

Abstract
Lysine specific demethylase 1 (LSD1, also known as KDM1) is a histone modifying enzyme that regulates the expression of many genes important in cancer progression and proliferation. It is present in various transcriptional complexes including those containing the estrogen receptor (ER). Indeed, inhibition of LSD1 activity and or expression has been shown to attenuate estrogen signaling in breast cancer cells in vitro implicating this protein in the pathogenesis of cancer. Herein we describe experiments that utilize small molecule inhibitors, phenylcyclopropylamines, along with small interfering RNA to probe the role of LSD1 in breast cancer proliferation and in estrogen-dependent gene transcription. Surprisingly, whereas we have confirmed that inhibition of LSD1 strongly inhibits proliferation of breast cancer cells, we have determined that the cytostatic actions of LSD1 inhibition are not impacted by ER-status. These data suggest that LSD1 may be a useful therapeutic target in several types of breast cancer; most notably inhibitors of LSD1 may have utility in the treatment of ER-negative cancers for which there are minimal therapeutic options.
Keywords: Lysine demethylase, phenylcyclopropylamines, estrogen receptor, breast cancer
The estrogen receptors (ER) are ligand-inducible transcription factors that belong to the nuclear receptor superfamily. The predominant ER subtype (ERα) is expressed in the majority of breast tumors where it enables the mitogenic actions of estrogens. As such, therefore, ERα has emerged as a major target for breast cancer therapeutics.(1) Unfortunately, up to one-third of breast carcinomas lack ER at the time of diagnosis, and a fraction of cancers that are initially ER positive apparently lose the expression of the receptor during tumor progression. The evolution of this phenotype obviously reduces the options for anti-estrogen treatment and ultimately negatively impacts survival rates. These trends are compounded by the large number of new breast cancers identified each year, predicted in 2011 to be greater than 230,000 diagnoses and 40,000 fatalities.(2) Collectively these data illustrate the pressing need to understand the pathophysiology of breast cancer and to discover and exploit new targets for all forms of breast cancers.
The complexity of gene regulation can be attributed not only to primary transcription factors but also to the large number of co-regulatory complexes that bind to and manipulate the chromatin landscape. The core histones, subject to many post-translational modifications including methylation, acetylation, phosphorylation, and ubiquitination, supply binding sites for numerous transcription factors. These modifications are present in different combinations within histones and have a powerful effect on chromatin structure. This combinatorial patterning of histone post-translational modifications influences the recruitment of factors that regulate gene expression, thus contributing to the ‘histone code’ hypothesis of trancriptional regulation.(3, 4) Understanding the role(s) of the enzymes involved in establishing and maintaining the histone code is an essential step in defining the role of epigenetic gene regulation in disease pathology and will be instructive with respect to new drug discovery. The likely impact of this class of targets has been highlighted by the success of inhibitors of histone deacetylases in leukemias and in select solid tumors.(5, 6)
Methylation of histone lysine residues within chromatin leads to dichotomous responses, in that depending on the specific histone modified and the extent to which modification occurs (mono, di, tri-methylation) transcriptional activity can be enhanced or decreased.(7, 8) Histone methylation is catalyzed by histone methyltransferases (HMTs) and methyl marks are removed by the catalytic activity of enzymes such as histone lysine-specific demethylases (LSD1/KDM1 and LSD2) and Jumonji C domain-containing histone demethylases.(9–11) The activity of LSD1 is essential for mammalian development and implicated in many important cellular processes.(12) Through interactions with various transcription factors including the androgen receptor, ER, and corepressor complexes, LSD1 impacts transcription by demethylating histone H3 lysine 4 (H3K4) or lysine 9 (H3K9) and non-histone substrates such as p53 and DMNT1.(13–18) Furthermore, the high expression of LSD1 in breast cancer, coupled with known roles of the enzyme in transcriptional activation, together have heightened interest in LSD1 as a potential therapeutic target for breast cancer.(19) LSD2 has similar substrate specificity to LSD1, but does not form stable complexes with CoREST or HDAC1/2 and is presumed to be found in distinctive chromatin remodeling complexes.(11, 20) Additionally, LSD2, unlike LSD1, has been found to associate with mitotic chromosomes.(20)
LSD1/2 are members of the flavin adenine dinucleotide-dependent (FAD) amine oxidase class of enzymes that also includes monoamine oxidase (MAO) A and B, and polyamine oxidase (PAO). Three chemotypes of small molecule inhibitors are known to target these enzymes and have been used to evaluate the biological activity of LSD1. First, Casero and Woster showed that bisguanidine and biguanide polyamine analogues, which also inhibit PAO, are effective LSD1 inhibitors and result in re-expression of silenced tumor suppressor genes in colon cancer.(21, 22) Very recently, these polyamine analogs have been shown to modulate gene expression in breast cancer cells.(23) Secondly, our group first identified that 2-phenylcyclopropylamine (2-PCPA, also known as tranylcypromine), a non-selective MAO inhibitor previously used clinically as an antidepressant, was a mechanism-based inactivator of LSD1 in vitro and in cellulo at physiologically-relevant concentrations.(24, 25) Cole and others showed that inhibition occurs via covalent modification of its flavin cofactor.(26–28) Various analogues of 2-PCPA have been synthesized and tested against LSD1 in vitro.(27, 29, 30) Some of these compounds have been shown to induce differentiation of promyelocytic leukemia cells(31) and to slow the growth of prostate cancer cell lines.(32) The third representative chemotype, propargylamines such as the isoform-selective MAO-B inhibitor pargyline was reported to inhibit LSD1-catalyzed demethylation of H3K9 in the presence of the androgen receptor at the relatively high concentration of 5 mM, a concentration known to only weakly inhibit LSD1 in vitro.(16) However, it is unclear that the antiproliferative effects of pargyline at these concentrations on prostate cancer cells are due to inhibition of LSD1 activity or off-target effects.
Motivated by recent evidence suggesting that LSD1 may play a critical role in hormone-dependent gene expression and cellular proliferation processes,(13, 19, 33) we demonstrate here the pathological importance of LSD1 demethylation chemistry in cellular models of ER-dependent and ER-independent breast cancer. Using a combination of siRNA knockdown and LSD1 chemical inhibition by designed 2-PCPA derivatives, we show that LSD1 enzymatic activity is required for ER function. Here, we also determine that inhibition of LSD1 demethylation abrogates estrogen-liganded ER recruitment to promotors of estrogen responsive genes and exhibits a strong antiproliferative effects on breast cancer cells. We observe anticipated effects on H3K4 and H3K9 histone demethylation patterns. Comparison of 2-PCPA inhibitors to a different structural class, the propargylamine MAO-B selective inhibitor pargyline, indicates that while both compound classes exhibit antiproliferative effects on breast cancer cells, only 2-PCPA had pronounced LSD1 inhibition at physiologically-relevant concentrations. These data suggest that caution may be prudent while interpreting data from proliferation experiments in pargyline-treated cells due to off target effects. This work connects nuclear hormone signaling with epigenetic transcriptional regulation and provides additional experimental support for LSD1 as a potential target for breast cancer therapeutics, especially from those cancers that are ER-independent.
