

Abstract
Background and Purpose
Bone marrow-derived cells (BMDCs) home to vascular endothelial growth factor (VEGF)-induced brain angiogenic foci, and VEGF induces cerebrovascular dysplasia in adult endoglin heterozygous (Eng+/−) mice. We hypothesized that Eng+/− BMDCs cause cerebrovascular dysplasia in the adult mouse after VEGF stimulation.
Methods
BM transplantation was performed using adult wild-type (WT) and Eng+/− mice as donors/recipients. An adeno-associated viral vector expressing VEGF (AAV-VEGF) was injected into the basal ganglia 4 weeks after transplantation. Vascular density, dysplasia index (vessels >15 μm/100 vessels), and BMDCs in the angiogenic foci were analyzed.
Results
The dysplasia index of WT/Eng+/− BM mice was higher than WT/WT BM mice (p<0.001) and was similar to Eng+/−/Eng+/− BM mice (p=0.2). Dysplasia in Eng+/− mice was partially rescued by WT BM (p<0.001). WT/WT BM and WT/Eng+/− BM mice had similar numbers of BMDCs in the angiogenic foci (p=0.4), most of which were CD68+. Eng+/− monocytes/macrophages expressed less matrix metalloproteinase-9 and Notch1.
Conclusions
ENG-deficient BMDCs are sufficient for VEGF to induce vascular dysplasia in the adult mouse brain. Our data support a previously unrecognized role of BM in the development of cerebrovascular malformations.
Keywords: arteriovenous malformation, adult mouse, brain angiogenesis
Introduction
Mutations in endoglin cause Hereditary Hemorrhagic Telangiectasia 1 (HHT1). Telangiectases and arteriovenous malformations (AVM) have been viewed as a disorder of the extant endothelium.1 VEGF induced cerebrovascular dysplasia in adult Eng+/− mice;2 the majority of BMDCs in the angiogenic foci were monocytes/macrophages (Mø),3 which contribute to vascular repair and angiogenesis.4 We hypothesized that Eng-deficiency in BMDCs causes cerebrovascular abnormalities in mice after VEGF stimulation.
Methods
After institutional approval, the design and groups listed in Supplemental Figures S1 and S2, and methods described in the online-only Data Supplement were used.
Results
AAV-VEGF induced brain angiogenesis in all groups and caused abnormal cerebrovascular morphology in mice with Eng+/− BM (Figure 1). Vascular densities (mean±SD) were: 820±153 (WT/WT BM), 720±150 (Eng+/−/WT BM), 653±120 (WT/Eng+/− BM), and 674±76 vessels/mm2 (Eng+/−/Eng+/− BM). Mice carrying Eng+/− somatic or BM cells showed a trend towards lower vascular density compared to WT/WT BM mice (p=0.06, Figure1B). WT/Eng+/− BM mice had more than five-fold greater dysplasia index than WT/WT BM mice (1.7±0.3 vs. 0.3±0.3, p<0.001, Figure 1C), comparable with the dysplasia index of Eng+//Eng+/− BM mice (1.9±0.4, p=0.2). Transplantation of WT BM to Eng+/− mice partially rescued dysplasia (p<0.001, Figure 1C).
Figure 1. VEGF induced cerebrovascular dysplasia in mice with Eng+/− BM.

(A) Representative images of VEGF-induced brain angiogenic foci. Arrows indicate dysplastic vessels. Scale bar: 50μm. Quantifications of (B) vascular density and (C) dysplasia index. *: p<0.001 compared to WT/WT BM group. #: p<0.001 compared to Eng+/−/WT BM group. Data: mean±SD. n=6 per group.
Using EGFP expressing donors, WT/WT BM and WT/Eng+/− BM mice had similar BMDC counts in the angiogenic foci (400±125 vs. 339±112/mm2, p=0.4) (Supplemental Figures S3A and S3B). The majority BMDCs was CD68+ (WT/WT BM: 67%±8 vs WT/Eng+/− BM: 64%±10, p = 0.6) (Figures 2A and 2C, Supplemental Figure S3C). About 7% of BMDCs in both groups were CD31+ endothelial cells (Figure 2B and 2D).
Figure 2. Eng-deficiency did not alter BMDC homing ability.

(A) & (C) Most of recruited GFP+ BMDCs were CD68+ Mø (arrows). (B) & (D) Few GFP+ BMDCs were CD31+ ECs (arrows). Scale bars: 50μm in (A), 20μm in (B). Data: mean±SD. n=6 per group.
