

Abstract
Background
Single nucleotide polymorphisms (SNPs) are the most common form of genetic variation in human DNA. The sequence of SNPs in each of the two copies of a given chromosome in a diploid organism is referred to as a haplotype. Haplotype information has many applications such as gene disease diagnoses, drug design, etc. The haplotype assembly problem is defined as follows: Given a set of fragments sequenced from the two copies of a chromosome of a single individual, and their locations in the chromosome, which can be pre-determined by aligning the fragments to a reference DNA sequence, the goal here is to reconstruct two haplotypes (h1, h2) from the input fragments. Existing algorithms do not work well when the error rate of fragments is high. Here we design an algorithm that can give accurate solutions, even if the error rate of fragments is high.
Results
We first give a dynamic programming algorithm that can give exact solutions to the haplotype assembly problem. The time complexity of the algorithm is O(n × 2t × t), where n is the number of SNPs, and t is the maximum coverage of a SNP site. The algorithm is slow when t is large. To solve the problem when t is large, we further propose a heuristic algorithm on the basis of the dynamic programming algorithm. Experiments show that our heuristic algorithm can give very accurate solutions.
Conclusions
We have tested our algorithm on a set of benchmark datasets. Experiments show that our algorithm can give very accurate solutions. It outperforms most of the existing programs when the error rate of the input fragments is high.
Background
The recognition of genetic variations is an important topic in bioinformatics. Single nucleotide polymorphisms (SNPs) are the most common form of genetic variation in human DNA. Humans are diploid organisms. There are two copies of each chromosome (except the sex chromosomes), one from each parent. The sequence of SNPs in a given chromosome copy is referred to as a haplotype. Haplotype information is useful in many applications, such as gene disease diagnoses [1,2], drug design, etc. Due to their essential importance in many biological analysis, haplotypes have been attracting great attention in recent years [3-7]. Since experimental methods for direct sequencing of haplotypes are both expensive and time consuming, computational methods are usually much more promising.
Currently, computational methods for computing haplotypes often fall into two categories: population haplotyping [8-11] and haplotype assembly (also known as single individual haplotyping) [12-15]. The former tries to compute haplotypes based on the genotype data from a sample of individuals in a population. Many software packages have been published in this field, e.g., PHASE [10]. An obvious drawback of population haplotyping lies in its weakness in recognizing rare and novel SNPs [16]. Contrary to population haplotyping, haplotype assembly is more efficient and has received more attention in recent years. The input to the haplotype assembly problem is a set of fragments sequenced from the two copies of a chromosome of a single individual, and their locations in the chromosome, which can be pre-determined by aligning the fragments to a reference DNA sequence. The task here is to reconstruct two haplotypes from the input fragments. In this paper, we focus on the haplotype assembly problem.
The haplotype assembly problem was first introduced by Lancia et al. [17]. In [17], the authors proposed three optimization criteria for solving this problem, i.e. minimum fragment removal (MFR), minimum SNP removal (MSR) and longest haplotype reconstruction (LHR). Some polynomial time algorithms have been designed to solve some versions of such optimization problems [18,19]. Lippert et al. [19] summarized the models in [17] and proposed some new models. Among these models, the most difficult and realistic one is minimum error correction (MEC), where we want to minimize the total number of conflicts (errors) between the fragments and the constructed haplotypes (h1, h2). The haplotype assembly problem with MEC is NP-hard [5,19] even for gapless fragments.
Levy et al. [20] designed a greedy heuristic algorithm that concatenates the fragments with minimum conflicts. The greedy heuristic algorithm is very fast but not very accurate when the error rate of fragments is high. Later, Bansal and Bafna [21] developed a software package HapCUT and the algorithm is based on the idea of building a graph from the sequenced fragments, in which each SNP site corresponds to a node in the graph and two nodes are connected by an edge if there exists a fragment that covers both SNP sites (which correspond to the two nodes). It then tries to minimize the MEC cost of the reconstructed haplotypes by iteratively finding max-cuts in the associated graph. Bansal et al. [22] designed a Markov Chain Monte Carlo (MCMC) algorithm, HASH. Both HASH and HapCUT have better performance than the greedy heuristic algorithm proposed in [20].
Recently, He et al. [16] gave a dynamic programming algorithm that can give the optimal solution to the haplotype assembly problem with MEC. The time complexity of the algorithm is O(m × 2k × n), where m is the number of fragments, n is the number of SNP sites, and k is the length of the longest fragments. This algorithm works well for k ≤ 15. However, it becomes impractical when k is large.
In this paper, we propose a heuristic algorithm for the haplotype assembly problem with MEC. It is worth mentioning that in HapCUT [21] and the dynamic programming algorithm proposed in [16], the authors assumed that the two constructed haplotypes are complementary with each other, i.e. there are only 2 choices at a SNP site in the reconstructed haplotypes. We drop this assumption in our heuristic algorithm. As a result, there are 4 choices at a SNP site in the reconstructed haplotypes. We have tested our algorithm on a set of benchmark datasets and compare it with several state-of-the-art algorithms. Experiments show that our algorithm is highly accurate. It outperforms most of the existing programs when the error rate of input fragments is high.
