

Abstract
Hepatitis C virus (HCV) is a major cause of liver cirrhosis and hepatocellular carcinoma. Here, we report that infection of hepatic cells by HCV stimulates NFκB-dependent production of thymic stromal lymphopoietin (TSLP). Hepatocyte-derived TSLP in turn conditions DCs to drive Th17 differentiation. The TSLP secreted by HCV-infected hepatoma cells is capable of activating human monocyte-derived DCs by upregulating the expression of CD40, CD86, CCL17, CCL22, and CCL20 which is activating marker of DCs. In addition, the production of key cytokines for Th17 differentiation, TGF-β, IL-6 and IL-21, is enhanced by human monocytes upon co-culture with HCV-infected cells. Importantly, the blockade of TSLP using neutralizing antibody prevented the activation and maturation of DCs as well as the production of Th17 differentiation cytokines. DCs conditioning by TSLP secreted from HCV-infected cells activated naïve CD4+ T lymphocytes, resulting in Th17 differentiation. Furthermore, we can detect substantial levels of hepatocyte TSLP in fibrotic liver tissue from chronic HCV patients. Thus, blockade of TSLP released by HCV-infected hepatocytes may suppress the induction/maintenance of hepatic Th17 responses and halt the progression of chronic liver disease to fibrosis and liver failure.
Conclusion
Hepatocyte-derived TSLP conditions DCs to drive Th17 differentiation. Treatment of TSLP neutralizing antibody in HCV-infected hepatocyte/DC coculture abrogates DC conditioning and thereby inhibits Th17 differentiation.
Introduction
Hepatitis C virus (HCV) is a serious worldwide health problem, with more than 170 million people infected globally. HCV establishes persistent infection in 70% of infected individuals, leading to chronic liver inflammation, fibrosis, and cirrhosis (1). The outcome of HCV infection is primarily dictated by the magnitude and character of T cell response to infection. CD4+ T cell responses play a critical role in the resolution of infection (2, 3), impaired HCV-specific CD4+ T cell responses are observed in chronic HCV (3, 4). However, it is not known how HCV impairs CD4+ T cell responses regarding the magnitude or alteration of differentiation of T cells and effector activity in the infected liver. Because of fenestrations in the liver sinusoidal enodothelial cells, liver parenchymal cells (hepatocytes) are not separated from the vascular compartment by a basal membrane, and consequently HCV-infected hepatocytes have the potential to directly interact with innate immune cells such as liver resident dendritic cells (DCs). As cells of the innate immune system play a pivotal role in inducing and shaping the character of adaptive immune responses, the encounter of HCV-infected hepatocytes with liver DCs are likely to affect the activation state and properties of DCs and thereby influence the quality and effector function of T cell responses to HCV.
Recently, IL-17-producing Th17 cells have been reported to trigger tissue inflammation and damage (5) and there is accumulating evidence that Th17 cells are important contributors to hepatic inflammation and liver cirrhosis (6, 7). During viral infection (8), IL-17 is produced by monocytes/DCs through recognition of viral PAMP such as TLR3 ligands (9). In addition to the ability of HCV to trigger the TLR3 pathway (10, 11), the increased number of Th17 cells appears to be associated with the severity of liver inflammation in chronic HCV patients, and treatment of infected patients with pegylated IFN-α and ribavirin reduced the level of Th17-related cytokines (ref). As one of crucial factors for Th17 differentiation, thymic stromal lymphopoietin (TSLP), a member of the common γ-chain cytokine, is capable of activating (conditioning) DCs, thereby stimulating naïve T cells to differentiate into Th2 cells (12). In addition, DCs treated with both TSLP and poly (I:C) activate naïve T cells and differentiate into Th2 and Th17 cells (9, 13). Thus, TSLP-activated DCs, which are known to be strong inducers of Th2 responses, can simultaneously induce Th17 cells under certain pathological conditions.
In this report, we demonstrate that the infection of hepatic cells in vitro by HCV triggers robust TSLP production and this HCV-induced production of TSLP is regulated in an NFκB-dependent manner. TSLP secreted by HCV-infected cells activates and conditions human monocyte-derived DCs to enhance the production of Th17 differentiating cytokines, TGF-β, IL-6 and IL-21, by the DCs. Moreover, the addition of TSLP neutralizing antibody to the coculture of monocytes/DCs with HCV-infected hepatocytes blocks the production of these cytokines. Consistent with these data, we find that the hepatocyte-derived TSLP is readily detected in liver biopsies from chronic HCV patients. Our studies suggest a novel role for the hepatocyte-derived TSLP in the generation of CD4+ Th17 effector T-cells through its capacity to condition DCs to drive CD4+ Th17 differentiation. The potential implications of these findings in the development of HCV-induced chronic progressive liver diseases are discussed.
Materials and Methods
Subject and Cell preparation
Human hepatoma cell lines, Huh 7.5.1 were maintained in DMEM with 10% FBS and penicillin/streptomycin (100 μg/ml). THP-1 cells purchased from ATCC were cultured in RPMI 1640 and supplements as recommended by ATCC. Liver biopsies and peripheral blood samples from chronic HCV or control patients were obtained Dr. Hugo Rosen (University of Colorado). Blood samples were also obtained from Virginia Blood Services. All information of age, gender, and HCV genotype are previously described (14, 15). For infection of cells with secreted HCV, Huh 7.5.1 permissive cells were seeded at 3 × 106 cells in a T75 plate for 24 hours. Cells were infected with 4 × 104 FFU (MOI of 0.01) of JFH-1 producing cell supernatant, and cultured for 10 days in DMEM-10% FCS media. Determination of infectivity was performed as previously described (16). To serve as a control, supernatant from JFH-1-infected hepatocytes (UV-JFH-1sup) were irradiated with 3.6 J/cm2 for 25 min at 254 nm using an UV cross-linker MT-10 (UVP, Upland, CA). JFH-1 was kindly provided by Dr. Wakita (Tokyo Metropolitan institute).
