

Abstract
Hyperoxia contributes to the development of bronchopulmonary dysplasia (BPD) in premature infants. New BPD is characterized as having alveolar simplification. We reported previously that aryl hydrocarbon receptor (AhR) deficiency increased susceptibility to hyperoxic lung injury in adult mice, and this was associated with decreased expression of cytochrome P450 1A enzymes and increased lung inflammation. Whether AhR protects newborn mice against hyperoxia-induced alveolar simplification is unknown. Thus, we tested the hypothesis that decreased activation of the pulmonary AhR augments hyperoxia-induced alveolar simplification and lung inflammation in newborn mice. Experimental groups included one-day old wild type (WT) and AhR dysfunctional (AhRd) mice exposed to 21% O2 (air) or 85% O2 (hyperoxia) for 14 d. Exposure of newborn WT mice to hyperoxia resulted in increased protein, enzyme and mRNA expression of the AhR-regulated lung cytochrome P450 1A1, NAD(P)H quinone oxidoreductase-1, and microsomal glutathione S-transferase 1 enzymes, suggesting that hyperoxia increases activation of the pulmonary AhR. On the other hand, in the AhRd mice, hyperoxia induced the AhR-regulated enzymes to a lesser extent probably due to the dysfunctional AhR in these mice. Alveolar simplification and lung inflammation was increased in mice exposed to hyperoxia compared to those exposed to air, and AhRd mice were more susceptible to hyperoxia-induced alveolar simplification and lung inflammation compared to WT mice. These findings suggest that decreased activation of the pulmonary AhR in newborn AhRd mice augments hyperoxia-induced alveolar simplification and lung inflammation in these mice.
Keywords: Aryl hydrocarbon receptor, hyperoxia, alveolar simplification, lung inflammation
Introduction
Bronchopulmonary dysplasia (BPD) is a chronic lung disease whose primary structural and functional defect is alveolar simplification (Husain et al., 1998; Jobe, 1999). Despite significant improvements in neonatal intensive care management of premature infants, BPD remains the most prevalent complication in these patients affecting approximately 52% of the extremely low birth weight infants (< 1000 g birthweight) (Natarajan et al., 2012). Infants developing BPD are more likely to have long-term pulmonary problems, increased re-hospitalizations during the first year of life, and abnormal neurodevelopment compared to infants of similar birth weights and gestation that do not develop BPD (Short et al., 2003; Fanaroff et al., 2007). Thus, there is an urgent need for improved therapies in the prevention and treatment of BPD.
Supplemental oxygen is commonly administered as an important and life-saving measure in patients with impaired lung function. Although delivery of enriched oxygen relieves the immediate life-threatening consequences of hypoxemia, it may also exacerbate lung injury (Thiel et al., 2005). Excessive oxygen exposure and lung stretching leads to increased reactive oxygen species (ROS) production and expression of proinflammatory cytokines (Jobe et al., 2008). ROS react with nearby molecules (e.g., protein, lipids, DNA, and RNA) and modify their structure and function (Bhandari, 2010), and alter signal transduction pathways all of which frequently results in chronic pulmonary toxicity such as BPD. In addition, the antioxidant defense system develops late in gestation, making preterm neonates highly susceptible to oxidative stress (Vina et al., 1995; Asikainen and White, 2005). Evidence implicates oxidative stress (Bhandari, 2010; Saugstad, 2010) and inflammation (Wright and Kirpalani, 2011) as major contributors to the development of BPD and its sequelae. However, the molecular mechanisms by which oxidative stress and inflammation causes BPD remain poorly understood.
The aryl hydrocarbon receptor (AhR) is a member of basic-helix-loop-helix/PER-ARNT-SIM family of transcriptional regulators (Burbach et al., 1992). The AhR is expressed in all mouse tissues (Abbott et al., 1995), and in humans, AhR is highly expressed in the lungs, thymus, kidney, and liver (Tirona and Kim, 2005). AhR activation results in the translocation of the cytosolic AhR to the nucleus, where it dimerizes with the AhR nuclear translocator to form a heterodimeric transcription factor. The heterodimeric transcription factor activates the transcription of many phase I and phase II detoxification enzymes such as cytochrome P450 (CYP) 1A1, CYP1A2, glutathione S-transferase-α (GST-α), NAD(P)H quinone oxidoreductase-1 (NQO1), UDP glucuronosyl transferase (UDPGT), and aldehyde dehydrogenase (ALDH), which are encoded by the Ah gene locus (Rushmore et al., 1990; Favreau and Pickett, 1991; Emi et al., 1996). AhR is of particular interest to toxicologists and extensive research has been conducted on its role in the bioactivation of polycyclic and aromatic hydrocarbons leading to carcinogenesis (Nebert et al., 2004). Transgenic mice with AhR deficiencies have provided insight into the potential role(s) that AhR might play in normal physiological homeostasis (Bock and Kohle, 2009; Fujii-Kuriyama and Kawajiri, 2010). We reported earlier that adult mice deficient in AhR are more susceptible to hyperoxic lung injury compared to wild type controls and this phenomenon was associated with marked decreases in the expression of pulmonary and hepatic CYP1A subfamily of enzymes that have been reported to detoxify lipid hydroperoxides generated by reactive oxygen species (ROS) (Couroucli et al., 2002; Jiang et al., 2004). Recently, the AhR has been shown to attenuate tobacco smoke-induced inflammation in the lungs (Thatcher et al., 2007; Baglole et al., 2008), suggesting that AhR is a suppressor of lung inflammation. However, whether AhR attenuates hyperoxia-induced inflammation and alveolar simplification in the newborn lungs are unknown, and the current study was done to address this gap. Hence, the objective of our study was to elucidate the mechanistic role of AhR in hyperoxia-induced alveolar simplification and lung inflammation in newborn mice. We pursued our objective by testing the hypothesis that decreased activation of the pulmonary AhR augments hyperoxia-induced alveolar simplification and lung inflammation in newborn mice.