RESULTS AND DISCUSSION
LSD1 expression is elevated in breast cancer cells and plays a role in cellular proliferation
LSD1 has been shown to be overexpressed in some breast cancers and may function as a biomarker of the aggressiveness of the disease.(19) LSD1 is a FAD-dependent amine oxidase and previously we have shown that the concentration of monoamine oxidase propargylamine inactivators (pargyline and clorgyline) necessary to inhibit the specific demethylation of histone substrates in vitro are not likely to be achievable in vivo or in cell culture.(25) However, there are several recent examples in the literature where propargylamines are used at very high concentrations to probe LSD1 function in a variety of cellular environments.(16, 34, 35) As compounds in this class are nonselective, off-target actions may be predominant at concentrations above 1 mM; therefore, we set out to develop more potent LSD1 inhibitors and use these inhibitors to help understand the role of LSD1 demethylation catalysis in ER signaling to aid in defining the utility of this enzyme as a cancer therapeutic.
In preparation for these studies, we first investigated the mRNA expression levels of all of the FAD-dependent amine oxidases in established cellular models of breast cancer to determine which members of this class of enzymes may be useful targets and to define the best model system(s) to study LSD1 action. To this end, the relative expression levels of nine different FAD-dependent amine oxidases were assessed in published array data derived from a panel of 51 breast cancer cell lines (dataset GSE12777(36)). The data obtained in this manner are presented as a heatmap (Figure 1A) and indicate that LSD1 and LSD2 are the most highly expressed across all cell lines. A similar analysis was performed in a breast cancer tumor dataset of 347 primary invasive breast tumors (GSE4922 combining both U133A with the U133B chips(37)). As observed in cell lines, LSD1 and LSD2 were consistently expressed at much higher levels than the other FAD-dependent amine oxidases (Figure 1B). The high expression levels of LSD1 and LSD2 across all types of breast cancer suggest that, if proven effective, inhibitors of these enzymes may be useful in the treatment of both ER-positive and ER-negative breast cancers. Most significant was the observation that LSD1 was highly expressed in cellular models of the difficult to treat triple negative breast cancers (MDA-MB-231, HCC1143 and HCC1937 cells (Supplemental Figure 1)). These expression data indicate that LSD1 is likely to be a useful therapeutic target, and considering expression alone, significant off-target activities on the structurally-related LSD2 enzyme may be observed.
Figure 1.

A. Heatmap illustrating expression levels of FAD-dependent amine oxidases in breast cancer cells lines. B. Heatmap illustrating expression levels of FAD-dependent amine oxidases in breast cancer tumors. Red indicates high expression and blue indicates low expression.
The roles of LSD1 and LSD2 in the proliferation of ERα-positive and triple negative breast cancer cells was assessed following knockdown of their expression using small interfering RNAs (siRNAs). Using this approach we were able to accomplish a knockdown of LSD1 and LSD2 in both MCF7 and MDA-MB-231 cells using two distinct siRNAs (Figure 2A–D and Supplemental Figure 2A). As shown in Figure 2E–F, knockdown of LSD1 dramatically inhibited proliferation of both MCF7 and MDA-MB-231 cells, respectively. This was primarily a cytostatic activity. Conversely, knockdown of LSD2 expression using the same approach was without effect on proliferation (Supplemental Figure 2B–C). These data suggest that LSD1, but not LSD2, is required for proliferation in these cell models; a result that highlights the utility of targeting this enzyme in breast cancer.
Figure 2.

Knockdown of LSD1 after transfection with either two unique siRNA duplexes to LSD1 (siLSD1 A and siLSD1 B) or siRNA control (siMED). A. mRNA levels measured after treatment for 18 h with either vehicle or 100 nM E2 in MCF7 cells. Data presented as ±SEM. B. mRNA levels measured in MDA-MB-231 cells. Data presented as ±SEM. C. LSD1 protein levels in MCF7 cells. D. LSD1 protein levels in MDA-MB-231 cells. E. MCF7 cell proliferation as measured by total DNA content after knockdown of LSD1. F. MDA-MB-231 cell proliferation as measured by total DNA content after knockdown of LSD1.
Tranylcypromine derivatives are effective inhibitors of LSD1
Because siRNA knockdown does not distinguish between the effects due to the reduction of LSD1 protein levels versus a specific requirement for demethylase enzymatic activity, we set out to determine the degree to which catalysis was required using mechanism-based small molecule inactivators of LSD1 in cells. Phenylcyclopropylamines, similar to 2-PCPA (tranylcypromine, brand name Parnate®) have been shown to inhibit LSD1 both in vitro and more recently in cellular environments.(24, 25, 27, 29) Our group, in addition to others, has synthesized derivatives of 2-PCPA that show selectivity towards LSD1 over MAO A/B enzymes. Phenylpropargylamines, such as pargyline, have been observed to affect global methylation levels in prostate cancer cells,(16) but concentrations greater than 5 mM are required to inhibit LSD1 when assayed in vitro.(25) Therefore, we sought to generate analogues of both classes of compounds and comparatively evaluate their efficacy in enzymatic assays and on breast cancer cell proliferation. By examining both structural classes of small molecules in the same study, we believed that we could shed light on discrepancies within the literature about the use of high concentrations of pargyline as a probe of LSD1 function in cells. The inhibitors (compounds 1a-1f and 2a-2c) were synthesized in good to excellent yields using methods developed previously in our laboratory (29) (see Supplemental Methods). Previously, we identified the para-methoxy derivative of 2-PCPA (Table 1, compound 1a) as a potent inhibitor within this class (29). This increased activity may result from improved binding to the LSD1 active site perhaps by hydrogen bonding to Thr355 within the LSD1 active site (29) or the electron-donating capability of the oxygen and its effect on the redox potential of the arylcyclopropylamine. Thus additional inhibitors were designed to contain a similar para-substituted ether or thioether to preserve these beneficial effects on activity (Table 1). Compounds 1a-1f and 2a-2c were assayed for their ability to inhibit the enzymatic activity of recombinant LSD1 utilizing a horseradish peroxidase coupled assay for the detection of hydrogen peroxide formed in the demethylase catalytic cycle.(29) All of the 2-PCPA-based inhibitors exhibit similar kinact values but the KI increased as the steric bulk in the para-position of the aryl ring increased (Table 1, compounds 1a-1f). Additionally, 2-PCPA and compounds 1a-1d are capable of inactivating MAO B (Supplemental Table 1). Compounds 1a-1d are not as potent against MAO B as 2-PCPA, with 1c being the least effective in vitro inhibitor of MAO B. Although these compounds are not completely selective for LSD1, progress has been made in decreasing activity against MAO B. To our knowledge, a completely selective LSD1 has not been synthesized, although other groups have made progress in decreasing inhibition against MAO B. However, as expected these compounds do have reactivity toward LSD2 as they target covalent inactivation of the flavin following oxidation.(27, 30) In contrast, pargyline and derivatives 2a-2c did not inhibit LSD1 in vitro except at concentrations greater than 5 mM. In fact, treating LSD1 with concentrations of pargyline and derivatives 2a-2c as high as 10 mM only resulted in weak partial inhibition of the enzyme, with greater than 70% of the enzymatic activity remaining after prolonged exposure to the inhibitor. Thus, we conclude that when probing LSD1 function in cellular environments, small molecule suicide inhibitors derived from phenylcyclopropyl amines may be more advantageous than inhibitors derived from propargylamines.
