

Abstract
Specific members of the inward rectifier potassium (Kir) channel family are postulated drug targets for a variety of disorders, including hypertension, atrial fibrillation, and pain1,2. For the most part, however, progress toward understanding their therapeutic potential or even basic physiological functions has been slowed by the lack of good pharmacological tools. Indeed, the molecular pharmacology of the inward rectifier family has lagged far behind that of the S4 superfamily of voltage-gated potassium (Kv) channels, for which a number of nanomolar-affinity and highly selective peptide toxin modulators have been discovered3. The bee venom toxin tertiapin and its derivatives are potent inhibitors of Kir1.1 and Kir3 channels4,5, but peptides are of limited use therapeutically as well as experimentally due to their antigenic properties and poor bioavailability, metabolic stability and tissue penetrance. The development of potent and selective small-molecule probes with improved pharmacological properties will be a key to fully understanding the physiology and therapeutic potential of Kir channels.
The Molecular Libraries Probes Production Center Network (MLPCN) supported by the National Institutes of Health (NIH) Common Fund has created opportunities for academic scientists to initiate probe discovery campaigns for molecular targets and signaling pathways in need of better pharmacology6. The MLPCN provides researchers access to industry-scale screening centers and medicinal chemistry and informatics support to develop small-molecule probes to elucidate the function of genes and gene networks. The critical step in gaining entry to the MLPCN is the development of a robust target- or pathway-specific assay that is amenable for high-throughput screening (HTS).
Here, we describe how to develop a fluorescence-based thallium (Tl+) flux assay of Kir channel function for high-throughput compound screening7,8,9,10.The assay is based on the permeability of the K+ channel pore to the K+ congener Tl+. A commercially available fluorescent Tl+ reporter dye is used to detect transmembrane flux of Tl+ through the pore. There are at least three commercially available dyes that are suitable for Tl+ flux assays: BTC, FluoZin-2, and FluxOR7,8. This protocol describes assay development using FluoZin-2. Although originally developed and marketed as a zinc indicator, FluoZin-2 exhibits a robust and dose-dependent increase in fluorescence emission upon Tl+ binding. We began working with FluoZin-2 before FluxOR was available7,8 and have continued to do so9,10. However, the steps in assay development are essentially identical for all three dyes, and users should determine which dye is most appropriate for their specific needs. We also discuss the assay's performance benchmarks that must be reached to be considered for entry to the MLPCN. Since Tl+ readily permeates most K+ channels, the assay should be adaptable to most K+ channel targets.
Keywords: Biochemistry, Issue 71, Molecular Biology, Chemistry, Cellular Biology, Chemical Biology, Pharmacology, Molecular Pharmacology, Potassium channels, drug discovery, drug screening, high throughput, small molecules, fluorescence, thallium flux, checkerboard analysis, DMSO, cell lines, screen, assay, assay development
Protocol
1. Generation of Stable Polyclonal Cell Lines
The establishment of a high quality stable cell line expressing the Kir channel of interest is an important first step toward developing a robust high-throughput screening assay. Constitutive K+ channel overexpression can lead to activation of cell death pathways, stable cell line degeneration and loss of assay performance. To avoid these potential problems and provide a convenient internal control for assay development (see below), a tetracycline-inducible expression system is recommended8.
Culture the parental T-REx-HEK293 cells using standard techniques in B-medium (DMEM growth medium containing 10% FBS, 50 U/ml Penicillin, 50 μg/ml Streptomycin and 5 μg/ml Blasticidin S). Use early passage cells (e.g. 3-4 passages since thawing from liquid nitrogen storage) for transfection and stable clone selection.
Plate 4 million T-REx-HEK293 cells in a 75 cm2 flask so that the flask is approximately 80% confluent the following day. Culture overnight in a 5% CO2 incubator at 37 °C.
Transfect the cells using 10-15 μg of pcDNA5/TO-Kir DNA and Lipofectamine LTX/Plus reagent according to manufacturer's protocol. After 5 hours, replace the transfection medium with B-medium.
24 hr after the transfection, replace the B-medium with B-medium containing 250 μg/ml Hygromycin (BH-medium) to begin stable clone selection. Feed the cells every 2-3 days with fresh BH-medium.
Greater than 90% of the cells should die over the next 7 days, leaving behind small colonies of stably transfected cells. Allow the colonies to grow for an additional 10-14 days before splitting to a 175 cm2 flask for expansion.
For cryopreservation, freeze 3 x 106 cells/ml in media containing 45% conditioned BH-medium, 45% antibiotic-free, serum-containing DMEM and 10% DMSO. Freeze the cells overnight at -80 °C in a cell freezing container and then move to liquid nitrogen for long-term storage. Thaw cells from liquid nitrogen using Invitrogen's recommended protocol. Note that the viability of the cells will be lower if they are frozen from a flask that is greater than 75% confluent at the time of processing.
2. Generation of Stable Monoclonal Cell Lines
Wash a sub-confluent 75 cm2 flask of stable polyclonal cells with divalent-free HBSS. Add 1 ml of trypsin and incubate for 3-5 min in a 5% CO2 incubator at 37 °C. Add 5 ml of BH-medium to the flask to inhibit trypsin activity and triturate repeatedly (e.g., 5 times) to fully dissociate the cells. Carefully inspect the cells under a microscope to ensure the suspension consists almost entirely of single cells. This will increase the likelihood of obtaining monoclonal cell lines.