Materials and Methods
Animals
This study was approved and conducted in strict accordance with the federal guidelines for the humane care and use of laboratory animals by the Institutional Animal Care and Use Committee of Baylor College of Medicine (Protocol number: AN-5631). C57BL6/J wild type (WT) and aryl hydrocarbon dysfunctional B6.D2N-Ahrd/J (AhRd) mice were obtained from Charles River laboratories (Wilmington) and Jackson laboratories (Bar Harbor, ME), respectively. Dr. Daniel Nebert (University of Cincinnati, Cincinnati, OH) initially backcrossed Ahrd allele from DBA/2N onto C57BL/6N via a backcross-intercross breeding scheme and transferred this congenic to Dr. Alan Poland (University of Wisconsin, Madison, WI) at generation N13, who then backcrossed the Ahrd allele onto C57BL/6J, again via a backcross-intercross breeding scheme. The resulting homozygotes at or beyond generation N17 were maintained at the Jackson laboratory by sibling intercross. The AhR dysfunction in AhRd mice is due to decreased affinity of the AhRd receptor for its ligand. There are 10 nucleotide differences in the coding regions between the AhRb allele present in WT mice and the AhRd allele present in AhRd mice. The structural changes in the AhRd receptor associated with these nucleotide differences is thought to be responsible for the differential agonist affinity between the AhRb and AhRd receptors (Chang et al., 1993). We maintained active colonies of WT and AhRd mice by breeding them in the animal facility at Texas Children’s Hospital’s Feigin Center. Time pregnant WT and AhRd mice raised in our animal facility were used for the experiments.
Exposure
Within 12 h of birth, pups from multiple litters were pooled before being randomly and equally redistributed to the dams, following which they were immediately exposed to either 21% O2 (air) or 85% O2 (hyperoxia) for 14 d as described earlier (Park et al., 2007). The dams were rotated between air- and hyperoxia-exposed litters every 24 h to prevent oxygen toxicity in the dams and to eliminate maternal effects between the groups. Oxygen exposures were conducted in Plexiglas chambers, into which O2 was delivered through an oxygen blender to achieve a constant level of 85% O2. Animals were monitored every 12 h for evidence of adverse lung symptoms or mortality.
Analyses of the pulmonary AhR activation
It is reported that functional activation of the AhR results in the expression of many phase I and II enzymes. So, we determined the functional activation of the pulmonary AhR by analyzing the expression of pulmonary CYP1A1 (phase I), and NQO1 and microsomal glutathione S-transferase 1 (MGST1) (phase II) enzymes.
Lung tissue preparations for analyses of the AhR activation
Following exposure, animals were euthanized with i.p. injections of 200 mg/kg of sodium pentobarbital and their lungs (n=6/group) were stored at − 80°C for isolation of total RNA. The lungs from a separate set of animals (n=10/group) were snap frozen in liquid nitrogen for subsequent isolation of nuclear and cytosolic proteins.
Preparation of nuclear and cytosolic protein
A mortar and pestle was used to homogenize the lung tissue in a buffer containing 50mM Tris-HCL (pH 7.5), 0.5M KCL, 1M MgCL, and 0.5M EDTA. The homogenate was centrifuged at 2400 g for 5 min at 4°C. The supernatant (cytoplasmic fraction) was stored at − 80°C. The pellet was resuspended in a lysis buffer containing 50mM Tris-HCL (pH 7.5), 2.1M NaCL, 1M MgCL, 0.5M EDTA, and 25% sucrose, incubated on ice for 20 min, and centrifuged at 19000 g at 4°C for 5 min. The resulting supernatant (nuclear fraction) was stored at − 80°C until further use.
Enzyme assays
CYP1A1 and NQO1 enzyme activities were measured in the cytosolic fraction according to the published protocols (Benson et al., 1980; Preusch et al., 1991; Moorthy et al., 2000). GST enzyme activity was quantified by using a GST assay kit according to the manufacturer’s protocol (Sigma-Aldrich, St. Louis, MO; CS0410).
Western blot assays
Ten or 20 μg of lung cytosolic protein extracts were separated by 10% SDS-polyacrylamide gel electrophoresis for detection of CYP1A1, NQO1, and MGST1 apoproteins, and transferred to polyvinylidene difluoride membranes. The membranes were incubated overnight at 4°C with the following primary antibodies: anti-CYP1A1 antibody (gift from P.E. Thomas, Rutgers University, Piscataway, NJ, USA; dilution 1:1500), anti-NQO1 antibody (Santa Cruz Biotechnologies; sc-16464, dilution 1:500), anti-MGST1 antibody (Santa Cruz; sc-17003, dilution 1:500) and anti-β-actin antibody (Sigma-Aldrich; A5316, dilution 1:5000). The primary antibodies were detected by incubation with the appropriate horseradish peroxidase-conjugated secondary antibodies. The immunoreactive bands were detected by chemiluminescence methods and the band density was analyzed by Kodak 1D 3.6 imaging software (Eastman Kodak Co., Rochester, NY, USA).
Quantitative real-time RT-PCR assays
Total RNA extracted from frozen lung tissues using Trizol reagent (Invitrogen) were treated with RQ1 RNase-free DNase I (Promega, Madison, WI) to eliminate genomic DNA contamination. RNA (50 ng), isolated as above, was subjected to one-step real-time quantitative TaqMan RT-PCR. Gene specific primers (CYP1A1-Mm00487218_m1; NQO1-Mm01253561_m1; MGST1-Mm00498294_m1; and 18S-Hs99999901_s1) in the presence of TaqMan reverse transcription reagents were used to reverse transcribe RNA, and TaqMan Gene Expression probes and TaqMan Universal PCR Master Mix (Applied Biosystems) were used for PCR amplification. The 18S was used as the reference gene. After an RT hold for 30 min at 48°C, the samples were denatured at 95°C for 10 min. The thermal cycling step was for 40 cycles at 95°C for 15s and 40 cycles at 60°C for 1 min. The ΔΔCt method was used to calculate the fold change in mRNA expression (Jiang et al., 2004).
Analyses of alveolarization
Tissue preparation for lung morphometry and immunohistochemistry
After completion of gas exposures, a subset of pups were euthanized and their lungs were inflated and fixed via the trachea with 10% formalin at 25 cm H2O pressure for at least 10 min. Serial five-micrometer sections of the paraffin embedded lungs were obtained perpendicular to the lung base (apical-basal axis) to attain portions of all the lobes of both the lungs for analysis. A systematic, uniform, random sampling principle (Hsia et al., 2010) was used to evaluate the sections for lung morphometry.