Real-time quantitative PCR
Total RNA was extracted with the RNeasy Mini kit (Sigma-Aldrich), and reverse-transcribed to cDNA using the SuperScript III (Invirogen). The real-time RT-PCR was performed on an ABI Prism sequence Detection System (Applied Biosystems). The primer sequences were as follows: hIL-17A (sense, 5′-GGTCCTCAGATTACTACAACCG-3′; antisense, 5′-GTTCCCATCAGCGTTGATGCAG-3′), hIL-17F (sense, 5′-CCATGTCACGTAACATCGAG-3′; antisense, 5′-GTTCCTACACTGGGCCTGTA-3′). The sense and antisense primer sequence for RORc, CCR6, TSLP, CCL17, CCL22, CCL20, GAPDH were synthesized as previously described(9, 17).
Western blot analysis
Cells were cultured in 6-well plates, and treated with JFH-1 as described in the figure legend. Western blot analysis was performed as previously described (17).
ELISA
Level of IL-17, TGF-β, IL-6, IL-21, and IL-12 (eBiosience), and TSLP (R&D System) in culture supernatants were quantified by ELISA kit, according to the manufacturer’s instructions.
Transfection and Luciferase assay
Huh 7.5.1 cells (5 × 105)were seeded into 6-well plates and transfected using the Mirus transfection reagent (Mirus Bio Corp.) with various plasmids (see figure legends for details), and pRL-TK was used as the normalization control. The cells were cultured for 30 hours and then infected with JFH-1 for 11 hours. Cells were harvested and lysed in 100 μl lysis buffer. Luciferase activity was measured as described (18).
Generation and activation of human monocyte-derived DCs
To obtain DCs, PBMCs were allowed to adhere at 5 × 106 cells/mL onto 6-well plates in RPMI-10 culture medium. After 24 hours of incubation, non-adherent cells were removed by stringent rinsing and adherent cells were cultured in DC culture medium supplemented with GM-CSF (50 ng/mL) and IL-4 (400 U/mL). On day 6, the culture consisted uniformly of CD11c+ CD14− HLA-DR+ CD86+ CD83− immature DCs. Cell differentiation was monitored by light microscopy, and the expression of cell surface molecules was analyzed by FACS after 6 days of culture. To determine the activation status, DCs were cultured in medium, supernatant from JFH-1-infected hepatocytes (JFH-1sup), rTSLP, or JFH-1sup plus anti-TSLP mAb for 24 hours and stained with PE-conjugated mouse antibodies to CD40, CD80 and CD86. The activation was analyzed on FACS.
DCs-CD4+ T cell co-culture and intracellular staining
Purified DCs were plated at 2 × 104 cells/well in 96 well plates and cultured in the presence of IL-1 (10 ng/mL), IL-6 (20 ng/mL), IL-23 (20 ng/mL), JFH-1sup, or JFH-1sup plus anti-TSLP mAb (10 μg/mL) for 24 hours. These DCs were cocultured with freshly isolated allogeneic CD4+ T cells (4 ×104 cells/well). After 7 days of culture, cells were harvested and stimulated with 0.1μg/ml PMA and 0.5 μg/mL ionomycin (Sigma-Aldrich) for 6 hours, with the last 4 hours in the presense of GolgiStop (BD Bioscience). Cells were stained for surface antigen CD4, fixed, and permeabilized using Cytofix/Cytoperm (BD Bioscience), followed by intracellular IL-17 and IFNγ staining. Flow cytometry was performed as previously described (19).
Confocal microscope analysis
Liver tissues from normal and chronic HCV patient were kindly obtained from Dr. Hugo R. Rosen. Frozen sections (5 mm thickness) of OCT-embedded tissues were treated with 0.3% triton X-100, blocked in horse and donkey serum, and incubated with anti-TSLP Ab followed by donkey A-546-anti-goat IgG Ab and A-488-cytokeratin mAb. Control goat IgG and A-488-IgG1 were used for staining. Sheep anti-TSLP Ab was purchased from R&D systems. Mouse anti-cytokeratin pan mAb (Clone C-11; Sigma) and biotinylated mouse anti-human collagen IV mAb (Cedarlane) followed by Alexa dye 555-conjugated streptavidin. A-546-conjugated donkey anti-goat Ab (Invitrogen) was absorbed with mouse and rat serum before use. Confocal images were captured on a Zeiss LSM-700 confocal microscope and analyzed by the software ZEN.
Statistical analysis
Student t test (two-tailed) was used for statistical analysis of differences between two groups. P values are depicted as *, P ≤ 0.05. All data were analyzed using Prism software (GraphPad Prism4).
Results
TSLP secretion by HCV-infected hepatoma cells and detection of TSLP-expressing hepatocytes from chronic HCV patients
Since TSLP plays a critical role in triggering inflammatory responses and promotes Th2 and Th17 differentiation in response to microbial infection (12), we examined whether HCV infection of hepatic cells stimulates TSLP production. To this end, we analyzed the impact of in vitro infection of Huh 7.5.1-derived cell lines with a replicating JFH-1 HCV strain. We first infected Huh 7.5.1 cells with HCV (JFH-1) virus for 10 days and infection was then confirmed by immunofluorescence through the expression of HCV core protein (Fig. 1a). We next examined the tempo of TSLP mRNA induction in Huh 7.5.1 cells following JFH-1sup (supernatant from JFH-1-infected hepatocytes) infection. TSLP message was first detected early in infection, from about 4 to 8 hours, and reached maximal levels at 12 hours p.i. (Fig. 1b). TSLP message also enhanced the TSLP protein release at 24 hours, which showed significantly higher fold increase of TSLP production by HCV-infected cells compared to control cells (Fig. 1c). In contrast, TSLP induction was significantly decreased in cells infected with UV-irradiated JFH-1sup (Fig. 1b, c). These results demonstrate that human TSLP is induced in hepatocytes by HCV infection.
