

Abstract
As part of our efforts aimed at searching for new antiparasitic agents, 2-alkylmercaptoethyl-1,1-bisphosphonate derivatives were synthesized and evaluated against Trypanosoma cruzi, the etiologic agent of Chagas disease, and Toxoplasma gondii, the responsible agent for toxoplasmosis. Many of these sulfur-containing bisphosphonates were potent inhibitors against the intracellular form of T. cruzi, the clinically more relevant replicative form of this parasite, and tachyzoites of T. gondii targeting T. cruzi or T. gondii farnesyl diphosphate synthases (FPPSs), which constitute valid targets for the chemotherapy of these parasitic diseases. Interestingly, long chain length sulfur-containing bisphosphonates emerged as relevant antiparasitic agents. Taking compounds 37, 38, and 39 as representative members of this class of drugs, they exhibited ED50 values of 15.8 μM, 12.8 μM, and 22.4 μM, respectively, against amastigotes of T. cruzi. These cellular activities matched the inhibition of the enzymatic activity of the target enzyme (TcFPPS) having IC50 values of 6.4 μM, 1.7 μM, and 0.097 μM, respectively. In addition, these compounds were potent anti-Toxoplasma agents. They had ED50 values of 2.6 μM, 1.2 μM, and 1.8 μM, respectively, against T. gondii tachyzoites, while they exhibited a very potent inhibitory action against the target enzyme (TgFPPS) showing IC50 values of 0.024 μM, 0.025 μM, and 0.021 μM, respectively. Bisphosphonates bearing a sulfoxide unit at C-3 were also potent anti-Toxoplasma agents, particularly those bearing long aliphatic chains such as 43–45, which were also potent antiproliferative drugs against tachyzoites of T. gondii. These compounds inhibited the enzymatic activity of the target enzyme (TgFPPS) at the very low nanomolar range. These bisphosphonic acids have very good prospective not only as lead drugs but also as potential chemotherapeutic agents.
Introduction
The isosteric replacement of the oxygen atom bridge of inorganic pyrophosphate (1) with substituted methylene groups gives rise to a class of drugs known as bisphosphonates (2) [1], which became compounds of pharmacological importance since calcification studies carried out many decades ago [2–4]-Several bisphosphonates such as, pamidronate (3), alendronate (4) and risedronate (5) are in clinical use for the treatment and prevention of osteoclast-mediated bone resorption associated with various bone disorders (Figure 1) [5–8].
Figure 1.
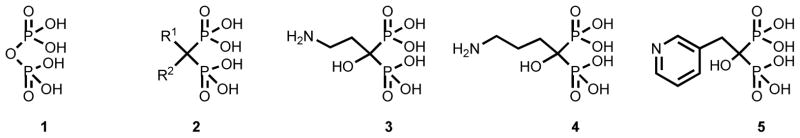
General formula and chemical structure of representative FDA-approved bisphosphonates clinically employed for the treatment of bone disorders.
Besides their use in long-term treatment of different bone disorders, bisphosphonates exhibit a wide range of biological activities, such as antibacterial agents [9], anticancer agents [10–13], as selective inhibitors of acid sphingomyelinase [14], in stimulation of γδ T cells [15], and, particularly, as antiparasitic agents [16–20]. Some years ago, selected bisphosphonates, comprising the FDA-approved pamidronate (3) and alendronate (4), were found to be potent inhibitors of T. cruzi proliferation in in vitro and in vivo assays without toxicity to the host cells [21]. Based on the previous findings, other bisphosphonates were found to be potent antiproliferative agents against other trypanosomatids such as T. brucei rhodesiense, Leishmania donovani, and L. mexicana, and Apicomplexans such as Toxoplasma gondii and Plasmodium falciparum [17–20].
Bone mineral has a similar mineral composition than acidocalcisomes, which are acidic organelles of high-density with a high concentration of phosphorus present as pyrophosphate and polyphosphate, which is associated to calcium and other cations. Then, it is reasonable to anticipate that accumulation of bisphosphonates in these organelles facilitates their antiparasitic action [22,23] FPPS catalyzes the two committed biosynthetic steps to form farnesyl diphosphate from dimethylallyl and isopentenyl diphosphates [20,21].
Bisphosphonates derived from fatty acids are promising antiparasitic agents, in particular, 2-alkyl(amino)ethyl derivatives. These compounds exhibit cellular activity against intracellular T. cruzi, which is one of the clinically relevant forms of this parasite, having IC50 values at the low nanomolar level against the target enzyme [24,25]. In addition, 1-hydroxy-, 1-alkyl-, and 1-amino-bisphosphonates such as 6–9 have been mainly useful in SAR studies as antiparasitic agents [26–29]. For example, bisphosphonate 6 is a potent growth inhibitor against T. cruzi (amastigotes) [26] and also against T. gondii (tachyzoites) [29,30], while 7 is effective against P. falciparum [30]. Compounds 10 and 12 have cellular activity against T. gondii, the latter one being unusually effective against the target enzyme (IC50 = 93 nM) [30,31]. In addition, in contrast to what would be expected, α-fluoro-1,1-bisphosphonates are devoid of activity against T. cruzi cells and TcFPPS regardless of the chain length [31]. However, these compounds behave as extremely potent inhibitors of the enzymatic activity of T. gondii FPPS [31]. Actually, 13 and 14 possess IC50 values of 35 nM and 60 nM, respectively, towards TgFPPS, that is, they are even more effective than risedronate (IC50 = 74 nM) used as positive control (Figure 2) [31]. The high selectivity observed by these drugs towards TgFPPS versus TcFPPS is not surprising bearing in mind that the amino acid sequences of these enzymes have less than 50% identity [20].
Figure 2.
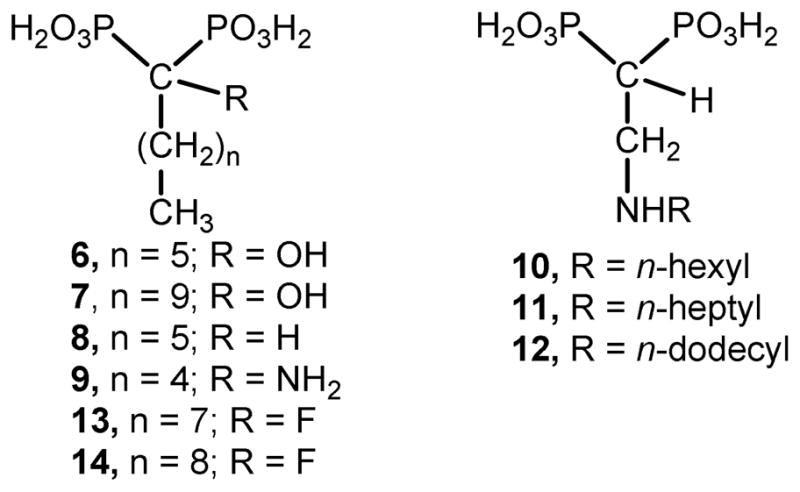
Chemical structure of representative members of bisphosphonic acids derived from fatty acids.