Lung morphometry
Alveolar development on selected mice (n=6/group) was evaluated by radial alveolar counts (RAC) and mean linear intercepts (MLI). The observers performing the measurements were masked to the slide identity. 1. Radial alveolar counts: RAC was determined as described by Cooney and Thurlbeck (Cooney and Thurlbeck, 1982). RAC measurements were made by dropping a perpendicular line from the center of a respiratory bronchiole to the edge of the septum or pleura and counting the number of alveoli traversed by this line. 2. Mean linear intercepts: MLIs were assessed as described previously (van Eijl et al., 2011). Briefly, grids of horizontal and vertical lines were superimposed on an image and the number of times the lines intersected with the tissue was counted. The total length of the grid lines was then divided by the number of intersections, to provide the mean linear intercept in μm. Photographs from at least 10 random nonoverlapping lung fields (10x magnification) were taken from each animal for RAC and MLI measurements.
Analyses of lung inflammation
Lung inflammation was assessed by 1. Immunostaining for macrophages: Deparaffinized lung sections (5 μM) were stained with rat anti-mouse Mac-3 antibody (553322, BD Pharmingen; dilution 1: 750), followed by staining with the appropriate biotinylated secondary antibody (Vector Laboratories, Burlingame, CA). The number of macrophages in the alveolar air spaces was counted from at least 10 random nonoverlapping lung fields (x400 magnification) per animal (n=6/group). The observers performing the macrophage counts were masked to the slide identity. 2. Monocyte chemoattractant protein 1 (MCP-1) mRNA expression: Total RNA extracted from lung tissues was used to quantify MCP-1 mRNA expression by real-time RT-PCR using a specific primer (MCP1-Mm00441242_m1, Applied Biosystems Inc., Foster City, CA) as described in analyses of CYP1A1, NQO1, and MGST1 mRNA expression.
Statistical Analyses
Statistical analyses were performed using SPSS 19.0. Data are expressed as means ± SEM. For the mRNA analysis, the fold change in mRNA expression was calculated by normalization to the air-exposed animals. The effects of genotype, exposure and their associated interactions for the outcome variables were assessed using ANOVA techniques. Multiple comparison testing by the posthoc Bonferroni test was performed if statistical significance of either variable or interaction was noted by ANOVA. The results were considered statistically significant if the p value of the posthoc Bonferroni test was less than 0.05.
Results
In this study, we investigated the role of AhR in hyperoxia-induced alveolar simplification and inflammation in newborn mice.
Hyperoxia Enhances Pulmonary CYP1A1, NQO1, and MGST1 mRNA Expression in WT Mice
To examine the effects of hyperoxia on functional activation of the AhR, we determined the expression of pulmonary CYP1A1, NQO1 and MGST1 mRNA, which are the phase I and II enzymes that are induced upon activation of the AhR. Quantification of mRNA levels by RT-PCR analysis in WT mice demonstrated that exposure to hyperoxia increased pulmonary CYP1A1 (Fig 1A), NQO1 (Fig 1B), and MGST1 (Fig 1C) mRNA levels by approximately 3-, 4-, 2.5-fold, respectively, when compared to corresponding air-breathing animals.
Figure 1. Hyperoxia increases pulmonary CYP1A1, NQO1, and MGST 1 mRNA expression.
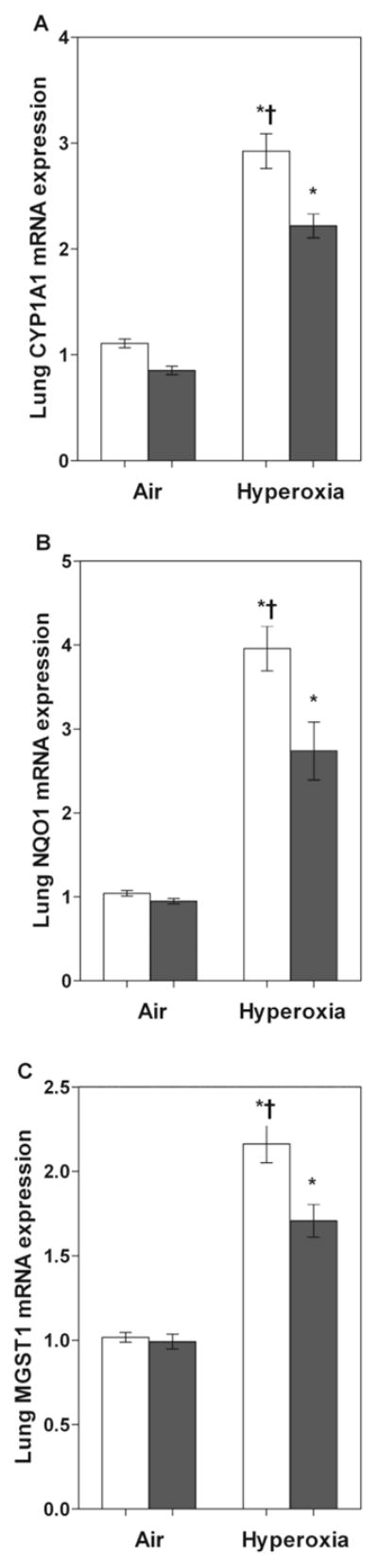
Total RNA, isolated from the lungs of WT and AhRd mice exposed to air or hyperoxia for 14 d, was subjected to real-time RT-PCR analysis for CYP1A1 (A), NQO1 (B), and MGST-1 (C) mRNA expression. Values are means ± S.E.M. from at least five individual WT (open bars) and AhRd (closed bars) animals in air or hyperoxia groups. Two-way ANOVA showed an effect of hyperoxia and genotype and a two-way interaction. Significant differences between air-breathing and hyperoxia-exposed animals are indicated by *, p < 0.05. Significant differences between hyperoxia-exposed WT and AhRd mice are indicated by †, p < 0.05.
Hyperoxia Enhances Pulmonary CYP1A1, NQO1, and MGST1 Protein Expression in WT Mice
Next, we determined the effect of hyperoxia on pulmonary CYP1A1, NQO1, and MGST1 apoprotein expression by western blotting. The pulmonary CYP1A1 (Fig 2A, B), NQO1 (Fig 2C, D), and MGST1 (Fig 2E, F) apoprotein expression was significantly increased in WT mice exposed to 14 d of hyperoxia compared to air-breathing animals. This was consistent with the effects of hyperoxia on the mRNA expression of the AhR-regulated phase I and II enzymes.
Figure 2. Hyperoxia increases pulmonary CYP1A1, NQO1, and MGST 1 protein expression.