Determine the cell density and dilute the suspension to a concentration of 0.7 cells per 20 μl. Using a multi-channel pipettor, pipette 20 μl of the cell suspension into each well of BD PureCoat amine-coated (or equivalent poly-D-lysine coated) 384-well plates. In principle, 70% of wells should receive one cell with these plating conditions. Thus, a 384-well plate should contain more than 200 clones to analyze. Continue culturing the cells in a 5% CO2 incubator at 37 °C.
After a week, inspect the plates under a microscope for wells containing single colonies. Note their position on the lid with a permanent marker for future reference. Be sure to also note and exclude wells containing multiple colonies. These are most easily recognized by the appearance of colonies growing from multiple sides of the well. Be aware that evaporation tends to occur more quickly from wells near the edge of the plate. Add BH-medium to wells where significant evaporation has occurred. Otherwise, it is unnecessary to feed the cells.
Monitor the wells more frequently during the next 7-10 days. The cells will be ready to split when the wells of interest are at least 50% confluent.
Split the cells to duplicate 384-well plates; one plate for assaying with Tl+ flux and the other for continuing the monoclonal lines in culture. Aspirate the medium from the wells containing the cell lines of interest. Wash the cells with divalent-free HBSS, add 20 μl of trypsin to each well and transfer the plate to a 37 °C incubator for 15-20 min. After moving the plate back to the cell culture hood, add 20 μl of BH-medium to each well and triturate several times to dissociate the cells. Inspect the wells under a microscope to ensure the cells are fully dissociated. This may require multiple rounds of pipetting, as the cells will be tightly adherent. Once the cells are dissociated, transfer 10 μl of each cell suspension to duplicate wells of a new BD PureCoat amine-coated 384-well plate. Be sure to note the relationship between the source well and duplicate destination wells so that the clones exhibiting robust Kir channel activity (see below) can be traced back to the original well. Add 20 μl of BH-medium to the original source well to feed the remaining cells and continue them in culture in 5% CO2 incubator at 37 °C.
Allow the cells in the destination plate to adhere and grow for up to a week. Once most of the clones reach at least 50% confluence, they can be assessed for Kir channel activity using the Tl+ flux assay described below.
The day before the assay, aspirate the culture medium and replace it with BH medium containing 10% dialyzed FBS. This prevents inadvertent channel expression that could arise from tetracycline contaminated serum. Induce Kir channel expression in only one of the duplicate wells for each clone by including 1 μg/ml tetracycline. The duplicate uninduced well will serve as a "background" control. Perform the Tl+ flux assays as described next.
3. General Tl+ Flux Assay Procedure
The day before a Tl+ flux experiment, dissociate the cells and quantitate the density of the cell suspension as described in sections 2.1 and 2.2. Plate 20,000 monoclonal cells stably transfected with a Kir channel gene of interest in each well of a BD PureCoat amine-coated 384-well plate using a Thermo Multidrop Combi or a multi-channel pipettor. Use BH medium containing 10% dialyzed FBS for plating. Note that some cells will be cultured overnight with 1 μg/ml tetracycline to induce Kir channel expression, whereas others will not. The location of induced and uninduced cells will differ for each type of experiment and are shown in Figure 1.
The next day, inspect the plates under a microscope to ensure that the cells are adherent and evenly distributed across the bottom of the wells. The wells should be 80-90% confluent.
Use an ELx405 Microplate Washer to replace the cell culture medium with 20 μl per well of HBSS assay buffer containing 20 mM HEPES and buffered to pH 7.3 with NaOH. Alternatively, one can use a "flick and slam" method, wherein the plate is inverted and snapped down sharply to eject the medium into a waste container, and then patted onto stacked paper towels to remove the remaining media. Immediately add back HBSS assay buffer to the plate to prevent the cells from desiccating.
Prepare the FluoZin-2, AM dye loading solution. After briefly centrifuging the tube, dissolve 50 μg of FluoZin-2 powder in 100 μl of anhydrous DMSO. At this point, aliquots of the 1,000x stock solution can be stored at -20 °C for later use. When ready to use, add 50 μl of a 20% weight per volume Pluronic F-127/DMSO solution to the dye and mix with gentle pipetting. Do not freeze the dye once Pluronic F-127 has been added, as low temperature may cause the surfactant to precipitate out of solution. Add the 150 μl volume of FluoZin-2, AM/Pluronic-F127 to 100 ml of HBSS assay buffer and mix gently to make the dye loading buffer. Using a Multidrop Combi or multi-channel pipettor, add 20 μl of dye loading buffer to each well already containing 20 μl of HBSS assay buffer. Incubate the cells at room temperature for approximately 1 hr (typically incubation has been done in the dark, although there is no direct evidence that it is necessary).
