

Abstract
Carboxylesterases (CEs) are ubiquitously expressed proteins that are responsible for the detoxification of xenobiotics. They tend to be expressed in tissues likely to be exposed to such agents (e.g., lung and gut epithelia, liver) and can hydrolyze numerous agents, including many clinically used drugs. Due to the considerable structural similarity between cholinesterases (ChE) and CEs, we have assessed the ability of a series of ChE inhibitors to modulate the activity of the human liver (hCE1) and the human intestinal CE (hiCE) isoforms, We observed inhibition of hCE1 and hiCE by carbamate-containing small molecules, including those used for the treatment of Alzheimer’s disease. For example, rivastigmine resulted in greater than 95% inhibition of hiCE that was irreversible under the conditions used. Hence, the administration of esterified drugs, in combination with these carbamates, may inadvertently result in decreased hydrolysis of the former, thereby limiting their efficacy. Therefore drug:drug interactions should be carefully evaluated in individuals receiving ChE inhibitors.
Keywords: Carboxylesterase, cholinesterase, inhibitor, carbamate
1. INTRODUCTION
Carboxylesterases (CEs1) hydrolyze carboxylesters into their corresponding alcohol and carboxylic acid [12]. Given that no endogenous substrates have been identified for these enzymes, and they are expressed in virtually all organisms ranging from bacteria to man, it is presumed that CEs have a protective function. Interestingly, while many lower mammals have very high levels of CE in the blood, little or no expression is observed in this fluid in higher primates, including man. In humans, two CEs have been extensively characterized. hCE1 (CES1) is primarily expressed in the liver, and demonstrates substrate specificity for small, relatively rigid molecules [4, 11]. hiCE (CES2) is present within the small intestine and the liver, and has a much more flexible active site, allowing for the hydrolysis of larger, more bulky esters [7, 13]. Recently we have identified several different classes of selective CE inhibitors [6, 16, 17]. These compounds are potent (Ki values in the low nM range), can inhibit CE activity intracellularly, and can modulate the metabolism of esterified drugs mediated by these enzymes. None of these compounds demonstrate any activity toward human acetyl-or butyrylcholinesterase (AChE, BChE). Inhibitors of AChE have been developed for use in Alzheimer’s disease [9] and to date, 4 drugs are approved for clinical use. These include Razadyne (galantamine), Aricept (donepezil), Cognex (tacrine) and Exelon (rivastigmine). Therefore in these studies, we have determined the structural similarity of CEs and ChE and assessed the ability of AChE and BChE inhibitors to modulate CE activity.
2. MATERIAL AND METHODS
2.1 Enzymes and inhibitors
hCE1 (UniprotKB accession number P23141) and hiCE (UniprotKB accession number O00748) were prepared from baculovirus infected Sf21 cells as previously described [5, 8]. Enzyme purity was greater than 98% as confirmed by gel electrophoresis and MALDI-TOF/TOF analyses. Human AChE from human erythrocytes was obtained from Sigma Biochemicals (St. Louis, MO) and BChE (purified from human plasma) was a generous gift from Dr. Charles Millard (US Army Medical Research and Materiel Command, Frederick, MD). Benzil, tacrine, acridine, 9-amino-6-chloro-2-methoxyacridine (ACMA), 5,9-diamino-2-ethoxyacridine (DEA) and bis(4-nitrophenyl) phosphate (BNPP) were all obtained from Sigma Biochemicals. Bis(7)-tacrine was from Cayman Chemical Co (Ann Arbor, MI); rivastigmine and galantamine were purchased from Toronto Research Chemicals (Toronto, Canada); donepezil was obtained from the St. Jude pharmacy; and tolserine and phenethylcymserine were generously provided by Dr. Nigel Greig (NIA, Bethesda, MD).