T. cruzi and T. gondii are the etiologic agents of American trypanosomiasis (Chagas disease) and toxoplasmosis, respectively, two major parasitic diseases according to the World Health Organization [20,21]. Chemotherapy for this two parasitic diseases, based on empirically discovered drugs, is still a challenge [23,32–34]. T. cruzi has a complex life cycle involving blood-sucking Reduviid insects and mammals [35]. This parasite has four main morphological forms and the amastigote form is the more relevant replicative form of the parasite [35]. This blood-sucking activity is the main way of dissemination of Chagas disease, while infection via the placenta or by blood transfusion is the mechanism responsible where this disease is not endemic [36]. The opportunistic parasite T. gondii is able to infect humans (basically all warm-blooded mammals) by contact with feces of infected cats, by eating undercooked meat or via the placenta from pregnant women [37,38]. Two asexual forms are able to affect humans: the tachyzoite form can invade cells and multiplies leading to host cell death, while the bradyzoite form proliferates slowly and forms cysts in muscle [39]. The main goal in toxoplasmosis is to develop a drug that is able to eliminate the cyst stage of the parasite to avoid recrudescence of the disease [20].
Rationale
In the last years, many efforts have been made to understand how bisphosphonic acids inhibit FPPS at the molecular level [40–42]. Recently, we were able to determine that TcFPPS inhibitors 10 and 11 bind to the allylic site of the enzyme [43] with the phosphates group of the bisphosphonate moiety coordinating three Mg2+ atoms that bridge the compound to the enzyme in a similar way that was observed for the physiological substrates [44,45]. The nitrogen atom at the C-3 position is very important to maintain a high degree of biological activity.
Analyses of the 2-alkylaminoethyl-1,1-bisphosphonates–TcFPPS complexes have indicated that methyl substitution at the N-linked carbon of the alkyl chain would be favorable for binding [43]. Then, 18 was envisioned for this purpose (Scheme 1). In addition, in order to study a potential synergistic effect, it was considered to add a hydroxyl group at C-1, present in many pharmacological important bisphosphonic acids, in the reference structure 11 to afford the 2-alkylaminoethyl-1-hydroxy-1,1-bisphosphonic acid 21.
Scheme 1.
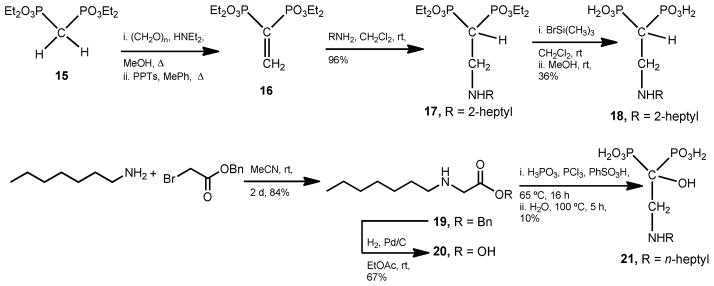
Synthetic approach for the preparation of modified alkylaminoethyl bisphosphonates.
To assess the necessity of the amine group for inhibitory activity against T. cruzi or T. gondii, as well as their corresponding target enzymes TcFPPS and TgFPPS, we decided to replace it for a sulfide, sulfoxide, sulfone and methylalkylsulfonium group.
Results and Discussion
Preparation of the methyl analogue of the lead structure 10 (compound 18) was conducted according to previously published procedures [24,25]. Briefly, the versatile Michael Acceptor 16 [46–48], which was straightforwardly obtained from commercially available tetraethyl methylenebis(phosphonate) (15), was reacted with 2-heptylamine in methylene chloride to afford the Michael adduct 17. This compound was hydrolyzed by treatment with bromotrimethylsilane in methylene chloride followed by digestion with methanol [49] to afford the free bisphosphonic acid 18. Additionally, the 1-[(n-alkylamino)ethyl]-1-hydroxy-1,1-bisphosphonic acid derivative 21 was readily prepared from n-heptylamine. Coupling reaction between this compound and benzyl bromoacetate in acetonitrile [50] afforded the expected benzyl n-alkylaminoacetate 19 in 84% yield, which was hydrogenated employing palladium on charcoal as catalyst to yield the free acid 20 in 67% yield, which was the substrate to form the title compound 21. Then, on treatment with phosphorous acid and phosphorous trichloride employing benzenesulfonic acid as a solvent at 65 °C followed by hydrolysis, 20 was converted into 21 according to the widely employed method for the preparation of 1-hydroxy-1,1-bisphosphonic acids [51].
Based on the ability of mercaptane derivatives to undergo 1,4-conjugate Michael-type addition reactions on a number of α,β-unsaturated carbonyl compounds [52–55], the synthetic precursors of the sulfur-containing 1,1-bisphosphonic acids (22–30) were successfully prepared via this 1,4-conjugated addition among commercial n-alkyl mercaptanes and the acceptor 16, in the presence of triethylamine. Reaction yields ranged 68–94%. Hydrolysis of these tetraethyl intermediates by treatment with bromotrimethylsilane in methylene chloride followed by digestion with methanol afforded the title compounds 31–39 in good yields (Scheme 2).
Scheme 2.
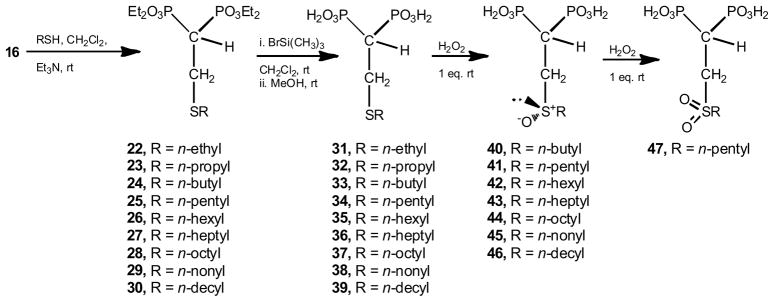
Synthetic approach to access to sulfur-containing bisphosphonates.
In order to obtain the sulfoxide derivatives of these compounds, it was first considered starting from the tetraethyl esters 22–30. The controlled oxidation reaction of the corresponding thioethers is the most widely employed method of preparation of sulfoxides [56]. However, contrarily to what had been depicted in closely related compounds [57], in our hands all the attempts to oxidize any of the sulfides 22–30 by using sodium metaperiodate [58–60], hydrogen peroxide [61,62], or m-chloroperoxybenzoic acid [63] underwent a retro-Michael reaction. These results are in agreement with published data where alkylsulfides bonded at the β-position of aldehydes and ketones experienced a retro-Michael reaction when treated with an oxidizing agent affording the α,β-unsaturated carbonyl compounds and the corresponding alkylsulfanol [64–66].
Oxidation reaction on the free bisphosphonic acids 33–39 would not undergo retro-Michael addition. In order to test this hypothesis, the reaction of 34 with hydrogen peroxide was monitored by 1H and 31P NMR spectroscopy. On treatment with hydrogen peroxide (one equivalent) compound 34 was converted rapidly into sulfoxide 41. No overoxidation was observed. Addition of a second equivalent of hydrogen peroxide gave rise to sulfone 47. Therefore, sulfides 33–39 were transformed into sulfoxides 40–46 as illustrated in Scheme 2.
The methyl(octyl)sulfonium derivative 48 was succesfully prepared by treatment of the respective free bisphosphonic acid 37 bearing a sulfide moeity at C-3, with methyl iodide and silver tetrafluoroborate [67] in acetronitrile affording 48 in very good yield. Attempts to methylate 28 failed due to a retro-Michael type reaction occurred instead of formation of the expected sulfonium derivative as illustrated in Scheme 3.
