200. [DOI] [PubMed] [Google Scholar]
- 21.Guo J, Cong L, Rybin VO, Gertsberg Z, Steinberg SF. Protein kinase C-δ regulates the subcellular localization of Shc in H2O2-treated cardiomyocytes. Am J Physiol Cell Physiol. 2010;299:C770–C778. doi: 10.1152/ajpcell.00170.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo J, Gertsberg Z, Ozgen N, Sabri A, Steinberg SF. Protein kinase D isoforms are activated in an agonist-specific manner in cardiomyocytes. J Biol Chem. 2011;286:6500–6509. doi: 10.1074/jbc.M110.208058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo J, Sabri A, Elouardighi H, Rybin V, Steinberg SF. Alpha1-adrenergic receptors activate AKT via a Pyk2/PDK-1 pathway that is tonically inhibited by novel protein kinase C isoforms in cardiomyocytes. Circ Res. 2006;99:1367–1375. doi: 10.1161/01.RES.0000252830.01581.fd. [DOI] [PubMed] [Google Scholar]
- 24.Harrison BC, Kim MS, van Rooij E, Plato CF, Papst PJ, Vega RB, McAnally JA, Richardson JA, Bassel-Duby R, Olson EN, McKinsey TA. Regulation of cardiac stress signaling by protein kinase D1. Mol Cell Biol. 2006;26:3875–3888. doi: 10.1128/MCB.26.10.3875-3888.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heidkamp MC, Bayer AL, Martin JL, Samarel AM. Differential Activation of Mitogen-Activated Protein Kinase Cascades and Apoptosis by Protein Kinase C ε and δ in Neonatal Rat Ventricular Myocytes. Circ Res. 2001;89:882–890. doi: 10.1161/hh2201.099434. [DOI] [PubMed] [Google Scholar]
- 26.Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. PKC phosphorylation of titin’s PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res. 2009;105:631–8. 17. doi: 10.1161/CIRCRESAHA.109.198465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoshi N, Langeberg LK, Gould CM, Newton AC, Scott JD. Interaction with AKAP79 modifies the cellular pharmacology of PKC. Mol Cell. 2010;37:541–550. doi: 10.1016/j.molcel.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang L, Wolska BM, Montgomery DE, Burkart EM, Buttrick PM, Solaro RJ. Increased contractility and altered Ca2+ transients of mouse heart myocytes conditionally expressing PKCβ. Am J Physiol Cell Physiol. 2001;280:C1114–C1120. doi: 10.1152/ajpcell.2001.280.5.C1114. [DOI] [PubMed] [Google Scholar]
- 29.Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, Rezaee M, Yock PG, Murphy E, Mochly-Rosen D. Inhibition of δ-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation. 2003;108:2304–2307. doi: 10.1161/01.CIR.0000101682.24138.36. [DOI] [PubMed] [Google Scholar]
- 30.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform βII and diacylglycerol levels in the aorta and heart of diabetic rats: Differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci USA. 1992;89:11059–11063. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang MY, Zhang Y, Matkovich SJ, Diwan A, Chishti AH, Dorn GW. Receptor-independent cardiac protein kinase Cα activation by calpain-mediated truncation of regulatory domains. Circ Res. 2010;107:903–912. doi: 10.1161/CIRCRESAHA.110.220772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase Cε mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kheifets V, Bright R, Inagaki K, Schechtman D, Mochly-Rosen D. Protein kinase Cδ-annexin V interaction: a required step in δPKC translocation and function. J Biol Chem. 2006;281:23218–23226. doi: 10.1074/jbc.M602075200. [DOI] [PubMed] [Google Scholar]
- 34.Kheifets V, Mochly-Rosen D. Insight into intra- and inter-molecular interactions of PKC: design of specific modulators of kinase function. Pharmacol Res. 2007;55:467–476. doi: 10.1016/j.phrs.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kishimoto A, Mikawa K, Hashimoto K, Yasuda I, Tanaka S, Tominaga M, Kuroda T, Nishizuka Y. Limited proteolysis of protein kinase C subspecies by calcium-dependent neutral protease (calpain) J Biol Chem. 1989;264:4088–4092. [PubMed] [Google Scholar]