Figure 1. HCV increases TSLP production in JFH-1-infected human hepatoma cells.
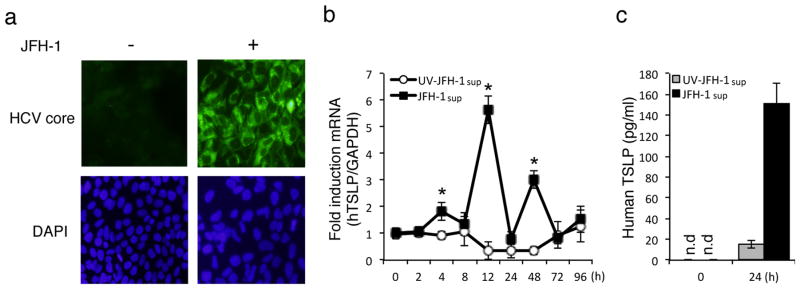
a) Huh7.5.1 cells infected with JFH-1 (MOI of 0.1) for 10 days and confirmed by immunofluorescence using antibodies; primary antibody was mouse anti-HCV core mAb, and secondary antibody was goat anti-mouse Alexa Fluor 488 antibody. DAPI was added to the staining to monitor the nuclear structure. b) Huh7.5.1 cells were inoculated by UV-irradiated JFH-1sup or JFH-1 (MOI of 0.1) for the indicated time course, and human TSLP mRNA levels were quantified by real-time PCR. TSLP mRNA levels were normalized with GAPDH (b) and TSLP protein levels were analyzed by ELISA (c). Data represent means ± SD of three independent experiments. Similar results were obtained for five independent experiments (b, c). n.d; not detected.
To determine if HCV infection of hepatocytes in situ within the infected liver stimulated TSLP production, we analyzed TSLP expression in liver tissues from chronic HCV patients. In keeping with our in vitro data on TSLP expression by HCV-infected hepatocytes (Fig. 1b, c), we detected significant upregulation of TSLP mRNA level in liver tissues from chronic HCV patients relative to those in healthy individuals (Fig. 2a). Of particular interest, we found that the expression of RORc, a key transcription factor in the Th17 cell lineage, was also significantly upregulated in liver tissue from chronic HCV patients (Fig. 2b). To precisely define the cellular source of TSLP, we carried out immunofluorescence staining of liver tissues from chronic HCV patients. As shown in Fig. 2c (panels P1 to P4), there was extensive fibrosis (indicated by collagen-red-staining) in the interlobular regions of the liver biopsies from HCV patients as well as intense cellular infiltration in the areas of fibrosis. As expected, in biopsies from individuals without chronic disease, there was staining of collagen reticulin fibers and minimal collagen deposition in liver stromal elements (Fig. 2c, N1 and N2, arrows). Cytokeratins are proteins of keratin-containing intermediate filaments found in the intracytoplasmic cytoskeleton of epithelial tissue. Human TSLP was found to be expressed by epithelial cells in the peripheral mucosal-associated lymphoid tissue, where it activates myeloid dendritic cells to induce a strong TH2 response in vivo and in vitro (20). Strikingly, a significant production of TSLP was found in the liver of HCV patients but not in those of normal individuals (Fig. 2d). TSLP staining was largely, if not exclusively, localized to hepatocytes since TSLP staining was found within hepatic lobules and colocalized with cytokeratin, a hepatocyte marker (arrows). Minimal TSLP staining was found in the fibrotic interlobular septa. In constrast, staining of normal and patient samples with control Ab showed little staining, indicating that the staining is specific for TSLP. Taken together, these results indicate that TSLP is indeed produced by hepatocytes in patients with chronic HCV infection. Furthermore, TSLP production in this tissue might be responsible for inducing the expression of Th17 differentiating cytokines and a transcription regulator, RORc, associated with CD4+ Th17 differentiation.
Figure 2. Increased TSLP is located in hepatocytes of liver tissues of HCV patients.

(a, b) Total RNA was extracted from liver tissues of healthy and chronic HCV patients. The mRNA expression for TSLP or RORc was quantified by real-time PCR. c) Fibrosis in HCV patient livers. Tissue sections (4 μm thickness) of normals (N1 and N2) and HCV patients (P1 to P4) were stained with Masson’s trichrome stain. Regions with collagen deposition in patients (red) were marked by asterisks (*) and small collagenous structures in normals were marked by arrows. Hp marks hepatocyte regions in the lobules. Liver histology from 2 normals and 4 patients are presented. Micrographs are captured at 200 X on a LSM700 confocal microscope and bars are 200 mm. *, fibrotic areas; Hp, hepatocytes. d) The confocal images of normal and chronic HCV patient samples are shown. Sections stained with control Ab showed low background staining (a). Low magnification images showed staining of TSLP in hepatocytes in patients but not in normal hepatocytes (b). Higher magnification showed the presence of TSLP staining of HCV-infected liver but minimal staining in normal liver (c). TSLP staining colocalizes with cytokeratin which stains hepatocytes (d). Arrows show the colocalized staining. The widths of the bars are in μm. A total of 3 normal and 5 HCV patient samples have been analyzed and the results are similar.