Scheme 3.
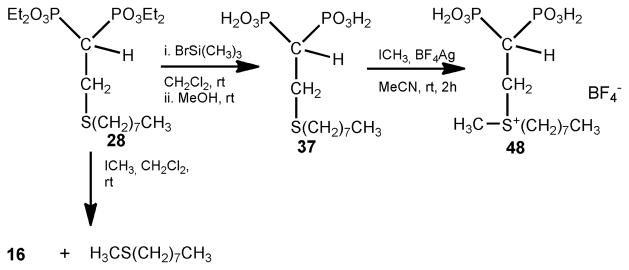
Method of preparation of the methyl sulfonium derivative 48.
Biological evaluation of the title compounds 18, 21, 31–48 resulted to be very interesting. Both nitrogen-containing bisphosphonic acids 18 and 21 were almost devoid of antiparasitic activity against the amastigote form of T. cruzi and also towards the target enzyme TcFPPS. Compound 18 exhibited moderate potency against tachyzoites of T. gondii having an EC50 value of 11.4 μM. In addition, sulfur-containing bisphosphonic acids proved to be very potent antiparasitic agents. Certainly, alkylthioethyl derivatives with relatively long aliphatic chains were very effective against either T. cruzi or T. gondii, with ED50 values of 15.8 μM (37), 12.8 μM (38), and 22.4 μM (39) against amastigotes forms of the former. These activities are in accordance with the strong inhibition of TcFPPS observed for these analogues (IC50 values of 6.4 μM, 1.7 μM, and 0.097μM, respectively). Besides their action against T. cruzi, these compounds were potent anti-Toxoplasma agents. In fact, they had ED50 values of 2.6 μM, 1.2 μM, and 1.8 μM, respectively against tachyzoites of T. gondii, while they exhibited a very potent inhibitory action against the target enzyme (TgFPPS) showing IC50 values of 0.024 μM, 0.025 μM, and 0.021 μM, respectively. With the exception of 37, all these compounds had equivalent potency to risedronate (ED50 = 2.4 μM against T. gondii and IC50 = 0.074 μM against TgFPPS) used as positive control. Compound 36 maintained the anti-Toxoplasma activity exhibited by 37–39 (ED50 = 0.97 μM against tachyzoites of T. gondii; ED50 = 0.069 μM against TgFPPS), but was devoid of anti-T. cruzi activity against either cells or TcFPPS. On the other hand, short chain length derivatives 31–35 exhibited some antiparasitic activity but to a lesser extent than 36–39.
Bisphosphonic acid derivatives bearing a sulfoxide moeity at the C-3 position were also potent anti-Toxoplasma agents, particularly those possessing long aliphatic chains such as 43–45, which were potent antiproliferative drugs against tachyzoites of T. gondii. They had EC50 values of 5.0 μM, 3.0 μM, and 1.4 μM, respectively. 45 showed a similar efficacy than risedronate under the same assay conditions. Above all, these compounds efficiently inhibited the enzymatic activity of the target enzyme (TgFPPS) at the very low nanomolar range showing IC50 values of 0.009 μM, 0.016 μM, and 0.056 μM, respectively. Except sulfoxide 45, which had moderate potency against intracellular T. cruzi (ED50 = 19.4 μM), the rest of lineal synthetic sulfoxides were devoid of biological activity against both T. cruzi cells and TcFPPS. The lack of biological activity against T. cruzi was somewhat unusual taking into account the inhibitory action shown by lineal closely related bisphosphonic acids.24–31 Sulfone 47 was just active against intracellular T. cruzi, while methylsulfonium 48 has proven to be an interesting antiparasitc agent exhibiting moderate antiproliferative action against both T. cruzi and T. gondii cells (ED50 = 32.0 μM and 7.0 μM, respectively, but behaved as a very potent drug against the target enzymes TcFPPS and TgFPPS (IC50 = 0.040 μM and 0.013 μM, respectively). Taking into account that the inhibitory activity does not always match with the ED50 values, it is necessary to consider that for cellular activity a compound has to cross the mammalian cell membrane, then, the parasite cell membrane, and finally has to reach the enzyme located in an organelle. At the concentrations used, no toxicity was associated to the title compounds. Assays were done with tissue culture cells infected with T. cruzi and toxicity on tissue culture cells could be seen easily if it happened (cells detach or show signs of necrosis). This was not observed and therefore the compounds have low or no toxicity against Vero cells. In addition, cytotoxicity studies (AlamarBlue™) of the most potent anti-Toxoplasma agents such as 36–39, 45 against hTerT cells, used as hosts of T. gondii tachyzoites, indicated that these compounds were almost devoid of toxicity. In fact, all of them showed high selectivity. Biological data are shown in Table 1.
Table.
Biological activity of sulfur containing 1,1-bisphosphonic acids against TcFPPS, TgFPPS, T. cruzi (amastigotes), and tachyzoites of T. gondii.
| Compound | TcFPPS IC50 (μM) | ED50 T. cruzi amastigotes | TgFPPS IC50 (μM) | ED50 T. gondii (μM) | Citotoxicity hTerT cells (μM)* |
|---|---|---|---|---|---|
| 18 | > 10 | > 20 | < 1.0 | 11.4 | |
| 21 | > 10 | > 20 | > 1.0 | > 10 | |
| 31 | 0.627 ± 0.433 | > 20 | 1.834 ± 0.145 | > 10 | |
| 32 | 0.925 ± 0.639 | > 20 | 0.066 ± 0.027 | > 10 | |
| 33 | 0.789 ± 0.531 | > 20 | 0.048 ± 0.016 | > 10 | |
| 34 | > 10 | > 20 | 0.110 ± 0.037 | > 10 | |
| 35 | > 10 | > 20 | 0.307 ± 0.202 | 6.5 | |
| 36 | > 10 | > 20 | 0.069 ± 0.046 | 0.97 | > 200 |
| 37 | 6.386 ± 0.847 | 15.8 | 0.024 ± 0.016 | 2.6 | > 1000 |
| 38 | 1.70 ± 0.186 | 12.8 | 0.025 ± 0.015 | 1.2 | ≥ 500 |
| 39 | 0.097 ± 1.494 | > 20 | 0.021 ± 0.014 | 1.8 | > 200 |
| 40 | > 10 | > 20 | > 10 | > 10 | |
| 41 | > 10 | > 20 | > 10 | > 10 | |
| 42 | > 10 | > 20 | > 10 | > 10 | |
| 43 | > 10 | > 20 | 0.009 ± 0.004 | 5.0 | |
| 44 | > 10 | > 20 | 0.016 ± 0.002 | 3.0 | |
| 45 | > 10 | 19.4 | 0.056 ± 0.028 | 1.4 | > 1000 |
| 46 | > 10 | > 20 | > 10 | 7.6 | |
| 47 | > 10 | > 20 | > 10 | > 10 | |
| 48 | 0.040 ± 0.016 | > 20 | 0.013 ± 0.006 | 7.0 | |
| Benznidazole | NDa | 1.7 ± 1.03 | NDa | NDa | |
| Risedronate | 0.027 ± 0.01 [68] | 55.0 ± 5.0 [68] | 0.074 ± 0.017 [30] | 2.4 ± 0.7 [29] |
ND = not determined
Values are maximal concentrations at which no toxicity was observed. DMSO controls showed toxicity at a concentration of ≥ 0.25% under similar conditions.