The lung homogenates of WT and AhRd mice exposed to air or hyperoxia for 14 d were separated into cytosolic and nuclear fractions. The cytosolic protein was subjected to western blotting using anti-CYP1A1, anti-NQO1, anti-MGST1, or anti-β-actin antibodies. A, C, and E are representative western blots showing the expression of CYP1A1, NQO1, and MGST1 proteins, respectively. PC: positive control (3-methylcholanthrene treated lungs). B, D, and F represent the relative amounts of CYP1A1, NQO1, and MGST1 proteins, respectively, that are normalized to β-actin. Values are means ± S.E.M. from at least five individual WT (open bars) and AhRd (closed bars) animals in air or hyperoxia groups. Two-way ANOVA showed an effect of hyperoxia and genotype and a two-way interaction. Significant differences between air-breathing and hyperoxia-exposed animals are indicated by *, p < 0.05. Significant differences between hyperoxia-exposed WT and AhRd mice are indicated by †, p < 0.05.
Hyperoxia Enhances Pulmonary CYP1A1, NQO1, and GST Enzyme Activities in WT Mice
Finally, we examined the effects of hyperoxia on the pulmonary CYP1A1, NQO1 and MGST1 enzyme activity levels. WT mice exposed to 14 d of hyperoxia had significantly increased AhR-regulated phase I CYP1A1 enzyme activity (Fig 3A), and phase II NQO1 (Fig 3B) and GST (Fig 3C) enzyme activities compared to air-breathing WT mice.
Figure 3. Hyperoxia increases pulmonary CYP1A1, NQO1, and GST enzyme activities.
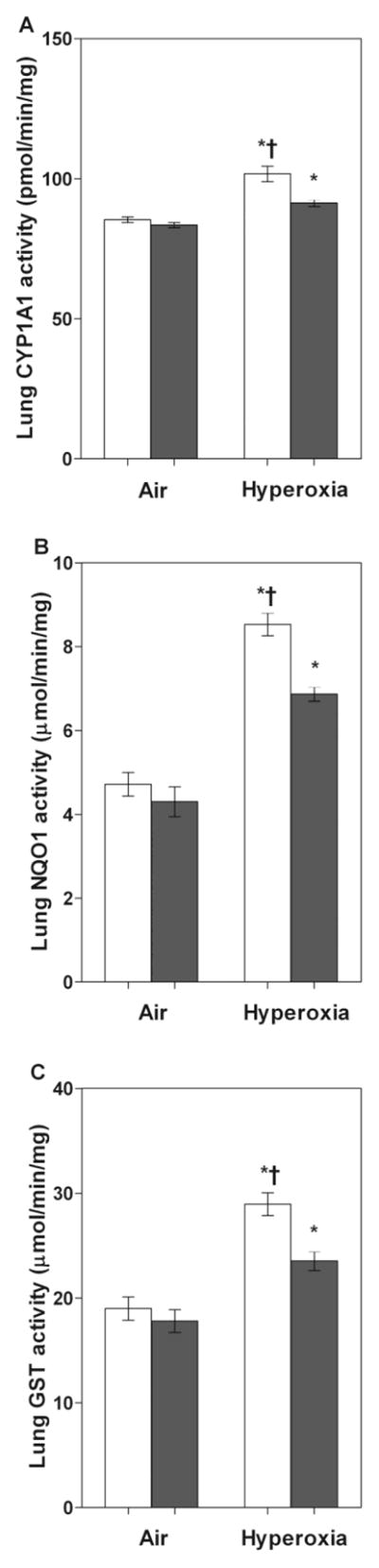
WT and AhRd mice were exposed to air or hyperoxia for 14 d, following which pulmonary CYP1A1 (A), NQO1 (B), and GST (C) enzyme activities were measured as described in materials and methods. Values are means ± S.E.M. from at least four individual WT (open bars) and AhRd (closed bars) animals in air or hyperoxia groups. Two-way ANOVA showed an effect of hyperoxia and genotype and a two-way interaction. Significant differences between air-breathing and hyperoxia-exposed animals are indicated by *, p < 0.05. Significant differences between hyperoxia-exposed WT and AhRd mice are indicated by †, p < 0.05.
Hyperoxia-Induced Expression of Pulmonary CYP1A1, NQO1, and MGST1 Enzymes were Attenuated in AhRd mice
To validate that AhR was dysfunctional in AhRd mice, we determined the effects of hyperoxia on the expression of pulmonary CYP1A1, NQO1, and MGST1 enzymes in AhRd mice. Not surprisingly, hyperoxia-induced expression of pulmonary CYP1A1, NQO1, and MGST1 enzymes were attenuated at the mRNA (Fig 1), protein (Fig 2), and enzyme (Fig 3) levels in AhRd mice compared to similarly exposed WT mice.
AhRd Mice had Increased Hyperoxia-Induced Alveolar Simplification
To investigate the mechanism(s) by which hyperoxia contributes to alveolar simplification, we studied the impact of AhR on alveolar simplification by exposing newborn WT and AhRD mice to hyperoxia. Alveolar simplification was determined by RAC and MLI. Exposure of WT and AhRD mice to 14 d of hyperoxia resulted in a significant decrease in RAC (Fig 4B, D, E) indicating that their alveoli were fewer in number compared to corresponding air-breathing animals (Fig 4A, C, E). In addition, hyperoxia-exposed WT and AhRd mice had significant increases in MLI (Fig 4B, D, F) indicating that their alveoli were also larger compared to corresponding air-breathing animals (Fig 4A, C, F). Interestingly, hyperoxia-induced alveolar simplification was significantly increased in AhRd mice compared to similarly exposed WT mice (Fig 4B, D, E, F). Statistical analyses indicated a significant two-way interaction between the genotype and hyperoxia exposure on alveolarization. In air-breathing animals, there was no significant difference in alveolarization between WT and AhRd mice (Fig 4A, C, E, F).
Figure 4. AhRd mice have increased hyperoxia-induced alveolar simplification.

AD: Representative hematoxylin and eosin-stained lung sections obtained from WT (A and B) and AhRd (C and D) mice exposed to air (A and C) or hyperoxia (B and D) for 14 d. Alveolarization was quantified by radial alveolar count (E) and mean linear intercept (F). Values are means ± S.E.M. from at least five individual WT (open bars) and AhRd (closed bars) animals in air or hyperoxia groups. Two-way ANOVA showed an effect of hyperoxia and genotype and a two-way interaction. Significant differences between air-breathing and hyperoxia-exposed animals are indicated by *, p < 0.05. Significant differences between hyperoxia-exposed WT and AhRd mice are indicated by †, p < 0.05.