Table 1.
Inhibition kinetics of LSD1 by tranylcypromine and derivatives (1a-f) and pargyline and derivatives (2a-c).
| inhibitor | structure | kinact (s−1) | KI (μM) | kinact/KI (M−1 s−1) |
|---|---|---|---|---|
| 2-PCPA |
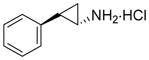
|
0.029 ± 0.004 | 550 ± 150 | 53 |
| 1a |
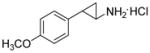
|
0.013 ± 0.001 | 173 ± 37 | 75 |
| 1b |
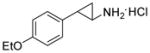
|
0.020 ± 0.002 | 456 ± 86 | 45 |
| 1c |
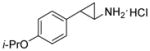
|
0.019 ± 0.002 | 352 ± 76 | 54 |
| 1d |
|
0.018 ± 0.002 | 296 ± 77 | 59 |
| 1e |
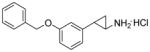
|
0.025 ± 0.005 | 760 ± 280 | 33 |
| 1f |
|
0.022 ± 0.003 | 540 ± 160 | 40 |
|
| ||||
| pargyline |
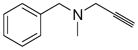
|
N.D. | N.D. | < 0.2 |
| 2a |
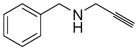
|
N.D. | N.D. | < 0.6 |
| 2b |
|
N.D. | N.D. | < 0.8 |
| 2c |
|
N.D. | N.D. | < 0.3 |
LSD1 enzymatic function is necessary for ERα-dependent transcription of genes
Recent studies by Hu and coworkers have shown that LSD1 is an essential mediator of the interchromosomal interactions necessary for estrogen (E2)-dependent ERα-mediated transcription.(13) Thus, we set out to elucidate if the role of LSD1 on ERα regulated transcription was dependent on the physical presence of LSD1 as a scaffolding protein or could be expanded to include a requirement for its demethylase enzymatic activity. We used compounds 1a-1c as probes of LSD1 function in breast cancer cells. Treatment of MCF7 cells for 24 hours with these compounds (0.2 μM and 1 mM) did not significantly impact viability enabling us to evaluate gene expression without the confounding influence of cell death or apoptosis (data not shown).
In order to confirm the role of LSD1 in E2-regulated transcription, ERα-positive MCF7 cells were treated with the LSD1 inhibitors or siRNAs to accomplish knockdown of LSD1. Both knockdown (siLSD1) and small molecule inhibition (compounds 1a-1c, 250 μM) of LSD1 resulted in decreased expression of pS2, a marker for hormone-dependent breast cancer, GREB1, a gene important in hormone-responsive cancer and PR, the gene encoding the progesterone receptor (Figure 3A–C). Similarly, inhibition of the ER-targets genes MCM2, AMyb, CatD, WISP2, SDF1, and Siah2 was also observed (data not shown). The products of many of these genes have been causally linked to breast cancer pathogenesis.(38) By contrast, other ERα target genes including Erbb4, Smad2, MYC, IL1-R1, and Notch3 were not affected by depletion of LSD1 activity (data not shown). Additionally, levels of EGR1 (Figure 3D), an ER unresponsive gene, were not significantly impacted by inhibition or knockdown of LSD1. It was noted that treatment with the 1a-1c also resulted in changes in basal levels of transcription of some target genes (data not shown). Collectively, these data point towards a role of LSD1 in estrogen-independent processes. Interestingly, when MCF7 cells were depleted of LSD2 using siRNA, the mRNA levels of LSD1 and ERα target genes were not changed (Supplemental Figure 2D–E). This implies that the LSD1 (and not LSD2) is involved in ER-dependent gene transcription.
Figure 3.

Inhibition of LSD1 expression of enzymatic activity compromises ERα transcriptional activity. MCF7 cells were transfected with either of two unique siRNA duplexes to LSD1 (siLSD1 A or siLSD1 B) or siRNA control (siMED). After 2 d, the cells were treated for 18 h with either vehicle or 100 nM 17β-estradiol (E2). Activation of genes was also examined for treatment with 1a-c for 6 h followed by vehicle or 100 nM E2 for 18 h. mRNA was analyzed by quantitative RT-PCR for A. pS2, B. GREB1, C. PR and D. EGR1. Data is presented as ±SEM.