In summary, lineal sulfur-containing-1,1-bisphosphonic acids seem to be promising antiparasitic drugs that were able to inhibit efficiently the enzymatic activity of T. gondii farnesyl diphosphate synthase as well as T. gondii cells and to a lesser extent against TcFPPS and intracellular T. cruzi cells. This effect was more noticeable in compounds having a relative long aliphatic chain. Particularly, 38 and 39, which contain twelve and thirteen atoms in their lineal aliphatic chains, including one sulfur atom at C-3, exhibited potent activity against the target enzymes T. gondii and T. cruzi FPPS. As a consequence of these enzymatic activities, these bisphosphonic acid derivatives had the ability to control T. gondii (tachyzoites) and T. cruzi (amastigotes) proliferation. Sulfoxide derivatives were more selective towards TgFPPS and T. gondii cells as it was the case of 45. Compound 43 was an extremely potent inhibitor towards TgFPPS having an IC50 as low as 9 nM. Among the designed sulfur-containing 1,1-bisphosphonic acids, the methylsulfonium 48 was the most potent inhibitor of the enzymatic activity of the T. cruzi enzyme and also very efficient towards TgFPPS, both at the low nanomolar range. In order to optimize this new family of bisphosphonic acids, structural modifications at the aliphatic chain including branching and conformational restriction tools will be considered as the next step. Work aimed at exploiting the potential value of these sulfur-containing bisphosphonic acids is currently being pursued in our laboratory.
Experimental Section
The glassware used in air and/or moisture sensitive reactions was flame-dried and carried out under a dry argon atmosphere. Unless otherwise noted, chemicals were commercially available and used without further purification. Solvents were distilled before use. Methylene chloride and acetonitrile were distilled from phosphorus pentoxide.
Nuclear magnetic resonance spectra were recorded using a Bruker AM-500 MHz spectrometer. Chemical shifts are reported in parts per million (δ) relative to tetramethylsilane. Coupling constants are reported in Hertz. 13C NMR spectra were fully decoupled. Splitting patterns are designated as s, singlet; d, doublet; t, triplet; q, quartet.
High-resolution mass spectra were obtained using a Bruker micrOTOF-Q II spectrometer, which is a hybrid quadrupole time of flight mass spectrometer with MS/MS capability.
Melting points were determined using a Fisher-Johns apparatus and are uncorrected. IR spectra were recorded using a Nicolet Magna 550 spectrometer.
Column chromatography was performed with E. Merck silica gel plates (Kieselgel 60, 230–400 mesh). Analytical thin layer chromatography was performed employing 0.2 mm coated commercial silica gel plates (E. Merck, DC-Aluminum sheets, Kieselgel 60 F254).
As judged from the homogeneity of the 1H, 13C, 31P NMR spectra of the title compounds 31–48 and HPLC analyses of the committed intermediates 22–30 employing a Beckmann Ultrasphere ODS-2 column 5 μM, 250 × 10 mm eluting with acetonitrile–water (1:1) at 3.00 mL/min with a refractive index detector indicated a purity >97%.
1-[(n-Hept-2-ylamino)ethyl] 1,1-bisphosphonic Acid (18)
A solution of compound 16 (300 mg, 1.0 mmol) in anhydrous methylene chloride (10 mL) was treated with the 2-heptylamine (115 mg, 1.1 mmol) under an argon atmosphere. The reaction mixture was stirred at room temperature overnight. The solvent was evaporated and the residue was purified by column chromatography (silica gel) employing hexane–EtOAc (17:3) as eluent to afford tetraethyl ester 17. To a solution of this product in anhydrous methylene chloride (10 mL) was added dropwise trimethylsilyl bromide (12 equivalents) in an argon atmosphere. The reaction mixture was stirred at room temperature for 48 h. After cooling at 0 °C, anhydrous methanol (10 mL) was added, and the resulting mixture was allowed to reach room temperature. The solution was then concentrated under reduced pressure. The residue was dissolved in anhydrous methanol (10 mL) and subsequently concentrated under reduced pressure twice. The solvent was evaporated and the residue was crystallized from ethanol–water to afford pure 18 as a white solid: mp 165–166 °C; 1H NMR (500.13 MHz, D2O) δ 0.78 (t, J = 7.1 Hz, 3H, H-9), 1.21 (m, 4H, H-7, H-8), 1.22 (d, J = 6.6 Hz, 3H, CH3 at C-4), 1.30 (m, 2H, H-6), 1.50 (m, 1H, H-5a), 1.62 (m, 1H, H-5b), 2.35 (tt, J = 21.4, 7.3 Hz, 1H, H-1), 3.25 (dt, J = 13.2, 6.6 Hz, 2H, H-2), 3.41 (m, 2H, H-4); 13C NMR (125.77 MHz, D2O) δ 13.2 (C-9), 15.5 (CH3 at C-4), 21.7 (C-8), 24.0 (C-7), 30.6 (C-6), 32.7 (C-5), 36.1 (t, J = 120.8 Hz, C-1), 42.1 (C-2), 54.7 (C-4); 31P NMR (D2O) δ 16.01 mAB. HRMS (ESI) calcd for C9H24O6NP2 [M+H]+ 304.1079; found: 304.1062. Anal. Calcd. for (C9H23O6NP2.1.25H2O): C, 33.18; H, 7.89; N, 4.30. Found C, 33.13; H, 7.27; N, 3.98.
