

Abstract
Dermal exposure to sulfur mustard causes inflammation and tissue injury. This is associated with changes in expression of antioxidants and eicosanoids which contribute to oxidative stress and toxicity. In the present studies we analyzed mechanisms regulating expression of these mediators using an in vitro skin construct model in which mouse keratinocytes were grown at an air-liquid interface and exposed directly to 2-chloroethyl ethyl sulfide (CEES), a model sulfur mustard vesicant. CEES (100-1000 μM) was found to cause marked increases in keratinocyte protein carbonyls, a marker of oxidative stress. This was correlated with increases in expression of Cu,Zn superoxide dismutase, catalase, thioredoxin reductase and the glutathione-S-transferases, GSTA1-2, GSTP1 and mGST2. CEES also upregulated several enzymes important in the synthesis of prostaglandins and leukotrienes including cyclooxygenase-2 (COX-2), microsomal prostaglandin E synthase-2 (mPGES-2), prostaglandin D synthase (PGDS), 5-lipoxygenase (5-LOX), leukotriene A4 (LTA4) hydrolase and leukotriene C4 (LTC4) synthase. CEES readily activated keratinocyte JNK and p38 MAP kinases, signaling pathways which are known to regulate expression of antioxidants, as well as prostaglandin and leukotriene synthases. Inhibition of p38 MAP kinase suppressed CEES-induced expression of GSTA1-2, COX-2, mPGES-2, PGDS, 5-LOX, LTA4 hydrolase and LTC4 synthase, while JNK inhibition blocked PGDS and GSTP1. These data indicate that CEES modulates expression of antioxidants and enzymes producing inflammatory mediators by distinct mechanisms. Increases in antioxidants may be an adaptive process to limit tissue damage. Inhibiting the capacity of keratinocytes to generate eicosanoids may be important in limiting inflammation and protecting the skin from vesicant-induced oxidative stress and injury.
Keywords: CEES, sulfur mustard, inflammation, skin
Introduction
Sulfur mustard (bis[2-chloroethyl] sulfide) is a bifunctional alkylating agent and a potent skin vesicant (Dacre and Goldman, 1996). It readily penetrates the skin, causing an early inflammatory response followed by blistering and persistent tissue damage (Dacre and Goldman, 1996; Rice, 2003). Oxidative stress is known to initiate inflammatory responses in the skin (Trouba et al., 2002). Sulfur mustard and related analogs including, 2,2′-dichloro-N-methyldiethylamine (nitrogen mustard) and 2-chloroethyl ethyl sulfide (CEES) or half mustard, have been reported to induce oxidative stress by depleting cells of intracellular antioxidants including glutathione and thioredoxin (Smith et al., 1995; Paromov et al., 2007). CEES has also been shown to cause changes in mitochondrial membrane potential resulting in increased production of reactive oxygen species (ROS) (Gould et al., 2009). Moreover, topical application of CEES to mouse skin results in the generation of 8-oxo-2-deoxyguanosine DNA adducts and increased lipid peroxidation and protein oxidation, markers of ROS and intracellular oxidative stress (Pal et al., 2009). In the guinea pig model, superoxide dismutase (SOD) protects against sulfur mustard-induced skin burns, while desferrioxamine, an iron chelator that prevents the formation of hydroxyl radicals, mitigates nitrogen mustard-induced skin toxicity (Eldad et al., 1998; Karayilanoglu et al., 2003). Sulphoraphane, an antioxidant enzyme inducer, as well as glutathione, also promote keratinocyte survival in culture following treatment with sulfur mustard (Smith et al., 1997; Gross et al., 2006). These findings demonstrate that ROS and oxidative stress play important roles in the toxicity of sulfur mustard and related analogs.
In the skin, a complex network of antioxidant enzymes functions to protect against oxidative stress (see Fig. 1 for summary of ROS detoxification pathways) (Darr and Fridovich, 1994; Sander et al., 2004). These enzymes include ROS scavengers such as SOD, catalase, and thioredoxin reductase (TrxR), as well as glutathione S-transferases (GST), a family of glutathione-metabolizing enzymes that detoxify electrophilic compounds (Hayes and McLellan, 1999b; Nordberg and Arner, 2001). Mechanisms mediating expression of vesicant-induced oxidative stress were analyzed in the present studies using a skin construct model consisting of mouse keratinocytes grown at an air-liquid interface. In this system, chemicals are applied directly to the air surface of the cells in order to simulate realistic exposure scenarios. Using the skin construct, CEES was found to cause marked increases in keratinocyte protein oxidation and alterations in expression of several key intracellular antioxidants. Moreover, this was associated with increased expression of enzymes important in the synthesis of prostaglandins and leukotrienes, a response dependent upon JNK and p38 MAP kinase signaling. These data demonstrate that keratinocytes grown in the skin construct system are highly responsive to vesicants, and that this is a useful model for analyzing mechanisms of sulfur mustard-induced skin toxicity. Currently, few effective countermeasures against vesicant-induced dermal toxicity are available and a more complete understanding of the mechanism of action of sulfur mustard using skin models is key to identifying potential therapeutic targets.
Figure 1. Summary of ROS detoxification pathways and eicosanoid biosynthetic pathways.
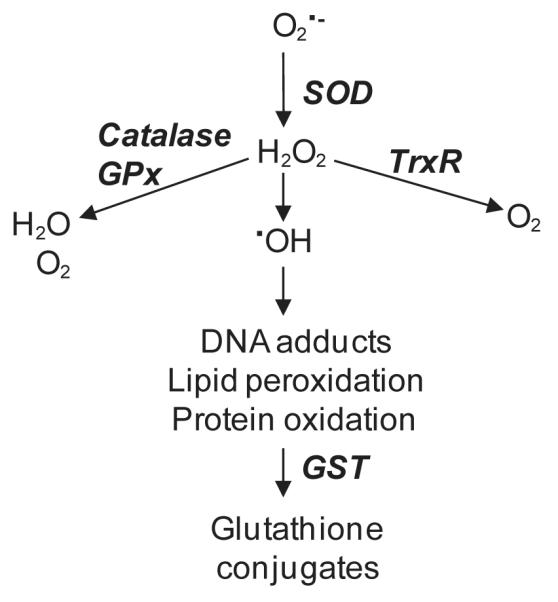

Panel A: ROS detoxification pathways. Superoxide anion is metabolized to hydrogen peroxide by SOD. Hydrogen peroxide is then detoxified by catalase and/or various peroxidases and reductases including TrxR. In the presence of transition metals, hydrogen peroxide is converted to highly reactive hydroxyl radicals. Oxidized proteins and lipids are conjugated to glutathione by the GST enzymes to facilitate cellular elimination. Panel B: Eicosanoid biosynthetic pathways: Arachidonic acid,mobilized directly from membrane phospholipids, is oxidized to form various eicosanoids by cyclooxygenases (COX), lipoxygenases (LOX) or cytochrome P450 monooxygenases. For prostaglandin metabolism, arachidonic acid is metabolized via cyclooxygenases (COX-1 and COX-2) to produce an intermediate prostaglandin, PGH2. Further metabolism by the prostanoid synthases, microsomal prostaglandin E2 synthase (mPGES), prostaglandin F2 synthase (PGFS), prostaglandin D2 synthase (PGDS), prostaglandin I2 synthase (PGIS) or thromboxane A2 synthase (TXAS), forms prostaglandins (PGE2, PGF2α and PGD2), prostacyclin (PGI2) or thromboxane (TXA2), respectively. For leukotriene biosynthesis, arachidonic acid is metabolized via 5-lipoxygenase (5-LOX), in association with the 5-LOX activating protein (FLAP), generating a series of intermediate metabolites, 5-hydroperoxyeicosatetraenoic acid (5-HPETE), 5-hydroxyeicosatetraenoic acid (5-HETE) and leukotriene A4 (LTA4). LTA4 is further metabolized by either LTA4 hydrolase to form leukotriene B4 (LTB4) or conjugated with glutathione by LTC4 synthase to form leukotriene C4 (LTC4).