Upon binding estrogen, ERα interacts with specific estrogen response elements (EREs) located within the regulatory regions of target genes where it nucleates the assembly of large multi-protein complexes that influence gene transcription. Using chromatin immunoprecipitation (ChIP) analyzes, we examined if recruitment of ERα to target genes was effected by the catalytic demethylase activity of LSD1, or rather if LSD1 served as a scaffolding protein. ChIP analysis showed after E2 treatment both ERα (Figure 4A–B) and LSD1 (Figure 4E) are clearly recruited to the ERE of pS2. However, when LSD1 expression is ablated using siRNA (Figure 4B) or the catalytic demethylase activity of the enzyme is inhibited by 1c (Figure 4A), recruitment of ERα to the pS2 ERE is markedly reduced. This reduction in recruitment coincides with a significant diminution of target gene transcription. This phenomenon is also observed at two validated EREs within the PR promoter (Figure 4C–D). These data suggest that LSD1 may regulate the DNA binding activity of the ERα-transcription factor complex or that LSD1 dependent modification of chromatin at the target ERE interferes with receptor binding. Although at this time there is not enough evidence to support the former hypothesis, we have been able to show that the methylation status of histones at or close to the pS2 and PR EREs were influenced by LSD1. Specifically, using ChIP, it was determined that E2 treated cells possessed histone H3K4 dimethylation levels that were markedly decreased as compared to untreated cells at the EREs examined. This demethylation event was not observed at histone H3K9 (data not shown). When the catalytic function of LSD1 is inhibited by small molecule inhibitors, or the LSD1 enzyme levels are reduced via siRNA knockdown, E2-induced demethylation of histone H3K4 does not occur. This suggests that LSD1 may be the primary demethylase acting at these sites (see Supplemental Figure 3). Although a role for LDS1 as a scaffold protein has been suggested to be important in ER action (13), our results confirm that the catalytic activity of LSD1 is also required for the transcriptional activity of ERα at some target genes. We believe that this may reflect a requirement for histone H3K4 methylation at or about the ERα enhancer.
Figure 4.

Chromatin Immunoprecipitation shows that after treatment with 1a or 1c or siRNA to LSD1 ERα is recruitment ERE promoters is decreased. A, B. Recruitment of ERα to pS2 ERE and distal promoter. C, D. Recruitment of ERα to two PR EREs and distal promoter. E. Recruitment of LSD1 to pS2 ERE and not distal promoter upon E2 treatment. F. Recruitment of LSD1 to PR ERE and not distal promoter upon E2 treatment. In all cases, IgG is used as a negative control. Data is presented as ±SEM.
Inhibition of LSD1 using 2-PCPA derivatives or siRNA-mediated knockdown inhibits breast cancer cell proliferation
Whereas LSD1 is important for estrogen-dependent gene transcription, we observed that the siRNA-mediated knockdown of LSD1 resulted in a decrease in the proliferation rate of both ER-positive and ER-negative cell lines, a result that highlighted a more fundamental role of this enzyme in breast cancer biology (Figure 2C–D). These data provided the impetus to explore whether or not LSD1 inhibitors such as 2-PCPA and derivatives impacted cell growth and proliferation of multiple breast cancer cell lines. To this end, MCF7, MDA-MB-231, HCC1143, and HCC1937 breast cancer cells lines were treated with 250 μM of each of the LSD1 inhibitors 2-PCPA and analogues 1a-1c every other day for 10 days. Similar to what was observed in cells treated with siRNAs directed against LSD1, small molecule LSD1 inhibitors significantly decreased cell proliferation over the course of the 10 day study (Figure 5A–D). In addition, the reduced rate of cellular proliferation was shown to be inhibitor dose-dependent when examined at concentrations between 10 μM and 250 μM (Supplemental Figure 4). Significantly, we observed that long term treatment with compounds 1b and 1c resulted in cell death between days 6 and 8. Interestingly, inhibitor 1c consistently showed the most dramatic effect on proliferation under all conditions examined. This is intriguing because in vitro this inhibitor did not have the highest inhibitory potency. The improved efficacy of 1c most likely is due to improved cellular bioavailability of the compound as it is predicted to be more hydrophobic and may have improved translocation across the cellular membrane. Taken together, these studies reveal that inactivation of LSD1 by the small molecule inhibitors significantly influences the proliferation of breast cancer cells, regardless of the levels of ERα.
Figure 5.

Proliferation of breast cancer cells after repeated treatment with DMSO control or 250 μM 2-PCPA and 1a-1c. A. ERα-positive MCF7 cells. B. Triple negative MDA-MB-231 cells. C. Triple negative HCC1143 cells. D. Triple negative HCC1937 cells.
It is important to note that treatment of either MCF7 or MDA-MB-231 cells for 24 h with 2-PCPA or compounds 1a-1c at drug concentrations up to 500 μM did not induce apoptosis (data not shown). However, MCF7 cells, treated with 250 μM 2-PCPA or compounds 1a-1c for 24 hours, were growth arrested as evidenced by the accumulation of cells in G1 and G2/M and a decrease in the number of cells in S phase (Figure 6A). As expected, inhibitor 1c has the most dramatic effect on cell cycle arrest. A similar, albeit less robust, response was observed in MCF7 cells following siRNA mediated knockdown of LSD1 (Figure 6B). Interestingly, the 2-PCPA derived inhibitors 1a-1c appear to have anti-proliferative effects similar to those observed when cells are treated with histone deacetylase (HDAC) inhibitors which also induce G1 and G2 cell cycle arrest.(39) Given that LSD1 and HDAC1/2 are found together in transcriptional complexes(9), it is not unreasonable that inhibitors of either protein may work in cooperation on some substrates. Interestingly, Huang and coworkers recently observed that treatment of breast cancer cells with HDAC inhibitors leads to an increase in methylation marks at some LSD1 target genes; a result that highlights the functional link between these two enzymes.(34) Other studies however, have shown that the catalytic activities of HDAC1/2 and LSD1 on some genes are distinct. This is important in light of our finding that inhibition of LSD1 using 2-PCPA was not associated with changes in the acetylation state of H3 in bulk nucleosomes(25). Thus, the role of LSD1 may differ between cells and on different promoters.
Figure 6.
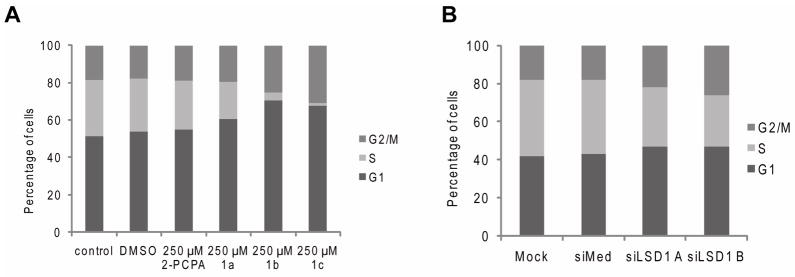
Cell cycle analysis after treatment with A. 2-PCPA or 1a-c or B. siRNA to LSD1 in MCF7 cells indicates G1 and G2/M arrest.