1-[(n-Heptylamino)ethyl]-1-hydroxy-1,1-bisphosphonic Acid (21)
To a solution of heptylamine (1.00 g, 8.7 mmol) in acetonitrile (15 mL) cooled at 0 °C was added dropwise benzyl bromoacetate (1.99 g, 8.9 mmol). Then, triethylamine (2.4 mL, 17.3 mmol) and the reaction mixture was stirred overnight. The solvent was evaporated and the residue was purified by column chromatography (sílica gel) eluting with a mixture of hexane–EtOAc (19:1) to afford 1.919 g (84% yield) of pure 19 as a colorless oil. A solution of benzyl ester 19 (1.919 g, 7.2 mmol) in ethyl acetate (50 mL) in the presence of palladium on charcoal (50 mg) was treated with hydrogen at 3 atm in a Parr apparatus. The reaction mixture was shaken for 6 h and the mixture was filtered through a fritted glass funnel. The solvent was evaporated to give 847 mg (67% yield) of pure 20 as a white solid that was used in the next step without further purification: mp = 187–191 °C; 1H NMR (500.13 MHz, CD3OD) δ 0.91 (t, J = 7.0 Hz, 3H, H-10), 1.32 (m, 4H, H-8, H-9), 1.37 (m, 4H, H-6, H-7), 1.67 (p, J = 7.5 Hz, 2H, H-5), 2.97 (m, 2H, H-4); 3.46 (s, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.6 (C-10), 22.2 (C-9), 28.5 (C-8), 28.7 (C-7), 29.4 (C-6), 31.3 (C-5), 47.4 (C-4), 49.2 (C-2), 169.5 (C-1). Anal. Calcd. for (C9H19O2N): C, 62.39; H, 11.05; N, 8.08. Found C, 62.05; H, 10.62; N, 7.74. To a flame dried 100 mL three neck flask having an addition funnel and a reflux condenser through which was circulated water at 0 °C was added the carboxylic acid 20 (500 mg, 2.9 mmol), H3PO3 acid (273 mg, 2.9 mmol), and anhydrous benzenesulfonic acid (1.0 g, 6.3 mmol) under argon atmosphere. The reaction mixture was heated to 65 °C, then PCl3 (500 μL, 5.8 mmol) was added dropwise with vigorous stirring. The reaction was stirred at 65 °C for 16 h. The reaction was allowed to cool to room temperature. Cold water (60 mL) was added and the reaction was stirred at 100 °C for 5 h. The reaction was cooled to room temperature and the pH was adjust to 4.3 with a 50% aqueous NaOH solution. Acetone (20 mL) was added, and the resulting mixture was cooled to 0 °C for 24 h. The product was filtrated and crystallized with water-ethanol. mp 155–159 °C; 1H NMR (500.13 MHz, CDCl3) δ 0.90 (t, J = 7.0 Hz, 3H, H-10), 1.18 (m, 6H, H-7, H-8, H-9), 1.24 (m, 2H, H-6), 1.60 (p, J = 7.4 Hz, 2H, H-5), 3.01 (t, J = 7.6 Hz, 2H, H-4), 3.39 (t, J = 11.7 Hz, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.3 (C-10), 21.8 (C-9), 25.3 (C-8), 25.5 (C-7), 27.8 (C-6), 30.7 (C-5), 48.3 (C-2), 49.9 (C-4), 70.3 (t, J = 137.7 Hz, C-1); 31P NMR (D2O) δ 15.31. HRMS (ESI) calcd for C9H24O7NP20 [M+H]+ 320.1030; found: 320.1037. Anal. Calcd. for (C9H23O7NP2.1.50H2O): C, 31.22; H, 7.57; N, 4.05. Found C, 31.53; H, 7.75; N, 4.36.
Synthesis of tetraethyl 2-[(alkylthio)ethyl] 1,1-bisphosphonates
General procedure
To a solution of tetraethyl ethenylidenbisphosphonate (16; 300 mg, 1 mmol) in anhydrous dichloromethane (10 mL) was added triethylamine (1 mmol) and the corresponding alkylmercaptane (1 mmol). The reaction mixture was stirred at room temperature for 1h. Water (20 mL) was added, and the mixture was extracted with dichloromethane (3 × 10 mL). The combined organic layers were washed with brine (20 mL), dried on sodium sulfate and the solvent was evaporated.
Tetraethyl 1-[(Ethylthio)ethyl] 1,1-bisphosphonate (22)
98% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 1.29 (t, J = 7.3 Hz, 3H, H-5), 1.37 (t, J = 7.0 Hz, 12H, H-2′), 2.61 (q, J = 7.3 Hz, 2H, H-4), 2.62 (tt, J = 23.9, 5.9 Hz, 1H, H-1), 3.07 (dt, J = 16.3, 5.7 Hz, 2H, H-2), 4.23 (m, 8H, H-2′); 13C NMR (125.77 MHz, CDCl3) δ 14.5 (C-5), 16.4 (d, J = 6.8 Hz, C-2′), 26.9 (C-4), 27.2 (t, J = 4.9 Hz, C-2), 39.1 (t, J = 131.6 Hz, C-1), 62.8 (dd, J = 18.6, 6.8 Hz, C-1′); 31P NMR (202.46 MHz, CDCl3) δ 21.74. HRMS (ESI) calcd for C12H28O6P2S [M+H]+ 363.1160; found: 363.1163.
Tetraethyl 1-[(n-Prop-1-ylthio)ethyl] 1,1-bisphosphonate (23)
95% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.99 (t, J = 7.3 Hz, 3H, H-6), 1.35 (t, J = 7.1 Hz, 12H, H-2′), 1.63 (sext, 2H, H-5), 2.55 (t, J = 7.3 Hz, 2H, H-4), 2.59 (tt, J = 24.0, 5.9 Hz, 1H, H-1), 3.04 (dt, J = 16.3, 5.9 Hz, 2H, H-2), 4.20 (m, 8H, H-2′); 13C NMR (125.77 MHz, CDCl3) δ 13.4 (C-6), 16.3 (d, J = 5.9 Hz, C-2′), 22.7 (C-5), 27.7 (t, J = 5.4 Hz, C-2), 35.1 (C-4), 39.1 (t, J = 131.1 Hz, C-1), 62.8 (dd, J = 18.6, 6.8 Hz, C-1′); 31P NMR (202.46 MHz, CDCl3) δ 21.75. HRMS (ESI) calcd for C13H30O6P2S [M+H]+ 377.1317; found: 377.1326.
Tetraethyl 1-[(n-But-1-ylthio)ethyl] 1,1-bisphosphonate (24)
88% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.91 (t, J = 7.4 Hz, 3H, C-7), 1.35 (t, J = 7.1 Hz, 12H, H-2′), 1.40 (sext, J = 7.4 Hz, 2H, H-6), 1.58 (p, J = 7.4 Hz, 2H, H-5), 2.57 (t, J = 7.5 Hz, 2H, H-4), 2.59 (tt, J = 24.0, 6.0 Hz, 1H, H-1), 3.04 (dt, J = 16.3, 5.8 Hz, 2H, H-2), 4.20 (m, 8H, H-2′); 13C NMR (125.77 MHz, CDCl3) δ 13.6 (C-7), 16.4 (d, J = 6.6 Hz, C-2′), 21.9 (C-6), 27.7 (t, J = 4.9 Hz, C-2), 31.5 (C-5), 32.8 (C-4), 39.1 (t, J = 131.6 Hz, C-1), 62.8 (dd, J = 18.6, 6.8 Hz, C-1′); 31P NMR (202.46 MHz, CDCl3) δ 21.8. HRMS (ESI) calcd for C14H32O6P2S [M+H]+ 391.1473; found: 391.1476.
Tetraethyl 1-[(n-Pent-1-ylthio)ethyl] 1,1-bisphosphonate (25)
72% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.90 (t, J = 7.1 Hz, 3H, H-8), 1.32 (m, 4H, H-6, H-7), 1.35 (t, J = 7.1 Hz, 12H, H-2′), 1.60 (p, J = 7.4 Hz, 2H, H-5), 2.56 (t, J = 7.4 Hz, 2H, H-4), 2.59 (tt, J = 23.7, 5.6 Hz, 1H, H-1), 3.04 (dt, J = 16.3, 5.9 Hz, 2H, H-2), 4.20 (m, 8H, H-2′); 13C NMR (125.77 MHz, CDCl3) δ 13.9 (C-8), 16.4 (d, J = 5.9 Hz, C-2′), 22.3 (C-7), 27.7 (t, J = 5.0 Hz, C-2), 29.1 (C-6), 31.0 (C-5), 33.1 (C-4), 39.1 (t, J = 131.2 Hz, C-1), 62.8 (dd, J = 18.7, 6.8 Hz, C-1′); 31P NMR (202.46 MHz, CDCl3) δ 21.77. HRMS (ESI) calcd for C15H34O6P2S [M+Na]+ 427.1449; found: 427.1457.