Preliminaries
The input to the haplotype assembly problem is a set of fragments sequenced from the two copies of a chromosome of a single individual. Each fragment covers some SNP sites. We assume that all the fragments have been pre-aligned to a reference DNA sequence. As a result, we can organize the input fragments as an m × n matrix M (called fragment matrix), where m is the number of fragments and n is the number of SNP sites. Each row of M corresponds to a fragment that can be represented as a string on the alphabet ∑ = {a, c, g, t, -}, where '-' indicates a space when the SNP site is not covered by the fragment or the SNP value cannot be determined with enough confidence. The start (respectively, end) position of a fragment is defined as the first (respectively, last) position in the corresponding row that is not a '-'. In the middle of a fragment, '-'s are allowed due to data missing or paired-end fragments. Throughout the remainder of this paper, we will use the two notations, i.e. fragments and rows of M interchangeably when there is no ambiguity. Moreover, we will use columns and SNP sites interchangeably when there is no ambiguity.
It is accepted that there are at most two distinct nucleotides at a SNP site. We assume that a column with more than two distinct nucleotides in M must contain errors. In this case, we keep the two distinct nucleotides that appear the most at this column and replace the rest of them with a '-'. After removing errors, M can be converted into , in which each entry is encoded by a character from the alphabet . Figure 1(a) gives an example of an original input matrix M containing errors in some columns, Figure 1(b) is the matrix after error correction. is given in Figure 1(c).
Figure 1.
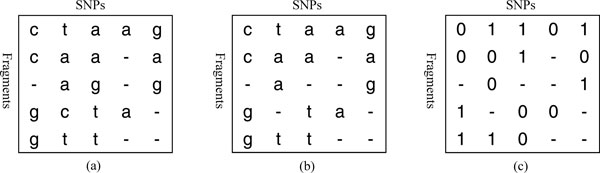
Illustration of the preprocessing on the input fragment matrix. (a) The original fragment matrix M. (b) The matrix obtained from (a) by removing possible errors. (c) The obtained matrix .
We say that row i covers column j in M if Mi,j is not a '-' or there are two integers p and q with p < j < q such that Mi,p ≠ - and Mi,q = -. The number of rows covering column i in M is referred to as the coverage of column i.
Two rows p and q in M are in conflict if there exists a column j such that Mp,j ≠ Mq,j, Mp,j ≠ - and Mq,j ≠ -. Obviously, for error-free data, two rows from the same copy of a chromosome should not conflict with each other, and two rows which conflict with each other must come from different copies of a chromosome. The distance between two rows i and j, denoted by D(i, j), is defined as the generalized hamming distance as follows:
| (1) |
where
| (2) |
Minimum error correction (MEC) is a commonly used model for the haplotype assembly problem. For the haplotype assembly problem with MEC, the input is a fragment matrix M, the task is to partition the fragments in M into two groups and construct two haplotypes (h1, h2), one from each group, such that the total number of conflicts (errors) between the fragments and the constructed haplotypes (h1, h2) is minimized.
Methods
In this section, we will describe the algorithms used to solve the problem. We first design a dynamic programming algorithm that gives an exact solution and runs in O(n × 2t × t) time, where n is the number of columns in M, and t is the maximum coverage of a column in M. The dynamic programming algorithm will be very slow when t is large. We then design a heuristic algorithm that first computes an initial pair of haplotypes by using the dynamic programming algorithm on only a subset of M. This initial pair of haplotypes can be viewed as an approximation to the optimal solution. To obtain a better solution, we further introduce some techniques to refine the initial solution.
A dynamic programming algorithm
Recall that the goal of the haplotype assembly problem is to partition the rows of the input fragment matrix M into two groups, each of which determining a haplotype. To obtain an optimal partition, a naive approach is to enumerate all possible partitions on the rows of M, among which we then choose the one minimizing MEC. For an instance with m rows, there are 2m total partitions, and thus the approach does not work in practice. Here we introduce a dynamic programming algorithm for the haplotype assembly problem with MEC that runs in O(n × 2t × t) time, where n is the number of columns in M, and t is the maximum coverage of a column in M.
Before we give the details of the dynamic programming algorithm, we first define some basic notations that will be used later:
•Ri: the set of rows covering column i in M.
•Pj(i): the j-th partition on Ri.
•Qj(i): the j-th partition on Ri ∩ Ri+1.
•Pj(i)|Ri∩Ri+1: the partition on Ri ∩ Ri+1 obtained from Pj(i) by restriction on the rows in Ri ∩ Ri+1.
•QQj(i): the set of partitions Pk(i) such that Pk(i)|Ri∩Ri+1 = Qj(i).
•C(Pj(i)): the minimum number of corrections to be made in column i of M when the partition on Ri is indicated by Pj(i).
•MEC(i, Pj(i)): the optimal cost for the first i columns in M such that column i has a partition Pj(i).
In order to compute MEC(i + 1, Pj(i + 1)) efficiently, we define
| (3) |
Let Pj(i + 1) be the j-th partition on Ri+1, Qk(i) = Pj(i + 1)|Ri∩Ri+1. The recursion formula of the dynamic programming algorithm is illustrated as follows:
| (4) |
Based on Pj(i + 1), we can get Qk(i) in O(t) time. Furthermore, we know the majority value (0 or 1) at column (i + 1) in each group. To compute C(Pj(i + 1)), we can simply count the number of minorities in each group (at column (i + 1)) separately, and then add them up. Thus, it takes O(t) time to compute C(Pj(i + 1)).
The optimal MEC cost for partitioning all the rows of M is the smallest MEC(n, Pj(n)) over all possible Pj(n), where n is the number of columns in M. A standard backtracking process can be used to obtain the optimal solution.