AhRd Mice had Increased Hyperoxia-Induced Lung Inflammation
We performed real-time RT-PCR analysis of lung MCP-1 mRNA, and immunohistochemistry on fixed lung sections using anti-Mac-3 antibodies to ascertain if AhR altered hyperoxia-induced lung inflammatory response. Real-time RT-PCR analysis and the immunohistochemistry study revealed that hyperoxia increased MCP-1 mRNA expression (Fig 5F) and accumulation of macrophages (Fig 5B, D, E) in the lungs of both WT and AhRd mice. However, the effects of hyperoxia-induced lung MCP-1 expression and macrophage influx were augmented in AhRd mice (Fig 5B, D, E, F) compared to similarly exposed WT mice. Statistical analyses indicated a significant two-way interaction between the genotype and hyperoxia exposure on lung inflammation. In air-breathing animals, there was no significant difference in lung MCP-1 expression and macrophage counts between WT and AhRd mice (Fig 5A, C, E, F).
Figure 5. AhRd mice have increased hyperoxia-induced lung inflammation.

A–D: Representative Mac 3 antibody immunostained lung sections obtained from WT (A and B) and AhRd (C and D) mice exposed to air (A and C) or hyperoxia (B and D) for 14 d. E. Quantitative analysis of macrophages per high power field. F. Real-time RT-PCR analysis showing the lung MCP-1 mRNA expression. Values are means ± S.E.M. from at least five individual WT (open bars) and AhRd (closed bars) animals in air or hyperoxia groups. Two-way ANOVA showed an effect of hyperoxia and genotype and a two-way interaction. Significant differences between air-breathing and hyperoxia-exposed animals are indicated by *, p < 0.05. Significant differences between hyperoxia-exposed WT and AhRd mice are indicated by †, p < 0.05.
Discussion
This study demonstrates that hyperoxia exposed AhR deficient newborn mice have an increased susceptibility to alveolar simplification, which is associated with an augmented lung inflammatory response. In newborn mice exposed to hyperoxia, alveolar simplification and lung inflammation correlated inversely with the activation of the pulmonary AhR and the expression of its downstream target genes, CYP1A1, NQO1, and MGST1.
To investigate the molecular mechanisms associated with hyperoxia-induced alveolar simplification, we examined the lungs of hyperoxia-exposed newborn AhRd mice. The AhR is a versatile transcription factor that has important physiological functions in addition to its widely established role in induction of a battery of genes involved in the metabolism of xenobiotics. Studies from our laboratory and others have reported that AhR may be a crucial regulator of oxidant stress and inflammation through induction of several detoxifying phase I and II enzymes or via cross talk with other signal transduction pathways. In adult mice, AhR deficiency has been shown to be associated with increased hyperoxic lung injury (Couroucli et al., 2002; Jiang et al., 2004). Furthermore, we demonstrated that activation of AhR by omeprazole in adult mice (Shivanna et al., 2011b) and in the adult human lung-derived H441 cells (Shivanna et al., 2011a) attenuates hyperoxic injury. Whether AhR and its target genes are responsible for protecting newborn lung against hyperoxic lung injury is unknown. The goal of this study thus was to determine whether AhR was involved in hyperoxia-induced alveolar simplification and lung inflammation in newborn mice.
Initially, we studied the interaction between hyperoxia and AhR. Functional activation of the AhR results in transcriptional activation of various target genes referred to as the AhR gene battery, some of which includes CYP1A1, NQO1, and MGST1. Hence, we analyzed the expression of pulmonary CYP1A1, NQO1, and MGST1 to determine the activation of the pulmonary AhR. Strikingly, hyperoxia activated pulmonary AhR as evident by increased expression of pulmonary CYP1A1, NQO1, and MGST1 in WT mice. However, we did notice a discrepancy between CYP1A1 activity and CYP1A1 mRNA and protein levels, which we believe may be due to the interference in the ethoxyresorufin deethylase activity by other heme proteins. Enhanced pulmonary CYP1A1, NQO1, and MGST1 mRNA expression that is seen in parallel with corresponding increases in enzyme activities and apoprotein contents provides evidence that hyperoxia induces these enzymes by a transcriptional mechanism. On the contrary, hyperoxia had a minimal induction of pulmonary CYP1A1, NQO1, and MGST1 genes in newborn AhRd mice. This suggests that these enzymes are downstream target molecules that are transcriptionally activated by the pulmonary AhR. However, the molecular mechanisms by which hyperoxia activate pulmonary AhR remain unknown at the present time.
Hyperoxia-induced expression of the phase I and II enzymes have been observed by several other investigators both in adult (Moorthy et al., 1997; Cho et al., 2002; Couroucli et al., 2002; Jiang et al., 2004) and newborn rodents (McGrath-Morrow et al., 2009; Couroucli et al., 2011). This phenomenon might be a protective responsive to oxidative stress because of its striking resemblance to the effects of hyperoxia on the “classic” antioxidant enzymes such as superoxide dismutase, glutathione peroxidase, glutathione reductase, and catalase (Ho et al., 1996; Clerch, 2000). In our study, there were no significant differences in expression of these enzymes between air-breathing WT and AhRd mice. These findings suggest that the constitutive expression of these enzymes might be equally regulated by other transcription factors such as Nrf2 in addition to the AhR.
Next, we studied the effects of AhR on hyperoxia-induced alveolar simplification and lung inflammation. Hyperoxia is known to cause alveolar simplification both in preterm infants (Husain et al., 1998) and newborn mice (Warner et al., 1998). A similar finding was noticed in our experimental animals exposed to hyperoxia for 2 weeks. However, the increased alveolar simplification noticed in hyperoxia-exposed AhRd mice compared to similarly exposed WT mice indicates that AhR protects newborn mice against hyperoxia-induced alveolar simplification.
Previous studies have demonstrated that ROS and inflammatory responses are the key mediators in the pathogenesis of hyperoxia-induced lung disorders such as BPD in preterm infants. MCP-1 is a CC chemokine that is increased in the bronchoalveolar lavage fluid and plasma of infants with oxidant injury who later develop BPD (Ambalavanan et al., 2009). Likewise, lung macrophage infiltration is an important marker of a chronic inflammatory state that is associated with BPD (Clement et al., 1988). Hence, we determined the lung MCP-1 expression and macrophage infiltration to determine the effects of AhR on hyperoxia-induced lung inflammatory response. Our observation of an increased hyperoxia-induced lung inflammatory response in AhRd mice suggests that AhR might mitigate alveolar simplification by decreasing lung inflammation, although we only demonstrated an association not necessarily a cause and effect relationship.