NFκB activation mediates TSLP gene expression in HCV-infected hepatoma cells
In host response to HCV infection, IPS-1 has been reported to localize in the mitochondria and play a critical role in the activation of IFN regulatory factor-3 (IRF-3) and NFκB. To understand the mechanism of TSLP induction by JFH-1-infected cells, we first assessed the ability of IPS-1 to trigger the TSLP promoter, which is located 4.0-kb upstream at the start of transcription (pGL3-b+hTSLP-full) in human Huh 7.5.1 cells. Overexpression of wild-type IPS-1 led to enhanced TSLP promoter activity following JFH-1sup infection (Fig. 3a). We next examined TSLP promoter induction by overexpression of wild-type IRF-3. No difference was found in the ability of IRF-3 to express TSLP in response to JFH-1sup (Fig. 3b). We also investigated the effect of overexpression of wild-type NFκB or a dominant-negative mutant of IκB kinase (IKKβ). Overexpression of the NFκB p65 subunit resulted in increased expression of the TSLP promoter in the absence of JFH-1sup infection. In addition, infection of the cells with JFH-1sup further enhanced expression (Fig. 3c). Conversely, overexpression of cDNA encoding a dominant-negative mutant of IKKβ inhibited JFH-1-mediated transcription in a dose-dependent fashion (Fig. 3d). The human TSLP gene promoter contains two NFκB binding sites, 3.8- and 0.2-kb upstream of the start of transcription (18). Single mutation in any of the two motifs caused notable decreases in TSLP expression. Furthermore, double mutations led to more profound decreases than a single mutation (Fig. 3e). We confirmed that NFκB was activated following infection with JFH-1sup. As expected, JFH-1 induced phosphorylation of NFκB in hepatoma-derived cells (Fig. 3f). NFκB is a ubiquitously expressed transcription factor known to mediate the expression of many inflammatory mediators including cytokines, adhesion molecules, chemokines, and growth factors. These results indicate that HCV-induced TSLP production occurs through the activation of NFκB, a crucial mediator in the innate immune pathway.
Figure 3. NFκB-dependent transcriptional activation of human TSLP by HCV-infected hepatoma cells.

a–e) Induction of human TSLP promoter in cells co-transfected with 1ug of wild-type human TSLP promoter plasmid or the indicated plasmids. At 30 h after transfection, cells were infected with JFH-1 virus (MOI of 0.1) for 11 h. Cell lysates were prepared and promoter activity was measured by luciferase activity. Huh7.5.1 cells were transiently transfected with control plasmid pGL3-basic, mixture of luciferase constructs containing only the human TSLP promoter plasmid (hTSLP-full), or containing a combination of hTSLP-full and expression vectors for IPS-1 expression plasmid (IPS-1) (0.05 or 0.5 μg) (a), IRF-3 expression plasmid (IRF-3) (10 or 50 ng) (b), NFκB expression plasmid (NFκB) (10 or 50 ng) (c), dominant-negative mutant of IκB plasmid (DN-IκB) (50 or 100 ng) (d), or promoter NFκB-site mutants (m1 = deletion of -3.2-kb site, m2 = deletion of -0.2 kb site) (1 μg) (e). Luciferase activity in the whole cell lysate was normalized to Renilla luciferase activity. Low doses are represented as + and high doses as ++. Data represent mean ± SD of triplicate data points of one experment and are representative of five independent experiments. f) Huh7.5.1 cells were infected with JFH-1sup (MOI of 0.1) for the indicated time course and the cell lysates were subject to immunoblotting by using phosphorylated NFκB or NFκB antibody.
TSLP released by HCV-infected cells activates DCs and produce factors crucial for Th17-differentiation
TSLP is a potent stimulator of DC activation to increase expression of MHC II, costimulatory molecules (i.e. CD40, CD80 and CD86), cytokines, and chemokines (i.e. CCL17, CCL22 CCR4+ T cell recruitment) (20) and CCL20 (i.e. recruitment for CCR6+ Th17 cells) (21). So, we examined the effect of TSLP secreted from JFH-1-infected hepatocytes on activating DCs by co-culturing monocyte-derived DCs with recombinant TSLP, JFH-1sup, JFH-1sup plus neutralizing anti-TSLP antibodies, or untreated control culture media. As shown in Fig. 4a, the expression of costimulatory molecules (i.e. CD40, CD86) was increased in DCs after exposure to JFH-1sup, to the level comparable to that of rTSLP-treated DCs. Addition of anti-TSLP neutralizing antibodies to JFH-1sup inhibited their ability to activate costimulatory molecules on DCs. We also wanted to know whether TSLP receptor is expressed these DCs and DCs activation blockable by anti-TSLP antibody. TSLP receptor express DCs and futher upregulated following culture in JFH-1sup. In contrast, TSLP receptor expression was decreased by neutralization of TSLP receptor (data not shown). In addition, CCL17, CCL22, and CCL20 were expressed at higher levels in JFH-1sup stimulated DCs than that in media control-treated DCs, and neutralization of TSLP suppressed chemokine production by these DCs (Fig. 4b). Taken together, these data demonstrated that TSLP produced by hepatocytes infected with HCV can support DC activation/maturation.
Figure 4. TSLP released by JFH-1-infected cells activates human monocyte-derived DCs by inducing the production of TGF-β, IL-6, and IL-21 cytokines.