Let us look at the time complexity of the dynamic programming algorithm. To compute each MEC(i + 1, Pj(i + 1)) in Equation (4), it requires O(t) time to compute C(Pj(i + 1)). Thus, it takes O(n × 2t × t) time to compute all C(Pj(i + 1))s for all the n columns in M. Now, let us look at the way to compute M E(i, Qk(i))s. For each partition Pj(i) on Ri, we can get Qk(i) = Pj(i)|Ri∩Ri+1 in O(t) time. We then update ME(i, Qk(i)) if the current value of ME(i, Qk(i)) is greater than MEC(i, Pj(i)). There are at most 2t Pj(i)s on Ri. Thus, it takes O(t × 2t) time to compute all ME(i, Qk(i))s on Ri. Since there are n columns in M, the total time required for computing all ME(i, Qk(i))s is O(n × 2t × t).
Theorem 1 Given a fragment matrix M, there is an O(n × 2t × t) time algorithm to compute an optimal solution for the haplotype assembly problem with MEC, where n is the number of columns in M, and t is the maximum coverage of a column in M.
Obtaining an initial solution via randomized sampling
The dynamic programming algorithm works well when t is relatively small. However, it will be very slow when t is large. To solve the problem when t is large, we look at each column i at a time, randomly select a fixed number of rows, say, boundOfCoverage, from the set of rows covering it and delete the characters in the remaining rows at all the columns after i - 1. After that, the coverage of each column in the newly obtained submatrix is at most boundOfCoverage. We then run the dynamic programming algorithm on the submatrix. The resulting pair of haplotypes, which is referred to as the initial solution, can be viewed as an approximation to the optimal solution.
The detailed procedure for obtaining a submatrix from M via the randomized sampling approach is as follows:
1. Compute the coverage ci for each column i in M.
2. For i = 1 to n, perform the following steps.
3. If ci ≤ boundOfCoverage, do nothing and goto the next column. Otherwise, goto step 4.
4. Randomly choose boundOfCoverage rows from the set of rows covering column i. Let be the set of rows covering column i but are not chosen during this process.
5. For each row , cut r from column i such that it no longer covers any column larger than i (including i). Accordingly, we need to reduce cj by 1 for each i ≤ j ≤ k, where k is the end position of r before being cut.
By employing this randomized sampling strategy, we can always make sure that the maximum coverage is bounded by the threshold boundOfCoverage in the selected submatrix. How to choose a proper value for boundOfCoverage? Actually, there is a tradeoff between the running time and the quality of the initial solution output by the dynamic programming algorithm. On one hand, reducing boundOfCoverage can reduce the running time of the algorithm. However, on the other hand, increasing boundOfCoverage can maintain more information from M. As a result, the initial solution output by the dynamic programming algorithm has a higher chance to be close to the optimal solution. In practice, boundOfCoverage is generally no larger than 15, which is feasible in terms of running time and is large enough to sample sufficient information from M. See Section Experiments for a detailed discussion on how the size of boundOfCoverage affects the initial solution.
Refining the initial solution with all fragments
In the newly obtained submatrix, it is possible that (1) some columns are not covered by any rows, thus leaving the haplotype values at these SNP sites undetermined in the initial solution, (2) the haplotype values at some SNP sites in the initial solution are wrongly determined due to the lack of sufficient information sampled from M during the randomized sampling process. In this subsection, we try to refine the initial solution with all input fragments, aiming to fill haplotype values that are left undetermined and correct those that are wrongly determined.
The refining procedure contains several iterations. In each iteration, we take two haplotypes as its input and output a new pair of haplotypes. Initially, the two haplotypes in the initial solution are used as the input to the first iteration. The haplotypes output in an iteration are then used as the input to the subsequent iteration. In each iteration, we try to reassign the rows of M into two groups based on the two input haplotypes. More specifically, for each row r of M, we first compute the generalized hamming distance between r and the two haplotypes. Then, we assign r to the group associated with the haplotype that has the smaller (generalized hamming) distance with r. After reassigning all rows of M into two groups, we can compute a haplotype from each of the two groups by majority rule. At the same time, we can also obtain the corresponding MEC cost.
The refining procedure stops when, at the end of some iteration, the obtained haplotypes no longer change, or when a certain number of iterations have been finished. The two haplotypes output in the last iteration are the output of the refining procedure.
Voting procedure
To further reduce the effect of randomness caused by the randomized sampling process, we try to obtain several different submatrices from M by repeating the randomized sampling process several times. Accordingly, we can obtain several initial solutions, one derived from each submatrix. Furthermore, we can refine these initial solutions with all fragments. Given a set of solutions, each of which containing a pair of haplotypes, the goal here is to compute a single pair of haplotypes by adopting a voting procedure.
In the voting procedure, the two haplotypes are computed separately. We next see how to compute one of the two haplotypes. The other case is similar. Let S be the set of solutions used for voting. First, we find a set of haplotypes (denoted by S1), one from each solution in S, such that the haplotypes in S1 all correspond to the same copy of a chromosome. With S1, we can then compute a haplotype by majority rule. Simply speaking, at each SNP site, we count the number of 0s and 1s at the given SNP site over the haplotypes in S1. If we have more 0s, the resulting haplotype takes 0 at the SNP site, otherwise, it takes 1.