Materials and Methods
Chemicals and reagents
Rabbit polyclonal antibodies to p38, phospho-p38, JNK, phospho-JNK, ERK 1/2, and phospho-ERK 1/2 were from Cell Signaling Technology (Beverly, MA). Goat polyclonal antibodies to COX-2, rabbit polyclonal antibodies to β-catenin and Cu,Zn-SOD, and horseradish peroxidase-labeled donkey anti-goat secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal antibodies to 5-LOX and LTA4 hydrolase were from Cayman Chemical (Ann Arbor, MI). Horse radish peroxidase-labeled goat anti-rabbit secondary antibodies and detergent-compatible protein assay reagents were from Bio-Rad Laboratories (Hercules, CA). Superscript III Reverse Transcriptase and the MTS CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay were from Promega (Madison, WI), and the Versagene RNA purification kit from Gentra Systems (Minneapolis, MN). The Western Lightning enhanced chemiluminescence kit (ECL) was from Perkin Elmer Life Sciences, Inc. (Boston, MA), the OxyBlot Protein Oxidation Detection kit from Chemicon International (Temecula, CA) and precast gels were from Pierce Biotechnology (Rockford, IL). SYBR Green Master Mix and other PCR reagents were from Applied Biosystems (Foster City, CA). Annexin V-APC and other apoptosis reagents were from BD Pharmigen (San Diego, CA). Cell culture reagents and 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) mixed isomers were from Invitrogen Corp (Carlsbad, CA), polyester Transwell permeable supports from Corning Life Sciences Inc. (Acton, MA) and SP600125 from Calbiochem (La Jolla, CA). CEES, SB203580, protease inhibitor cocktail, and all other chemicals were from Sigma-Aldrich (St. Louis, MO).
Cells and treatments
PAM212 mouose keratinocytes were grown in DMEM supplemented with 10% fetal bovine serum (Yuspa et al., 1980). A three-dimensional skin-like construct was generated by plating the cells 1 × 106/well on 24 mm Transwell semi-permeable polyester membrane supports (0.4 μm pore size) in 6-well cell culture plates (Aufderheide et al., 2003). After reaching confluence, the medium was removed from the top portion of the Transwell support such that the keratinocytes were exposed to air at the apical surface and medium at the basolateral surface. After 24 hr, one ml of vehicle control or CEES was added to the apical surface of the construct. A stock CEES solution (100 mM) was prepared fresh in absolute ethanol immediately before use and diluted to the appropriate concentrations in PBS. Cells were incubated for 2 hr at 37°C in a humidified CO2 incubator. The Transwells were then removed, washed in PBS and immediately replaced in the same 6-well culture plates for 24 hr unless otherwise indicated. In some experiments, one ml PBS/Transwell containing the p38 MAP kinase inhibitor, SB203580 (10 μM), the JNK kinase inhibitor, SP600125 (20 μM), or DMSO control were added to the apical surface, and the cells incubated at 37°C for 3 hr prior to CEES treatment.
Western blotting
Cell lysates were prepared using an SDS-lysis buffer (10 mM Tris-base and 1% SDS, pH 7.6 supplemented with 10 μl protease inhibitor cocktail consisting of 4-(2-aminoethyl)benzenesulfonyl fluoride, aprotinin, bestatin hydrochloride, N-(trans-epoxysuccinyl)-L-leucine 4-guanidinobutylamide, EDTA and leupeptin). Proteins (20 μg) from lysates were separated on 10% SDS-polyacrylamide gels and then transferred to nitrocellulose membranes. After incubation in blocking buffer (5% dry milk Tris-buffered saline containing 0.1% Tween 20) for 1 hr at room temperature or overnight at 4°C, the membranes were incubated for 2 hr at room temperature or overnight at 4°C with primary antibodies followed by horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 hr at room temperature. Protein expression was visualized using enhanced chemiluminescence reagents. Equal loading of protein was confirmed by expression of β-catenin.
Densitometric analysis of western blots were performed using a Fluor Chem SP imaging system (Alpha Innotech Corp., San Leandro, CA), and absolute values for each protein were normalized with the values for β-catenin. Results are reported in arbitrary units, comparing each value with that obtained from each respective β-catenin measurement.
Determination of cell viability and apoptosis
Cell viability was assessed using an MTS cell viability assay as described by the manufacturer. In this assay, viable cells convert MTS to an aqueous formazan product, which can be quantified by absorbance at 492 nm. Briefly, the cells grown on Transwell supports were treated with CEES (100-1000 μM) as described above. After 24 hr, cells were treated with the MTS reagent (20 μl/Transwell support) and incubated for 2 hr at 37°C. The supernatants were then collected, transferred to a 96-well plate and analyzed using a Molecular Devices SpectraMax M2 spectrophotometer (Sunnyvale, CA). Cell viability was determined to be > 80% for all treatments.
For analysis of keratinocyte apoptosis, the cells treated with CEES (100-1000 μM) as described above. After 24 hr, the cells were removed from the Transwell supports with trypsin, incubated for 15 min at room temperature with Annexin V-APC followed by 10 min with 7-amino-actinomycin (7-AAD). The cells were then analyzed by flow cytometry on a Becton Dickinson FACSArray Bioanalyzer (San Diego, CA). Apoptotic keratinocyte populations were gated electronically and data analyzed using quadrant statistics based on relative Annexin V-APC and 7-AAD fluorescence (Lecoeur et al., 2002). Approximately 13% of the control cells were apoptotic while 13%, 17% and 36% were apoptotic after treatment with 100, 300 and 1000 μM CEES, respectively.
Measurement of ROS production
Intracellular hydrogen peroxide production was measured using the hydrogen peroxide sensitive dye, CM-H2DCFDA, in conjunction with flow cytometry as previously described (Black et al., 2008b). Briefly, cells grown on Transwell supports were incubated with CM-H2DCFDA (5 μM) in PBS for 15 min and then treated with CEES (100-1000 μM) or control for 30 min. The cells were then immediately analyzed for DCF fluorescence on a Beckman Coulter Cytomics FC 500 flow cytometer. Data were analyzed using the Beckman Coulter CXP software.
Protein oxidation assay
Protein oxidation was assessed by the formation of carbonyl groups on protein side chains using an OxyBlot Protein Oxidation Detection kit (Chevion et al., 2000). Keratinocyte lysates, prepared as described above with the addition of 20 μl β-mercatoethanol per sample, were incubated with 2,4-dinitrophenylhydrazine to derivatize the carbonyl groups on proteins to 2,4-dinitrophenylhydrazone-tagged products. Nonderivatized samples were used as a control. Samples were then separated on pre-cast 4-12% gradient SDS-polyacrylamide gels and transferred to nitrocellulose membranes. After blocking, the membranes were incubated with primary antibodies to dinitrophenylhydrazone-modified carbonyl groups, followed by HRP-conjugated secondary antibodies. Protein expression was visualized using ECL reagents.
Real-time polymerase chain reaction
For these experiments, three constructs (n = 3) were treated with each CEES concentration or control. RNA was isolated from the cells using the Versagene RNA purification kit following the manufacturer’s protocol. RNA was converted to cDNA using Superscript reverse transcriptase. The cDNA was diluted 1:10 in RNase-DNase-free water for PCR analysis. For each gene to be tested, a standard curve composed of a serial dilution of pooled cDNA from the samples was used as a reference. All values were normalized to GAPDH. The control was assigned a value of one and treated samples calculated relative to control. Real-time PCR was performed on an ABI Prism 7900 Sequence Detection System using 96-well optical reaction plates. SYBR-Green was used for detection of fluorescent signal and the standard curve method was used for relative quantification analysis. The primer sequences for the genes were generated using Primer Express software (Applied Biosystems) and the oligonucleotides were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA). The forward and reverse sequences used are listed in Table 1.