- 36.Klein G, Schaefer A, Hilfiker-Kleiner D, Oppermann D, Shukla P, Quint A, Podewski E, Hilfiker A, Schroder F, Leitges M, Drexler H. Increased collagen deposition and diastolic dysfunction but preserved myocardial hypertrophy after pressure overload in mice lacking PKCε. Circ Res. 2005;96:748–755. doi: 10.1161/01.RES.0000161999.86198.1e. [DOI] [PubMed] [Google Scholar]
- 37.Konishi H, Tanaka M, Takemura Y, Matsuzaki H, Ono Y, Kikkawa U, Nishizuka Y. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc Natl Acad Sci U S A. 1997;94:11233–11237. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konishi H, Yamauchi E, Taniguchi H, Yamamoto T, Matsuzaki H, Takemura Y, Ohmae K, Kikkawa U, Nishizuka Y. Phosphorylation sites of protein kinase Cδ in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proc Natl Acad Sci U S A. 2001;98:6587–6592. doi: 10.1073/pnas.111158798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kooij V, Boontje N, Zaremba R, Jaquet K, Dos RC, Stienen GJ, van d V. Protein kinase Cα and ε phosphorylation of troponin and myosin binding protein C reduce Ca2+ sensitivity in human myocardium. Basic Res Cardiol. 2010;105:289–300. doi: 10.1007/s00395-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kostelecky B, Saurin AT, Purkiss A, Parker PJ, McDonald NQ. Recognition of an intra-chain tandem 14-3-3 binding site within PKCε. EMBO Rep. 2009;10:983–989. doi: 10.1038/embor.2009.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JY, Chiu YH, Asara J, Cantley LC. Inhibition of PI3K binding to activators by serine phosphorylation of PI3K regulatory subunit p85α Src homology-2 domains. Proc Natl Acad Sci U S A. 2011;108:14157–14162. doi: 10.1073/pnas.1107747108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leitges M, Gimborn K, Elis W, Kalesnikoff J, Hughes MR, Krystal G, Huber M. Protein kinase C-δ is a negative regulator of antigen-induced mast cell degranulation. Mol Cell Biol. 2002;22:3970–3980. doi: 10.1128/MCB.22.12.3970-3980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leitges M, Mayr M, Braun U, Mayr U, Li C, Pfister G, Ghaffari-Tabrizi N, Baier G, Hu Y, Xu Q. Exacerbated vein graft arteriosclerosis in protein kinase Cδ-null mice. J Clin Invest. 2001;108:1505–1512. doi: 10.1172/JCI12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Q, Chen X, MacDonnell SM, Kranias EG, Lorenz JN, Leitges M, Houser SR, Molkentin JD. Protein kinase Cα, but not PKCβ or PKCγ, regulates contractility and heart failure susceptibility: implications for ruboxistaurin as a novel therapeutic approach. Circ Res. 2009;105:194–200. doi: 10.1161/CIRCRESAHA.109.195313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu W, Lee HK, Xiang C, Finniss S, Brodie C. The phosphorylation of tyrosine 332 is necessary for the caspase 3-dependent cleavage of PKCδ and the regulation of cell apoptosis. Cell Signal. 2007;19:2165–2173. doi: 10.1016/j.cellsig.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 46.Maeno Y, Li Q, Park K, Rask-Madsen C, Gao B, Matsumoto M, Liu Y, Wu IH, White MF, Feener EP, King GL. Inhibition of insulin signaling in endothelial cells by protein kinase C induced phosphorylation of p85 subunit of PI3-kinase. J Biol Chem. 2011 doi: 10.1074/jbc.M111.286591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malhotra R, D’Souza KM, Staron ML, Birukov KG, Bodi I, Akhter SA. Gαq-mediated activation of GRK2 by mechanical stretch in cardiac myocytes: the role of protein kinase C. J Biol Chem. 2010;285:13748–13760. doi: 10.1074/jbc.M110.109272. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Markou T, Yong CS, Sugden PH, Clerk A. Regulation of protein kinase C δ by phorbol ester, endothelin-1, and platelet-derived growth factor in cardiac myocytes. J Biol Chem. 2006;281:8321–8331. doi: 10.1074/jbc.M508398200. [DOI] [PubMed] [Google Scholar]
- 49.Mayr M, Metzler B, Chung YL, McGregor E, Mayr U, Troy H, Hu Y, Leitges M, Pachinger O, Griffiths JR, Dunn MJ, Xu Q. Ischemic preconditioning exaggerates cardiac damage in PKC-δ null mice. Am J Physiol. 2004;287:H946–H956. doi: 10.1152/ajpheart.00878.2003. [DOI] [PubMed] [Google Scholar]