The activity of pargyline and derivatives in transcription and cell proliferation are unlikely to be related to inhibitory effects of LSD1
Interestingly, pargyline and derivatives 2a-2b, although exhibiting no inhibition of LSD1 in vitro at concentrations below 5 mM, slowed proliferation of MCF7 and MDA-MB-231 cells after treatment at a concentration of 250 μM (Figure 7A–B). At this time, the exact mechanism of anti-proliferative effects of pargyline is unknown but is clear that at concentrations below 5 mM it is not likely due to LSD1 inactivation. It is possible that it is the result of irreversible inhibition of other flavoenzymes or of reversible inhibition of other proteins or enzymes found in the cells. We arrived at this conclusion by evaluating the activity of pargyline in a manner similar to that which was described above for the 2-PCPA and its derivatives. In cells treated with pargyline (up to 250 μM), we did not observe changes in E2-dependent transcription (pS2 or PR) (Supplemental Figure 5). Pargyline has been used as a probe of LSD1 function in breast cancer cells(19), prostate cancer cells(16), and herpes infection(35). However, as highlighted by our results the activities of this class of compounds are less pronounced than those observed with LSD1 knockdown or with the 2-PCPA derived compounds. More importantly, we have shown that the KI for pargyline is greater than 5mM for LSD1 in vitro, suggesting that it is unlikely that the anti-proliferative activities of this compound and its derivatives are related to their ability inhibit LSD1. As such, we suggest that caution should be taken in the interpretation of studies that have relied solely on the use of pargyline to implicate LSD1 in various processes.
Figure 7.
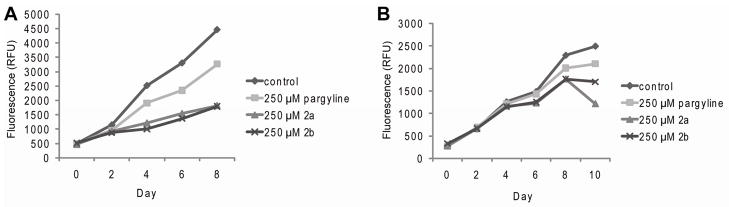
Proliferation of breast cancer cells after repeated treatment with 250 μM pargyline, 2a and 2b. A. ERα-positive MCF7 cells. B. ERα-negative MDA-MB-231 cells.
Global histone H3K4-Me2 is increased in breast cancer cell lines after LSD1 inhibition
Having demonstrated substantial effects of 2-PCPA and derivatives and siRNA-mediated knockdown on transcription and proliferation, we next established the impact of these manipulations on histone H3K4 and histone H3K9 methylation. Towards this end, MCF7 and MDA-MB-231 breast cancer cell lines were treated with 2-PCPA and the derivatives 1a-1c, and the levels of H3K4 and H3K9 dimethylation were examined by Western blot analysis. In the ERα-positive MCF7 cells, the level of histone H3K4-Me2 was increased after treatment with 2-PCPA or compounds 1a-1c (Figure 8A). However, under the same conditions we did not observe significant changes in the levels of histone H3K9-Me2. In contrast, we observed robust increases in global dimethylation of both histone H3K4 and histone H3K9 in MDA-MB-231 cells following treatment with the 2-PCPA and compounds 1a-1c (Figure 8B). Interestingly, siRNA mediated knockdown of LSD1 expression resulted in an increase in global H3K4-Me2 levels in both cell lines although the level of H3K9-Me2 levels were unchanged (Figure 8C–D). These results highlight a previously unappreciated complexity in the mechanisms that impact the activity and or target gene specificity of LSD1 action. One of the most intriguing results observed is that inhibition of LSD1 using either 2-PCPA or siLSD1 resulted in an increase in histone H3K4-Me2 but not histone H3K9-Me2. However, increases in both marks were observed in MDA-MB-231 cells. It must be stressed that these differences were observed in several independent experiments. One interpretation of these data is that an unidentified methyl transferase responsible for the histone H3K9-Me2 mark in MCF-7 cells is highly active and/or the ability of the histone H3K9 mark to recruit LSD1 in these cells is compromised. Alternatively, this could reflect an off target effect of the compounds on a H3K9 demethylase that is expressed in the MDA-MB-231 cells but not MCF7 cells. At the enzyme level, LSD1 overexpression in MDA-MB-231 cells may contribute to increased demethylation at a secondary site, such as histone H3K9, albeit to a lower efficiency than the preferred histone H3K4 site. At this time we do not know the identity of other factors that might influence LSD1 specificity among these two breast cancer cell lines. As such, future work will extend these studies to other cells to see which specific processes can be associated with and/or be responsible for regulating the specificity of LSD1 for the two positions, H3K4-Me2 and H3K9-Me2.
Figure 8.
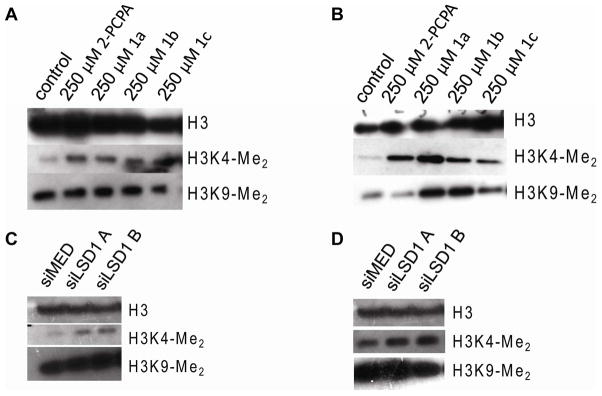
Global dimethylation levels of histones from nuclear extracts after treatment with 250 μM 2-PCPA or 1a-1c for 24 h or after knockdown of LSD1 with siRNA. A. MCF7 cells after treatment with inhibitors. B. MDA-MB-231 cells after treatment with inhibitors. C. MCF7 cells after knockdown. D. MDA-MB-231 cells after knockdown.
LSD1 inhibition as a potential breast cancer therapy
We have determined that LSD1 is essential for the proliferation of both ERα-positive and negative breast cancer cells. It appears that 2-PCPA derivatives 1a-1c have a similar but slightly more profound effect on the proliferation of the breast cancer cells (Figure 5A–B) than the knockdown of LSD1 by siRNA (Figure 2A–B). This could be attributed to a few factors. First, the knockdown approach we have developed does not completely deplete the cellular levels of LSD1 protein, and therefore, active enzyme may be present, albeit at lower levels. Thus, the residual levels could be expected to carry out the essential functions of LSD1. Second, the 2-PCPA derived compounds may be inhibiting other enzymes that are also crucial to breast cancer cell proliferation and survival. However, among the many breast cancer cell lines we found to be sensitive to 2-PCPA, LSD1 and LSD2 were the only two FAD dependent amine oxidases highly expressed.