While the cells are loading with dye, prepare a Tl+ stimulus plate. Freshly dissolve 0.5 g of sodium bicarbonate in 50 ml of 5x Tl+ stimulus buffer containing 1 mM magnesium sulfate, 1.8 mM calcium sulfate, 5 mM glucose, 10 mM HEPES and 12 mM Tl+ sulfate. Alternatively, sodium bicarbonate can be replaced with sodium gluconate if, for example, the solution pH must be kept within a very narrow range and loss of carbon dioxide is a concern. Cap the tube tightly to limit the escape of carbon dioxide and invert several times until the sodium bicarbonate goes into solution. Using a multi-channel pipettor, add 50 μl of the solution to each well of a polypropylene 384-well plate (Table 1).
After the cells have been loaded with FluoZin-2, AM, wash the plates with an ELx405 Microplate Washer or using the "flick and slam" method, as described above in section 3.3. Depending on the type of Tl+ flux experiment to be performed, add back 20 μl or 40 μl of HBSS assay buffer to each well. The plates are now ready for experiments.
The cell and Tl+ plates into a Hamamatsu Functional Drug Screening System (FDSS) or equivalent kinetic imaging plate reader with integrated liquid dispensing capabilities. Use appropriate filters for fluorescein/fluorescein-based dyes such as Fluo-4.
Set up a single-add protocol so that 10 μl the 5x Tl+ series is added to the corresponding wells of the cell plate containing 40 μl of HBSS assay buffer. Record baseline fluorescence at a 1 Hz sampling frequency for at least 10 sec. The well-to-well fluorescence should be uniform and stable across the plate once optimal cell plating, washing and dye loading conditions are established. An integrated 384-channel pipettor is used to simultaneously add the Tl+ stimulus buffer to each well.
Record for at least 2 min so that the rate and peak of the Tl+-induced increase in fluorescence is captured for off-line analysis.
4. Determination of Optimal Tl+ Concentration
Tl+ readily permeates most inward rectifier K+ channels. To ensure that Tl+ concentrations do not exceed the dynamic range of the Tl+ reporter dye FluoZin-2, one should empirically determine the optimal Tl+ concentration to be used in a high-throughput screen. We recommend a Tl+ concentration that evokes 80% of the maximal fluorescence response (EC80) under conditions where the channel is maximally activated.
Plate, dye load and wash the cells in BD PureCoat amine-coated or equivalent poly-D-lysine-coated 384-well plates as described above in section 3, leaving 40 μl of HBSS assay buffer in each well. As shown in the plate map in Figure 1A, column 1 and rows A1-A23 should contain cells that have not been induced with tetracycline and therefore do not express the Kir channel of interest. The FluoZin-2 fluorescence signals from the uninduced wells will be used to determine the level of Tl+ flux through endogenous pathways, such as the Na+- K+ -ATPase pump and voltage gated K+ channels, which are normally expressed in HEK-293 cells.
Use an Agilent Bravo Automated Liquid Handling Platform or manual pipetting to prepare an eleven-point Tl+ concentration dilution series in HBSS assay buffer. Typically, a 3-fold serial dilution series ranging from 100% to 0.002% Tl+ is evaluated in the assay. The series should be made up at a 5x concentration because the Tl+ solutions will be diluted 1:5 in the final assay. Prepare the dilution series by diluting the standard 5x Tl+ buffer described in section 3 with a 5x Tl+-free buffer containing 1 mM magnesium sulfate, 1.8 mM calcium sulfate, 5 mM glucose and 10 mM HEPES. Again, freshly dissolve 0.5 g of sodium bicarbonate in 50 ml of the final Tl+ buffer immediately before plating in a polypropylene 384-well plate according to the plate map shown in Figure 2A.
Load the cell and Tl+ stimulus plates into the FDSS. Set up a single-add protocol so that 10 μl the 5x Tl+ series is added to the corresponding wells of the cell plate containing 40 μl of HBSS assay buffer. Record baseline FluoZin-2 fluorescence for 10 sec before adding Tl+ to the plate. Repeat the experiment on 3 separate days to establish the reproducibility of the results.
5. Determination of Assay Sensitivity to DMSO
The small molecules interrogated in a high-throughput screen are dissolved in the organic solvent dimethyl sulfoxide (DMSO), which itself can affect the performance of the assay. Therefore, the assay's sensitivity to DMSO must first be examined to establish the highest DMSO concentration allowable in the screen.
Plate, dye load and wash the cells in BD PureCoat amine-coated 384-well plates as described above in section 3, leaving 20 μl of HBSS assay buffer in each well. The entire plate should contain cells that have been induced with tetracycline (Figure 3A).
Use a Bravo Automated Liquid Handling Platform or manual pipetting to prepare an eleven-point DMSO concentration dilution series in HBSS assay buffer. Typically, a 2-fold dilution series ranging from 10% to 0.01% v/v DMSO is evaluated for activity in the assay. The series should be made up at a 2x concentration because the DMSO solutions will be diluted by half in the final assay. The dilution series should be plated in a polypropylene 384-well plate, according to the plate map shown in Figure 3A.
Prepare a 5x Tl+ stimulus plate based on the EC80 or optimal Tl+ concentration determined in section 4.
Load the cell, DMSO and Tl+ stimulus plates into the FDSS. Set up a two-add protocol so that 20 μl of the 2x concentration DMSO series is added to the corresponding wells of the cell plate containing 20 μl of HBSS assay buffer. Leave the DMSO on the cells for the same amount of time the cells will be exposed to small molecule vehicle during a screen. Record baseline FluoZin-2 fluorescence for at least 10 sec before adding 10 μl of 5x Tl+ buffer to each well of the cell plate. Repeat the experiment on 3 separate days to establish the reproducibility of the results.