a) FACS analysis of DCs after 24 h incubation with the indicated stimuli: rTSLP, JFH-1sup, JFH-1sup plus anti-TSLP mAb. Filled histogram represents staining for DCs cultured with media alone and open histogram represents staining for DCs cultured with indicated stimuli. The expression levels of DC activation markers CD40, CD80, and CD86 were determined by flow cytometry. b) The expression level of CCL17, CCL22, and CCL20 chemokines relative to GAPDH was determined by real time PCR. Data shown are the mean ± SD of three independent experiments (b) or one experiment representative of three independent experiments with similar results (a). Both the JFH-1-infected and non-infected HepG2 cells were co-cultured with THP-1 cells in the transwell system at a 1:1 ratio for the indicated time course in the presence or absence of anti-TSLP antibody (15 ng/ml). The levels of TGF-β, IL-6, IL-21, and IL-12 production were measured in the culture supernatant by ELISA (c). Data represent mean ± SD of three independent experiments. These results were reproducible from five independent experiments.
To determine whether HCV-infected hepatocyte-derived TSLP might also condition antigen presenting cells (e.g. monocytes, macrophages, DCs) to stimulate Th17 differentiation, we examined the capacity of mononuclear cells exposed to HCV-infected hepatic cells or their supernatants to release inflammatory cytokines, in particular, TGF-β, IL-6 and IL-21, which supports the differentiation and expansion of human Th17 cells. The production of these cytokines was determined from the co-culture of the human monocytic cell line, THP-1, with either JFH-1-infected or uninfected hepatoma cells using the transwell system. Notably, JFH-1-infected HepG2 cells stimulated a statistically significant increase in the secretion of TGF-β, IL-6, IL-21, but not IL-12, by THP-1 cells in a transwell membrane system (Fig. 4c). However, cultures of THP-1 cells alone produced low levels of TGF-β, IL-6, and IL-21 cytokines regardless of the presence of JFH-1sup (datanot shown). Importantly, the addition of anti-TSLP-neutralizing antibodies led to a decrease of Th17 specific cytokines (Fig. 4c). These results suggest that monocytes/DCs conditioned by TSLP secreted from HCV-infected hepatocytes produce Th17 differentiating cytokines which could support the induction of CD4+ Th17 responses.
DCs conditioned by TSLP released from HCV-infected cells promote CD4+ T cell differentiation into Th17 cells
Based on the role of IL-1, IL-6 and IL-21 production in Th17 polarization, we evaluated the effect of hepatocyte-derived TSLP on Th17 differentiation in co-culture of naïve T cells with DCs activated by IL-1/IL-6/IL-23, JFH-1sup, or JFH-1sup plus anti-TSLP antibodies. Following stimulation with PMA/ionomycin, the production of intracellular cytokines (IFN-γ, IL-17A) by CD4+ T cells was assessed using flow cytometry. As expected, IL-1/IL-6/IL-23-treated DCs, used as positive control, produced more IL-17 cells compared to control cells (5.09 ± 0.6% vs 0.91 ± 0.08%). Notably, the percentage of IL-17-producing cells increased after co-culture of CD4+ T cells with JFH-1sup treated DCs (4.65 ± 0.55%), which then significantly decreased upon the addition of anti-TSLP mAbs (1.21 ± 0.1%) (Fig. 5a, b). While there was no significantly difference in the percentage of IFN-γ production from JFH-1sup treated DCs precense or absence of anti-TSLP antibody (Fig. 5a, b). This result was further verified by the detection of IL-17 release using ELISA. The enhancement of IL-17-producing T cells by JFH-1sup treated DCs was significantly inhibited by neutralization of TSLP (Fig. 5c). It suggest sthat TSLP released from infected hepatocytes activates and conditions DCs to drive the differentiation of activated CD4+ T cells into Th17 cells.
Figure 5. DC conditioned by TSLP released from HCV-infected cells contributes to Th17 differentiation.

a) CD4+ T cells were co-cultured with monocyte-derived DCs in the presence or absence of JFH-1sup for 7 days. After 7 days of co-culture, T cells were restimulated with PMA and ionomycin for 6 h, with the last 4 h in the presence of GolgiStop, and production of IL-17 and IFNγ was determined by intracellular cytokine staining (a). b) Bar graphs show percentage of IL-17, IFNγ-producing cells, measured by Flow cytometry. c) IL-17A production was measured in the culture supernatant by ELISA. Data shown are the mean ± SD of three independent experiments (b, c) or one experiment representative of three independent experiments with similar results (a).
Addition of both TSLP and NS3/5 enhances the production of Th17 cytokines by CD4+ T cells from chronic HCV patients
To further examine the effect of TSLP on promoting Th17 differentiation during HCV infection, we assessed the capacity of Th17 cell generation by CD4+ T cells from PBMC with chronic HCV patient. As shown in Fig. 6a, there is a significant increase of Th17 lineage-specific transcription factor (i.e. RORc) and markers (i.e. CCR6 and CD161) which is required for CD4+ T cells from chronic HCV patients as compared to those in healthy individuals. We next determined the role of HCV-specific antigen in induction of Th17 CD4+ T cells. HCV NS3/5 proteins have been reported to induce Th17 response (22). Th1/Th17 cells differentiations were compared using intracellular staining of IFNγ and IL-17, respectively. The results indicated that Th17 cells were significantly increased in response to NS3/NS5 compared to normal control (5.18 ± 1.09% vs 1.21 ± 0.90%) (Fig. 6b, c). We also observed the higher percentage of IL-4-producing cells in addition to IL-17-producing cells, but not IL-10-producing cells (data not shown). In addition, NS3/5 treatment also resulted in significant induction of IL-17 production, while they reduced the production of IFN-γ (Fig. 6d). Collectively, these data indicate that IL-17-producing CD4+ T cells are induced during HCV infection.