While the specific mechanisms underlying sensitivity to LSD1 inhibition remain to be defined, it is clear from the results of our studies that the anti-proliferative activities of the compounds we have developed is not secondary to inhibition of ER-transcriptional activity and that this enzyme is involved in additional processes fundamental for proliferation. Despite the important role of LSD1, our studies demonstrate that LSD2 may have a different cellular responsibility in breast cancer or be less important for contributing to proliferative effects in breast cancers. Although both enzymes catalyze similar chemical reactions, their sequences and the presence of conserved protein-protein interaction domains (such as the Tower domain in LSD1 that recruits CoREST) suggests that there are clearly structural differences that might contribute to differing functional roles in the cell. Also, there is a strong possibility that additional components of respective LSD1 and LSD2 complexes may facilitate target selection and specificity. Nonetheless it is clear that LSD1 plays a critical role in ERα signaling.
Molecules that block the function of LSD1 selectively may prove to be effective anti-cancer therapeutics. However, because of the similarity to other amine oxidases and the large active of the enzyme, selective inhibitors have been very difficult to identify. We predict that targeting the transcriptional complexes that LSD1 associated with, such as ERα or CoREST, may allow for selective inhibition without targeting the active site.
MATERIALS AND METHODS
Bioinformatic Analysis
The breast cancer cell line dataset, GSE12777(36) was downloaded from the Gene Expression Omnibus (GEO) at “http://www.ncbi.nlm.nih.gov/geo/”. For expression analysis the CEL files were normalized using R/Bioconductor(40–42) with RMA and individual probe expression values for each gene were obtained. The FAD-dependent amine-oxidase probes were then subset from this dataset, and the expression data was converted into a heatmap using the gplots package. To analyze the expression of FAD-dependent amine-oxidase genes in tumor datasets, we used GSE4922(37), combining both U133A with the U133B chips into a single dataset and normalized as above. For purposes of identifying relative expression within each tumor, the rows consisting of tumor samples were scaled and displayed as a heatmap.
Enzymatic Assays
LSD1 overexpression, purification and horseradish peroxidase coupled enzyme assay were performed as previously described. (24, 29, 43) Inhibitors were prepared as 100 mM stocks in dimethylsulfoxide (DMSO) before dilution into assay reagents at appropriate concentrations. Progress curves for time-dependent enzyme inactivation were fit to Equation 1
| (1) |
to obtain values of kobs as a function of inhibitor concentration, which were then fit to Equation 2 to obtain values of kinact and KI.
| (2) |
Cell Culture
MCF7 cells were maintained in DMEM/F12 (Gibco) supplemented with 8% fetal bovine serum (FBS) (Sigma), 1 mM sodium pyruvate and 0.1 mM non-essential amino acids. MDA-MB 231 cells were maintained in DMEM (Cellgro) supplemented with 8% FBS, 1 mM sodium pyruvate and 0.1 mM non-essential amino acids. HCC1937 and HCC1143 cells were maintained in RPMI 1640 (Gibco) supplemented with 8% FBS, 1 mM sodium pyruvate and 0.1 mM non-essential amino acids. All cells were grown in a 37 °C incubator with 5% carbon dioxide.
Transfection assays
For siRNA transfections, MCF7 cells were plated in phenol red-free media containing 8% charcoal-stripped FBS (Hyclone laboratories), 1 mM sodium pyruvate and 0.1 mM non-essential amino acids into either 150 mm dishes (for ChIP), 12-well plates (for mRNA levels) or 6-well plates (for Western blot) and were transfected with DharmaFECT 1 (Invitrogen) according to the supplier’s protocol. MDA-MB-231 cells were plated in DMEM (Gibco) supplemented with 8% FBS, 1 mM sodium pyruvate and 0.1 mM non-essential amino acids into either 12-well plates (for mRNA levels) or 6-well plates (for Western blot) and were transfected with DharmaFECT 1 (Invitrogen) according to the supplier’s protocol.
RNA isolation and Real-Time PCR
For RNA analysis, MCF7 cells were seeded in 12-well plates in phenol red-free media containing 8% charcoal-stripped serum, 1 mM sodium pyruvate, and 1 mM non-essential amino acids. After 4 d, the cells were treated with the inhibitors (250 μM). After 6 h, the cells were treated with ethanol (no treatment) or 100 nM E2 for 18 h and then were harvested. Total RNA was isolated using the Aurum Total RNA Mini Kit (Bio-Rad). One half microgram of RNA was reverse transcribed using the iScript cDNA sysnthesis kit (Bio-Rad). The Bio-Rad iCycler Realtime PCR System was used to amplify and quantify levels of target gene cDNA. qRT-PCR were performed with 8 UL cDNA, 0.4 UM specific primers and iQ SYBR Green Supermix (Bio-Rad). Data are normalized to the 36B4 housekeeping gene and presented as fold induction over control. Data are presented as the mean ± SEM for triplicate amplification reactions from one representative experiment. Each experiment was repeated at least three independent times with nearly identical results.
ChIP assays
MCF7 cells were grown to 90% confluence in 15 cm dishes in phenol red-free media containing 8% charcoal-stripped FBS, 1 mM sodium pyruvate, and 0.1 mM non-essential amino acids for 3 d, after which the cells were serum starved for 24 h. After treatment with vehicle or E2 (100 mM) for 45 min, the cells were fixed with 1% formaldehyde for 10 min at rt. The reaction was stopped with glycine (250 nM final concentration) by incubation at rt for 5 min. The cells were washed with ice-cold PBS, harvested in PBS, and centrifuged for 1 min. The cells were frozen (−80 °C) until ready to lyse. The cells were lysed in 1 mL sonication buffer (50 mM HEPES, pH 7.8, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 1X protease inhibitor) by sonication (13 X 13 sec at 9–10 W). The lysate was clarified by centrifugation (15 min, 4 °C, 17000 xg) and the supernatant collected, diluted with RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.05% SDS, 1 mM EDTA, 1X protease inhibitor), and precleared in 100 UL Protein A/G Agarose beads (50% slurry in 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 200 Ug sonicated salmon sperm DNA, and 500 Ug Bovine serum albumin) for 30 min at 4 °C. Immunoprecipitation was performed for 4–6 hr at 4 °C with antibodies as described below. After immunoprecipitation, 100 UL Protein A/G Agarose beads (50% slurry in PBS) was added and allowed to incubate overnight at 4 °C. Precipitates were washed sequentially twice with sonication buffer, buffer A (50 mM HEPES, pH 7.8, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS, 1X protease inhibitor), buffer B (20 mM Tris, pH 8.0, 1 mM EDTA, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1X protease inhibitor), and TE (10 mM Tris, pH 8.0, 1 mM EDTA). The precipitates were eluted twice with 50 mM Tris, pH 8.0, 1 mM EDTA, 1% SDS at 65 °C for 10 min. Cross-linking was reversed by addition of NaCl (final concentration 230 mM) and incubation overnight at 65 °C. Protein was removed by incubation with EDTA (final concentration 4.5 mM) and proteinase K (final concentration 45 Ug mL−1) for 1 h at 42 °C. DNA was isolated with a QIA-quick PCR Purification kit (Qiagen). qRT-PCR was performed with immunoprecipitated DNA, specific primers, and iQ SYBR Green Supermix (Bio-Rad). Data were normalized to the input for the immunoprecipitation.