Table 1.
Primers used to analyze expression of antioxidants and inflammatory mediators1.
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| catalase | ACCAGGGCATCAAAAACTTG | GCCCTGAAGCTTTTTGTCAG |
| COX-1 | GTCCCAGAACCAGGGTGTCT | CACTGGTAGTTGTCGAGGCCA |
| COX-2 | CATTCTTTGCCCAGCACTTCAC | GACCAGGCACCAGACCAAAGAC |
| FLAP | GGTGGAGCATGAAAGCAAGG | CCGGTCCTCTGGAAGCTTC |
| GAPDH | TGAAGCAGGCATCTGAGGG | CGAAGGTGGAAGAGTGGGAG |
| GSTA1-2 | CAGAGTCCGGAAGATTTGGA | CAAGGCAGTCTTGGCTTCTC |
| GSTA3 | GCAAGCCTTGCCAAGATCAA | GGCAGGGAAGTAACGGTTCC |
| GSTA4 | CCCTTGGTTGAAATCGATGG | GAGGATGGCCCTGGTCTGT |
| GSTM1 | CCTACATGAAGAGTAGCCGCTAC | TAGTGAGTGCCCGTGTAGCAA |
| GSTP1 | CCTTGGCCGCTCTTTGG | GGCCTTCACGTAGTCATTCTTAC |
| mGST1 | GCTTTGGCAAGGGAGAGAATG | CCTTCTCGTCAGTGCGAACA |
| mGST2 | TGCAGCCTGTCTGGGTCTC | CAGAAATACTTGTGACGGGCG |
| mGST3 | GGAGGTGTACCCTCCCTTCC | TGGTAAACACCTCCCACCGT |
| LTA4 | CTCACGGTCCAGTCACAGGA | TGTGTCCAAAGTCAGGCTGC |
| LTC4 synthase | GGACGAAGTGGCTCTTCTGG | TGCAACAGAACTCCCACGAG |
| 5-LOX | AGGGAGAAGCTGTCCGAGT | GCAGAGGCCGTGAAGATCAC |
| 8-LOX | TGTTTGCACACTGGCAGGAA | CATTTAGGAACTGGGAGGCG |
| 12-LOX epi | AAGTTCCTTGGCAGACGCC | TCTTTATGCTGCCCCAGGG |
| 12-LOX plt | ACCAGCAAGGACGACGTGAC | ATCAGGTAGCGACCCCATCA |
| 15-LOX | TCGGAGGCAGAATTCAAGGT | CAGCAGTGGCCCAAGGTATT |
| PGDS | CTGCTCTGAGCAAATGGCTG | AGGACCAAACCCATCCACAG |
| mPGES-1 | GGCCTTTCTGCTCTGCAGC | GCCACCGCGTACATCTTGAT |
| mPGES-2 | AGCCCCTGGAAGAGGTCATC | CATTCATGGCCTTCATGGGT |
| PGFS | GAGGAAGTAGGGCTGGCCAT | CCTCACAGTGCCATCAGCAA |
| PGIS | ATCTGCTGCTCCCCAAACTG | CTTTATCCCCCGCTGACAAG |
| Cu,Zn-SOD | ACCAGTGCAGGACCTCATTTTAA | TCTCCAACATGCCTCTCTTCATC |
| Mn-SOD | CACATTAACGCGCAGATCATG | CCAGAGCCTCGTGGTACTTCTC |
| TrxR | TTAGAGACCGTGGGCGTGA | GACGGGTATCTTTCCGGTTTTT |
| TXAS | CCCCAAGCCTTCTCCTTTTG | AAACCCTGGCGGAAAAACAT |
Primer sequences were designed using Primer Express software (Applied Biosystems).
Statistical analysis
Data are expressed as mean ± SEM. Statistical differences between the means were determined using two-way ANOVA and were considered statistically significant at p < 0.05.
Results
Effects of CEES on keratinocyte oxidative stress and expression of antioxidant enzymes
In initial studies, we determined if CEES induces oxidative stress in keratinocytes in the skin construct model by quantifying the generation of intracellular hydrogen peroxide (Fig. 2A). We found that the cells constitutively generated low levels of hydrogen peroxide. Small increases in hydrogen peroxide production were noted following treatment with 100 μM CEES, while 5-10-fold increases were found after treatment with 300 and 1000 μM CEES.
Figure 2. CEES stimulates intracellular hydrogen peroxide production and protein oxidation.

Panel A: Keratinocytes were incubated with DCFH-DA (5 μM) for 15 min.. The cells were then treated with CEES (100, 300 or 1000 μM) or control and analyzed by flow cytometry. Data are presented on a three decade log scale. Panel B: Keratinocytes were treated with CEES (100, 300 or 1000 μM) or control. After 24 hr, lysates were prepared and proteins derivatized with 2,4-dinitrophenylhydrazine to form 2,4-dinitrophenylhydrazone-modified carbonyl groups. After separation on SDS-polyacrylamide gels, modified proteins were quantified by Western blotting using antibodies to dinitrophenylhydrazone-tagged carbonyl groups.
CEES treatment was also found to cause increased protein oxidation in the keratinocytes, as measured by protein carbonyl formation. Low constitutive levels of oxidized proteins were detected in control cells, as evidenced by the presence of an oxidized protein with a molecular weight of approximately 75 kDa (Fig. 2B). Treatment of the cells with CEES (100 – 1000 μM) caused a marked increase in oxidation of this protein, and the appearance of additional oxidized proteins with molecular weights of approximately 36, 43, 50, 66 and 70 kDa. Lower levels of oxidized proteins with molecular weights of 24 and 34 kDa were also noted in CEES-treated cells. CEES-induced protein oxidation was associated with a 4-6 fold increase in mRNA expression of the primary antioxidant enzymes, Cu,Zn-SOD, catalase and thioredoxin reductase (Fig. 3). Cu,Zn-SOD protein expression was also upregulated by CEES (Fig. 4A). In contrast, Mn-SOD mRNA expression decreased (Fig. 3). GST’s, including GSTA1-2 (14-fold), GSTP1 (3-fold) and mGST2 (2-fold), were also upregulated in keratinocytes after CEES treatment while no major changes in expression of GSTA3, GSTA4, GSTM1, mGST1 or mGST3 were observed (Fig. 5).
Figure 3. Effects of CEES on keratinocyte expression of antioxidants.

Cells were treated with CEES (100, 300 or 1000 μM) or control. After 24 hr, expression of Cu,Zn-SOD, Mn-SOD, catalase, and TrxR mRNA was analyzed by real-time PCR. Data are presented as fold change in gene expression relative to control. *Significantly (p < 0.05) different from control (p < 0.05).
Figure 4. Effects of CEES on protein expression of antioxidants, eicosanoid biosynthetic enzymes and MAP kinases.

Cells were treated with CEES (100-1000 μM) or control and then analyzed for protein expression by Western blotting. β-catenin was analyzed as a control for equal protein loading. Panel A: Protein expression was analyzed 24 hr after CEES treatment. Panel B: Densitometric analysis of Western blot expression normalized to β-catenin expression. Panel C: MAP kinases were analyzed 5 min after CEES treatment.
Figure 5. Effects of CEES on keratinocyte expression of glutathione S-transferases.

Cells were treated with CEES (100, 300 or 1000 μM) or control. After 24 hr, expression of cytosolic and microsomal glutathione S-transferases was analyzed by real-time PCR. Data are presented as fold change in gene expression relative to control. *Significantly (p < 0.05) different from control (p < 0.05).