Figure 6. Direct action of TSLP enhance IL-17 production by PBMC from HCV-infected patients.

(a) CD4+ T cells were isolated from PBMC of healthy and chronic HCV patients. Expression levels of mRNA encoding IL-17A, IL-17F, RORc, and CCR6 were quantified using real-time PCR. (b, c, d) CD4+ T cells were cocultured for 7 days by DCs activated with 5 μg/ml mixed-NS3/NS5 or medium alone. T cells were restimulated with PMA plus ionomycin after 7 days of coculture. Production of IL-17, and IFNγ was determined by intracellular cytokine staining (b). c) Bar graph show the percentage of IL-17-producing cells in total CD4+ cells. d) Cytokine production was measured in the culture supernatant by ELISA. Data shown are the mean ± SD of three independent experiments (c, d) or one experiment representative of three independent experiments with similar results (b). Purified CD4+ T cells from PBMC of HCV-infected patients were stimulated with 5 μg/ml mixed-NS3/NS5 or medium alone, either presense or absense of recombinant TSLP antibody (5 ng/ml). Cells were harvested after 72 hours culture. Expression levels of mRNA encoding IL-17A, IL-17F were quantified using real-time PCR (e, f). Production of IL-17 was measured in the culture supernatant using ELISA (g). Data represent mean ± SD of three independent experiments.
We next examined the possibillity that the direct action of TSLP on CD4+ T cells is involved the differentiation of Th17 cells. CD4+ T cells from PBMC of HCV-infected patients were stimulated NS3/NS5 in the presense or absense of TSLP for 3 days. Stimulation with TSLP or NS3/5 significantly enhanced IL-17 mRNA and protein compared to control cells cultured medium only (Fig. 6 e, g, f). Moreover, differentiation of IL-17 cells was increased following combined stimulation of TSLP and NS3/5 compared to either TSLP or NS3/5 alone. These results clearly indicate that TSLP is an important factor to differentiation of IL-17-producing CD4+ T cells during HCV infection and might play a role in the development of chronic liver diseases.
Discussion
In this report, we demonstrate that HCV infection of hepatocytes induces NFκB dependent TSLP gene expression and protein production. Furthermore, TSLP mRNA and protein was increased selectively in liver tissues from chronic HCV patients. Intriguingly, TSLP released from HCV-infected hepatocytes activates human monocyte-derived DCs and conditions DCs to support the polarization of CD4+ T cells toward Th17 cells. The blockade of TSLP action by neutralizing antibody suppresses differentiation of Th17 cells. These results suggest a novel mechanism to account for the infiltration of TH17 cells in the liver is likely as a result of the role of TSLP in promoting Th17 differenitation. However, it remains unclear whether hepatic TSLP accounts for facilitating the recruitment of Th17 infiltration in the infected liver. These results also raise the intriguing possibility that the crosstalk between HCV-infected hepatocytes and local (liver and/or liver draining lymph node) DCs may be a pivotal mechanism both in a defective antiviral (Th1) CD4+ T cell response and also in an enhanced expression of an injury-provoking (Th17) CD4+ T cell response. To our knowledge, our findings represent the first report identifying hepatic-secreted TSLP as a regulator of Th17 differentiation.
Although CD4+ T cells have been reported to be critical to the antiviral function of CD8+ T cells in chronic infection, failure of CD4+ T cell help is associated with the inability to clear HCV infection and CD4+ T cells responses in chronically infected HCV patients leans toward Th2 deviation and Treg cells. Based on the report that the frequency of IFN-γ-producing CD4+ T cells in the liver was significantly reduced compared to those in the peripheral blood, it is likely that the function of cells within the liver may be subject to be influenced by local immunoregulatory factor(s). Our studies in this report implicate that circulating CD4+ T cells from HCV-infected individuals are skewed toward Th2 and Th17 cell differentiation via secretion of TSLP from HCV-infected hepatocytes (Fig. 1). In addition, combination of TSLP with HCV proteins (i.e. NS3/5) increases Th17 differentiation (Fig. 6). Interestingly, other group showed that TGF-β and IL-10, which are induced by the HCV NS4 protein, suppress Th1 and Th17 responses in HCV-infected patients (23). Given that the above study used the NS4 protein alone, it is possible that this viral protein alone is able to dampen the immune response via production of IL-10, which, in turn counteracts Th17 responses. However, our study is based on the whole virus, JFH-1, containing all HCV proteins and thereby DCs in exposure to JFH-1 HCV virus produce TSLP. Moreover, the production of TSLP depends on TGF-β and has been shown to antagonize IL-10 production. Recent studies have reported that HCV-specific IL-17-producing CD8+ T cells are detectable in blood and liver of chronically HCV-infected patients (8, 24). In addition, IL-17-producing CD4+ T cells have also been shown to be present in chronic HCV-infected patients (22, 23, 25). These results suggest that HCV-specific IL-17-producing T cells are not limited to the CD4+ T cells alone, but also CD8+ T cells. Nevertheless, the molecular and cellular mechanisms underlying the generation of these Th17 cell responses in HCV infection were not elucidated. Our results suggest a mechanistic link between TSLP derived from HCV-infected hepatocytes and the infiltration of IL-17 producing Th17 T-cells into the HCV-infected liver. HCV-derived proteins play a role in inducing TGF-β, IL-6 and IL-21 production from monocyte-differentiated DCs (26). In terms of a potential relationship between TSLP and these cytokines, there is no any existing report for a direct effect of these cytokines in TSLP induction. However, it is worthwhile to point that IL-6 is well known to activate STAT3 and TSLP is also able to induce STAT3 activation (27). Thus, there is the convergence of intracellular pathways downstream of TSLP and IL-6 and thereby these cytokines act in similar ways leading to skew T cell responses towards Th17 cells. Our studies in this report demonstrate that HCV infection of hepatocytes induce TSLP production by these cells, resulting in the activation and maturation of DCs with increased expression of CCL17, CCL22, and CCL20 chemokines. Importantly, the blockade of TSLP action by neutralizing antibodies prevents DC activation/maturation conditioned by HCV-infected hepatocytes (Fig. 4). Thus, there may be at least two different pathways for triggering DC activation and maturation during viral infection: 1) direct sensing of viral PAMP by DCs, and 2) crosstalk between virus-infected cells and DCs. While plasmacytoid DCs are activated by direct sensing of HCV via TLR7 and produce Type 1 IFN (28), TSLP acts as a key molecule in linking HIV-exposed epithelial cells to activation of myeloid DCs and these activated myeloid DCs, in turn, promote CD4+ T cell proliferation and increase HIV replication (29).