Cell Viability Assays
MCF7 cells were seeded at 8000 cells per well in 96 well plates in phenol red-free media containing 8% charcoal-stripped FBS, 1 mM sodium pyruvate, and 0.1 mM non-essential amino acids. MDA-MB-231 cells were seeded at 8000 cells per well in 96 well plates in DMEM media containing 8% FBS, 1 mM sodium pyruvate and 0.1 mM non-essential amino acids. After 4 d, cells were given fresh media and treated with inhibitors at various concentrations (0.2 μM – 1 mM) for 24 h. Cells were incubated for 2 h after addition of CellTiter-Blue reagent (Promega), and then the fluorescence was measured (excitation 535 nm, emission 630 nm) using SpectraMax Gemini EM plate (Molecular Devices). The data were background corrected using a ‘no cell’ control.
Cell Cycle Analysis Assays
MCF7 cells were seeded at 400,000 cells per well in 6 well plates in DMEM/F12 (Invitrogen) containing 8% FBS, 1 mM sodium pyruvate and 0.1 mM non-essential amino acids. After 24 h, the cells were treated with fresh media containing the 250 μM inhibitor. After 48 h of treatment, the cells were pulsed for 2 h with BrdU (10 Ug mL−1). The cells were trypsinized and collected using cold IFA buffer (3 mL) (4% charcoal stripped FBS, 150 mM sodium chloride, 10 mM HEPES, pH 7.5). They were washed with PBS, resuspended in 1 mL of PBS and fixed with 70% ethanol. The cells were incubated for 30 min on ice and then stored at −20 °C. The cells were washed with PBS containing 0.5% BSA. The pellet was denatured with 2 M hydrochloric acid containing 0.5% BSA. The residual acid was neutralized using 0.1M sodium borate, pH 8.5. The cell pellet was resuspended in dilute anti-BrdU-alexa fluor-488 antibody (Molecular Probes) (PBS + 0.5% Tween 20 + 0.5% BSA + 5% antibody). After washing excess away, the cell pellets were resuspended in PI + RNase A (10 Ug mL−1 propidium iodide and 10 Ug mL−1 RNase A in PBS). The cells were vortexed and analyzed using flow cytometry (Accuri C6).
Cell Proliferation Assays
MDA-MB 231 or MCF7 cells were seeded at 3000 cells per well in 96-well plates. After 2 d, the cells were treated with fresh media containing the inhibitors at the concentrations indicated. Every 2 d, for a total of 6 or 10 d, respectively, the cells were treated similarly. Total DNA content was measured by fluorescence using Hoechst 33258 dye (Sigma, λex = 360 nm, λem = 460 nm). Data are presented as the mean ±SEM for triplicate wells in one representative experiment. Each experiment was repeated at least two independent times, with nearly identical results.
Western Blot Analysis
MCF7 cells or MDA-MB 231 cells were seeded in six-well plates. Cells were treated after 48 h with inhibitor for 24 h. Whole-cell extracts were isolated using RIPA buffer [50 mM Tris (pH 8.0), 200 mM NaCl, 1.5 mM MgCl2, 1% Nonidet P-40 (NP40), 1 mM EGTA, 10% glycerol, 50 mM NaF, 2 mM Na3VO4 and 1x protease inhibitor mixture]. Crude histones were extracted from the lysate pellet by resuspending in water and precipitating with 25% TCA. The pellets were washed with acetone and then resuspended. Concentration of whole-cell lysate or resuspended histones was determined using Bio-Rad Bradford reagent using BSA for standard curve. For each sample, proteins were resolved by SDS-PAGE and transferred to a PVDF membrane (Biorad).
Antibodies
H3K4-Me2 was detected using polyclonal rabbit antibody (Millipore 07-030). H3K9-Me2 was detected using a monoclonal mouse antibody (Abcam ab1220). Total H3 was detected using a polyclonal rabbit antibody (Abcam ab1791). LSD1 was detected using a monoclonal rabbit antibody (Millipore 05-939) or polyclonal rabbit antibody (Abcam ab17721). ERα (D12) was detected using monoclonal mouse antibody (Santa Cruz sc-8005). GAPDH was detected using polyclonal goat antibody (Santa Cruz sc-20357). Secondary antibodies were purchased from Bio-Rad.
Supplementary Material
Acknowledgments
This research was kindly supported by National Institutes of Health research grants 1R01GM087566 to DGM and 5R37 DK048807 to DPM. J.A.P. is recipient of a Burroughs Wellcome Predoctoral Fellowship in Synthetic Organic Chemistry.