Tetraethyl 1-[(n-Hex-1-ylthio)ethyl] 1,1-bisphosphonate (26)
94% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.88 (t, J = 7.0 Hz, 3H, H-9), 1.29 (m, 6H, H-6, H-7, H-8), 1.35 (t, J = 7.0 Hz, 12H, H-2′), 1.59 (p, J = 7.4 Hz, 2H, H-5), 2.56 (t, J = 7.4 Hz, 2H, H-4), 2.59 (tt, J = 24.0, 6.0 Hz, 1H, H-1), 3.04 (dt, J = 16.3, 5.8 Hz, 2H, H-2), 4.20 (m, 8H, H-2′); 13C NMR (125.77 MHz, CDCl3) δ 14.0 (C-9), 16.4 (d, J = 5.9 Hz, C-2′), 22.5 (C-8), 27.8 (t, J = 4.9 Hz, C-2), 28.5 (C-7), 29.4 (C-6), 31.4 (C-5), 33.2 (C-4), 39.1 (t, J = 131.4 Hz, C-1), 62.8 (dd, J = 18.8, 6.7 Hz, C-1′); 31P NMR (202.46 MHz, CDCl3) δ 21.77. HRMS (ESI) calcd for C16H37O6P2S [M+H]+ 419.1786; found 419.1792.
Tetraethyl 1-[(n-Hept-1-ylthio)ethyl] 1,1-bisphosphonate (27)
87% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.88 (t, J = 7.0 Hz, 3H, H-10), 1.29 (m, 8H, H-6, H-7, H-8, H-9), 1.35 (t, J = 7.1 Hz, 12H, H-2′), 1.59 (p, J = 7.4 Hz, 2H, H-5), 2.56 (t, J = 7.5 Hz, 2H, H-4), 2.59 (tt, J = 24.2, 6.0 Hz, 1H, H-1), 3.04 (dt, J = 16.2, 5.6 Hz, 2H, H-2), 4.21 (m, 8H, H-2′); 13C NMR (125.77 MHz, CDCl3) δ 14.1 (C-10), 16.4 (d, J = 5.9 Hz, C-2′), 22.6 (C-9), 27.8 (t, J = 4.9 Hz, C-2), 28.8 (C-8), 28.9 (C-7), 29.4 (C-6), 31.7 (C-5), 33.2 (C-4), 39.1 (t, J = 133.0 Hz, C-1), 62.8 (dd, J = 19.4, 6.3 Hz, C-1′); 31P NMR (202.46 MHz, CDCl3) δ 21.77. HRMS (ESI) calcd for C17H39O6P2S [M+H]+ 433.1943; found 433.1952.
Tetraethyl 1-[(n-Oct-1-ylthio)ethyl] 1,1-bisphosphonate (284)
83% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.88 (t, J = 7.0 Hz, 3H, H-11), 1.28 (m, 10H, H-6, H-7, H-8, H-9, H-10), 1.35 (t, J = 7.1 Hz, 12H, H-2′), 1.59 (p, J = 7.4 Hz, 2H, H-5), 2.56 (t, J = 7.5 Hz, 2H, H-4), 2.59 (tt, J = 24.0, 6.0 Hz, 1H, H-1), 3.04 (dt, J = 16.2, 5.9 Hz, 2H, H-2), 4.21 (m, 8H, H-2′); 13C NMR (125.77 MHz, CDCl3) δ 14.1 (C-11), 16.4 (d, J = 5.9 Hz, C-2′), 22.6 (C-10), 27.8 (t, J = 4.9 Hz, C-2), 28.9 (C-9), 29.2 (C-7, C-8), 29.4 (C-6), 31.8 (C-5), 33.2 (C-4), 39.1 (t, J = 130.1 Hz, C-1), 62.8 (dd, J = 19.0, 6.7 Hz, C-1′); 31P NMR (202.46 MHz, CDCl3) δ 21.77. HRMS (ESI) calcd. for C18H41O6P2S [M+H]+ 447.2099; found 447.2107.
Tetraethyl 1-[(n-Non-1-ylthio)ethyl] 1,1-bisphosphonate (29)
90% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.89 (t, J = 7.0 Hz, 3H, H-12), 1.26 (m, 12H, -CH2-), 1.35 (t, J = 7.1 Hz, 12H, H-2′), 1.59 (p, J = 7.4 Hz, 2H, H-5), 2.55 (t, J = 7.4 Hz, 2H, H-4), 2.59 (tt, J = 23.9, 5.8 Hz, 1H, H-1), 3.04 (dt, J = 16.3, 5.8 Hz, 2H, H-2), 4.20 (m, 8H, H-2′); 13C NMR (125.77 MHz, CDCl3) δ 14.1 (C-12), 16.4 (d, J = 5.9 Hz, C-2′), 22.7 (C-11), 27.8 (t, J = 4.9 Hz, C-2), 28.9 (C-10), 29.2 (C-8, C-9), 29.4 (C-7), 29.5 (C-6), 31.8 (C-5), 33.2 (C-4), 39.1 (t, J = 131.1 Hz, C-1), 62.8 (dd, J = 18.6, 6.8 Hz, C-1′); 31P NMR (202.46 MHz, CDCl3) δ 21.77. HRMS (ESI) calcd for C19H43O6P2S [M+H]+ 461.2256; found 461.2256.
Tetraethyl 1-[(n-Dec-1-ylthio)ethyl] 1,1-bisphosphonate (30)
68% yield; colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.88 (t, J = 7.0 Hz, 3H, H-13), 1.26 (m, 14H, -CH2-), 1.35 (t, J = 7.1 Hz, 12H, H-2′), 1.59 (p, J = 7.5 Hz, 2H, H-5), 2.56 (t, J = 7.3 Hz, 2H, H-4), 2.59 (tt, J = 23.9, 5.8 Hz, 1H, H-1), 3.04 (dt, J = 16.3, 5.8 Hz, 2H, H-2), 4.21 (m, 8H, H-2′); 13C NMR (125.77 MHz, CDCl3) δ 14.1 (C-13), 16.4 (d, J = 5.9 Hz, C-2′), 22.7 (C-12), 27.8 (t, J = 4.9 Hz, C-2), 28.9 (C-11), 29.2 (C-10), 29.3 C-9), 29.4 (C-8), 29.53 (C-7), 29.54 (C-6), 31.9 (C-5), 33.2 (C-4), 39.1 (t, J = 131.1 Hz, C-1), 62.8 (dd, J = 18.6, 6.8 Hz, C-1′); 31P NMR (202.46 MHz, CDCl3) δ 21.78. HRMS (ESI) calcd for C20H45O6P2S [M+H]+ 475.2412; found 475.2412.
Synthesis of 2-(alkylthio)ethyl-1,1-bisphosphonic acids (31–39)
General procedure
A solution of the corresponding tetraethyl 2-[(alkylthio)ethyl] 1,1-biphosphonate (1 mmol) in anhydrous methylene chloride (10 mL) was treated with trimethylsilyl bromide (10 equiv.) under an argon atmosphere. The reaction mixture was stirred at room temperature for 48 h. Then, methanol (1.0 mL) was added and the solvent was evaporated. The residue was dissolved in methanol (8 mL) and the mixture was stirred at room temperature for 24 h. The solvent was evaporated and the residue redissolved/evaporated in methanol four times, to complete the hydrolysis of remaining trimethylsilyl bromide and eliminate the hydrobromic acid created. The residue was purified by column chromatography on reverse phase with a mixture of water–methanol as eluent and the pure compound was obtained after lyophilization. Yields are reported relatively to compound 16.