How to find S1? First, we need to clarify that the two haplotypes in each solution in S are unordered. That is, given a solution H = (h1, h2), we do not know which chromosome copy h1 (or h2) corresponds to. So, we should first find the correspondence between the haplotypes in different solutions. Let be the set of solutions in S. Without loss of generality, assume that the MEC cost associated with H1 is the smallest among all the y solutions. We use H1 as our reference and try to find the correspondence between haplotypes in H1 and other solutions. For each i (1 < i ≤ y), we first compute two generalized hamming distances and . If , we claim that corresponds to the same chromosome copy as . Otherwise, corresponds to the same chromosome copy as . As a result, the set of haplotypes in S that correspond to the same chromosome copy as is the S1 we want to find.
Assume that at the beginning of this procedure, we obtain x solutions by repeating the randomized sampling process along with the refining procedure x times. It is worth mentioning that in the voting procedure, we only use part of the solutions, say, the first y (y ≤ x) solutions with the highest quality. Given two solutions A and B, we say that A has higher quality than B if the MEC cost associated with A is smaller than that of B. In this case, we assume that A is much closer to the optimal solution and contains less noises than B. To reduce the sideeffect of noises and improve the quality of the solution output by the voting procedure, it is helpful to use only solutions with high quality in the voting procedure.
Summarization of the algorithm
Generally speaking, given an input fragment matrix M, our heuristic algorithm can be summarized as the following four steps.
Step 1: We first perform a preprocessing on M to detect possible errors in it. After removing errors from M, we further convert it into in which each entry is encoded by a character from the alphabet . See Section Preliminaries for more details. is used as the input to the following steps.
Step 2: We compute an initial solution by running the dynamic programming algorithm on a subset of . The submatrix is computed by using the randomized sampling approach.
Step 3: Refine the initial solution with all the fragments in , instead of the submatrix that is used to generate the initial solution in Step 2.
Step 4: To further reduce the effect of randomness caused by the randomized sampling process, we repeat Step 2 and Step 3 several times. Each repeat ends with a solution, from which we then compute a single pair of haplotypes by adopting the voting procedure. The resulting pair of haplotypes is the output of our algorithm.
Results
We have tested our algorithm on a set of benchmark datasets and compare its performance with several other algorithms. The main purpose here is to evaluate how accurately our algorithm can reconstruct haplotypes from input fragments. All the tests have been done on a Windows-XP (32 bits) desktop PC with 3.16 GHz CPU and 4GB RAM.
The benchmark we use was created by Geraci in [23]. It was generated by using real human haplotype data from the HapMap project [4]. There are three parameters associated with the benchmark, i.e haplotype length, error rate and coverage rate, denoted by l, e, c, respectively. Each parameter has several different values, l = 100, 350, and 700, e = 0.0, 0.1, 0.2 and 0.3, c = 3, 5, 8 and 10. Note that unlike the "coverage" defined in Section Preliminaries, the coverage rate c defined in this benchmark refers to the number of times each of the two haplotypes replicates when generating the dataset. In other words, given an instance in the benchmark, i.e. a fragment matrix, there are up to 2c rows which take non '-' value at each column in the matrix. For each combination of the three parameters, there are 100 instances in the benchmark. As for the details on how to generate the benchmark, the reader is referred to [23].
Throughout our experiments, we measure the performance of our algorithm by the reconstruction rate, a frequently used criterion in the haplotype assembly problem. Given a problem instance in the benchmark, the reconstruction rate is defined as follows:
| (5) |
where H = (h1, h2) is the pair of correct haplotypes that is used to generate the problem instance, and is thus known a prior, is the pair of haplotypes output by the algorithm, n is the length of the haplotypes, and is the hamming distance between two haplotypes. More specifically, is defined as follows:
| (6) |
where
| (7) |
Intuitively speaking, the reconstruction rate measures the ability of an algorithm to reconstruct the correct haplotypes.
Recall that in Step 2 of our algorithm, we try to compute an initial solution by using only a subset of the input matrix. The initial solution forms the basis for the following steps of our algorithm and is closely related to the parameter boundOfCoverage. Briefly speaking, boundOfCoverage is the maximum coverage of a column in the submatrix selected during this step. For a formal description of boundOfCoverage, we refer you to Section Methods. In this experiment, we first evaluate how the size of boundOfCoverage affects the initial solution. As aforementioned earlier, boundOfCoverage is generally no larger than 15. Here we consider three different sizes of boundOfCoverage, i.e. 10, 12 and 15. Given a problem instance, we can obtain three initial solutions by using the three different sizes of boundOfCoverage, respectively. As an example, we choose the set of benchmark datasets with l = 350 and e = 0.2. For each combination of the coverage rate c and boundOfCoverage, there are 100 instances and we compute the average of the reconstruction rates over the 100 instances. The results are listed in Table 1.
Table 1.
Evaluation of how the size of boundOfCoverage affects the initial solution.
| c = 3 | c = 5 | c = 8 | c = 10 | |
|---|---|---|---|---|
| 10 | 0.708(<0.1) | 0.753(<0.1) | 0.764(0.14) | 0.774(0.18) |
| 12 | 0.728(<0.1) | 0.785(<0.1) | 0.794(0.15) | 0.797(0.21) |
| 15 | 0.776(0.30) | 0.837(0.33) | 0.841(0.36) | 0.857(0.45) |
There are 3 different sizes of boundOfCoverage, i.e. 10, 12 and 15, and 4 different coverage rates c, i.e. 3, 5, 8 and 10. The number outside each bracket refers to the average reconstruction rate under the corresponding parameter setting, while the one enclosed in the bracket refers to the average running time in seconds.