Effects of CEES on keratinocyte expression of enzymes mediating the generation of eicosanoids
In further studies, we analyzed the effects of CEES on enzymes mediating the production of prostaglandins and leukotrienes, which are important in the development of inflammatory responses in the skin (Fogh and Kragballe, 2000). CEES (100-1000 μM) was found to upregulate mRNA expression of COX-2 (4-fold), mPGES-2 (4-fold) and PGDS (8-fold) (Fig. 6A) in keratinocyte skin constructs. Maximal increases in expression of COX-2 and PGDS were observed with 300 μM CEES, while mPGES-2 expression increased continuously over all concentrations tested. CEES also upregulated COX-2 protein expression (Fig. 4A). In contrast, a concentration-dependent decrease in mPGES-1 mRNA expression was observed following CEES treatment, while no major changes were noted in mRNA expression of COX-1, PGFS, PGIS or TXAS (Fig 6A).
Figure 6. Effects of CEES on expression of prostaglandin and leukotriene biosynthetic enzymes.

Cells were treated with CEES (100, 300 or 1000 μM) or control. After 24 hr, expression of eicosanoid biosynthetic enzymes was analyzed by real-time PCR. Panel A: Keratinocyte expression of prostaglandin biosynthetic enzymes. Panel B: Keratinocyte expression of leukotriene biosynthetic enzymes. Data are presented as fold change in gene expression relative to control. *Significantly (p < 0.05) different from control (p < 0.05).
CEES was also found to upregulate expression of 5-LOX, LTA4 hydrolase and LTC4 synthase mRNA, but not FLAP in keratinocytes (Fig. 6B), CEES also increased 5-LOX and LTA4 hydrolase protein expression in the cells (Fig. 4A). In contrast, CEES had no effect on expression of enzymes mediating production of HETEs, including 8-LOX, epidermal and platelet-type 12-LOX, and 15-LOX (Fig. 6B).
Role of MAP kinases in CEES-induced alterations in expression of antioxidants and eicosanoid generating enzymes
In our next series of studies, we analyzed the role of MAP kinases in CEES-induced alterations in gene expression. Treatment of keratinocytes with CEES resulted in a concentration-dependent activation of JNK and p38 MAP kinase expression in keratinocytes, as measured by increases in the phosphorylated forms of the enzymes (Fig. 4C). In contrast, only small increases in activated ERK1/2 kinase were detected at the highest concentration of CEES tested (1000 μM) (Fig. 4C). Keratinocytes also constitutively expressed activated phosphorylated forms of p38 and ERK1/2 kinases, but not JNK kinase.
To determine if JNK and p38 MAP kinases mediated CEES-induced changes in mRNA expression in keratinocytes, we used specific inhibitors of these enzymes. Treatment of the cells with the p38 MAP kinase inhibitor, SB203580, was found to markedly suppress CEES-induced expression of COX-2, mPGES-2, PGDS, 5-LOX, LTA4 hydrolase and LTC4 synthase mRNA, as well as GSTA1-2 mRNA (Fig. 7). In contrast, inhibition of JNK kinase using SP600125 was only partially effective in suppressing expression of PGDS and GSTA1-2. No major effects of kinase inhibition on CEES-induced expression of Cu,Zn-SOD, catalase, thioredoxin reductase, GSTP1 or mGST2 were noted (Fig. 7 and not shown).
Figure 7. Effects of MAP kinase inhibitors on CEES-induced expression of antioxidants and eicosanoid biosynthetic enzymes.

Cells were incubated with the p38 MAP kinase inhibitor or JNK MAP kinase inhibitor or control (CTL) for 3 hr and then with CEES (100-1000 μM) or control. After 24 hr, gene expression was analyzed by real-time PCR. Data are presented as fold change in gene expression relative to control. aSignificantly (p < 0.05) different from control (p38 inhibitor); bSignificantly different (p < 0.05) from control (JNK inhibitor).
Discussion
Three dimensional in vitro skin constructs have been developed as models to assess skin toxicity (Andreadis et al., 2001). By growing cells at an air-liquid interface, an epidermal-like structure is created in which chemicals can be applied directly to the air surface (Nakamura et al., 1990; Greenberg et al., 2006). Some advantages of using skin constructs as models for toxicity testing are that this system provides an alternative to animal experiments, as well as a practical method for mechanistic investigation of large numbers of chemicals and their biological effects (Nakamura et al., 1990). Previous studies with human skin constructs have shown that these models can also be used to evaluate morphological changes in basal keratinocytes and microblister formation in response to CEES and sulfur mustard (Blaha et al., 2000; Blaha et al., 2001; Hayden et al., 2009). The present studies demonstrate that mouse keratinocytes in a construct model are also highly responsive to CEES. Thus, treatment of cells with CEES induced oxidative stress, activated signal transduction pathways, and modulated expression of antioxidants and enzymes that generate eicosanoids, responses known to occur in sulfur mustard-treated rodent and human skin (Papirmeister et al., 1991; Kehe et al., 2009; Shakarjian et al., 2010). Our findings that these responses are dependent on JNK and p38 MAP kinases are novel and suggest a potential therapeutic target for mitigating dermal toxicity induced by vesicants.
Vesicants such as sulfur mustard have been shown to induce oxidative stress in the skin or in skin-derived cells (Papirmeister et al., 1991; Trouba et al., 2002; Bickers and Athar, 2006). This process is the result of excessive production of ROS which can damage lipids, proteins and nucleic acids (Ryter et al., 2007). Using an in vivo mouse skin model, Pal et al. (2009) have recently demonstrated that topical application of CEES results in oxidative stress, as evidenced by the appearance of several key biomarkers including 4-hydroxynonenal-protein adducts, protein radical adducts, and protein and DNA oxidation products. 4-hydroxynonenal is a highly reactive aldehyde generated following lipid peroxidation (Niki, 2009); in mouse skin, CEES induces the formation of 4-hydroxynonenal protein adducts with molecular weights ranging from 6-92 kDa (Pal et al., 2009). This was correlated with the generation of protein carbonyls, products resulting from the oxidation of amino acids such as lysine, arginine, proline, threonine and cysteine (Stadtman, 1993). In our skin construct model, CEES treatment was found to induce oxidative stress. It readily stimulated hydrogen peroxide production in the keratinocytes and resulted in the formation of several distinct carbonyl protein products with molecular weights ranging from 36-75 kDa. These findings suggest that the skin construct model can be used to recapitulate the process of vesicant-induced oxidative stress. The identity of the oxidized proteins generated by keratinocytes in the skin construct model in response to CEES, and their role in vesicant-induced toxicity remains to be determined. Oxidized proteins and their degradation products can regulate cell growth and differentiation, processes known to be important in the response of the skin to sulfur mustard (Papirmeister et al., 1991). In this regard, oxidative stress has been shown to result in carbonylation of several cytoskeletal proteins important in controlling cell growth including β-actin and tubulin, annexin A1, a protein regulating apoptosis, and mitochondrial ATP synthase, which is critical for cellular energy production (Magi et al., 2004; Je et al., 2008; Wong et al., 2008).