The assay should be tolerant to DMSO concentrations of at least 0.1% v/v for a typical high-throughput screen in which compounds are tested at a concentration of 10 μM.
6. Determination of Assay Sensitivity to Known Pharmacological Modulators
After determining the optimal Tl+ concentration and DMSO tolerance of the assay, the effects of known pharmacologically active agents on Kir channel-mediated Tl+ flux should be examined. This series of experiments will assess the assay's sensitivity, ability to rank-order compounds based on their potency, ability to categorize compounds based on their mode of efficacy (e.g. activator, inhibitor), and identify well-behaved control compounds to be used later in the assay development and screen.
Plate, dye load and wash the cells in BD PureCoat amine-coated 384-well plates as described above in section 3, leaving 20 μl of HBSS assay buffer in each well. Column 1 and rows A1-A23 should contain cells that have not been induced with tetracycline (Figure 4A).
Use a Bravo Automated Liquid Handling Platform or manual pipetting to prepare an eleven-point concentration dilution series of known modulators in HBSS assay buffer. Typically, a 3-fold dilution series ranging from 100 μM to 2 nM is evaluated for activity in the assay. The series should be made up at a 2x concentration because the DMSO solutions will be diluted by half in the final assay. The dilution series should be plated in triplicate in a polypropylene 384-well plate, according to the plate map shown in Figure 4A. Be sure that the dilutions are made such that the final DMSO concentrations are the same between drug treatments and less than or equal to the maximum allowable DMSO concentration defined in section 5.
Prepare a 5x Tl+ stimulus plate based on the optimal Tl+ concentration determined in section 4.
Load the cell, compound and Tl+ stimulus plates into the FDSS. Set up a two-add protocol so that 20 μl the 2x concentration compound series is added to the corresponding wells of the cell plate containing 20 μl of HBSS assay buffer. Add the compounds to the cell plate and incubate for up to 20 min. Note that the optimal incubation time for the compounds to detect the best signal to background ratio can be determined by running a series of experiments where couple different incubation times are chosen. Record baseline FluoZin-2 fluorescence for at least 10 sec before adding 10 μl of 5x Tl+ buffer to each well of the cell plate. Repeat the experiment on 3 separate days to establish the reproducibility of the results.
7. Checkerboard Analysis
In the next series of experiments, the uniformity and reproducibility of the assay will be evaluated using a "checkerboard" analysis. Typically, a control inhibitor is plated in every other well of each column and row of a 384-well plate, as shown in Figure 5A. To rigorously assess the noise in the assay, an EC80 concentration of inhibitor should be used. DMSO is added to the other wells as a vehicle control. Tl+ flux in DMSO- and drug-treated cells is used to calculate a value for Z prime, a statistical measure of well-to-well variability among the two populations of wells. Z prime is calculated using the formula: Z prime = 1- (3SDp + 3SDn)/|meanp + meann | where SD is standard deviation, p is uninhibited flux and n is fully inhibited flux values. An assay with Z′ values greater than or equal to 0.5 on 3 separate days is considered suitable for high-throughput screening.
Plate, dye load and wash the cells in BD PureCoat amine-coated 384-well plates as described above in section 3, leaving 20 μl of HBSS assay buffer in each well. Note that the entire plate should be induced with tetracycline.
Make a compound/DMSO plate by pipetting into a 384-well polypropylene plate using a multi-channel pipettor, 80 μl/well of a known inhibitor of the target Kir channel at the concentration determined in section 6.5, and 0.1% v/v DMSO vehicle as shown in Figure 5A. Add the known inhibitor starting in well A1 using the multi-channel pipettor and alternate to well B2 and so on up to well B24. Repeat this procedure with DMSO starting in well B1 and so on up to well A24. The final layout of the plate should match that of a checkerboard.
Prepare a 5x Tl+ stimulus plate based on the optimal Tl+ determined in section 4.
Load the cell, compound and Tl+ stimulus plates into the FDSS. Set up a two-add protocol so that 20 μl the 2x concentration compound series is added to the corresponding wells of the cell plate containing 20 μl of HBSS assay buffer. Add the compounds to the cell plate and incubate for up to 20 min at room temperature. Record baseline FluoZin-2 fluorescence for at least 10 sec before adding 10 μl of 5x Tl+ buffer to each well of the cell plate. Repeat the experiment on 3 separate days to establish the reproducibility of the results.
8. Pilot Screen
In the final stage of assay development, perform a pilot screen of a few thousand compounds to evaluate the assay's performance under conditions that will be used eventually in the large-scale high-throughput screen.
Plate, dye load and wash the cells in BD PureCoat amine-coated 384-well plates as described above in section 3), leaving 20 μl of HBSS assay buffer in each well. Note that the entire plate should be cultured overnight with tetracycline to induce Kir channel expression.