2.2 Enzyme assays
CE activity was determined using o-nitrophenylacetate as a substrate in a spectrophotometric assay as previously described [10]. AChE and BChE activity was determined using either acetylthiocholine or butyrylthiocholine as substrates, respectively, with detection by Ellman’s reagent [1].
2.3 Enzyme inhibition
Enzyme inhibition was determined by comparing activity in the presence or absence of inhibitor. IC50 values were then calculated using Cheng-Prusoff equation:
where Ki is the binding affinity of the inhibitor, [S] is substrate concentration and Km is the concentration of substrate at which enzyme activity is at half maximal. Irreversible enzyme inhibition was assessed by pre-incubating enzyme with the desired inhibitor at a concentration equivalent to 5× IC50 value or 200μM (where IC50 values were not available). After 60 mins, the small molecule was removed by centrifugal filter devices (10,000 Da cutoff) and samples were then assayed for CE activity. Data was expressed as the percentage of enzyme activity lost as compared to a DMSO-containing, control sample. Greater than 75% loss in enzyme activity was considered as irreversible in these assays.
2.4 Molecular modeling
Modeling was performed using ICM Pro software (Molsoft LLC, San Diego, CA) using the coordinates 1MX5 and 3LII for hCE1 and human AChE, respectively. Briefly, structures were overlaid using the default ‘Homology’ subroutine of the program and active site residues were highlighted. Global searches of structural databases were performed using DALI (http://ekhidna.biocenter.helsinki.fi/dali_server). In these studies, the 3D structure of hCE1 (1MX5) was compared to all reported structures, and those demonstrating the greatest statistical significance (Z score) were ranked. Small molecule analysis was undertaken using the flexible alignment subroutine present within MOE 2011.10 software (Chemical Computing Group, Montreal, Canada).
3. RESULTS
3.1 Structural homology between AChE, BChE and hCE1
To assess the structural homology between CEs and ChEs, the x-ray coordinates for hCE1 (1MX5) were used to search the RCSB database using DALI. Over 1000 statistically significant matches were obtained with Z-scores ranging from 78.7 to 6.4. Structures ranked 2–71 were previously reported hCE1 coordinates and entry 72 was a rabbit liver CE. The entries for ChEs starting at #73 are presented in Table 1. As indicated, structural similarity between the CE and ChEs was highly significant, yielding Z-scores as high as 56. A graphical example of the overlays that were typically observed is shown in Figure 1. This demonstrates the homology between hCE1 (1MX5) and hAChE ((3LII); entry 213 in Table 1). All major domains within the proteins demonstrate considerable structural identity, including architecture that delineates the α,β-hydrolase fold, the catalytic gorges, as well as the juxtaposition and orientation of the catalytic amino acids. Indeed, the latter are almost completely super-imposable (Figure 1D). The only major differences occur within regions that form the entrances to the active site gorges.
Table 1.
Structural similarity between hCE1 and cholinesterases as demonstrated using DALI. Data are ranked based upon Z score, with only the highest scoring cholinesterase sequences listed.
| Rank | Name | Za | rmsd (Å) | Number of residues | Identity (%) | Description | Species |
|---|---|---|---|---|---|---|---|
| 1 | 1MX5 | 78.7 | 0 | 532 | 100 | Liver CE | Human |
| 73 | 2J4C | 56.7 | 1.9 | 524 | 34 | BChE | Human |
| 74 | 2WIL | 56 | 1.9 | 526 | 34 | BChE | Human |
| 75 | 2Y1K | 56.5 | 1.9 | 525 | 34 | BChE | Human |
| 76 | 2WIG | 56.2 | 1.9 | 526 | 34 | BChE | Human |
| 77 | 1P0P | 56.2 | 1.9 | 522 | 34 | BChE | Human |
| 78 | 2WIF | 56.1 | 1.9 | 527 | 34 | BChE | Human |
| 79 | 2ACK | 56.1 | 2.0 | 527 | 35 | AChE | Torpedo californica |
| 80 | 2ACE | 56.1 | 2.0 | 527 | 35 | AChE | Torpedo californica |
| 213 | 3LII | 53.4 | 2.1 | 534 | 34 | AChE | Human |
| 339 | 1B41 | 46.5 | 2.0 | 531 | 34 | AChE | Human |
| 342 | 1F8U | 46.4 | 2.0 | 531 | 34 | AChE | Human |
| 343 | 2X8B | 46.1 | 2.1 | 537 | 34 | AChE | Human |
Z-scores represent number of standard deviations from the mean, and values less than 2 are not considered statistically significant.