It is well recognized that cells respond to increased oxidative stress with changes in expression of antioxidant enzymes, which is key to limiting cellular damage (Adler et al., 1999; Droge, 2002; Scandalios, 2005). Using monolayer cultures of mouse keratinocytes, we previously showed that paraquat-induced oxidative stress was associated with increased expression of Cu,Zn-SOD, catalase and heme oxygenase-1, as well as the glutathione-S-transferases GSTA1-2, GSTP1, GSTA3 and GSTA4, while UVB light-induced oxidative stress resulted in increased glutathione peroxidase and GSTA1-2 (Black et al., 2008a; Black et al., 2008b). In our skin construct model, CEES-induced protein carbonylation was associated with upregulation of mRNA for catalase, TrxR and Cu,Zn-SOD. These findings suggest that the antioxidant response to oxidative stress varies with the inducer. Catalase is a critical enzyme for detoxifying hydrogen peroxide (Chen et al., 2004; Shim et al., 2005). Increased catalase activity has been reported in the livers of rats following injection of sulfur mustard (Jafari, 2007) and liposomes containing catalase provide significant protection from CEES-induced lung damage in rats (McClintock et al., 2006). These results suggest a key role for hydrogen peroxide in vesicant-induced toxicity. TrxR is a selenocysteine containing enzyme important in retaining thioredoxin in its reduced state, and in maintaining cellular redox balance (Schallreuter and Wood, 2001). CEES has been reported to inhibit TrxR enzyme activity; reducing intracellular production of thioredoxin by inhibiting TrxR may be a trigger for increasing synthesis of the enzyme (Jan et al., 2009). Increased total SOD activity has been described previously in rodents following exposure to sulfur mustard or CEES (Mukhopadhyay et al., 2006; Jafari, 2007); in guinea pig lung, CEES induced mRNA for Cu,Zn-SOD (Mukhopadhyay et al., 2006). Our findings in the present studies that Cu,Zn-SOD increased in keratinocytes are consistent with these reports. In contrast, Mn-SOD mRNA expression decreased in keratinocytes after CEES treatment. Cu,Zn-SOD is a cytosolic enzyme, whereas Mn-SOD is a mitochondrial matrix and inner membrane enzyme; thus, these antioxidants protect different subcellular compartments from oxidative stress and are known to be regulated by distinct mechanisms (Fridovich, 1978). Increases in Cu,Zn-SOD are most likely the result of CEES-induced generation of ROS in keratinocyte cytosol. Sulfur mustard and CEES damage mitochondria, resulting in mitochondrial DNA adducts and altered mitochondrial membrane potential (Shahin et al., 2001; Gould et al., 2009) and this is likely to contribute to reduced expression of Mn-SOD.
CEES was also found to upregulate mRNA expression of GSTA1-2, GSTP1 and mGST2. As members of a large superfamily of cytosolic and microsomal enzymes, the GSTs primarily function in the detoxification of xenobiotics via conjugation to glutathione (Hayes et al., 2004). Previous studies have shown that sulfur mustard increases total GST activity in human keratinocytes (Gross et al., 2006). GSTA and GSTP enzymes disrupt lipid radical chain reactions, thereby enhancing cellular elimination of lipid and protein peroxidation products (Hayes et al., 2004). Microsomal GSTs including mGST2 are key for detoxifying membrane bound peroxidation products; sulfur mustard has been shown to induce lipid peroxidation in mouse lung (Elsayed and Omaye, 2004) and liver (Vijayaraghavan et al., 1991), and increases in expression of GSTs may be important in repairing damage to cellular membranes (Papirmeister et al., 1991).
Sulfur mustard-induced oxidative stress is correlated with the production of inflammatory mediators in skin including the lipid-derived mediators, prostaglandins and leukotrienes (Dacre and Goldman, 1996; Paromov et al., 2007). Sulfur mustard has been reported to induce the release of membrane-derived arachidonic acid, an important precursor for prostaglandin and leukotriene biosynthesis (Ray et al., 1995; Funk, 2001; Lefkowitz and Smith, 2002). Consistent with previous studies with sulfur mustard-treated mouse skin (Nyska et al., 2001), in our skin construct model, CEES increased expression of mRNA and protein for COX-2, which mediates the metabolism of arachidonic acid to prostaglandin precursors. Findings that sulfur mustard-induced skin inflammation and tissue damage are significantly reduced in COX-2 deficient mice demonstrate the importance of this enzyme in vesicant-induced dermatotoxicity (Wormser et al., 2004a). This is supported by reports that topical anti-inflammatory agents targeting cyclooxygenase are effective in reducing sulfur mustard-induced skin damage (Babin et al., 2000; Casillas et al., 2000; Dachir et al., 2002; Watanabe et al., 2003; Dachir et al., 2004; Wormser et al., 2004b). We also found that CEES treatment of keratinocytes resulted in increased mRNA expression of mPGES-2 and PGDS, two prostanoid synthases downstream of COX-2 that are responsible for the generation PGE2 and PGD2, respectively. PGE2 is known to mediate skin inflammation (Murakami et al., 2002) and sulfur mustard has been reported to stimulate PGE2 production in full-thickness human skin explants (Rikimaru et al., 1991) and in intact mouse skin (Dachir et al., 2004). Increases in PGDS in response to CEES suggest that PGD2 may also be formed in the skin. PGD2 is an important mediator of dermal allergic responses (Satoh et al., 2006), and it may play a similar role in the development of inflammation following exposure to sulfur mustard. Our observation that mPGES-1 is downregulated, while mPGES-2 is upregulated in keratinocytes following CEES treatment, indicates that the mechanisms mediating expression of these two enzymes are distinct, and that mPGES-2 may be more important for generating PGE2 in keratinocytes in response to vesicants.
Although the effects of sulfur mustard on the biosynthesis of leukotrienes is less well characterized, increased concentrations of these mediators have been measured in inflammatory lesions in the skin of sulfur mustard-treated rabbits (Tanaka et al., 1997). Our findings that CEES caused increases in 5-LOX, LTA4 hydrolase and LTC4 synthase, enzymes responsible for the synthesis of leukotriene B4 (LTB4) and leukotriene C4 (LTC4), are in agreement with these data. mGST2, like LTC4 synthase, conjugates glutathione to leukotriene A4 forming LTC4 (Hayes and McLellan, 1999a). Thus, the increases in mGST2 expression that we observed following CEES treatment of keratinocytes may be representative of a coordinated inflammatory response important not only in protecting against lipid peroxidation, but also in generating leukotrienes. Further studies correlating protein expression of the eicosanoid biosynthetic enzymes in the skin construct model will be important in elucidating their role in sulfur mustard-induced skin injury.
ROS are potent activators of MAP kinase signaling pathways which are key regulators of cell proliferation and cytotoxicity (Kyriakis and Avruch, 2001; Matsuzawa and Ichijo, 2005; McCubrey et al., 2006). MAP kinases have been reported to be activated following treatment of mouse skin or human and mouse keratinocytes in vitro with vesicants (Dillman et al., 2004; Rebholz et al., 2008; Pal et al., 2009). In SKH-1 hairless mouse skin, CEES activates ERK1/2, JNK and p38 MAP kinases (Pal et al., 2009). Similarly, in our skin construct model, CEES was found to activate JNK and p38 MAP kinases, and to a lesser extent ERK1/2. Apparent differences in MAP kinase activation in response to CEES between the mouse skin construct model and mouse skin may be due to differences in the post exposure times evaluated. Thus, in our studies, MAP kinases were induced within 5 min of CEES exposures, while 3-72 hr was required to activate MAP kinases in mouse skin. Using MAP kinase inhibitors, we demonstrated that p38 MAP kinase regulates keratinocyte expression of COX-2, mPGES-2, PGDS, 5-LOX, LTA4 hydrolase, and GSTA1-2 in the skin construct system, while JNK kinase regulates PGDS. Our data are largely in agreement with earlier studies showing that p38 MAP kinase regulates proinflammatory cytokine expression in keratinocytes post-sulfur mustard exposure (Dillman et al., 2004). These data indicate that CEES-induced expression of antioxidant enzymes and eicosanoid biosynthetic enzymes can occur via distinct mechanisms. Earlier studies showed that inhibition of p38 MAP kinase can effectively suppress UVB-induced inflammation in mouse skin including expression of proinflammatory cytokines and COX-2 (Hildesheim et al., 2004; Kim et al., 2005). It remains to be determined if inhibitors of this enzyme and/or JNK also suppress sulfur mustard-induced inflammation and skin injury.