- 50.Mima A, Ohshiro Y, Kitada M, Matsumoto M, Geraldes P, Li C, Li Q, White GS, Cahill C, Rask-Madsen C, King GL. Glomerular-specific protein kinase C-βinduced insulin receptor substrate-1 dysfunction and insulin resistance in rat models of diabetes and obesity. Kidney Int. 2011;79:883–896. doi: 10.1038/ki.2010.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly-Rosen D. Protein kinase Cδ activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J Biol Chem. 2004;279:47985–47991. doi: 10.1074/jbc.M405071200. [DOI] [PubMed] [Google Scholar]
- 52.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 54.Ozgen N, Obreztchikova M, Guo J, Elouardighi H, Dorn GW, Wilson BA, Steinberg SF. Protein kinase D links Gq-coupled peceptors to cAMP response element-binding protein (CREB)-Ser133 phosphorylation in the heart. J Biol Chem. 2008;283:17009–17019. doi: 10.1074/jbc.M709851200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patel NA, Apostolatos HS, Mebert K, Chalfant CE, Watson JE, Pillay TS, Sparks J, Cooper DR. Insulin regulates protein kinase CβII alternative splicing in multiple target tissues: development of a hormonally responsive heterologous minigene. Mol Endocrinol. 2004;18:899–911. doi: 10.1210/me.2003-0391. [DOI] [PubMed] [Google Scholar]
- 56.Ping P, Zhang J, Qiu Y, Tang XL, Manchikalapudi S, Cao X, Bolli R. Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ Res. 1997;81:404–414. doi: 10.1161/01.res.81.3.404. [DOI] [PubMed] [Google Scholar]
- 57.Porter MJ, Heidkamp MC, Scully BT, Patel N, Martin JL, Samarel AM. Isoenzyme-selective regulation of SERCA2 gene expression by protein kinase C in neonatal rat ventricular myocytes. Am J Physiol. 2003;285:C39–C47. doi: 10.1152/ajpcell.00461.2002. [DOI] [PubMed] [Google Scholar]
- 58.Robles-Flores M, Rendon-Huerta E, Gonzalez-Aguilar H, Mendoza-Hernandez G, Islas S, Mendoza V, Ponce-Castaneda MV, Gonzalez-Mariscal L, Lopez-Casillas F. p32 (gC1qBP) is a general protein kinase C (PKC)-binding protein. J Biol Chem. 2002;277:5247–5255. doi: 10.1074/jbc.M109333200. [DOI] [PubMed] [Google Scholar]
- 59.Roman BB, Geenen DL, Leitges M, Buttrick PM. PKCβ is not necessary for cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2001;280:H2264–H2270. doi: 10.1152/ajpheart.2001.280.5.H2264. [DOI] [PubMed] [Google Scholar]
- 60.Rybin VO, Guo J, Gertsberg Z, Elouardighi H, Steinberg SF. Protein kinase Cε and Src control PKCδ activation loop phosphorylation in cardiomyocytes. J Biol Chem. 2007;282:23631–23638. doi: 10.1074/jbc.M701676200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rybin VO, Guo J, Gertsberg Z, Feinmark SJ, Steinberg SF. Phorbol 12-myristate 13-acetate-dependent protein kinase C-δ-Tyr311 phosphorylation in cardiomyocyte caveolae. J Biol Chem. 2008;283:17777–17788. doi: 10.1074/jbc.M800333200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rybin VO, Guo J, Harleton E, Feinmark SJ, Steinberg SF. Regulatory autophosphorylation sites on protein kinase C-δ at Thr141 and Thr295. Biochemistry. 2009;48:4642–4651. doi: 10.1021/bi802171c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rybin VO, Guo J, Sabri A, Elouardighi H, Schaefer E, Steinberg SF. Stimulus-specific differences in protein kinase C-δ localization and activation mechanisms in cardiomyocytes. J Biol Chem. 2004;279:19350–19361. doi: 10.1074/jbc.M311096200. [DOI] [PubMed] [Google Scholar]
- 64.Rybin VO, Steinberg SF. Do adult rat ventricular myocytes express protein kinase Cα? Am J Physiol. 1997;272:H2485–H2491. doi: 10.1152/ajpheart.1997.272.5.H2485. [DOI] [PubMed] [Google Scholar]
- 65.Rybin VO, Steinberg SF. Protein kinase C isoform expression and regulation in the developing rat heart. Circ Res. 1994;74:299–309. doi: 10.1161/01.res.74.2.299. [DOI] [PubMed] [Google Scholar]
- 66.Stebbins EG, Mochly-Rosen D. Binding specificity for RACK1 resides in the V5 region of βII protein kinase C. J Biol Chem. 2001;276:29644–29650. doi: 10.1074/jbc.M101044200. [DOI] [PubMed] [Google Scholar]