Figure 1.

An overlay of the x-ray structures of hCE1 and human AChE.
Panels A and B represent the same structure with the latter image representing a 90 degree rotation to the right. Panel A depicts the view looking down into the active site gorge, whereas panel B has been ‘sliced’ in the horizontal plane to allow visualization of the catalytic amino acids. Panel C represents an enlargement of the gorge entrance and D indicates the overlay of the catalytic serine, histidine and glutamic acid. The white arrows in image B represent areas where the structural overlap is reduced and corresponds to the domains that form the entrance to the active site gorge.
3.2 Inhibition of carboxylesterases by cholinesterase inhibitors
Given that hCE1 and the ChEs demonstrate considerable structural homology, we assessed the ability of panel of known AChE and BChE inhibitors to modulate CE activity. As indicated in Table 2, none of the clinically used ChE inhibitors was able to reduce hydrolysis of o-NPA. However, phenethylcymserine, bis(7)-tacrine and the acridine analogues, ACMA and DEA, were weak CE inhibitors, exhibiting IC50 values in the mid micromolar range. However, because carbamate-containing compounds can irreversibly inhibit esterases [3], we evaluated the ability of a selected panel of these molecules to inactivate the human CEs. All of the compounds, with the exception of donepezil, demonstrated activity toward hiCE (Table 3). Indeed for tolserine and rivastigmine, significant loss of enzyme activity was observed. In contrast, only these two molecules were capable of irreversibly inhibiting hCE1.
Table 2.
Inhibition of carboxylesterases by cholinesterase inhibitors.
| Inhibitor | IC50 (μM) | |||
|---|---|---|---|---|
| hCE1 | hiCE | AChE | BChE | |
| Benzil | 0.37 ± 0.04 | 0.33 ± 0.03 | >100 | >100 |
| Tolserine | >100 | >100 | 1.4 ± 0.1 | 53.1 ± 20.1 |
| Phenethylcymserine | 20.5 ± 10.4 | 5.6 ± 2.7 | 1.0 ± 0.2 | 0.51 ± 0.06 |
| Donepezil | >100 | >100 | 0.022 ± 0.005 | 7.6 ± 1.3 |
| Tacrine | >100 | >100 | 0.079 ± 0.002 | 0.034 ± 0.001 |
| Rivastigmine | >100 | >100 | 299 ± 24.6 | 280 ± 110 |
| Galantamine | >100 | >100 | 1.2 ± 0.4 | 21.7 ± 4.3 |
| Bis(7)-tacrine | 40.5 ± 22.0 | 23.7 ± 3.5 | 0.0011 ± 0.0001 | 0.033 ± 0.003 |
| Acridine | >100 | >100 | 252 ± 80.9 | 6.9 ± 1.4 |
| ACMA | 111 ± 54.5 | 28.4 ± 11.4 | 2.0 ± 0.2 | 2.5 ± 0.9 |
| DEA | 99.3 ± 32.0 | 41.6 ± 15.2 | 6.9 ± 0.7 | 2.4 ± 0.5 |
Table 3.