In summary, our data demonstrate that CEES induces oxidative stress in a skin construct model as shown by increased keratinocyte protein oxidation. This is associated with increased mRNA expression of antioxidant enzymes including Cu,Zn-SOD, catalase, TrxR, GSTA1-2, GSTP1, and mGST2. At the same time, CEES stimulates expression of key enzymes important in the production of prostaglandins and leukotrienes, including, COX-2, mPGES-2, PGDS, 5-LOX, LTA4 hydrolase and LTC4 synthase. The MAP kinase signal transduction pathways appear to be important in mediating expression of several of these enzymes. Whether or not changes in expression of these enzymes mediate inflammation or protect the skin from sulfur mustard toxicity will depend on the levels of expression of functional proteins for these enzymes, as well as their roles in repairing or enhancing cellular damage. Our data also demonstrate that the skin construct model may be useful in understanding the responses of keratinocytes to sulfur mustard-induced toxicity.
Acknowledgements
This work was supported in part by National Institutes of Health grants CA100994, CA093798, CA132624, ES004738, ES005022, GM034310 and AR055073. This work was also supported in part by the National Institutes of Health CounterACT Program through the National Institute of Arthritis and Musculoskeletal and Skin Diseases (award #U54AR055073). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the federal government.
List of Abbreviations
- (CEES)
2-chloroethyl ethyl sulfide
- (COX-1)
cyclooxygenase-1
- (COX-2)
cyclooxygenase-2
- (GST)
glutathione S-transferase
- (mGST)
microsomal GST
- (LTA4)
leukotriene A4
- (LTC4)
leukotriene C4
- (5-LOX)
5-lipoxygenase
- (FLAP), )
5-LOX activating protein
- (8-LOX), )
8-lipoxygenase
- (12-LOX), )
12-lipoxygenase
- (15-LOX)
15-lipoxygenase
- (PGE2)
prostaglandin E2
- (mPGES-1)
microsomal PGE2 synthase-1
- (mPGES-2)
microsomal PGE2 synthase-2
- (PGD2)
prostaglandin D2
- (PGDS)
PGD2 synthase
- (PGF2)
prostaglandin F2
- (PGFS)
PGF2 synthase
- (PGH2)
prostaglandin H2
- (PGI2)
prostaglandin I2
- (PGIS)
PGI2 synthase
- (ROS)
reactive oxygen species
- (SOD)
superoxide dismutase
- (TrxR)
thioredoxin reductase
- (TXA2)
thromboxane A2
- (TXAS)
TXA2 synthase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler V, Yin Z, Tew KD, Ronai Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene. 1999;18:6104–6111. doi: 10.1038/sj.onc.1203128. [DOI] [PubMed] [Google Scholar]
- Andreadis ST, Hamoen KE, Yarmush ML, Morgan JR. Keratinocyte growth factor induces hyperproliferation and delays differentiation in a skin equivalent model system. FASEB J. 2001;15:898–906. doi: 10.1096/fj.00-0324com. [DOI] [PubMed] [Google Scholar]
- Aufderheide M, Knebel JW, Ritter D. Novel approaches for studying pulmonary toxicity in vitro. Toxicol Lett. 2003;140-141:205–211. doi: 10.1016/s0378-4274(02)00512-x. [DOI] [PubMed] [Google Scholar]
- Babin MC, Ricketts K, Skvorak JP, Gazaway M, Mitcheltree LW, Casillas RP. Systemic administration of candidate antivesicants to protect against topically applied sulfur mustard in the mouse ear vesicant model (MEVM) J Appl Toxicol. 2000;20(Suppl 1):S141–144. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat666>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Bickers DR, Athar M. Oxidative stress in the pathogenesis of skin disease. J Invest Dermatol. 2006;126:2565–2575. doi: 10.1038/sj.jid.5700340. [DOI] [PubMed] [Google Scholar]
- Black AT, Gray JP, Shakarjian MP, Laskin DL, Heck DE, Laskin JD. Distinct effects of ultraviolet B light on antioxidant expression in undifferentiated and differentiated mouse keratinocytes. Carcinogenesis. 2008a;29:219–225. doi: 10.1093/carcin/bgm242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black AT, Gray JP, Shakarjian MP, Laskin DL, Heck DE, Laskin JD. Increased oxidative stress and antioxidant expression in mouse keratinocytes following exposure to paraquat. Toxicol Appl Pharmacol. 2008b;231:384–392. doi: 10.1016/j.taap.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaha M, Bowers W, Jr., Kohl J, DuBose D, Walker J, Alkhyyat A, Wong G. Effects of CEES on inflammatory mediators, heat shock protein 70A, histology and ultrastructure in two skin models. J Appl Toxicol. 2000;20(Suppl 1):S101–108. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat672>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Blaha M, Kohl J, DuBose D, Bowers W, Jr., Walker J. Ultrastructural and histological effects of exposure to CEES or heat in a human epidermal model. In Vitr Mol Toxicol. 2001;14:15–23. doi: 10.1089/109793301316882513. [DOI] [PubMed] [Google Scholar]
- Casillas RP, Kiser RC, Truxall JA, Singer AW, Shumaker SM, Niemuth NA, Ricketts KM, Mitcheltree LW, Castrejon LR, Blank JA. Therapeutic approaches to dermatotoxicity by sulfur mustard. I. Modulaton of sulfur mustard-induced cutaneous injury in the mouse ear vesicant model. J Appl Toxicol. 2000;20(Suppl 1):S145–151. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat665>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Chen X, Liang H, Van Remmen H, Vijg J, Richardson A. Catalase transgenic mice: characterization and sensitivity to oxidative stress. Arch Biochem Biophys. 2004;422:197–210. doi: 10.1016/j.abb.2003.12.023. [DOI] [PubMed] [Google Scholar]
- Chevion M, Berenshtein E, Stadtman ER. Human studies related to protein oxidation: protein carbonyl content as a marker of damage. Free Radic Res. 2000;33(Suppl):S99–108. [PubMed] [Google Scholar]
- Dachir S, Fishbeine E, Meshulam Y, Sahar R, Amir A, Kadar T. Potential anti-inflammatory treatments against cutaneous sulfur mustard injury using the mouse ear vesicant model. Hum Exp Toxicol. 2002;21:197–203. doi: 10.1191/0960327102ht229oa. [DOI] [PubMed] [Google Scholar]
- Dachir S, Fishbeine E, Meshulam Y, Sahar R, Chapman S, Amir A, Kadar T. Amelioration of sulfur mustard skin injury following a topical treatment with a mixture of a steroid and a NSAID. J Appl Toxicol. 2004;24:107–113. doi: 10.1002/jat.955. [DOI] [PubMed] [Google Scholar]
- Dacre JC, Goldman M. Toxicology and pharmacology of the chemical warfare agent sulfur mustard. Pharmacol Rev. 1996;48:289–326. [PubMed] [Google Scholar]
- Darr D, Fridovich I. Free radicals in cutaneous biology. J Invest Dermatol. 1994;102:671–675. doi: 10.1111/1523-1747.ep12374036. [DOI] [PubMed] [Google Scholar]
- Dillman JF, 3rd, McGary KL, Schlager JJ. An inhibitor of p38 MAP kinase downregulates cytokine release induced by sulfur mustard exposure in human epidermal keratinocytes. Toxicol In Vitro. 2004;18:593–599. doi: 10.1016/j.tiv.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Eldad A, Ben Meir, P., Breiterman S, Chaouat M, Shafran A, Ben-Bassat H. Superoxide dismutase (SOD) for mustard gas burns. Burns. 1998;24:114–119. doi: 10.1016/s0305-4179(97)00085-5. [DOI] [PubMed] [Google Scholar]