- 67.Steinberg SF. Distinctive activation mechanisms and functions for protein kinase C-δ. Biochem J. 2004;384:449–459. doi: 10.1042/BJ20040704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stempka L, Girod A, Muller HJ, Rincke G, Marks F, Gschwendt M, Bossemeyer D. Phosphorylation of protein kinase Cδ at threonine 505 is not a prerequisite for enzymatic activity. J Biol Chem. 1997;272:6805–6811. doi: 10.1074/jbc.272.10.6805. [DOI] [PubMed] [Google Scholar]
- 70.Sumandea MP, Pyle WG, Kobayashi T, de Tombe PP, Solaro RJ. Identification of a functionally critical protein kinase C phosphorylation residue of cardiac troponin T. J Biol Chem. 2003;278:35135–35144. doi: 10.1074/jbc.M306325200. [DOI] [PubMed] [Google Scholar]
- 71.Sumandea MP, Rybin VO, Hinken AC, Wang C, Kobayashi T, Harleton E, Sievert G, Balke CW, Feinmark SJ, Solaro RJ, Steinberg SF. Tyrosine phosphorylation modifies PKCδ-dependent phosphorylation of cardiac troponin I. J Biol Chem. 2008;283:22680–22689. doi: 10.1074/jbc.M802396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takeishi Y, Chu G, Kirkpatrick DM, Li Z, Wakasaki H, Kranias EG, King GL, Walsh RA. In vivo phosphorylation of cardiac troponin I by protein kinase CβII decreases cardiomyocyte calcium responsiveness and contractility in transgenic mouse hearts. J Clin Invest. 1998;102:72–78. doi: 10.1172/JCI2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takeishi Y, Jalili T, Ball NA, Walsh RA. Responses of cardiac protein kinase C isoforms to distinct pathological stimuli are differentially regulated. Circ Res. 1999;85:264–271. doi: 10.1161/01.res.85.3.264. [DOI] [PubMed] [Google Scholar]
- 74.Takeishi Y, Ping P, Bolli R, Kirkpatrick DL, Hoit BD, Walsh RA. Transgenic overexpression of constitutively active protein kinase Cε causes concentric cardiac hypertrophy. Circ Res. 2000;86:1218–1223. doi: 10.1161/01.res.86.12.1218. [DOI] [PubMed] [Google Scholar]
- 75.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 76.Wakasaki H, Koya D, Schoen FJ, Jirousek MR, Ways DK, Hoit BD, Walsh RA, King GL. Targeted overexpression of protein kinase CβII isoform in myocardium causes cardiomyopathy. Proc Natl Acad Sci U S A. 1997;94:9320–9325. doi: 10.1073/pnas.94.17.9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Way KJ, Isshiki K, Suzuma K, Yokota T, Zvagelsky D, Schoen FJ, Sandusky GE, Pechous PA, Vlahos CJ, Wakasaki H, King GL. Expression of connective tissue growth factor is increased in injured myocardium associated with protein kinase C β2 activation and diabetes. Diabetes. 2002;51:2709–2718. doi: 10.2337/diabetes.51.9.2709. [DOI] [PubMed] [Google Scholar]
- 78.Yamakawa H, Banno Y, Nakashima S, Yoshimura S, Sawada M, Nishimura Y, Nozawa Y, Sakai N. Crucial role of calpain in hypoxic PC12 cell death: calpain, but not caspases, mediates degradation of cytoskeletal proteins and protein kinase C-alpha and -delta. Neurol Res. 2001;23:522–530. doi: 10.1179/016164101101198776. [DOI] [PubMed] [Google Scholar]
- 79.Yamamoto M, Acevedo-Duncan M, Chalfant CE, Patel NA, Watson JE, Cooper DR. The roles of protein kinase C-βI and -βII in vascular smooth muscle cell proliferation. Exp Cell Res. 1998;240:349–358. doi: 10.1006/excr.1998.3999. [DOI] [PubMed] [Google Scholar]
- 80.Yang L, Doshi D, Morrow J, Katchman A, Chen X, Marx SO. Protein kinase C isoforms differentially phosphorylate Cav1.2 α1c. Biochemistry. 2009;48:6674–6683. doi: 10.1021/bi900322a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang L, Liu G, Zakharov SI, Morrow JP, Rybin VO, Steinberg SF, Marx SO. S1928 is a common site for Cav1.2 phosphorylation by protein kinase C isoforms. J Biol Chem. 2005;280:207–214. doi: 10.1074/jbc.M410509200. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Y, Matkovich SJ, Duan X, Diwan A, Kang MY, Dorn GW. Receptor-independent protein kinase C-α signaling by calpain-generated free catalytic domains induces HDAC5 nuclear export and regulates cardiac transcription. J Biol Chem. 2011;286:26943–26951. doi: 10.1074/jbc.M111.234757. [DOI] [PMC free article] [PubMed] [Google Scholar]