Select approximately 2,000 to 3,000 structurally diverse compounds to be tested. It is often appropriate and convenient to use compounds collections with known activities potentially enriched for ion channel modulators. Such collections include the Spectrum Collection (MicroSource) and the LOPAC collection (Sigma). In addition, it is prudent to include known modulators of the Kir of interest in the pilot screen. Ideally these compounds will be added to wells in a blinded fashion in order to allow an unbiased testing of the hit picking methods described later. Prepare the compounds in 384-well polypropylene plates (destination plates) using a Labcyte Echo Liquid Handler or suitable pin tool to transfer an appropriate volume of selected compounds in DMSO from the MLPCN collection (source plates) to the destination plates Note that test compounds are only in columns 3-22; In every other well of columns 1, 2, 23, 24 add a known inhibitor at a concentration known to fully inhibit the Kir as determined in section 7. In the remaining wells of columns 1, 2, 23, and 24 add the appropriate volume of DMSO to produce DMSO concentration-matched vehicle controls. Dilute all wells with Assay Buffer using the Multidrop Combi. Typically test compounds will be 20 μM, 2-fold above their target screening concentration.
Make Tl+ stimulus plates based on the optimal Tl+ concentration determined in section 4.
Execute the pilot screen. Take care to stagger steps of the protocol's execution to maintain consistent timing between plates in the pilot screening run.
Once the pilot screen is completed, analyze the checkerboard wells from columns 1, 2, 23, and 24 using the Z-prime equation. Inspect plates that show Z-prime values of less than 0.5 to determine the source of poor separation of control populations. Hits can be selected using a number of methods. For pilot screens it is common to calculate the mean and standard deviation of the vehicle control population and pick hits based on wells producing values >/= 3 standard deviations from the mean of the vehicle controls.
Following hit picking, retest selected hits in duplicate in 2 sets of plates. One set of plates contains the stable Kir cell in the absence of tetracycline and the other contains the stable Kir cell line in the presence of tetracycline. Hits that retest positive in at least one of the two retest plates and fail to show significant activity in the uninduced plates may be considered verified hits. Examine the verified hit list to determine how many of the "hidden" control samples were detected. Failure to detect the controls in the pilot screen indicates the need for further optimization of screening or hit-picking parameters.
Representative Results
The use of a tetracycline-inducible expression system provides a convenient internal control for distinguishing Tl+ flux through endogenous pathways and the Kir channel of interest. Figure 1 shows some examples of cell plating maps used in different types of experiments. The positions of wells containing uninduced or tetracycline-induced cells are indicated with different colors. Figure 2A shows the source plate map used to determine the optimal Tl+ concentration for assay development and compound screening. The color gradient represents the 3-fold dilution series ranging from 100% to 0.002% Tl+. A representative fluorescence intensity map is shown in Figure 2B, with cool-to-hot colors indicating low-to-high Tl+ flux values, respectively. The cell plating map shown in Figure 1C was used for this experiment. A fit of a 4-parameter logistic function to the Tl+ CRC (Figure 2C) is used to determine an EC80 value of 15% Tl+. Figure 3A shows the source plate map used to determine the DMSO tolerance of an assay. Columns 1 and 24 contain assay buffer only, whereas the color gradient indicates the 2-fold dilution series ranging from 10% to 0.01% DMSO. A representative fluorescence intensity map is shown in Figure 3B, with low Tl+ flux values indicated with darker blue. The cell plating map shown in Figure 1B was used in this experiment. The average Tl+ flux values recorded from wells containing the indicated concentrations of DMSO are summarized in Figure 3C. The data are plotted as the percentage of Tl+ flux recorded in the absence of DMSO. For this particular Kir channel, DMSO concentrations up to 2.5% had no effect on Tl+ flux and can therefore be used in experiments. Figure 4A shows the source plate map used to establish concentration-response curves for known inhibitors of a Kir channel. Each compound is indicated with a different color and is typically plated as a 3-fold dilution series. A representative fluorescence intensity map is shown in Figure 4B. The cell plating map shown in Figure 1C was used for this experiment. A representative experiment showing dose-dependent inhibition of Tl+ flux by an inhibitor is shown in Figure 4C. The robustness of an assay is determined in so-called "checkerboard" assays, which are summarized in Figure 5. In the source plate map shown in Figure 5A, a maximally effective concentration of an inhibitor or 0.1% DMSO as a vehicle control are plated in alternating wells. Figure 5B shows a representative fluorescence intensity map. A scatter plot of the peak Tl+ flux recorded from individual wells is plotted in Figure 5C. The mean fluorescence values are indicated with solid line, whereas 3 standard deviations are indicated with a dotted line. The Z prime value, a statistical measure of how well separated the two cell populations are separated, calculated for this plate is 0.75, which is well above the 0.5 threshold required for high-throughput screening.
 Figure 1. Cell plating maps used for Tl+ flux assay development. (A) Cell plating map use to determine the optimal assay Tl+ concentration. The wells in column 1, rows A1-23, K1-12, F13-23, and P13-23 contain uninduced cells (-Tet). The remaining wells contain cells that were treated with tetracycline (+Tet) to induce Kir channel expression. (B) Cell plate map use to determine the assay DMSO tolerance and perform checkerboard analysis. Note that all the wells are treated with tetracycline (+Tet). (C) Cell plate map used to determine the assay sensitivity to known pharmacological modulators. The wells in column 1 and row A1-23 contain uninduced cells (-Tet). The remaining wells contain cells that were treated with tetracycline (+Tet). Click here to view larger figure.