The molecular mechanisms by which the pulmonary AhR protects against hyperoxic lung injury remains poorly defined. Interestingly, we observed significant upregulation of pulmonary CYP1A1, NQO1, and MGST1 enzymes in WT mice compared to AhRd mice upon exposure to hyperoxia. The protective effects of CYP1A enzymes against hyperoxic lung injury in rodents have been extensively documented, as evidenced by 1) attenuation of hyperoxic lung injury in rodents treated with CYP1A inducers, β-naphthoflavone or 3-methylcholanthrene (Mansour et al., 1988; Sinha et al., 2005; Moorthy, 2008; Couroucli et al., 2011); 2) potentiation of hyperoxic injury in rats treated with CYP1A inhibitor, 1-aminobenzotriazole (Moorthy et al., 2000); 3) increased susceptibility of rodents deficient in genes for AhR (Couroucli et al., 2002; Jiang et al., 2004) to hyperoxic lung injury. In addition, the phase II enzymes such as NQO1 and MGST1 have been shown to protect cells and tissues against oxidant injury induced by various toxic chemicals (O’Brien, 1991; Rahman et al., 1999; Johansson et al., 2010) and oxygen (Cho et al., 2002; Das et al., 2006; McGrath-Morrow et al., 2009). The protective mechanisms of these enzymes have been attributed to their ability to conjugate and excrete the reactive electrophiles and lipid peroxidation products generated by an oxidant injury (Cho et al., 2002; Johansson et al., 2010). AhR-mediated protection against hyperoxia-induced alveolar simplification may be attributed at least in part to these enzymes. It is important to note that AhR activation results in the induction of several detoxifying enzymes, which may be collectively more effective against an oxidant injury compared to the induction of a single enzyme. Furthermore, Baglole et al. (Baglole et al., 2008) recently observed that AhR decreases inflammation in lung cells by regulating the transcription factor, NF-κb. We have also conducted experiments to investigate the interaction between AhR and NF-kB in hyperoxic conditions. Our preliminary experiments suggest that NFkB activity is increased in hyperoxia-exposed AhRd mice (data not shown). However, we need to confirm this finding by conducting additional experiments. It is thus possible the protective effects of the AhR might also be mediated in part by its interaction with NF-κb.
We recognize the limitations of our study. Firstly, the expression of the pulmonary AhR could not be precisely regulated because our experimental animals were not conditional knockout mice. However, these results provide a background to conduct studies using transgenic animals that target the developing lungs. Secondly, we have shown structural defects associated with altered gene expression, but not the functional consequences. In future, we will determine the functional correlate by performing lung function tests in these animals.
In summary, we demonstrate that decreased activation of the pulmonary AhR augments hyperoxia-induced alveolar simplification and increased lung inflammation in newborn mice. We propose that the protective effects of AhR may be due to the induction of a battery of phase I and II enzymes that have potential antioxidant and anti-inflammatory properties. It is important to note that even with extensive back cross breeding, the AhRd mice are still not similar to AhR-null mice and they have some residual AhR function. Therefore, we do expect the effects of AhR on alveolarization and the expression of phase I and II enzymes to be more remarkable when we use AhR-null mice in our future studies.
The etiology of BPD is multifactorial, and therefore, therapies targeting a single inflammatory or oxidative stress pathway have largely been unsuccessful in the management of BPD (Wright and Kirpalani, 2011). Recent studies including our present one suggests that AhR decreases lung inflammation, and upregulates phase I and II enzymes, which are known to have antioxidant properties. AhR can thus be used to improve current therapies of BPD in preterm infants because activation of pulmonary AhR would target more than one pathway and mitigate both inflammation and oxidant stress, which are significant in the development of BPD.
Highlights.
AhR deficiency increases oxygen toxicity in newborn mice.
Hyperoxia-exposed AhRd mice have decreased expression of CYP1A1, NQO1, and MGST1 enzymes.
Functional deficiency of AhR increases hyperoxia-induced lung inflammation.
Functional deficiency of AhR increases hyperoxia-induced arrest in alveolarization.
Acknowledgments
This study was supported by Division of Neonatology, Texas Children’s Hospital. We thank Raul Gonzalez for the timely processing of histopathology and immunohistochemistry slides and Dr. Roberto Barrios for evaluating the lung sections.
Funding
This work was in part supported by funds from the Section of Neonatal-Perinatal Medicine, Texas Children’s Hospital to B.S.; and RO1 grants from National Institutes of Health [ES- 009132, HL-112516, HL-087174, and ES-019689] to B.M. and HL-088343 to XIC.
Abbreviations
- BPD
Bronchopulmonary dysplasia
- ROS
Reactive oxygen species
- AhR
Aryl hydrocarbon receptor
- CYP
Cytochrome P450
- NQO1
NAD(P)H quinone oxidoreductase-1
- MGST1
Microsomal glutathione S-transferase1
- WT mice
C57BL6/J wild type mice
- AhRd mice
Aryl hydrocarbon receptor dysfunctional mice
- RAC
Radial alveolar count
- MLI
Mean linear intercept
- MCP-1
Monocyte chemoattractant protein-1
- ANOVA
Analysis of variance
Footnotes
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott BD, Birnbaum LS, Perdew GH. Developmental expression of two members of a new class of transcription factors: I. Expression of aryl hydrocarbon receptor in the C57BL/6N mouse embryo. Dev Dyn. 1995;204:133–143. doi: 10.1002/aja.1002040204. [DOI] [PubMed] [Google Scholar]