Irreversible inhibition of human CEs by cholinesterase inhibitors
| Inhibitor | Inhibitiona (%) | |
|---|---|---|
| hCE1 | hiCE | |
| Benzil | 0 | 0 |
| BNPP | 99.9 | 99.2 |
| Tolserine | 43.4 | 82.4 |
| Phenethylcymserine | 0 | 64.5 |
| Donepezil | 0 | 0 |
| Tacrine | 0 | 58.7 |
| Rivastigmine | 78.8 | 97.5 |
| Galantamine | 0 | 44.1 |
Indicates the amount of enzyme inhibition after preincubation with inhibitor for 1 hour. Inhibitor was removed by centrifugation through 10kDa cutoff filters and residual enzyme activity determined using o-nitrophenyl acetate as a substrate.
3.3 Molecular similarity of cholinesterase inhibitors that inhibit human CEs
Having established that selected ChE inhibitors could irreversibly inhibit human CEs, we examined the chemical structures of these compounds. As indicated in Figure 2, rivastigmine, phenethylcymserine and tolserine are structurally very similar, with all molecules maintaining the 3-methylphenyl ethyl(methyl)carbamate moiety. Following alignment of these inhibitors, a very high degree of structural similarity was observed for these molecules (Figure 2). This included an almost direct overlay of the carbamate moieties, as well as the maintenance of planar geometry for both groups adjacent to the carbamate nitrogen and oxygen atoms.
Figure 2.
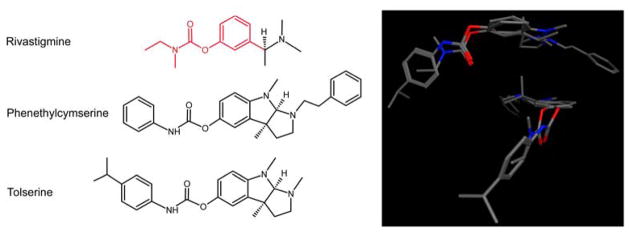
The structures of rivastigmine, phenethylcymserine and tolserine.
Right panel - The chemical structures are aligned to highlight their similarity, with the common atoms and bonds indicated in red (in rivastigmine).
Left panel - Two different orientations of molecular overlays are shown to demonstrate the significant overlap of the carbamate group and the co-planar nature of the substituents adjacent to this moiety.
4. DISCUSSION
Comparison of the X-ray coordinates of hCE1 with other structures present within the RCSB PDB database indicated numerous similar entries with exceedingly high statistically significance (Table 1). Indeed, the complete list of results generated by these analyses yielded over 1000 statistically significant matches from widely differing species. Furthermore, homology was observed in proteins with a diverse series of ‘esterase-like’ activities (e.g., triacylglycerol lipase, acetyl xylan esterase, S-formylglutathione hydrolase; data not shown). This demonstrates the ubiquitous nature of the α,β-hydrolase fold, and its application to the catalytic activity for numerous different substrates.
ChEs ranked as the first non-carboxylesterase class of proteins to yield the highest Z-scores in these analyses. However, the specificity of ChE inhibitors toward human CEs has not been assessed and due to the structural similarity that we observed (Table 1 and Figure 1), we hypothesized that these small molecules may demonstrate activity toward the latter. Therefore we obtained a panel of AChE and BChE inhibitors and assessed their activity toward CEs. We observed no inhibition of hCE1 or hiCE by clinically used AChE inhibitors in conventional substrate hydrolysis assays. However, we determined that carbamate-containing compounds could irreversibly inhibit CEs, leading to up to 97% loss of enzyme activity (Table 3). This was most apparent with rivastigmine, a ChE inhibitor that is currently used for the treatment of Alzheimer’s disease [9]. The irreversible mechanism of action of this inhibitor suggests that the catalytic serine within the CE active site attacks the carbonyl carbon atom of the carbamate moiety to yield a tetrahedral intermediate. This would decompose to release the [(dimethylamino)ethyl]phenol group (the alcohol) resulting in the carbamylation of the serine Oγ. This adduct is presumably relatively stable, limiting the next step in the hydrolytic reaction. Hence the apparent outcome for these experiments is the irreversible inhibition of the esterase.