Figure 1. Cell plating maps used for Tl+ flux assay development. (A) Cell plating map use to determine the optimal assay Tl+ concentration. The wells in column 1, rows A1-23, K1-12, F13-23, and P13-23 contain uninduced cells (-Tet). The remaining wells contain cells that were treated with tetracycline (+Tet) to induce Kir channel expression. (B) Cell plate map use to determine the assay DMSO tolerance and perform checkerboard analysis. Note that all the wells are treated with tetracycline (+Tet). (C) Cell plate map used to determine the assay sensitivity to known pharmacological modulators. The wells in column 1 and row A1-23 contain uninduced cells (-Tet). The remaining wells contain cells that were treated with tetracycline (+Tet). Click here to view larger figure.
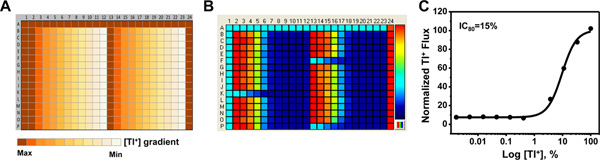 Figure 2. Determination of optimal assay Tl+ concentration. (A) Source plate map used to determine the Tl+ concentration that evokes approximately 80% of the maximal FluoZin-2 fluorescence increase (EC80). Row A and columns 1 and 24 (yellow) contain a 5x concentration of 12 mM Tl2SO4. The remaining rows contain a 3-fold dilution series ranging from 12 mM (100%) to 0.024 mM (0.002%) Tl2SO4. The series is repeated in columns 2-12 and 13-23. (B) Fluorescence intensity map depicting Tl+ flux for each well approximately 1 min after Tl+ addition. The pseudo-color bar on the right indicates the extent of Tl+ flux, with cooler and hotter colors representing low and high flux values, respectively. Note that the cooler colors in column 1, rows A1-23, K1-12, F13-23, and P13-23 are due to low Tl+ flux in uninduced cells. The remaining wells, including those in column 24, contain cells that were treated with tetracycline to induce Kir channel expression (see cell plating map in Figure 1A). (C) Mean ± SEM CRC for Tl+-dependent changes in fluorescence (n=3). Fitting a four-parameter logistic function to the data yielded an IC80 value of 15% Tl+. Click here to view larger figure.
Figure 2. Determination of optimal assay Tl+ concentration. (A) Source plate map used to determine the Tl+ concentration that evokes approximately 80% of the maximal FluoZin-2 fluorescence increase (EC80). Row A and columns 1 and 24 (yellow) contain a 5x concentration of 12 mM Tl2SO4. The remaining rows contain a 3-fold dilution series ranging from 12 mM (100%) to 0.024 mM (0.002%) Tl2SO4. The series is repeated in columns 2-12 and 13-23. (B) Fluorescence intensity map depicting Tl+ flux for each well approximately 1 min after Tl+ addition. The pseudo-color bar on the right indicates the extent of Tl+ flux, with cooler and hotter colors representing low and high flux values, respectively. Note that the cooler colors in column 1, rows A1-23, K1-12, F13-23, and P13-23 are due to low Tl+ flux in uninduced cells. The remaining wells, including those in column 24, contain cells that were treated with tetracycline to induce Kir channel expression (see cell plating map in Figure 1A). (C) Mean ± SEM CRC for Tl+-dependent changes in fluorescence (n=3). Fitting a four-parameter logistic function to the data yielded an IC80 value of 15% Tl+. Click here to view larger figure.
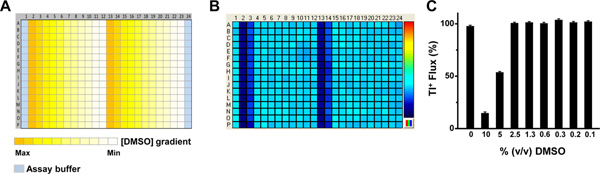 Figure 3. Assay tolerance to DMSO. (A) Source plate map used to determine assay tolerance to DMSO. Columns 1 and 24 contain HBSS assay buffer. The rows contain a 2x concentration of a 2-fold DMSO dilution series ranging from 10% to 0.01% v/v. (B) Representative fluorescence intensity map depicting Tl+ flux in each well recorded approximately 1 min after Tl+ addition. The pseudo-color bar on the right indicates the extent of Tl+ flux, with cooler and hotter colors representing low and high flux values, respectively. (C) Mean ± SEM (n=9) Tl+ flux normalized to that recorded in the presence of HBSS assay buffer alone. Click here to view larger figure.
Figure 3. Assay tolerance to DMSO. (A) Source plate map used to determine assay tolerance to DMSO. Columns 1 and 24 contain HBSS assay buffer. The rows contain a 2x concentration of a 2-fold DMSO dilution series ranging from 10% to 0.01% v/v. (B) Representative fluorescence intensity map depicting Tl+ flux in each well recorded approximately 1 min after Tl+ addition. The pseudo-color bar on the right indicates the extent of Tl+ flux, with cooler and hotter colors representing low and high flux values, respectively. (C) Mean ± SEM (n=9) Tl+ flux normalized to that recorded in the presence of HBSS assay buffer alone. Click here to view larger figure.