1-[(Ethylthio)ethyl]-1,1-bisphosphonic Acid (31)
10% yield; Amorphous solid; 1H NMR (500.13 MHz, D2O) δ 1.17 (t, J = 7.4 Hz, 3H, H-5), 2.32 (tt, J = 22.3, 6.6 Hz, 1H, H-1), 2.55 (q, J = 7.4 Hz, 2H, H-4), 2.95 (dt, J = 15.6, 6.9 Hz, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.7 (C-5), 25.9 (C-4), 26.8 (t, J = 3.9 Hz, C-2), 39.3 (t, J = 120.3 Hz, C-1), 31P NMR (202.46 MHz, CDCl3) δ 19.54. HRMS (ESI) calcd for C4H12O6P2S [M+Na]+ 272.9728; found 272.9728.
1-[(n-Propylthio)ethyl]-1,1-bisphosphonic Acid (32)
50% yield; Amorphous solid; 1H NMR (500.13 MHz, D2O) δ 0.87 (t, J = 7.3 Hz, 3H, H-6), 1.53 (sxt, J = 7.3 Hz, 2H, H-5), 2.42 (tt, J = 22.8, 6.5 Hz, 1H, H-1), 2.51 (t, J = 7.3 Hz, 2H, H-4), 2.94 (dt, J = 15.9, 6.5 Hz, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 12.7 (C-6), 22.0 (C-5), 27.1 (t, J = 4.5 Hz, C-2), 34.1 (C-4), 39.3 (t, J = 122.6 Hz, C-1); 31P NMR (202.46 MHz, D2O) δ 20.12. HRMS (ESI) calcd for C5H15O6P2S [M+H]+ 265.0065; found 265.0062.
1-[(n-Butylthio)ethyl]-1,1-bisphosphonic Acid (33)
Amorphous solid; 1H NMR (500.13 MHz, D2O) δ 0.81 (t, J = 7.4 Hz, 3H, H-7), 1.31 (sxt, J = 7.4 Hz, 2H, H-6), 1.51 (p, J = 7.4 Hz, 2H, H-5), 2.39 (tt, J = 22.6, 6.6 Hz, 1H, H-1), 2.55 (t, J = 7.4 Hz, 2H, H-4), 2.94 (dt, J = 15.8, 6.4 Hz, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 12.8 (C-7), 21.3 (C-6), 27.2 (t, J = 4.3 Hz, C-2), 30.6 (C-5), 31.7 (C-4), 39.3 (t, J = 121.0 Hz, C-1); 31P NMR (202.46 MHz, D2O) δ 19.97. HRMS. calcd for C6H16O6P2S [M+Na]+: 301.0041; found 301.0039.
1-[(n-Pentylthio)ethyl]-1,1-bisphosphonic Acid (34)
44% yield; Amorphous solid; 1H NMR (500.13 MHz, D2O) δ 0.79 (t, J = 7.3 Hz, 3H, H-8), 1.25 (m, 4H, -CH2-), 1.53 (p, J = 7.3 Hz, 2H, H-5), 2.42 (tt, J = 22.8, 6.3 Hz, 1H, H-1), 2.54 (t, J = 7.6 Hz, 2H, H-4), 2.94 (dt, J = 15.8, 6.6 Hz, 2H, H-2); 13C NMR (125.77 MHz, DMSO) δ 13.2 (C-8), 21.6 (C-7), 27.1 (t, J = 4.9 Hz, C-2), 28.2 (C-6), 30.3 (C-5), 32.0 (C-4), 39.3 (t, J = 121.8 Hz, C-1); 31P NMR (202.46 MHz, D2O) δ 20.12. HRMS (ESI) calcd for C7H18O6P2SNa [M+Na]+ 315.0197; found 315.0192.
1-[(n-Hexylthio)ethyl]-1,1-bisphosphonic Acid (35)
Purificartion by column chromatography (C-18 silica gel) eluting with methanol–water (9:1) afforded 129 mg of 35 (41% yield) as an amorphous solid: 1H NMR (500.13 MHz, D2O) δ 0.78 (t, J = 7.0 Hz, 3H, H-9), 1.21 (m, 4H, -CH2-), 1.30 (p, J = 7.1 Hz, 2H, H-6), 1.53 (p, J = 7.4 Hz, 2H, H-5), 2.41 (tt, J = 22.8, 6.5 Hz, 1H, H-1), 2.54 (t, J = 7.4 Hz, 2H, H-4), 2.94 (dt, J = 15.8, 6.6 Hz, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.3 (C9), 21.9 (C-8), 27.3 (t, J = 4.1 Hz, C-2), 27.7 (C-7), 28.5 (C-6), 30.6 (C-5), 32.0 (C-4), 39.3 (t, J = 121.7 Hz, C-1); 31P NMR (202.46 MHz, D2O) δ 20.06. HRMS (ESI) calcd for C8H21O6P2S [M+H]+ 307.0534; found 307.0522.
1-[(n-Heptylthio)ethyl]-1,1-bisphosphonic Acid (36)
Purificartion by column chromatography (C-18 silica gel) eluting with methanol–water (7:3) afforded 83 mg of pure 36 as an amorphous solid: 1H NMR (500.13 MHz, D2O) δ 0.78 (t, J = 7.0 Hz, 3H, H-10), 1.20 (m, 6H, -CH2-), 1.30 (p, J = 7.0 Hz, 2H, H-6), 1.54 (p, J = 7.4 Hz, 2H, H-5), 2.38 (m, 1H, H-1), 2.54 (t, J = 7.4 Hz, 2H, H-4), 2.94 (dt, J = 15.8, 6.5 Hz, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.4 (C-10), 22.0 (C-9), 27.2 (t, J = 3.9 Hz, C-2), 28.0 (C-7, C-8), 28.5 (C-6), 31.0 (C-5), 32.1 (C-4), 39.3 (t, J = 122.3 Hz, C-1); 31P NMR (202.46 MHz, D2O) δ 19.97. HRMS (ESI) calcd for C9H22O6P2SNa [M+Na]+ 343.0510; found 343.0591.
1-[(n-Octylthio)ethyl]-1,1-bisphosphonic Acid (37)
Purificartion by column chromatography (C-18 silica gel) eluting with methanol–water (7:3) afforded 65 mg of pure 37 as a syrup: 1H NMR (500.13 MHz, D2O) δ 0.78 (t, J = 7.0 Hz, 3H, H-11), 1.22 (m, 8H, -CH2-), 1.31 (p, J = 6.9 Hz, 2H, H-6), 1.53 (p, J = 7.5 Hz, 2H, H-5), 2.36 (tt, J = 22.5, 6.6 Hz, 1H, H-1), 2.48 (2.55 (t, J = 7.4 Hz, 2H, H-4), 2.94 (dt, J = 15.7, 6.6 Hz, 2H, H-2); 13C NMR (125.77 MHz, DMSO) δ 13.4 (C-11), 22.0 (C-10), 27.3 (t, J = 3.9 Hz, C-2), 28.0 (C-9), 28.25 (C-8). 28.32 (C-7), 28.5 (C-6), 31.1 (C-5), 32.1 (C-4), 39.4 (t, J = 119.4 Hz, C-1); 31P MR (202.46 MHz, D2O) δ 19.80. HRMS (ESI) calcd for C10H24O6P2SNa [M+Na]+ 357.0667; found 357.0671.