From Table 1, we can see that for a fixed coverage rate c, when increasing the size of boundOfCoverage, the reconstruction rate of the obtained initial solution gets higher, and the running time increases accordingly. Since the reconstruction rate in the case where boundOfCoverage = 12 is relatively high, and the running time is feasible, we will set boundOfCoverage to be 12 in the following experiments.
Next, to evaluate the performance of our algorithm, we have tested it on the set of benchmark datasets. The parameters we use are as follows: boundOfCoverage = 12, x = 100, and y = 11, where x is the number of initial solutions obtained in Step 4, i.e. the number of times we repeat Step 2 and Step 3, and y is the number of solutions used for voting in Step 4. The results for l = 100, 350 and 700 are given in the last column in Table 2 Table 3 and Table 4, respectively. In [23], Geraci compared the reconstruction rates of seven state-of-the-art algorithms on the same benchmark datasets. These seven algorithms are SpeedHap [24], Fast hare [25], 2d-mec [26], HapCUT [21], MLF [27], SHR-three [28] and DGS, the greedy heuristic proposed in [20]. For a full review of these seven algorithms, the reader is referred to [23]. For the sake of comparison, we list the reconstruction rates of the seven algorithms, see Columns 3 - 9 in Table 2, Table 3 and Table 4. Note that the results for the seven algorithms are directly taken from [23]. Each reconstruction rate shown in the three tables is the average over 100 instances under the same parameter setting.
Table 2.
Comparisons of the algorithms when l = 100.
| e | c | SpeedHap | Fast Hare | 2d-mec | HapCUT | MLF | SHR-three | DGS | Ours |
|---|---|---|---|---|---|---|---|---|---|
| 0.0 | 3 | 0.999 | 0.999 | 0.990 | 1.000 | 0.973 | 0.816 | 1.000 | 1.000 |
| 5 | 1.000 | 0.999 | 0.997 | 1.000 | 0.992 | 0.861 | 1.000 | 1.000 | |
| 8 | 1.000 | 1.000 | 1.000 | 1.000 | 0.997 | 0.912 | 1.000 | 1.000 | |
| 10 | 1.000 | 1.000 | 1.000 | 1.000 | 0.998 | 0.944 | 1.000 | 1.000 | |
| 0.1 | 3 | 0.895 | 0.919 | 0.912 | 0.929 | 0.889 | 0.696 | 0.930 | 0.973 |
| 5 | 0.967 | 0.965 | 0.951 | 0.920 | 0.970 | 0.738 | 0.985 | 0.996 | |
| 8 | 0.989 | 0.993 | 0.983 | 0.901 | 0.985 | 0.758 | 0.989 | 0.999 | |
| 10 | 0.990 | 0.998 | 0.988 | 0.892 | 0.995 | 0.762 | 0.997 | 1.000 | |
| 0.2 | 3 | 0.623 | 0.715 | 0.738 | 0.782 | 0.725 | 0.615 | 0.725 | 0.903 |
| 5 | 0.799 | 0.797 | 0.793 | 0.838 | 0.836 | 0.655 | 0.813 | 0.963 | |
| 8 | 0.852 | 0.881 | 0.873 | 0.864 | 0.918 | 0.681 | 0.878 | 0.990 | |
| 10 | 0.865 | 0.915 | 0.894 | 0.871 | 0.938 | 0.699 | 0.917 | 0.996 | |
| 0.3 | 3 | 0.480 | 0.617 | 0.623 | 0.602 | 0.618 | 0.557 | 0.611 | 0.776 |
| 5 | 0.637 | 0.639 | 0.640 | 0.629 | 0.653 | 0.599 | 0.647 | 0.874 | |
| 8 | 0.667 | 0.661 | 0.675 | 0.673 | 0.697 | 0.632 | 0.663 | 0.950 | |
| 10 | 0.676 | 0.675 | 0.678 | 0.709 | 0.715 | 0.632 | 0.688 | 0.972 |
The columns e and c refer to the error rate and coverage rate, respectively. Columns 3-9 represent the reconstruction rate of the seven algorithms, i.e. SpeedHap, Fast Hare, 2d-mec, HapCUT, MLF, SHR-three and DGS. For each combination of e and c, the best among the seven algorithms is highlighted in bold. The last column lists the reconstruction rate of our algorithm.
Table 3.
Comparisons of the algorithms when l = 350.