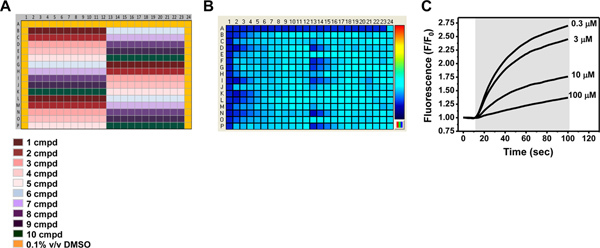 Figure 4. Assay sensitivity to known pharmacologically active compounds. (A) Source plate map used to assess the activity of known pharmacologically active compounds in the Tl+ flux assay. Row A and columns 1 and 24 contain 0.1% v/v DMSO. The plate layout allows for the testing of 10 compounds in triplicate in columns 2-23. The rows contain a 2x concentration of a 3-fold serial dilution series of compounds ranging from 100 μM to 2 nM (B) Fluorescence intensity map depicting Tl+ flux for each well approximately 1 min after Tl+ addition. The pseudo-color bar on the right indicates the extent of Tl+ flux, with cooler and hotter colors representing low and high flux values, respectively. Note that wells in column 1 and row A1-23 contain uninduced cells. The remaining wells contain tetracycline-induced cells expressing the Kir channel of interest (See cell plate map in Figure 1C). (C) Representative Tl+-induced changes in FluoZin-2 fluorescence in wells pre-treated with the indicated concentration of a Kir channel inhibitor. Click here to view larger figure.
Figure 4. Assay sensitivity to known pharmacologically active compounds. (A) Source plate map used to assess the activity of known pharmacologically active compounds in the Tl+ flux assay. Row A and columns 1 and 24 contain 0.1% v/v DMSO. The plate layout allows for the testing of 10 compounds in triplicate in columns 2-23. The rows contain a 2x concentration of a 3-fold serial dilution series of compounds ranging from 100 μM to 2 nM (B) Fluorescence intensity map depicting Tl+ flux for each well approximately 1 min after Tl+ addition. The pseudo-color bar on the right indicates the extent of Tl+ flux, with cooler and hotter colors representing low and high flux values, respectively. Note that wells in column 1 and row A1-23 contain uninduced cells. The remaining wells contain tetracycline-induced cells expressing the Kir channel of interest (See cell plate map in Figure 1C). (C) Representative Tl+-induced changes in FluoZin-2 fluorescence in wells pre-treated with the indicated concentration of a Kir channel inhibitor. Click here to view larger figure.
 Figure 5. Determination of assay suitability for HTS. (A) Source plate map used to evaluate the well-to-well variability among wells containing 0.1% DMSO (vehicle) or a maximally effective concentration of a known inhibitor. (B) Fluorescence intensity map depicting Tl+ flux in each well approximately 1 min after Tl+ addition. Note that all wells contain tetracycline-induced cells expressing the Kir channel of interest (See cell plate map in Figure 1B). (C) Representative scatter plot of steady-state fluorescence values obtained from vehicle- or inhibitor-treated wells. The mean fluorescence amplitude of each sample population is indicated with a solid line, 3 standard deviations from the mean is shown with a dashed line, and alternating samples for vehicle (VHL) and inhibitor (INH) are graphed as individual points. Click here to view larger figure.
Figure 5. Determination of assay suitability for HTS. (A) Source plate map used to evaluate the well-to-well variability among wells containing 0.1% DMSO (vehicle) or a maximally effective concentration of a known inhibitor. (B) Fluorescence intensity map depicting Tl+ flux in each well approximately 1 min after Tl+ addition. Note that all wells contain tetracycline-induced cells expressing the Kir channel of interest (See cell plate map in Figure 1B). (C) Representative scatter plot of steady-state fluorescence values obtained from vehicle- or inhibitor-treated wells. The mean fluorescence amplitude of each sample population is indicated with a solid line, 3 standard deviations from the mean is shown with a dashed line, and alternating samples for vehicle (VHL) and inhibitor (INH) are graphed as individual points. Click here to view larger figure.
Discussion
Data treatment: Once the data are collected, a common step in the analysis involves normalizing each well's fluorescence response, F, to its initial value at the beginning of the experiment, F0. This is commonly referred to as the "static ratio" and symbolized "F/F0". In cases where F0 is dominated by the indicator dye the static ratio operation will substantially correct for many factors such as disuniformities in illumination, signal collection, and cell number. In cases where the dye signal is weak or background fluorescence or reflections in the system are high, the static ratio will not be effective unless the background can be appropriately dealt with prior to calculating the static ratio. After data normalization, it is typical to reduce the fluorescence waveform to a single value that will be used to quantify activity and pick hits. Most commonly this will be done by fitting the data points in the 10 seconds following addition of Tl+ to obtain an initial slope of the evoked fluorescence increase. For hit picking, a popular approach is to assume that the vast majority of compounds in the test population are inactive (the null hypothesis is in force). A mean and standard deviation is calculated for the test population and hits are selected that are three standard deviations from the mean.