Although Th17 cells are participating in inducing pro-inflmmatory responses during viral infection, antiviral activity of Th17 cells to control HCV replication appears to be limited. Rather, Th17 cells appear to play a role in the progression of liver fibrosis and cirrhosis (22, 25). Indeed, histological studies show extensive infiltration of Th17 cells in the area of severe hepatic damage. Based on the role of TSLP in inducing differentiation of CD4+ Th17 effector T-cells, we speculate a potential mechanism(s) for HCV-induced Th17 differentiation through several stepwise processes of crosstalk between virus-infected cells and local DCs. Liver-resident DCs are conditioned by TSLP released from HCV-infected hepatocytes to favor, either at the site of viral infection and/or in the draining lymph node, the differentiation of Th17 cells, which then home to liver damage sites and possibly contribute to disease progression by interacting with other immune cells and non-immune cells. This points out a critical biological role of TSLP secreted by HCV-infected hepatocytes in activating DCs which result in Th17 differentiation.
Future studies are necessary to elucidate whether Th17 cells promote liver damage and are directly involved in enhanced virus survival or if they are simply less capable than Th1 cells in the induction of antiviral responses. Nevertheless, our studies point out that blockade of TSLP might be a new strategy for the treatment of chronic HCV patients with severe liver diseases. As TSLP is known for its role in inducing inflammation, efforts to block TSLP is being investigated for its potential as a treatment for asthma. The availability of therapeutic agent(s) targeting TSLP could accelerate the translation of these findings into clinical practices such as treatment of HCV-associated chronic liver diseases.
In summary, our findings suggest that TSLP produced by HCV-infected hepatocytes can enhance the development of potential injury-provoking CD4+ Th17 effector T cells through the ability of cytokines to condition DCs to drive the differentiation of CD4+ T cells towards Th17 effector status. If, as we believe, CD4+ Th17 effector T cell responses in the HCV-infected liver are important regulators of liver injury, then TSLP might represent a novel therapeutic target in HCV-infected liver with the potential to limit tissue injury and possibly promote virus clearance.
Acknowledgments
We thank Susan Landes for excellent technical assistance, Dr. Thomas J. Braciale and Dr. Taeg S. Kim for critical discussion of the manuscript prior to submission, and members of the Hahn laboratory for helpful discussions throughout the course of this work. This work was supported by NIH Grants (AI098126 to YSH, U19AI066328 to HR, YSH; U19AI083024 to SJS).
References
- 1.Dustin LB, Rice CM. Flying under the radar: the immunobiology of hepatitis C. Annu Rev Immunol. 2007;25:71–99. doi: 10.1146/annurev.immunol.25.022106.141602. [DOI] [PubMed] [Google Scholar]
- 2.Gerlach JT, Diepolder HM, Jung MC, Gruener NH, Schraut WW, Zachoval R, Hoffmann R, et al. Recurrence of hepatitis C virus after loss of virus-specific CD4(+) T-cell response in acute hepatitis C. Gastroenterology. 1999;117:933–941. doi: 10.1016/s0016-5085(99)70353-7. [DOI] [PubMed] [Google Scholar]
- 3.Kaplan DE, Sugimoto K, Newton K, Valiga ME, Ikeda F, Aytaman A, Nunes FA, et al. Discordant role of CD4 T-cell response relative to neutralizing antibody and CD8 T-cell responses in acute hepatitis C. Gastroenterology. 2007;132:654–666. doi: 10.1053/j.gastro.2006.11.044. [DOI] [PubMed] [Google Scholar]
- 4.Smyk-Pearson S, Tester IA, Klarquist J, Palmer BE, Pawlotsky JM, Golden-Mason L, Rosen HR. Spontaneous recovery in acute human hepatitis C virus infection: functional T-cell thresholds and relative importance of CD4 help. J Virol. 2008;82:1827–1837. doi: 10.1128/JVI.01581-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E, et al. Syk-and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 6.Lemmers A, Moreno C, Gustot T, Marechal R, Degre D, Demetter P, de Nadai P, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49:646–657. doi: 10.1002/hep.22680. [DOI] [PubMed] [Google Scholar]
- 7.Rong G, Zhou Y, Xiong Y, Zhou L, Geng H, Jiang T, Zhu Y, et al. Imbalance between T helper type 17 and T regulatory cells in patients with primary biliary cirrhosis: the serum cytokine profile and peripheral cell population. Clin Exp Immunol. 2009;156:217–225. doi: 10.1111/j.1365-2249.2009.03898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foster RG, Golden-Mason L, Rutebemberwa A, Rosen HR. Interleukin (IL)-17/IL-22-producing T cells enriched within the liver of patients with chronic hepatitis C viral (HCV) infection. Dig Dis Sci. 57:381–389. doi: 10.1007/s10620-011-1997-z. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka J, Watanabe N, Kido M, Saga K, Akamatsu T, Nishio A, Chiba T. Human TSLP andTLR3 ligands promote differentiation of Th17 cells with a central memory phenotype under Th2-polarizing conditions. Clin Exp Allergy. 2009;39:89–100. doi: 10.1111/j.1365-2222.2008.03151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci U S A. 2002;99:15669–15674. doi: 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bigger CB, Guerra B, Brasky KM, Hubbard G, Beard MR, Luxon BA, Lemon SM, et al. Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J Virol. 2004;78:13779–13792. doi: 10.1128/JVI.78.24.13779-13792.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ziegler SF, Liu YJ. Thymic stromal lymphopoietin in normal and pathogenic T cell development and function. Nat Immunol. 2006;7:709–714. doi: 10.1038/ni1360. [DOI] [PubMed] [Google Scholar]