- Ambalavanan N, Carlo WA, D’Angio CT, McDonald SA, Das A, Schendel D, Thorsen P, Higgins RD. Cytokines associated with bronchopulmonary dysplasia or death in extremely low birth weight infants. Pediatrics. 2009;123:1132–1141. doi: 10.1542/peds.2008-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asikainen TM, White CW. Antioxidant defenses in the preterm lung: role for hypoxia-inducible factors in BPD? Toxicol Appl Pharmacol. 2005;203:177–188. doi: 10.1016/j.taap.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Baglole CJ, Maggirwar SB, Gasiewicz TA, Thatcher TH, Phipps RP, Sime PJ. The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappaB family member RelB. J Biol Chem. 2008;283:28944–28957. doi: 10.1074/jbc.M800685200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson AM, Hunkeler MJ, Talalay P. Increase of NAD(P)H:quinone reductase by dietary antioxidants: possible role in protection against carcinogenesis and toxicity. Proc Natl Acad Sci U S A. 1980;77:5216–5220. doi: 10.1073/pnas.77.9.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari V. Hyperoxia-derived lung damage in preterm infants. Semin Fetal Neonatal Med. 2010;15:223–229. doi: 10.1016/j.siny.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock KW, Kohle C. The mammalian aryl hydrocarbon (Ah) receptor: from mediator of dioxin toxicity toward physiological functions in skin and liver. Biol Chem. 2009;390:1225–1235. doi: 10.1515/BC.2009.138. [DOI] [PubMed] [Google Scholar]
- Burbach KM, Poland A, Bradfield CA. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci U S A. 1992;89:8185–8189. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Smith DR, Prasad VS, Sidman CL, Nebert DW, Puga A. Ten nucleotide differences, five of which cause amino acid changes, are associated with the Ah receptor locus polymorphism of C57BL/6 and DBA/2 mice. Pharmacogenetics. 1993;3:312–321. doi: 10.1097/00008571-199312000-00005. [DOI] [PubMed] [Google Scholar]
- Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002;26:175–182. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- Clement A, Chadelat K, Sardet A, Grimfeld A, Tournier G. Alveolar macrophage status in bronchopulmonary dysplasia. Pediatr Res. 1988;23:470–473. doi: 10.1203/00006450-198805000-00007. [DOI] [PubMed] [Google Scholar]
- Clerch LB. Post-transcriptional regulation of lung antioxidant enzyme gene expression. Ann N Y Acad Sci. 2000;899:103–111. doi: 10.1111/j.1749-6632.2000.tb06179.x. [DOI] [PubMed] [Google Scholar]
- Cooney TP, Thurlbeck WM. The radial alveolar count method of Emery and Mithal: a reappraisal 1--postnatal lung growth. Thorax. 1982;37:572–579. doi: 10.1136/thx.37.8.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couroucli XI, Liang YH, Jiang W, Wang L, Barrios R, Yang P, Moorthy B. Prenatal administration of the cytochrome P4501A inducer, Beta-naphthoflavone (BNF), attenuates hyperoxic lung injury in newborn mice: implications for bronchopulmonary dysplasia (BPD) in premature infants. Toxicol Appl Pharmacol. 2011;256:83–94. doi: 10.1016/j.taap.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couroucli XI, Welty SE, Geske RS, Moorthy B. Regulation of pulmonary and hepatic cytochrome P4501A expression in the rat by hyperoxia: implications for hyperoxic lung injury. Mol Pharmacol. 2002;61:507–515. doi: 10.1124/mol.61.3.507. [DOI] [PubMed] [Google Scholar]
- Das A, Kole L, Wang L, Barrios R, Moorthy B, Jaiswal AK. BALT development and augmentation of hyperoxic lung injury in mice deficient in NQO1 and NQO2. Free Radic Biol Med. 2006;40:1843–1856. doi: 10.1016/j.freeradbiomed.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Emi Y, Ikushiro S, Iyanagi T. Xenobiotic responsive element-mediated transcriptional activation in the UDP-glucuronosyltransferase family 1 gene complex. J Biol Chem. 1996;271:3952–3958. doi: 10.1074/jbc.271.7.3952. [DOI] [PubMed] [Google Scholar]
- Fanaroff AA, Stoll BJ, Wright LL, Carlo WA, Ehrenkranz RA, Stark AR, Bauer CR, Donovan EF, Korones SB, Laptook AR, Lemons JA, Oh W, Papile LA, Shankaran S, Stevenson DK, Tyson JE, Poole WK. Trends in neonatal morbidity and mortality for very low birthweight infants. Am J Obstet Gynecol. 2007;196:147, e141–148. doi: 10.1016/j.ajog.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Favreau LV, Pickett CB. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene. Identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. J Biol Chem. 1991;266:4556–4561. [PubMed] [Google Scholar]
- Fujii-Kuriyama Y, Kawajiri K. Molecular mechanisms of the physiological functions of the aryl hydrocarbon (dioxin) receptor, a multifunctional regulator that senses and responds to environmental stimuli. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:40–53. doi: 10.2183/pjab.86.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YS, Dey MS, Crapo JD. Antioxidant enzyme expression in rat lungs during hyperoxia. Am J Physiol. 1996;270:L810–818. doi: 10.1152/ajplung.1996.270.5.L810. [DOI] [PubMed] [Google Scholar]
- Hsia CC, Hyde DM, Ochs M, Weibel ER. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med. 2010;181:394–418. doi: 10.1164/rccm.200809-1522ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain AN, Siddiqui NH, Stocker JT. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum Pathol. 1998;29:710–717. doi: 10.1016/s0046-8177(98)90280-5. [DOI] [PubMed] [Google Scholar]
- Jiang W, Welty SE, Couroucli XI, Barrios R, Kondraganti SR, Muthiah K, Yu L, Avery SE, Moorthy B. Disruption of the Ah receptor gene alters the susceptibility of mice to oxygen-mediated regulation of pulmonary and hepatic cytochromes P4501A expression and exacerbates hyperoxic lung injury. J Pharmacol Exp Ther. 2004;310:512–519. doi: 10.1124/jpet.103.059766. [DOI] [PubMed] [Google Scholar]
- Jobe AH, Hillman N, Polglase G, Kramer BW, Kallapur S, Pillow J. Injury and inflammation from resuscitation of the preterm infant. Neonatology. 2008;94:190–196. doi: 10.1159/000143721. [DOI] [PubMed] [Google Scholar]
- Jobe AJ. The new BPD: an arrest of lung development. Pediatr Res. 1999;46:641–643. doi: 10.1203/00006450-199912000-00007. [DOI] [PubMed] [Google Scholar]
- Johansson K, Jarvliden J, Gogvadze V, Morgenstern R. Multiple roles of microsomal glutathione transferase 1 in cellular protection: a mechanistic study. Free Radic Biol Med. 2010;49:1638–1645. doi: 10.1016/j.freeradbiomed.2010.08.013. [DOI] [PubMed] [Google Scholar]