| e | c | SpeedHap | Fast Hare | 2d-mec | HapCUT | MLF | SHR-three | DGS | Ours |
|---|---|---|---|---|---|---|---|---|---|
| 0.0 | 3 | 0.999 | 0.990 | 0.965 | 1.000 | 0.864 | 0.830 | 0.999 | 1.000 |
| 5 | 1.000 | 0.999 | 0.993 | 1.000 | 0.929 | 0.829 | 1.000 | 1.000 | |
| 8 | 1.000 | 1.000 | 0.998 | 1.000 | 0.969 | 0.895 | 1.000 | 1.000 | |
| 10 | 1.000 | 0.999 | 0.999 | 1.000 | 0.981 | 0.878 | 1.000 | 1.000 | |
| 0.1 | 3 | 0.819 | 0.871 | 0.837 | 0.930 | 0.752 | 0.682 | 0.926 | 0.970 |
| 5 | 0.959 | 0.945 | 0.913 | 0.913 | 0.858 | 0.724 | 0.978 | 0.993 | |
| 8 | 0.984 | 0.985 | 0.964 | 0.896 | 0.933 | 0.742 | 0.996 | 0.999 | |
| 10 | 0.984 | 0.995 | 0.978 | 0.888 | 0.962 | 0.728 | 0.998 | 1.000 | |
| 0.2 | 3 | 0.439 | 0.684 | 0.675 | 0.771 | 0.642 | 0.591 | 0.691 | 0.877 |
| 5 | 0.729 | 0.746 | 0.728 | 0.831 | 0.728 | 0.632 | 0.769 | 0.953 | |
| 8 | 0.825 | 0.853 | 0.791 | 0.862 | 0.798 | 0.670 | 0.842 | 0.988 | |
| 10 | 0.855 | 0.877 | 0.817 | 0.867 | 0.831 | 0.668 | 0.878 | 0.994 | |
| 0.3 | 3 | 0.251 | 0.590 | 0.593 | 0.565 | 0.581 | 0.548 | 0.578 | 0.725 |
| 5 | 0.578 | 0.602 | 0.606 | 0.582 | 0.606 | 0.557 | 0.609 | 0.833 | |
| 8 | 0.629 | 0.626 | 0.623 | 0.621 | 0.634 | 0.604 | 0.628 | 0.922 | |
| 10 | 0.638 | 0.644 | 0.634 | 0.664 | 0.641 | 0.619 | 0.641 | 0.951 |
The columns e and c refer to the error rate and coverage rate, respectively. Columns 3-9 represent the reconstruction rate of the seven algorithms, i.e. SpeedHap, Fast Hare, 2d-mec, HapCUT, MLF, SHR-three and DGS. For each combination of e and c, the best among the seven algorithms is highlighted in bold. The last column lists the reconstruction rate of our algorithm.
Table 4.
Comparisons of the algorithms when l = 700.
| e | c | SpeedHap | Fast Hare | 2d-mec | HapCUT | MLF | SHR-three | DGS | Ours |
|---|---|---|---|---|---|---|---|---|---|
| 0.0 | 3 | 0.999 | 0.988 | 0.946 | 1.000 | 0.787 | 0.781 | 0.999 | 0.997 |
| 5 | 1.000 | 0.999 | 0.976 | 1.000 | 0.854 | 0.832 | 1.000 | 0.999 | |
| 8 | 1.000 | 1.000 | 0.992 | 1.000 | 0.919 | 0.868 | 1.000 | 1.000 | |
| 10 | 1.000 | 0.999 | 0.997 | 1.000 | 0.933 | 0.898 | 1.000 | 1.000 | |
| 0.1 | 3 | 0.705 | 0.829 | 0.786 | 0.927 | 0.698 | 0.668 | 0.931 | 0.951 |
| 5 | 0.947 | 0.949 | 0.880 | 0.916 | 0.809 | 0.716 | 0.977 | 0.989 | |
| 8 | 0.985 | 0.986 | 0.948 | 0.896 | 0.863 | 0.743 | 0.987 | 0.997 | |
| 10 | 0.986 | 0.995 | 0.965 | 0.889 | 0.884 | 0.726 | 0.997 | 0.998 | |
| 0.2 | 3 | 0.199 | 0.652 | 0.647 | 0.753 | 0.624 | 0.591 | 0.669 | 0.837 |
| 5 | 0.681 | 0.712 | 0.697 | 0.825 | 0.682 | 0.617 | 0.741 | 0.927 | |
| 8 | 0.801 | 0.808 | 0.751 | 0.856 | 0.747 | 0.653 | 0.818 | 0.974 | |
| 10 | 0.813 | 0.872 | 0.778 | 0.861 | 0.765 | 0.675 | 0.861 | 0.982 | |
| 0.3 | 3 | 0.095 | 0.581 | 0.583 | 0.552 | 0.570 | 0.536 | 0.573 | 0.676 |
| 5 | 0.523 | 0.591 | 0.596 | 0.555 | 0.594 | 0.562 | 0.595 | 0.777 | |
| 8 | 0.616 | 0.615 | 0.613 | 0.597 | 0.614 | 0.611 | 0.614 | 0.876 | |
| 10 | 0.627 | 0.616 | 0.622 | 0.645 | 0.625 | 0.625 | 0.622 | 0.909 |
The columns e and c refer to the error rate and coverage rate, respectively. Columns 3-9 represent the reconstruction rate of the seven algorithms, i.e. SpeedHap, Fast Hare, 2d-mec, HapCUT, MLF, SHR-three and DGS. For each combination of e and c, the best among the seven algorithms is highlighted in bold. The last column lists the reconstruction rate of our algorithm.
Take a close look at the three tables, we can see that (1) each of the seven algorithms studied in [23] only works well in some cases, e.g., SpeedHap works well when the error rate is low (≤ 0.1), while MLF works well when the error rate is high (≥ 0.2); (2) the reconstruction rates of all the seven algorithms are relatively low when the error rate of input fragments is high. For example, in the case where l = 700, e = 0.3 and c = 10, the best reconstruction rate of the seven other algorithm is 0.645. Compared with its competitors, our algorithm can give solutions with high reconstruction rate. It outperforms its competitors in almost all cases, especially in cases in which the error rate of fragments is high (≥ 0.2). We also notice that when the error rate is 0, our algorithm may introduce some errors in the output solution, e.g., in the case where l = 700, c = 3, e = 0.0. However, even in this case, the reconstruction rate can still reach up to 0.997.
Discussion
In the first step of our algorithm, we perform a preprocessing on the input fragment matrix. This allows us to detect errors in the input. For example, for the benchmark datasets with l = 350 and e = 0.3, Step 1 of our algorithm can identify about 47%, 55%, 59% and 61% of the total errors for the cases c = 3, 5, 8 and 10, respectively. Thus, Step 1 has significant importance to the following steps of our algorithm.