- 13.Kato A, Favoreto S, Jr, Avila PC, Schleimer RP. TLR3-and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol. 2007;179:1080–1087. doi: 10.4049/jimmunol.179.2.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tacke RS, Lee HC, Goh C, Courtney J, Polyak SJ, Rosen HR, Hahn YS. Myeloid suppressor cells induced by hepatitis C virus suppress T-cell responses through the production of reactive oxygen species. Hepatology. 55:343–353. doi: 10.1002/hep.24700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McMahan RH, Golden-Mason L, Nishimura MI, McMahon BJ, Kemper M, Allen TM, Gretch DR, et al. Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viralpersistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. J Clin Invest. 120:4546–4557. doi: 10.1172/JCI43127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellecave P, Cazenave C, Rumi J, Staedel C, Cosnefroy O, Andreola ML, Ventura M, et al. Inhibition of hepatitis C virus (HCV) RNA polymeraseby DNA aptamers: mechanism of inhibition of in vitro RNA synthesis and effect on HCV-infected cells. Antimicrob Agents Chemother. 2008;52:2097–2110. doi: 10.1128/AAC.01227-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee HC, Headley MB, Iseki M, Ikuta K, Ziegler SF. Cutting edge: Inhibition of NF-kappaB-mediated TSLP expression by retinoid X receptor. J Immunol. 2008;181:5189–5193. doi: 10.4049/jimmunol.181.8.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HC, Ziegler SF. Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFkappaB. Proc Natl Acad Sci U S A. 2007;104:914–919. doi: 10.1073/pnas.0607305104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hall CH, Kassel R, Tacke RS, Hahn YS. HCV+ hepatocytes induce human regulatory CD4+ T cells through the production of TGF-beta. PLoS One. 5:e12154. doi: 10.1371/journal.pone.0012154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, Gilliet M, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3:673–680. doi: 10.1038/ni805. [DOI] [PubMed] [Google Scholar]
- 21.Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, et al. Preferential recruitment of CCR6-expressing Th17 cellsto inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. 2007;204:2803–2812. doi: 10.1084/jem.20071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang Q, Wang YK, Zhao Q, Wang CZ, Hu YZ, Wu BY. Th17 cells are increased with severity of liver inflammation in patients with chronic hepatitis C. J Gastroenterol Hepatol. doi: 10.1111/j.1440-1746.2011.06782.x. [DOI] [PubMed] [Google Scholar]
- 23.Rowan AG, Fletcher JM, Ryan EJ, Moran B, Hegarty JE, O’Farrelly C, Mills KH. Hepatitis C virus-specific Th17 cells are suppressed by virus-induced TGF-beta. J Immunol. 2008;181:4485–4494. doi: 10.4049/jimmunol.181.7.4485. [DOI] [PubMed] [Google Scholar]
- 24.Grafmueller S, Billerbeck E, Blum HE, Neumann-Haefelin C, Thimme R. Differential antigen specificity of hepatitis C virus-specific interleukin 17-and interferon gamma-producing CD8(+) T cells during chronic infection. J Infect Dis. 205:1142–1146. doi: 10.1093/infdis/jis018. [DOI] [PubMed] [Google Scholar]
- 25.Zhang JY, Zhang Z, Lin F, Zou ZS, Xu RN, Jin L, Fu JL, et al. Interleukin-17-producing CD4(+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology. 51:81–91. doi: 10.1002/hep.23273. [DOI] [PubMed] [Google Scholar]
- 26.Tacke RS, Tosello-Trampont A, Nguyen V, Mullins DW, Hahn YS. Extracellular hepatitis C virus core protein activates STAT3 in human monocytes/macrophages/dendritic cells via an IL-6 autocrine pathway. J Biol Chem. 286:10847–10855. doi: 10.1074/jbc.M110.217653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isaksen DE, Baumann H, Zhou B, Nivollet S, Farr AG, Levin SD, Ziegler SF. Uncoupling of proliferation and Stat5 activation in thymic stromal lymphopoietin-mediated signal transduction. J Immunol. 2002;168:3288–3294. doi: 10.4049/jimmunol.168.7.3288. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi K, Asabe S, Wieland S, Garaigorta U, Gastaminza P, Isogawa M, Chisari FV. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci U S A. 107:7431–7436. doi: 10.1073/pnas.1002301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fontenot D, He H, Hanabuchi S, Nehete PN, Zhang M, Chang M, Nehete B, et al. TSLP production by epithelial cells exposed to immunodeficiency virus triggers DC-mediatedmucosal infection of CD4+ T cells. Proc Natl Acad Sci U S A. 2009;106:16776–16781. doi: 10.1073/pnas.0907347106. [DOI] [PMC free article] [PubMed] [Google Scholar]