- Elsayed NM, Omaye ST. Biochemical changes in mouse lung after subcutaneous injection of the sulfur mustard 2-chloroethyl 4-chlorobutyl sulfide. Toxicology. 2004;199:195–206. doi: 10.1016/j.tox.2004.02.020. [DOI] [PubMed] [Google Scholar]
- Fogh K, Kragballe K. Eicosanoids in inflammatory skin diseases. Prostaglandins Other Lipid Mediat. 2000;63:43–54. doi: 10.1016/s0090-6980(00)00096-4. [DOI] [PubMed] [Google Scholar]
- Fridovich I. The biology of oxygen radicals. Science. 1978;201:875–880. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- Gould NS, White CW, Day BJ. A role for mitochondrial oxidative stress in sulfur mustard analog 2-chloroethyl ethyl sulfide-induced lung cell injury and antioxidant protection. J Pharmacol Exp Ther. 2009;328:732–739. doi: 10.1124/jpet.108.145037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg S, Kamath P, Petrali J, Hamilton T, Garfield J, Garlick JA. Characterization of the initial response of engineered human skin to sulfur mustard. Toxicol Sci. 2006;90:549–557. doi: 10.1093/toxsci/kfi306. [DOI] [PubMed] [Google Scholar]
- Gross CL, Nealley EW, Nipwoda MT, Smith WJ. Pretreatment of human epidermal keratinocytes with D,L-sulforaphane protects against sulfur mustard cytotoxicity. Cutan Ocul Toxicol. 2006;25:155–163. doi: 10.1080/15569520600859985. [DOI] [PubMed] [Google Scholar]
- Hayden PJ, Petrali JP, Stolper G, Hamilton TA, Jackson GR, Jr., Wertz PW, Ito S, Smith WJ, Klausner M. Microvesicating effects of sulfur mustard on an in vitro human skin model. Toxicol In Vitro. 2009;23:1396–1405. doi: 10.1016/j.tiv.2009.07.021. [DOI] [PubMed] [Google Scholar]
- Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2004;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McLellan L,I. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defense against oxidative stress. Free Radical Res. 1999a;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McLellan L,I. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defense against oxidative stress. Free Radic Res. 1999b;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- Hildesheim J, Awwad RT, Fornace AJ., Jr. p38 mitogen-activated protein kinase inhibitor protects the epidermis against the acute damaging effects of ultraviolet irradiation by blocking apoptosis and inflammatory response. J Invest Dermatol. 2004;122:497–502. doi: 10.1111/j.1523-1747.2004.22229.x. [DOI] [PubMed] [Google Scholar]
- Jafari M. Dose- and time-dependent effects of sulfur mustard on antioxidant system in liver and brain of rat. Toxicology. 2007;231:30–39. doi: 10.1016/j.tox.2006.11.048. [DOI] [PubMed] [Google Scholar]
- Jan Y, Gray JP, Gerecke DR, Zheng H, Casillas RP, Heck DE, Laskin DL, Laskin JD. Identification of selenocysteine adducts in thioredoxin reductase by 2-chloroethyl ethyl sulfide (CEES), a model sulfur mustard vesicant. The Toxicologist. 2009;108:128. [Google Scholar]
- Je JH, Lee TH, Kim DH, Cho YH, Lee JH, Kim SC, Lee SK, Lee J, Lee MG. Mitochondrial ATP synthase is a target for TNBS-induced protein carbonylation in XS-106 dendritic cells. Proteomics. 2008;8:2384–2393. doi: 10.1002/pmic.200700962. [DOI] [PubMed] [Google Scholar]
- Karayilanoglu T, Gunhan O, Kenar L, Kurt B. The protective and therapeutic effects of zinc chloride and desferrioxamine on skin exposed to nitrogen mustard. Mil Med. 2003;168:614–617. [PubMed] [Google Scholar]
- Kehe K, Balszuweit F, Steinritz D, Thiermann H. Molecular toxicology of sulfur mustard-induced cutaneous inflammation and blistering. Toxicology. 2009;263:12–19. doi: 10.1016/j.tox.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Kim AL, Labasi JM, Zhu Y, Tang X, McClure K, Gabel CA, Athar M, Bickers DR. Role of p38 MAPK in UVB-induced inflammatory responses in the skin of SKH-1 hairless mice. J Invest Dermatol. 2005;124:1318–1325. doi: 10.1111/j.0022-202X.2005.23747.x. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Lecoeur H, de Oliveira-Pinto LM, Gougeon ML. Multiparametric flow cytometric analysis of biochemical and functional events associated with apoptosis and oncosis using the 7-aminoactinomycin D assay. J Immunol Methods. 2002;265:81–96. doi: 10.1016/s0022-1759(02)00072-8. [DOI] [PubMed] [Google Scholar]
- Lefkowitz LJ, Smith WJ. Sulfur mustard-induced arachidonic acid release is mediated by phospholipase D in human keratinocytes. Biochem Biophys Res Commun. 2002;295:1062–1067. doi: 10.1016/s0006-291x(02)00811-2. [DOI] [PubMed] [Google Scholar]
- Magi B, Ettorre A, Liberatori S, Bini L, Andreassi M, Frosali S, Neri P, Pallini V, Di Stefano A. Selectivity of protein carbonylation in the apoptotic response to oxidative stress associated with photodynamic therapy: a cell biochemical and proteomic investigation. Cell Death Differ. 2004;11:842–852. doi: 10.1038/sj.cdd.4401427. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Ichijo H. Stress-responsive protein kinases in redox-regulated apoptosis signaling. Antioxid Redox Signal. 2005;7:472–481. doi: 10.1089/ars.2005.7.472. [DOI] [PubMed] [Google Scholar]
- McClintock SD, Hoesel LM, Das SK, Till GO, Neff T, Kunkel RG, Smith MG, Ward PA. Attenuation of half sulfur mustard gas-induced acute lung injury in rats. J Appl Toxicol. 2006;26:126–131. doi: 10.1002/jat.1115. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal. 2006;8:1775–1789. doi: 10.1089/ars.2006.8.1775. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Rajaratnam V, Mukherjee S, Smith M, Das SK. Modulation of the expression of superoxide dismutase gene in lung injury by 2-chloroethyl ethyl sulfide, a mustard analog. J Biochem Mol Toxicol. 2006;20:142–149. doi: 10.1002/jbt.20128. [DOI] [PubMed] [Google Scholar]
- Murakami M, Nakatani Y, Tanioka T, Kudo I. Prostaglandin E synthase. Prostaglandins Other Lipid Mediat. 2002;68-69:383–399. doi: 10.1016/s0090-6980(02)00043-6. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Rikimaru T, Yano T, Moore KG, Pula PJ, Schofield BH, Dannenberg AM., Jr. Full-thickness human skin explants for testing the toxicity of topically applied chemicals. J Invest Dermatol. 1990;95:325–332. doi: 10.1111/1523-1747.ep12485073. [DOI] [PubMed] [Google Scholar]
- Niki E. Lipid peroxidation: physiological levels and dual biological effects. Free Radic Biol Med. 2009;47:469–484. doi: 10.1016/j.freeradbiomed.2009.05.032. [DOI] [PubMed] [Google Scholar]
- Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- Nyska A, Lomnitski L, Maronpot R, Moomaw C, Brodsky B, Sintov A, Wormser U. Effects of iodine on inducible nitric oxide synthase and cyclooxygenase-2 expression in sulfur mustard-induced skin. Arch Toxicol. 2001;74:768–774. doi: 10.1007/s002040000199. [DOI] [PubMed] [Google Scholar]