Next, we further investigate how the voting procedure in Step 4 affects the performance of our algorithm. In Step 4, we first obtain x solutions, from which we then choose the first y (y ≤ x) solutions with the smallest MEC cost. The y solutions are then used in the voting procedure to compute the final solution of our algorithm. To demonstrate the effect of the voting procedure, we compare the final version of our algorithm with the one without the voting procedure. For the version without the voting procedure, we simply outputs the solution with the smallest MEC cost among all the x solutions in Step 4. As an example, we have tested both versions on the set of benchmark datasets with l = 350. The parameters are as follows: boundOfCoverage = 12, x = 100 and y = 11. Figure 2(a) (respectively, 2(b)) shows the results for the two versions in the case where e = 0.2 (respectively, e = 0.3). The results for e = 0.0 and 0.1 are similar as that of e = 0.2, and we omit it here.
Figure 2.
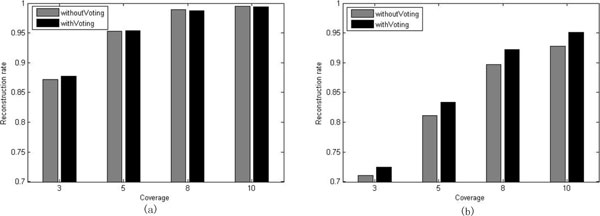
Illustration of the effect of the voting procedure. The reconstruction rates for the final version of our algorithm and the one without the voting procedure are depicted by black and gray bar, respectively. The error rate for the benchmark used in (a)(respectively, (b)) is 0.2 (respectively, 0.3).
From Figure 2(a), we can see that the two versions of our algorithm have almost the same reconstruction rate. However, when e = 0.3, the final version of our algorithm has higher reconstruction rate than the one without the voting procedure, see Figure 2(b). Thus, the voting procedure is essential for our algorithm.
To see how the size of the parameter x affects the reconstruction rate of our algorithm, we have tested our algorithm with three different sizes of x, i.e. 25, 50, 100. The values of boundOfCoverage and y are fixed to be 12 and 11, respectively. The tests are done on the set of benchmark datasets with l = 350. The results for e = 0.2 and 0.3 are shown in Figure 3(a) and 3(b), respectively. For e = 0.0 and 0.1, the reconstruction rates are almost the same in all the three cases, and we do not list it here. As can be seen from Figure 3(a), the reconstruction rate increases with the increasing of x in the cases where c = 3 and 5. For c = 8 and 10, the cases where x = 50 and 100 have almost the same reconstruction rate which is higher than that in the case where x = 25. As for Figure 3(b), it is much more obvious that the reconstruction rate increases as x gets larger.
Figure 3.
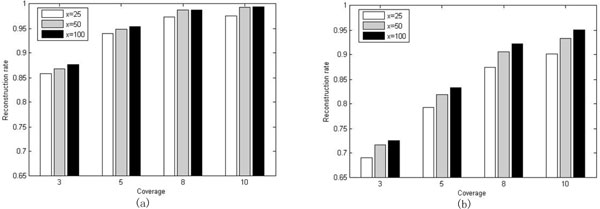
Evaluation of how the size of x affects the performance of our algorithm. The reconstruction rates for x = 25, 50 and 100 are depicted by white, gray and black bar, respectively. The error rate for the benchmark used in (a)(respectively, (b)) is 0.2 (respectively, 0.3).
Conclusion
In this paper, we propose a heuristic algorithm for the haplotype assembly problem. Experiments show that our algorithm is highly accurate. It outperforms most of the existing programs when the error rate of input fragments is high.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
FD participated in the design of the study, performed the experiments and drafted the manuscript. WC participated in the design of the study and helped to draft the manuscript. LW conceived the study, participated in its design and helped to draft the manuscript. All authors read and approved the final manuscript.
Declarations
The publication costs for this article were funded by the corresponding author's institution.
Contributor Information
Fei Deng, Email: fdeng4@student.cityu.edu.hk.
Wenjuan Cui, Email: wenjuacui2@student.cityu.edu.hk.
Lusheng Wang, Email: cswangl@cityu.edu.hk.
Acknowledgements
The authors would like to thank Filippo Geraci for kindly providing us with the set of benchmark datasets. This work is fully supported by a grant from City University of Hong Kong (Project No. 7002728).
This article has been published as part of BMC Genomics Volume 14 Supplement 2, 2013: Selected articles from ISCB-Asia 2012. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcgenomics/supplements/14/S2.
References
- Hoehe M, Köpke K, Wendel B, Rohde K, Flachmeier C, Kidd K, Berrettini W, Church G. Sequence variability and candidate gene analysis in complex disease: association of μ opioid receptor gene variation with substance dependence. Human Molecular Genetics. 2000;9(19):2895–2908. doi: 10.1093/hmg/9.19.2895. [DOI] [PubMed] [Google Scholar]
- Schaid D. Evaluating associations of haplotypes with traits. Genetic epidemiology. 2004;27(4):348–364. doi: 10.1002/gepi.20037. [DOI] [PubMed] [Google Scholar]
- Bonizzoni P, Della Vedova G, Dondi R, Li J. The haplotyping problem: an overview of computational models and solutions. Journal of Computer Science and Technology. 2003;18(6):675–688. doi: 10.1007/BF02945456. [DOI] [Google Scholar]
- Altshuler D, Wing T. et al. A haplotype map of the human genome. Nature. 2005;437(7063):1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilibrasi R, Van Iersel L, Kelk S, Tromp J. On the complexity of several haplotyping problems. Algorithms in Bioinformatics. 2005. pp. 128–139.