BM-derived Mø from WT and Eng+/− mice were cultured and treated with 4 doses of VEGF (0, 10, 50, and 100ng/ml) for 18 hours. Compared to WT, Eng expression was 50% lower in Eng+/− Mø (Supplemental Figure S4A). The presence of both Vegfr1/Flt1 and Vegfr2/Flk1/Kdr indicate that Mø can be stimulated by VEGF (Supplemental Figure S4B and C). Mmp9 was up-regulated in WT but not Eng+/− cells at 50ng/ml of VEGF (p=0.003, Figure 3A). Notch1 expression in Eng+/− Mø decreased at 100ng/ml VEGF treatment compared to WT (p<0.001, Figure 3B).
Figure 3. Mmp9 and Notch1 expression were reduced in Eng+/− monocytes/macrophages after VEGF stimulation.
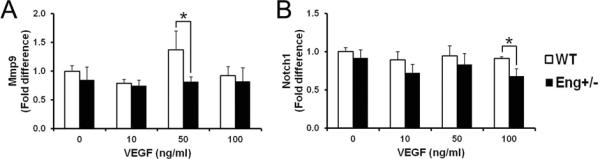
Quantification of (A) Mmp9 and (B) Notch1 expression. Expression levels are relative to that of WT-untreated cells. Data: mean±SD from three independent experiments (n=3 per group). *: p<0.05.
Discussion
This is the first demonstration that Eng-haploinsufficiency in BMDCs was sufficient to cause cerebrovascular dysplasia in the adult mouse after angiogenic stimulation. The abnormal angiogenic response was associated with altered expression of angiogenesis-related genes in Mø. These findings are consistent with prior work in Eng+/− myocardial infarction mice showing that transfusion of normal, but not HHT1, human mononuclear cells (MNCs) rescued the defect.4
TGF-β, VEGF, and Notch pathways act either synergistically with or antagonistically against each other during angiogenesis in a context-dependent manner.5 Notch signaling in Mø plays a critical role in angiogenesis and repair. Abrogation of monocytic Notch1 adversely affected repair after myocardial injury.6 Conditional deletion of Mø Notch1 caused abnormal anastomosis between angiogenic sprouts.7 Further study is needed to examine whether reduced Notch1 signaling in Eng+/− Mø contributes to a dysplastic phenotype.
VEGF dose-dependent effect on Mø depends on culture conditions. Chemotactic response of human Mø to VEGF peaked at 12ng/ml and decreased after 40ng/ml with 2-hour incubation.8 We found that 50ng/ml VEGF up-regulated Mmp9 in murine Mø, whereas neither 10 nor 100ng/ml had any effect. Possible explanations are: (1) human cells response to VEGF differently from mouse cells; and (2) various VEGF doses differentially trigger various signaling pathways to regulate diverse monocytic functions. Notch1 is induced by VEGF in arterial ECs.5 However, its expression in mouse Mø was not affected by VEGF (10–100ng/ml) in our study, possibly because only a subpopulation of Mø expresses Notch1 during angiogenesis.7
Growth factors and cytokines produced by BMDCs can affect local angiogenesis via systemic signaling. We showed that the mobilization of Mmp9−/− BMDCs into the circulation in response to VEGF was reduced, which resulted in less BMDC homing and brain angiogenesis.9 VEGF may affect macrophage polarization by effects on Notch signaling.10 Further studies should address the indirect/systemic effects of endoglin-deficiency on the BMDC function and the effect VEGF on Mø polarization.
Eng-deficiency in EC precursors may also play a role and deserves further study. In tumors, very few EC precursors are capable of triggering the angiogenic switch.11 Further, only a small number of homozygously Eng-deleted ECs (~ 1%) was sufficient to induce macroscopic cerebrovascular dysplasia after VEGF stimulation.12
In summary, one or more subpopulations of Eng+/− BMDCs are sufficient to induce an abnormal vascular response to brain angiogenic stimulation. Highly relevant to HHT1, it may be possible to envision development of a rescue strategy using BM transplantation therapy. The role of BMDCs in sporadic brain AVM needs further study, as Mø are associated with the lesion13 and EC precursors incorporate into the abnormal vascular structures.14 Consideration should also be given to the role of endoglin in other cerebrovascular diseases such as stroke.