- Pal A, Tewari-Singh N, Gu M, Agarwal C, Huang J, Day BJ, White CW, Agarwal R. Sulfur mustard analog induces oxidative stress and activates signaling cascades in the skin of SKH-1 hairless mice. Free Radic Biol Med. 2009;47:1640–1641. doi: 10.1016/j.freeradbiomed.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papirmeister B, Feister AJ, Robinson SI, Ford RD. Medical Defense Against Mustard Gas: Toxic Mechanisms and Pharmacological Implications. CRC Press, Inc.; Boca Raton, FL.: 1991. [Google Scholar]
- Paromov V, Suntres Z, Smith M, Stone WL. Sulfur mustard toxicity following dermal exposure. Role of oxidative stress, and antioxidant therapy. Journal of burns and wounds. 2007;7:60–85. [PMC free article] [PubMed] [Google Scholar]
- Ray R, Legere RH, Majerus BJ, Petrali JP. Sulfur mustard-induced increase in intracellular free calcium level and arachidonic acid release from cell membrane. Toxicol Appl Pharmacol. 1995;131:44–52. doi: 10.1006/taap.1995.1045. [DOI] [PubMed] [Google Scholar]
- Rebholz B, Kehe K, Ruzicka T, Rupec RA. Role of NF-kappaB/RelA and MAPK pathways in keratinocytes in response to sulfur mustard. J Invest Dermatol. 2008;128:1626–1632. doi: 10.1038/sj.jid.5701234. [DOI] [PubMed] [Google Scholar]
- Rice P. Sulphur mustard injuries of the skin. Pathophysiology and management. Toxicol Rev. 2003;22:111–118. doi: 10.2165/00139709-200322020-00006. [DOI] [PubMed] [Google Scholar]
- Rikimaru T, Nakamura M, Yano T, Beck G, Habicht GS, Rennie LL, Widra M, Hirshman CA, Boulay MG, Spannhake EW, et al. Mediators, initiating the inflammatory response, released in organ culture by full-thickness human skin explants exposed to the irritant, sulfur mustard. J Invest Dermatol. 1991;96:888–897. doi: 10.1111/1523-1747.ep12475292. [DOI] [PubMed] [Google Scholar]
- Ryter SR, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, Choi AM. Mechanisms of cell death in oxidative stress. Antiox Redox Signal. 2007;9:49–89. doi: 10.1089/ars.2007.9.49. [DOI] [PubMed] [Google Scholar]
- Sander CS, Chang H, Hamm F, Elsner P, Thiele JJ. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int J Dermatol. 2004;43:326–335. doi: 10.1111/j.1365-4632.2004.02222.x. [DOI] [PubMed] [Google Scholar]
- Satoh T, Moroi R, Aritake K, Urade Y, Kanai Y, Sumi K, Yokozeki H, Hirai H, Nagata K, Hara T, Utsuyama M, Hirokawa K, Sugamura K, Nishioka K, Nakamura M. Prostaglandin D2 plays an essential role in chronic allergic inflammation of the skin via CRTH2 receptor. J Immunol. 2006;177:2621–2629. doi: 10.4049/jimmunol.177.4.2621. [DOI] [PubMed] [Google Scholar]
- Scandalios JG. Oxidative stress: molecular perception and transduction of signals triggering antioxidant defenses. Braz J Med Biol Res. 2005;38:995–1014. doi: 10.1590/s0100-879x2005000700003. [DOI] [PubMed] [Google Scholar]
- Schallreuter KU, Wood JM. Thioredoxin reductase - its role in epidermal redox status. J Photochem Photobiol B. 2001;64:179–184. doi: 10.1016/s1011-1344(01)00235-4. [DOI] [PubMed] [Google Scholar]
- Shahin S, Cullinane C, Gray PJ. Mitochondrial and nuclear DNA damage induced by sulphur mustard in keratinocytes. Chem Biol Interact. 2001;138:231–245. doi: 10.1016/s0009-2797(01)00275-7. [DOI] [PubMed] [Google Scholar]
- Shakarjian MP, Heck DE, Gray JP, Sinko PJ, Gordon MK, Casillas RP, Heindel ND, Gerecke DR, Laskin DL, Laskin JD. Mechanisms Mediating the Vesicant Actions of Sulfur Mustard after Cutaneous Exposure. Toxicol Sci. 2010;114:5–19. doi: 10.1093/toxsci/kfp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim JH, Cho KJ, Lee RA, Kim SH, Myung PK, Choe YK, Yoon DY. E7-expressing HaCaT keratinocyte cells are resistant to oxidative stress-induced cell death via the induction of catalase. Proteomics. 2005;5:2112–2122. doi: 10.1002/pmic.200401106. [DOI] [PubMed] [Google Scholar]
- Smith CN, Lindsay CD, Upshall DG. Presence of methenamine/glutathione mixtures reduces the cytotoxic effect of sulphur mustard on cultured SVK-14 human keratinocytes in vitro. Hum Exp Toxicol. 1997;16:247–253. doi: 10.1177/096032719701600502. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Hurst CG, Moeller RB, Skelton HG, Sidell FR. Sulfur mustard: its continuing threat as a chemical warfare agent, the cutaneous lesions induced, progress in understanding its mechanism of action, its long-term health effects, and new developments for protection and therapy. J Am Acad Dermatol. 1995;32:765–776. doi: 10.1016/0190-9622(95)91457-9. [DOI] [PubMed] [Google Scholar]
- Stadtman ER. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu Rev Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- Tanaka F, Dannenberg AM, Jr., Higuchi K, Nakamura M, Pula PJ, Hugli TE, Discipio RG, Kreutzer DL. Chemotactic factors released in culture by intact developing and healing skin lesions produced in rabbits by the irritant sulfur mustard. Inflammation. 1997;21:251–267. doi: 10.1023/a:1027378422627. [DOI] [PubMed] [Google Scholar]
- Trouba KJ, Hamadeh HK, Amin RP, Germolec DR. Oxidative stress and its role in skin disease. Antioxid Redox Signal. 2002;4:665–673. doi: 10.1089/15230860260220175. [DOI] [PubMed] [Google Scholar]
- Vijayaraghavan R, Sugendran K, Pant SC, Husain K, Malhotra RC. Dermal intoxication of mice with bis(2-chloroethyl)sulphide and the protective effect of flavonoids. Toxicology. 1991;69:35–42. doi: 10.1016/0300-483x(91)90151-p. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Ohkubo H, Niwa H, Tanikawa N, Koda N, Ito S, Ohmiya Y. Essential 110Cys in active site of membrane-associated prostaglandin E synthase-2. Biochem Biophys Res Commun. 2003;306:577–581. doi: 10.1016/s0006-291x(03)01025-8. [DOI] [PubMed] [Google Scholar]
- Wong CM, Cheema AK, Zhang L, Suzuki YJ. Protein carbonylation as a novel mechanism in redox signaling. Circ Res. 2008;102:310–318. doi: 10.1161/CIRCRESAHA.107.159814. [DOI] [PubMed] [Google Scholar]
- Wormser U, Langenbach R, Peddada S, Sintov A, Brodsky B, Nyska A. Reduced sulfur mustard-induced skin toxicity in cyclooxygenase-2 knockout and celecoxib-treated mice. Toxicol Appl Pharmacol. 2004a;200:40–47. doi: 10.1016/j.taap.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Wormser U, Sintov A, Brodsky B, Casillas RP, Nyska A. Protective effect of topical iodine containing anti-inflammatory drugs against sulfur mustard-induced skin lesions. Arch Toxicol. 2004b;78:156–166. doi: 10.1007/s00204-003-0523-2. [DOI] [PubMed] [Google Scholar]
- Yuspa SH, Hawley-Nelson P, Koehler B, Stanley JR. A survey of transformation markers in differentiating epidermal cell lines in culture. Cancer Res. 1980;40:4694–4703. [PubMed] [Google Scholar]