- Schwartz R. Theory and algorithms for the haplotype assembly problem. Communications in Information and Systems. 2010;10:23–38. [Google Scholar]
- Zhang X, Wang R, Wu L, Chen L. Models and algorithms for haplotyping problem. Current Bioinformatics. 2006;1:104–114. [Google Scholar]
- Gusfield D. Proceedings of the Sixth Annual International Conference on Computational Biology. ACM; 2002. Haplotyping as perfect phylogeny: conceptual framework and efficient solutions; pp. 166–175. [Google Scholar]
- Wang L, Xu Y. Haplotype inference by maximum parsimony. Bioinformatics. 2003;19(14):1773–1780. doi: 10.1093/bioinformatics/btg239. [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith N, Donnelly P. A new statistical method for haplotype reconstruction from population data. The American Journal of Human Genetics. 2001;68(4):978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halperin E, Eskin E. Haplotype reconstruction from genotype data using imperfect phylogeny. Bioinformatics. 2004;20(12):1842–1849. doi: 10.1093/bioinformatics/bth149. [DOI] [PubMed] [Google Scholar]
- Duitama J, Huebsch T, McEwen G, Suk E, Hoehe M. ReFHap: A reliable and fast algorithm for single individual haplotyping. Proceedings of the First ACM International Conference on Bioinformatics and Computational Biology. 2010. pp. 160–169. ACM.
- Li L, Kim J, Waterman M. Haplotype reconstruction from SNP alignment. Journal of Computational Biology. 2004;11(2-3):505–516. doi: 10.1089/1066527041410454. [DOI] [PubMed] [Google Scholar]
- Xie M, Wang J, Chen J. A model of higher accuracy for the individual haplotyping problem based on weighted SNP fragments and genotype with errors. Bioinformatics. 2008;24(13):i105–i113. doi: 10.1093/bioinformatics/btn147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Wu L, Li Z, Zhang X. Haplotype reconstruction from SNP fragments by minimum error correction. Bioinformatics. 2005;21(10):2456–2462. doi: 10.1093/bioinformatics/bti352. [DOI] [PubMed] [Google Scholar]
- He D, Choi A, Pipatsrisawat K, Darwiche A, Eskin E. Optimal algorithms for haplotype assembly from whole-genome sequence data. Bioinformatics. 2010;26(12):i183–i190. doi: 10.1093/bioinformatics/btq215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancia G, Bafna V, Istrail S, Lippert R, Schwartz R. Proceedings of the 9th Annual European Symposium on Algorithms. Springer-Verlag; 2001. SNPs Problems, Complexity, and Algorithms; pp. 182–193. [Google Scholar]
- Rizzi R, Bafna V, Istrail S, Lancia G. Workshop on Algorithms in Bioinformatics. Springer; 2002. Practical Algorithms and Fixed-Parameter Tractability for the Single Individual SNP Haplotyping Problem; pp. 29–43. [Google Scholar]
- Lippert R, Schwartz R, Lancia G, Istrail S. Algorithmic strategies for the single nucleotide polymorphism haplotype assembly problem. Briefings in bioinformatics. 2002;3:23–31. doi: 10.1093/bib/3.1.23. [DOI] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng P, Feuk L, Halpern A, Walenz B, Axelrod N, Huang J, Kirkness E, Denisov G. et al. The diploid genome sequence of an individual human. PLoS biology. 2007;5(10):e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal V, Bafna V. HapCUT: an efficient and accurate algorithm for the haplotype assembly problem. Bioinformatics. 2008;24(16):i153–i159. doi: 10.1093/bioinformatics/btn298. [DOI] [PubMed] [Google Scholar]
- Bansal V, Halpern A, Axelrod N, Bafna V. An MCMC algorithm for haplotype assembly from whole-genome sequence data. Genome research. 2008;18(8):1336–1346. doi: 10.1101/gr.077065.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraci F. A comparison of several algorithms for the single individual SNP haplotyping reconstruction problem. Bioinformatics. 2010;26(18):2217–2225. doi: 10.1093/bioinformatics/btq411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese L, Geraci F, Pellegrini M. SpeedHap: an accurate heuristic for the single individual SNP haplotyping problem with many gaps, high reading error rate and low coverage. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2008;5(4):492–502. doi: 10.1109/TCBB.2008.67. [DOI] [PubMed] [Google Scholar]
- Panconesi A, Sozio M. Fast hare: A fast heuristic for single individual SNP haplotype reconstruction. Algorithms in Bioinformatics. 2004. pp. 266–277.
- Wang Y, Feng E, Wang R. A clustering algorithm based on two distance functions for MEC model. Computational biology and chemistry. 2007;31(2):148–150. doi: 10.1016/j.compbiolchem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Wu L, Zhang J, Wang R, Zhang X. Haplotype assembly from aligned weighted SNP fragments. Computational Biology and Chemistry. 2005;29(4):281–287. doi: 10.1016/j.compbiolchem.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Chen Z, Fu B, Schweller R, Yang B, Zhao Z, Zhu B. Linear time probabilistic algorithms for the singular haplotype reconstruction problem from SNP fragments. Journal of Computational Biology. 2008;15(5):535–546. doi: 10.1089/cmb.2008.0003. [DOI] [PubMed] [Google Scholar]