Splitting the Protocol for long compound incubations: For detecting Kir channel inhibitors, particularly those acting at intracellular binding sites, it may be valuable to incubate the cells with test compounds for an extended period of time (e.g. 20 min) prior to the addition of Tl+. If the compounds are added "offline" before the assay is introduced to the plate reader, the optical properties of test compounds (e.g. fluorescence) may not be easily recognized and can adversely affect commonly used data normalization approaches such as the "static ratio" F/F0 described above resulting in false positives and/or false negatives. To avoid this problem while simultaneously avoiding having to keep an assay plate in a reader for a long incubation, one solution is to utilize a "two read" protocol. The first read is a short (e.g. 30 sec) experiment where the compound is added after 10 seconds to allow capture of the pre-compound addition F0 and to assess the compounds' optical effects on the system. The plate is then removed from the reader and incubated for the desired period and returned to the reader for thallium addition. After both reads are completed, the first read F0 can be used to normalize the data from the second read thus avoiding many issues associated with adding compounds "offline" while allowing the opportunity to keep the reader busy collecting data thus improving screening throughput. The two read approach is most easily implemented when using an automated screening system. It is important to note that the two read approach will not eliminate problems caused by fluorescent compounds that saturate the detector and/or cause read-out artifacts in the CCD camera.
Configuring an assay for activators: Not all Kir channels are maximally activated under resting conditions, thus it may be possible to design an assay that can detect small-molecule activators. In these cases, selecting an EC80 concentration of Tl+ may not provide enough "headroom" for the assay to reliably detect activators. Thus, one may choose to use a lower concentration of Tl+ (e.g. EC20) in activator assays. In some cases, the assay may show low enough variability as to allow the use of an EC50 concentration of Tl+ and still provide suitable resolution of both activated and inhibited channel populations. In this case, the assay may be conducted in dual activator/inhibitor mode. When available, a known activator can be used to help determine the appropriate Tl+ concentration to maximize the chances of discovering channel activators.
Limitations: There are some limitations that should be considered during the development and execution of a Tl+ flux-based high-throughput screen. For example, the assay relies on an indirect measure of Kir channel activity using a fluorescent probe whose optical properties could be directly affected by compounds in a screen. Therefore, important observations from Tl+ flux experiments must be confirmed using voltage clamp electrophysiology, which is considered the "gold-standard" method for ion channel pharmacology. Furthermore, small-molecules may have direct effects on endogenous Tl+ flux pathways expressed in HEK-293 cells, leading to the identification of false-positive hits. The tetracycline-inducible system is particularly useful for distinguishing false-positive hits from Kir channel-directed modulators because the hits can be rapidly screened in uninduced cells for effects on endogenous pathways. Finally, the low solubility of Tl+ in chloride-containing buffers limit the concentration of Tl+ that can be used in an assay, requiring the use of a non-physiological Tl+ stimulus buffer that may augment the pharmacology of the target11. The compatibility of different buffers with the target of interest should be carefully evaluated during assay development. This can be done by establishing CRCs for known modulators using different Tl+ stimulus buffers and choosing conditions in which the pharmacology most closely matches that observed using physiological buffers.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by funding from National Institutes of Health grants 1R21NS073097-01 and 1R01DK082884 (J.S.D.) and Foundation for the National Institutes grant PIER11VCTR.
References
- Ehrlich JR. Inward rectifier potassium currents as a target for atrial fibrillation therapy. J. Cardiovasc. Pharmacol. 2008;52(2):129. doi: 10.1097/FJC.0b013e31816c4325. [DOI] [PubMed] [Google Scholar]
- Bhave G, Lonergan D, Chauder BA, Denton JS. Small-molecule modulators of inward rectifier K+ channels: recent advances and future possibilities. Future Med. Chem. 2010;2(5):757. doi: 10.4155/fmc.10.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz KJ. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon. 2007;49(2):213. doi: 10.1016/j.toxicon.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Lu Z. A novel high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry. 1998;37(38):13291. doi: 10.1021/bi981178p. [DOI] [PubMed] [Google Scholar]
- Jin W, Lu Z. Synthesis of a stable form of tertiapin: a high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry. 1999;38(43):14286. doi: 10.1021/bi991205r. [DOI] [PubMed] [Google Scholar]
- Roy A, McDonald PR, Sittampalam S, Chaguturu R. Open access high throughput drug discovery in the public domain: a Mount Everest in the making. Curr. Pharm. Biotechnol. 2010;11(7):764. doi: 10.2174/138920110792927757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CD, et al. A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells. J. Biomol. Screen. 2004;9(8):671. doi: 10.1177/1087057104268749. [DOI] [PubMed] [Google Scholar]
- Lewis LM, et al. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and. 2009;76(5):1094. doi: 10.1124/mol.109.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G, et al. Development of a selective small-molecule inhibitor of Kir1.1, the renal outer medullary potassium channel. Mol. Pharmacol. 2011;79(1):42. doi: 10.1124/mol.110.066928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphemot R, et al. Discovery, characterization, and structure-activity relationships of an inhibitor of inward rectifier potassium (Kir) channels with preference for Kir2.3, Kir3.x, and Kir7.1. Front Pharmacol. 2012;2:75. doi: 10.3389/fphar.2011.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, et al. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol. Pharmacol. 2008;73(4):1213. doi: 10.1124/mol.107.041053. [DOI] [PubMed] [Google Scholar]