Considering that numerous drugs are subject to activation and inactivation by CEs (e.g. Plavix, Tamiflu, Altace [2, 14, 15]) potentially, combination of rivastigmine with these esterified agents may result in reduced drug hydrolysis. More important however, is the fact that ChE inhibitors are given continuously, and hence prolonged suppression of CE activity might occur. In addition, due to the fact that we observed irreversible enzyme inhibition, either enzyme reactivation and/or new protein synthesis to replace the CE would be required. Presuming therefore that sufficient concentrations of the ChE inhibitors are present following administration, it is likely that modulation of esterified drug would occur. Studies to evaluate this are currently underway in plasma esterase-deficient mice.
5. CONCLUSIONS
We have determined that significant structural homology (similarity?) exists between the CEs and ChEs, and that inhibitors that target the latter have activity toward the former. While this is unlikely to have any obvious consequences in the case of mono-therapy (no endogenous substrates for CEs have been identified and plasma esterase deficient mice are viable and healthy), in the presence of an esterified agent, it is likely that reduced hydrolysis would be observed. Given that the ester function is commonly seen in drug molecules, principally to improve water solubility and bioavailability, potentially modulation of CE activity may impact a significant number of therapies. Hence drug:drug interactions should be monitored carefully in individuals who are receiving this combination of medications.
Acknowledgments
We thank Drs. Millard and Greig for the provision of reagents used in this research. This work was supported in part by an NIH Cancer Center Core Grant CA21765, and by the American Lebanese Syrian Associated Charities and St Jude Children’s Research Hospital (SJCRH).
Footnotes
Abbreviations: AChE – acetylcholinesterase; ACMA – 9-amino-6-chloro-2-methoxyacridine; BChE – butyrylcholinesterase; BNPP – bis(4-nitrophenyl)phosphate; CE – carboxylesterase; ChE – cholinesterase; DEA – 5,9-diamino-2-ethoxyacridine; hCE1 – human liver CE, CES1; hiCE human intestinal CE, CES2; o-NPA – o-nitrophenyl acetate.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ellman GL, Courtney KD, Anders V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 2.Grima M, Welsch C, Michel B, Barthelmebs M, Imbs JL. In vitro tissue potencies of converting enzyme inhibitors. Prodrug activation by kidney esterase. Hypertension. 1991;17:492–496. doi: 10.1161/01.hyp.17.4.492. [DOI] [PubMed] [Google Scholar]
- 3.Gupta RC, Dettbarn WD. Role of carboxylesterases in the prevention and potentiation of N-methylcarbamate toxicity. Chem Biol Interact. 1993;87:295–303. doi: 10.1016/0009-2797(93)90057-6. [DOI] [PubMed] [Google Scholar]
- 4.Hatfield MJ, Tsurkan L, Garrett M, Shaver T, Edwards CC, Hyatt JL, Hicks LD, Potter PM. Organ-specific carboxylesterase profiling identifies the small intestine and kidney as major contributors of activation of the anticancer prodrug CPT-11. Biochem Pharamcol. 2011;81:24–31. doi: 10.1016/j.bcp.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hatfield MJ, Tsurkan L, Hyatt JL, Yu X, Edwards CC, Hicks LD, Wadkins RM, Potter PM. Biochemical and molecular analysis of carboxylesterase-mediated hydrolysis of cocaine and heroin. Br J Pharmacol. 2010;160:1916–1928. doi: 10.1111/j.1476-5381.2010.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hyatt JL, Moak T, Hatfield JM, Tsurkan L, Edwards CC, Wierdl M, Danks MK, Wadkins RM, Potter PM. Selective inhibition of carboxylesterases by isatins, indole-2,3-diones. J Med Chem. 2007;50:1876–1885. doi: 10.1021/jm061471k. [DOI] [PubMed] [Google Scholar]