- Mansour H, Levacher M, Azoulay-Dupuis E, Moreau J, Marquetty C, Gougerot-Pocidalo MA. Genetic differences in response to pulmonary cytochrome P-450 inducers and oxygen toxicity. J Appl Physiol. 1988;64:1376–1381. doi: 10.1152/jappl.1988.64.4.1376. [DOI] [PubMed] [Google Scholar]
- McGrath-Morrow S, Lauer T, Yee M, Neptune E, Podowski M, Thimmulappa RK, O’Reilly M, Biswal S. Nrf2 increases survival and attenuates alveolar growth inhibition in neonatal mice exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2009;296:L565–573. doi: 10.1152/ajplung.90487.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorthy B. Cytochromes P450. In: CI, editor. Role in Drug Metabolism and Toxicity of Drugs and other Xenobiotics. RSC publishing; Cambridge: 2008. pp. 97–135. [Google Scholar]
- Moorthy B, Nguyen UT, Gupta S, Stewart KD, Welty SE, Smith CV. Induction and decline of hepatic cytochromes P4501A1 and 1A2 in rats exposed to hyperoxia are not paralleled by changes in glutathione S-transferase-alpha. Toxicol Lett. 1997;90:67–75. doi: 10.1016/s0378-4274(96)03832-5. [DOI] [PubMed] [Google Scholar]
- Moorthy B, Parker KM, Smith CV, Bend JR, Welty SE. Potentiation of oxygen-induced lung injury in rats by the mechanism-based cytochrome P-450 inhibitor, 1-aminobenzotriazole. J Pharmacol Exp Ther. 2000;292:553–560. [PubMed] [Google Scholar]
- Natarajan G, Pappas A, Shankaran S, Kendrick DE, Das A, Higgins RD, Laptook AR, Bell EF, Stoll BJ, Newman N, Hale EC, Bara R, Walsh MC. Outcomes of extremely low birth weight infants with bronchopulmonary dysplasia: Impact of the physiologic definition. Early Hum Dev. 2012 doi: 10.1016/j.earlhumdev.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279:23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- O’Brien PJ. Molecular mechanisms of quinone cytotoxicity. Chem Biol Interact. 1991;80:1–41. doi: 10.1016/0009-2797(91)90029-7. [DOI] [PubMed] [Google Scholar]
- Park MS, Rieger-Fackeldey E, Schanbacher BL, Cook AC, Bauer JA, Rogers LK, Hansen TN, Welty SE, Smith CV. Altered expressions of fibroblast growth factor receptors and alveolarization in neonatal mice exposed to 85% oxygen. Pediatr Res. 2007;62:652–657. doi: 10.1203/PDR.0b013e318159af61. [DOI] [PubMed] [Google Scholar]
- Preusch PC, Siegel D, Gibson NW, Ross D. A note on the inhibition of DT-diaphorase by dicoumarol. Free Radic Biol Med. 1991;11:77–80. doi: 10.1016/0891-5849(91)90191-5. [DOI] [PubMed] [Google Scholar]
- Rahman Q, Abidi P, Afaq F, Schiffmann D, Mossman BT, Kamp DW, Athar M. Glutathione redox system in oxidative lung injury. Crit Rev Toxicol. 1999;29:543–568. doi: 10.1080/10408449991349276. [DOI] [PubMed] [Google Scholar]
- Rushmore TH, King RG, Paulson KE, Pickett CB. Regulation of glutathione S-transferase Ya subunit gene expression: identification of a unique xenobiotic-responsive element controlling inducible expression by planar aromatic compounds. Proc Natl Acad Sci U S A. 1990;87:3826–3830. doi: 10.1073/pnas.87.10.3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saugstad OD. Oxygen and oxidative stress in bronchopulmonary dysplasia. J Perinat Med. 2010;38:571–577. doi: 10.1515/jpm.2010.108. [DOI] [PubMed] [Google Scholar]
- Shivanna B, Chu C, Welty SE, Jiang W, Wang L, Couroucli XI, Moorthy B. Omeprazole attenuates hyperoxic injury in H441 cells via the aryl hydrocarbon receptor. Free Radic Biol Med. 2011a doi: 10.1016/j.freeradbiomed.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivanna B, Jiang W, Wang L, Couroucli X, Moorthy B. Omeprazole Attenuates Hyperoxic Lung Injury in Mice via Aryl hydrocarbon Receptor Activation, and is Associated with Increased Expression of Cytochrome P4501A Enzymes. J Pharmacol Exp Ther. 2011b doi: 10.1124/jpet.111.182980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short EJ, Klein NK, Lewis BA, Fulton S, Eisengart S, Kercsmar C, Baley J, Singer LT. Cognitive and academic consequences of bronchopulmonary dysplasia and very low birth weight: 8-year-old outcomes. Pediatrics. 2003;112:e359. doi: 10.1542/peds.112.5.e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha A, Muthiah K, Jiang W, Couroucli X, Barrios R, Moorthy B. Attenuation of hyperoxic lung injury by the CYP1A inducer beta-naphthoflavone. Toxicol Sci. 2005;87:204–212. doi: 10.1093/toxsci/kfi226. [DOI] [PubMed] [Google Scholar]
- Thatcher TH, Maggirwar SB, Baglole CJ, Lakatos HF, Gasiewicz TA, Phipps RP, Sime PJ. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am J Pathol. 2007;170:855–864. doi: 10.2353/ajpath.2007.060391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel M, Chouker A, Ohta A, Jackson E, Caldwell C, Smith P, Lukashev D, Bittmann I, Sitkovsky MV. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 2005;3:e174. doi: 10.1371/journal.pbio.0030174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirona RG, Kim RB. Nuclear receptors and drug disposition gene regulation. J Pharm Sci. 2005;94:1169–1186. doi: 10.1002/jps.20324. [DOI] [PubMed] [Google Scholar]
- van Eijl S, Mortaz E, Versluis C, Nijkamp FP, Folkerts G, Bloksma N. A low vitamin A status increases the susceptibility to cigarette smoke-induced lung emphysema in C57BL/6J mice. J Physiol Pharmacol. 2011;62:175–182. [PubMed] [Google Scholar]
- Vina J, Vento M, Garcia-Sala F, Puertes IR, Gasco E, Sastre J, Asensi M, Pallardo FV. L-cysteine and glutathione metabolism are impaired in premature infants due to cystathionase deficiency. Am J Clin Nutr. 1995;61:1067–1069. doi: 10.1093/ajcn/61.4.1067. [DOI] [PubMed] [Google Scholar]
- Warner BB, Stuart LA, Papes RA, Wispe JR. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol. 1998;275:L110–117. doi: 10.1152/ajplung.1998.275.1.L110. [DOI] [PubMed] [Google Scholar]
- Wright CJ, Kirpalani H. Targeting inflammation to prevent bronchopulmonary dysplasia: can new insights be translated into therapies? Pediatrics. 2011;128:111–126. doi: 10.1542/peds.2010-3875. [DOI] [PMC free article] [PubMed] [Google Scholar]