Footnotes
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cosman F, Lindsay R. Selective estrogen receptor modulators: clinical spectrum. Endocr Rev. 1999;20:418–434. doi: 10.1210/edrv.20.3.0371. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 3.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 4.Schreiber SL, Bernstein BE. Signaling network model of chromatin. Cell. 2002;111:771–778. doi: 10.1016/s0092-8674(02)01196-0. [DOI] [PubMed] [Google Scholar]
- 5.Kelly WK, O’Connor OA, Marks PA. Histone deacetylase inhibitors: from target to clinical trials. Expert Opin Investig Drugs. 2002;11:1695–1713. doi: 10.1517/13543784.11.12.1695. [DOI] [PubMed] [Google Scholar]
- 6.Tan J, Cang S, Ma Y, Petrillo RL, Liu D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J Hematol Oncol. 2010;3:5–17. doi: 10.1186/1756-8722-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bannister AJ, Kouzarides T. Histone methylation: recognizing the methyl mark. Methods Enzymol. 2004;376:269–288. doi: 10.1016/S0076-6879(03)76018-2. [DOI] [PubMed] [Google Scholar]
- 8.Bannister AJ, Kouzarides T. Reversing histone methylation. Nature. 2005;436:1103–1106. doi: 10.1038/nature04048. [DOI] [PubMed] [Google Scholar]
- 9.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 11.Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C, Mattevi A. A novel mammalian flavin-dependent histone demethylase. J Biol Chem. 2009;284:17775–17782. doi: 10.1074/jbc.M109.003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, Liu F, Taylor H, Lozach J, Jayes FL, Korach KS, Glass CK, Fu XD, Rosenfeld MG. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446:882–887. doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- 13.Hu Q, Kwon YS, Nunez E, Cardamone MD, Hutt KR, Ohgi KA, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG, Fu XD. Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc Natl Acad Sci USA. 2008;105:19199–19204. doi: 10.1073/pnas.0810634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 15.Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, Berger SL. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- 16.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, Gaudet F, Li E, Chen T. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Bassets I, Kwon YS, Telese F, Prefontaine GG, Hutt KR, Cheng CS, Ju BG, Ohgi KA, Wang J, Escoubet-Lozach L, Rose DW, Glass CK, Fu XD, Rosenfeld MG. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell. 2007;128:505–518. doi: 10.1016/j.cell.2006.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, Kirfel J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis. 2010;31:512–520. doi: 10.1093/carcin/bgp324. [DOI] [PubMed] [Google Scholar]
- 20.Yang Z, Jiang J, Stewart DM, Qi S, Yamane K, Li J, Zhang Y, Wong J. AOF1 is a histone H3K4 demethylase possessingdemethylase activity-independent repression function. Cell Research. 2010;20:276–287. doi: 10.1038/cr.2010.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Y, Greene E, Murray Stewart T, Goodwin AC, Baylin SB, Woster PM, Casero RA., Jr Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci USA. 2007;104:8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Y, Stewart TM, Wu Y, Baylin SB, Marton LJ, Perkins B, Jones RJ, Woster PM, Casero RA., Jr Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin Cancer Res. 2009;15:7217–7228. doi: 10.1158/1078-0432.CCR-09-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Q, Huang Y, Marton LJ, Woster PM, Davidson NE, Casero RA., Jr Polyamine analogs modulate gene expression by inhibiting lysine-specific demethylase 1 (LSD1) and altering chromatin structure in human breast cancer cells. Amino Acids. 2012;42:887–898. doi: 10.1007/s00726-011-1004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt DM, McCafferty DG. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry. 2007;46:4408–4416. doi: 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- 25.Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol. 2006;13:563–567. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 26.Mimasu S, Sengoku T, Fukuzawa S, Umehara T, Yokoyama S. Crystal structure of histone demethylase LSD1 and tranylcypromine at 2.25 A. Biochemical and biophysical research communications. 2008;366:15–22. doi: 10.1016/j.bbrc.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 27.Mimasu S, Umezawa N, Sato S, Higuchi T, Umehara T, Yokoyama S. Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry. 2010;49:6494–6503. doi: 10.1021/bi100299r. [DOI] [PubMed] [Google Scholar]
- 28.Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M, Cole PA, Yu H. Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry. 2007;46:8058–8065. doi: 10.1021/bi700664y. [DOI] [PubMed] [Google Scholar]
- 29.Gooden DM, Schmidt DM, Pollock JA, Kabadi AM, McCafferty DG. Facile synthesis of substituted trans-2-arylcyclopropylamine inhibitors of the human histone demethylase LSD1 and monoamine oxidases A and B. Bioorg Med Chem Lett. 2008;18:3047–3051. doi: 10.1016/j.bmcl.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Binda C, Valente S, Romanenghi M, Pilotto S, Cirilli R, Karytinos A, Ciossani G, Botrugno OA, Forneris F, Tardugno M, Edmondson DE, Minucci S, Mattevi A, Mai A. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J Am Chem Soc. 2010;132:6827–6833. doi: 10.1021/ja101557k. [DOI] [PubMed] [Google Scholar]
- 31.Culhane JC, Wang D, Yen PM, Cole PA. Comparative analysis of small molecules and histone substrate analogues as LSD1 lysine demethylase inhibitors. J Am Chem Soc. 2010;132:3164–3176. doi: 10.1021/ja909996p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benelkebir H, Hodgkinson C, Duriez PJ, Hayden AL, Bulleid RA, Crabb SJ, Packham G, Ganesan A. Enantioselective synthesis of tranylcypromine analogues as lysine demethylase (LSD1) inhibitors. Bioorg Med Chem. 2011;19:3709–3716. doi: 10.1016/j.bmc.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 33.Scoumanne A, Chen X. The lysine-specific demethylase 1 is required for cell proliferation in both p53-dependent and -independent manners. J Biol Chem. 2007;282:15471–15475. doi: 10.1074/jbc.M701023200. [DOI] [PubMed] [Google Scholar]
- 34.Huang Y, Vasilatos SN, Boric L, Shaw PG, Davidson NE. Inhibitors of histone demethylation and histone deacetylation cooperate in regulating gene expression and inhibiting growth in human breast cancer cells. Breast Cancer Res Treat. 2012;131:777–789. doi: 10.1007/s10549-011-1480-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med. 2009;15:1312–1317. doi: 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoeflich KP, O’Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, Januario T, Savage H, Punnoose E, Truong T, Zhou W, Berry L, Murray L, Amler L, Belvin M, Friedman LS, Lackner MR. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009;15:4649–4664. doi: 10.1158/1078-0432.CCR-09-0317. [DOI] [PubMed] [Google Scholar]
- 37.Ivshina AV, George J, Senko O, Mow B, Putti TC, Smeds J, Lindahl T, Pawitan Y, Hall P, Nordgren H, Wong JE, Liu ET, Bergh J, Kuznetsov VA, Miller LD. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006;66:10292–10301. doi: 10.1158/0008-5472.CAN-05-4414. [DOI] [PubMed] [Google Scholar]
- 38.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinol. 2003;144:4562–4574. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 39.Sambucetti LC, Fischer DD, Zabludoff S, Kwon PO, Chamberlin H, Trogani N, Xu H, Cohen D. Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J Biol Chem. 1999;274:34940–34947. doi: 10.1074/jbc.274.49.34940. [DOI] [PubMed] [Google Scholar]
- 40.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Team, R. D. C. A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2011. [Google Scholar]
- 42.Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B. gplots: Various R programming tools for plotting data. 2.8.0. 2010. p R. package. [Google Scholar]
- 43.Gaweska H, Henderson Pozzi M, Schmidt DM, McCafferty DG, Fitzpatrick PF. Use of pH and kinetic isotope effects to establish chemistry as rate-limiting in oxidation of a peptide substrate by LSD1. Biochemistry. 2009;48:5440–5445. doi: 10.1021/bi900499w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.