- 7.Khanna R, Morton CL, Danks MK, Potter PM. Proficient metabolism of CPT-11 by a human intestinal carboxylesterase. Cancer Res. 2000;60:4725–4728. [PubMed] [Google Scholar]
- 8.Morton CL, Potter PM. Comparison of Escherichia coli, Saccharomyces cerevisiae, Pichia pastoris, Spodoptera frugiperda and COS7 cells for recombinant gene expression: Application to a rabbit liver carboxylesterase. Mol Biotechnol. 2000;16:193–202. doi: 10.1385/MB:16:3:193. [DOI] [PubMed] [Google Scholar]
- 9.Munoz-Torrero D. Acetylcholinesterase inhibitors as disease-modifying therapies for Alzheimer’s disease. Curr Med Chem. 2008;15:2433–2455. doi: 10.2174/092986708785909067. [DOI] [PubMed] [Google Scholar]
- 10.Potter PM, Pawlik CA, Morton CL, Naeve CW, Danks MK. Isolation and partial characterization of a cDNA encoding a rabbit liver carboxylesterase that activates the prodrug Irinotecan (CPT-11) Cancer Res. 1998;52:2646–2651. [PubMed] [Google Scholar]
- 11.Redinbo MR, Bencharit S, Potter PM. Human carboxylesterase 1: from drug metabolism to drug discovery. Biochem Soc Trans. 2003;31:620–624. doi: 10.1042/bst0310620. [DOI] [PubMed] [Google Scholar]
- 12.Redinbo MR, Potter PM. Mammalian Carboxylesterases: From drug targets to protein therapeutics. Drug Discov Today. 2005;10:313–325. doi: 10.1016/S1359-6446(05)03383-0. [DOI] [PubMed] [Google Scholar]
- 13.Schwer H, Langmann T, Daig R, Becker A, Aslanidis C, Schmitz G. Molecular cloning and characterization of a novel putative carboxylesterase, present in human intestine and liver. Biochem Biophys Res Comm. 1997;233:117–120. doi: 10.1006/bbrc.1997.6413. [DOI] [PubMed] [Google Scholar]
- 14.Shi D, Yang J, Yang D, LeCluyse EL, Black C, You L, Akhlaghi F, Yan B. Anti-influenza prodrug oseltamivir is activated by carboxylesterase human carboxylesterase 1, and the activation is inhibited by antiplatelet agent clopidogrel. J Pharmacol Exp Ther. 2006;319:1477–1484. doi: 10.1124/jpet.106.111807. [DOI] [PubMed] [Google Scholar]
- 15.Tang M, Mukundan M, Yang J, Charpentier N, LeCluyse EL, Black C, Yang D, Shi D, Yan B. Antiplatelet agents aspirin and clopidogrel are hydrolyzed by distinct carboxylesterases, and clopidogrel is transesterificated in the presence of ethyl alcohol. J Pharmacol Exp Ther. 2006;319:1467–1476. doi: 10.1124/jpet.106.110577. [DOI] [PubMed] [Google Scholar]
- 16.Wadkins RM, Hyatt JL, Wei X, Yoon KJ, Wierdl M, Edwards CC, Morton CL, Obenauer JC, Damodaran K, Beroza P, Danks MK, Potter PM. Identification and characterization of novel benzil (diphenylethane-1,2-dione) analogues as inhibitors of mammalian carboxylesterases. J Med Chem. 2005;48:2905–2915. doi: 10.1021/jm049011j. [DOI] [PubMed] [Google Scholar]
- 17.Wadkins RM, Hyatt JL, Yoon KJ, Morton CL, Lee RE, Damodaran K, Beroza P, Danks MK, Potter PM. Identification of novel selective human intestinal carboxylesterase inhibitors for the amelioration of irinotecan-induced diarrhea: Synthesis, quantitative structure-activity relationship analysis, and biological activity. Mol Pharmacol. 2004;65:1336–1343. doi: 10.1124/mol.65.6.1336. [DOI] [PubMed] [Google Scholar]