Supplementary Material
Acknowledgements
We thank Jeffrey Nelson for statistical consultation, Voltaire Gungab for manuscript preparation, and UCSF BAVM project members (http://avm.ucsf.edu) for support.
Sources of Funding Supported by grants from National Institutes of Health (R01NS027713 to W.L.Y., R21NS070153 to H.S., and P01NS044155 to W.L.Y., H.S.), American Heart Association (SDG0535018N to H.S.), Leslie Munzer Foundation (H.S.); Aneurysm and AVM Foundation (H.S.); and Michael Ryan Zodda Foundation (W.L.Y., J.P.S.).
Footnotes
Disclosures None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mahmoud M, Allinson KR, Zhai Z, Oakenfull R, Ghandi P, Adams RH, et al. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ Res. 2010;106:1425–1433. doi: 10.1161/CIRCRESAHA.109.211037. [DOI] [PubMed] [Google Scholar]
- 2.Hao Q, Zhu Y, Su H, Shen F, Yang GY, Kim H, et al. VEGF induces more severe cerebrovascular dysplasia in Endoglin+/− than in Alk1+/− mice. Transl Stroke Res. 2010;1:197–201. doi: 10.1007/s12975-010-0020-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hao Q, Liu J, Pappu R, Su H, Rola R, Gabriel RA, et al. Contribution of bone marrow-derived cells associated with brain angiogenesis is primarily through leucocytes and macrophages. Arterioscler Thromb Vasc Biol. 2008;28:2151–2157. doi: 10.1161/ATVBAHA.108.176297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Laake LW, van den Driesche S, Post S, Feijen A, Jansen MA, Driessens MH, et al. Endoglin has a crucial role in blood cell-mediated vascular repair. Circulation. 2006;114:2288–2297. doi: 10.1161/CIRCULATIONAHA.106.639161. [DOI] [PubMed] [Google Scholar]
- 5.Holderfield MT, Hughes CC. Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-beta in vascular morphogenesis. Circ Res. 2008;102:637–652. doi: 10.1161/CIRCRESAHA.107.167171. [DOI] [PubMed] [Google Scholar]
- 6.Li F, Lan Y, Wang Y, Wang J, Yang G, Meng F, et al. Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev Cell. 2011;20:291–302. doi: 10.1016/j.devcel.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 7.Outtz HH, Tattersall IW, Kofler NM, Steinbach N, Kitajewski J. Notch1 controls macrophage recruitment and Notch signaling is activated at sites of endothelial cell anastomosis during retinal angiogenesis in mice. Blood. 2011;118:3436–3439. doi: 10.1182/blood-2010-12-327015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sawano A, Iwai S, Sakurai Y, Ito M, Shitara K, Nakahata T, et al. Flt-1, vascular endothelial growth factor receptor 1, is a novel cell surface marker for the lineage of monocytemacrophages in humans. Blood. 2001;97:785–791. doi: 10.1182/blood.v97.3.785. [DOI] [PubMed] [Google Scholar]
- 9.Hao Q, Su H, Palmer D, Sun B, Gao P, Yang GY, et al. Bone marrow-derived cells contribute to vascular endothelial growth factor-induced angiogenesis in the adult mouse brain by supplying matrix metalloproteinase-9. Stroke. 2011;42:453–458. doi: 10.1161/STROKEAHA.110.596452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY, et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 2010;70:4840–4849. doi: 10.1158/0008-5472.CAN-10-0269. [DOI] [PubMed] [Google Scholar]
- 11.Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319:195–198. doi: 10.1126/science.1150224. [DOI] [PubMed] [Google Scholar]
- 12.Choi EJ, Walker EJ, Shen F, Oh SP, Arthur HM, Young WL, et al. Minimal homozygous endothelial deletion of Eng with VEGF stimulation is sufficient to cause cerebrovascular dysplasia in the adult mouse. Cerebrovasc Dis. 2012;33:540–547. doi: 10.1159/000337762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, Zhu W, Bollen AW, Lawton MT, Barbaro NM, Dowd CF, et al. Evidence of inflammatory cell involvement in brain arteriovenous malformations. Neurosurgery. 2008;62:1340–1349. doi: 10.1227/01.neu.0000333306.64683.b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao P, Chen Y, Lawton MT, Barbaro NM, Yang GY, Su H, et al. Evidence of endothelial progenitor cells in the human brain and spinal cord arteriovenous malformations. Neurosurgery. 2010;67:1029–1035. doi: 10.1227/NEU.0b013e3181ecc49e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.