1-[(n-Nonylthio)ethyl]-1,1-bisphosphonic Acid (38)
The product was purified by column chromatography (C-18 silica gel) eluting with methanol–water (1:1) to afford 66 mg (19% yield) of pure compound 38 as an amorphous solid: 1H NMR (500.13 MHz, CD3OD) δ 0.89 (t, J = 7.0 Hz, 3H, H-12), 1.30 (m, 10H, -CH2-), 1.40 (p, J = 7.2 Hz, 2H, H-6), 1.60 (p, J = 7.4 Hz, 2H, H-5), 2.44 (tt, J = 23.2, 6.0 Hz, 1H, H-1), 2.58 (t, J = 7.4 Hz, 2H, H-4), 3.03 (dt, J = 16.2, 6.2 Hz, 2H, H-2); 13C NMR (125.77 MHz, CD3OD) δ 14.4 (C-12), 23.7 (C-11), 28.3 (t, J = 4.4 Hz, C-2), 29.9 (C-10), 30.4 (C-9), 30.4 (C-8), 30.6 (C-7), 30.7 (C-6), 33.1 (C-5), 33.6 (C-4), 41.5 (t, J = 125.7 Hz, C-1); 31P NMR (202.46 MHz, D2O) δ 20.48. HRMS (ESI) calcd for C11H27O6P2S [M+H]+ 349.1004; found 349.1010.
1-[(n-Decylthio)ethyl]-1,1-bisphosphonic Acid (39)
Amorphous solid; 1H NMR (125.77 MHz, DMSO-d6) δ 0.84 (t, J = 7.0 Hz, 3H, H-13), 1.23 (m, 12H, -CH2-), 1.30 (p, J = 7.5 Hz, 2H, H-6), 1.49 (p, J = 7.3 Hz, 2H, H-5), 2.12 (tt, J = 22.5, 6.0 Hz, 1H, H-1), (t, J = 6.9 Hz, 2H, H-4), 2.85 (dt, J = 15.6, 6.1 Hz, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.9 (C-13), 22.7 (C-12), 27.5 (t, J = 3.9 Hz, C-2), 29.1 (C-10, C-11), 29.3 (C-8, C-9), 29.6 (C-7), 30.0 (C-6), 32.1 (C-5), 32.6 (C-4), 39.3 (t, J = 127.2 Hz, C-1); 31P NMR (202.46 MHz, D2O) δ 20.28. HRMS (ESI) calcd for C12H29O6P2S [M+H]+ 363.1160; found 363.1161.
Synthesis of 2-(alkylsulfinyl)ethyl-1,1-bisphosphonic acids (40–46)
General procedure
To a solution of the corresponding 2-(alkylthio)ethyl-1,1-biphosphonic acid (1 mmol) in deuterated water (2 mL) was added 30% hydrogen peroxide dropwise (1 mmol) and the mixture is stirred at room temperature. The reaction was monitored by proton NMR until the reaction was complete. The reaction mixture was freezed and lyophilized. The product was purified by column chromatography (reverse phase C-18 silica gel) eluting with water. Purity was determined not only by homogeneity (>95%) of the NMR data, but also by HPLC analysis eluting with a mixture of water methanol (4:1) employing a reversed phase column (250 × 10 mm).
1-[(n-Butylsulfinyl)ethyl]-1,1-bisphosphonic Acid (40)
Amorphous solid; 1H NMR (500.13 MHz, D2O) δ 0.85 (t, J = 7.4 Hz, 3H, H-7), 1.40 (m, 2H, H-6), 1.66 (p, J = 7.6 Hz, 2H, H-5), 2.59 (m, 1H, H-1), 2.79 (m, 1H, H-4a), 2.93 (m, 1H, H-4b), 3.18 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 12.8 (C-7), 21.2 (C-6), 23.9 (C-5), 33.3 (t, J = 123.2 Hz, C-1), 48.4 (t, J = 3.9 Hz, C-2), 51.2 (C-4); 31P NMR (202.46 MHz, D2O) δ 17.81 mAB. HRMS (ESI) calcd for C6H16O7P2SNa [M+Na]+ 316.9990; found 316.9985.
1-[(n-Pentylsulfinyl)ethyl]-1,1-bisphosphonic Acid (41)
Amorphous solid p; 1H NMR (500.13 MHz, D2O) δ 0.78 (t, J = 7.3 Hz, 3H, H-8), 1.24 (sxt, J = 7.2 Hz, 2H, H-7), 1.34 (m, 2H, H-6), 1.66 (m, 2H, H-5), 2.59 (m, 1H, H-1), 2.76 (ddd, J = 13.4, 8.1, 5.6 Hz, 1H, H-4a), 2.90 (ddd, J = 13.4, 8.5, 7.6 Hz, 1H, H-4b), 3.16 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.0 (C-8), 21.45 (C-7), 21.48 (C-5), 30.0 (C-6), 33.1 (t, J = 124.7 Hz, C-1), 48.2 (t, J = 3.8 Hz, C-2), 51.5 (C-4); 31P NMR (202.46 MHz, D2O) δ 18.02 mAB. HRMS (ESI) calcd for C7H18O7P2SNa [M+Na]+ 331.0146; found 331.0140.
1-[(n-Hexylsulfinyl)ethyl]-1,1-bisphosphonic Acid (42)
Amorphous solid; 1H NMR (500.13 MHz, D2O) δ 0.76 (t, J = 7.1 Hz, 3H, H-9), 1.21 (m, 4H, H-8, H-7), 1.37 (m, 2H, H-6), 1.65 (m, 2H, H-5), 2.61 (m, 1H, H-1), 2.78 (m, 1H, H-4a), 2.91 (m, 1H, H-4b), 3.16 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.2 (C-9), 21.7 (C-8), 21.8 (C-5), 27.4 (C-6), 30.4 (C-7), 33.2 (t, J = 124.1 Hz, C-1), 48.3 (t, J = 3.7 Hz, C-2), 51.6 (C-4); 31P NMR (202.46 MHz, D2O) δ 17.99 mAB. HRMS (ESI) calcd for C8H20O7P2SNa [M+Na]+ 345.0303; found 345.0291.
1-[(n-Heptylsulfinyl)ethyl]-1,1-bisphosphonic Acid (43)
Amorphous solid; 1H NMR (500.13 MHz, D2O) δ 0.79 (t, J = 6.9 Hz, 3H, H-10), 1.17 (m, 4H, -CH2-), 1.24 (p, J = 7.0 Hz, 2H, H-7), 1.35 (m, 2H, H-6), 1.65 (m, 2H, H-5), 2.61 (m, 1H, H-1), 2.76 (ddd, J = 13.6, 8.1, 5.8 Hz, 1H, H-4a), 2.89 (ddd, J = 13.5, 8.4, 7.7 Hz, 1H, H-4b), 3.15 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.3 (C-10), 21.79 (C-9), 21.84 (C-5), 27.6 (C-6), 27.8 (C-7), 30.7 (C-8), 33.1 (t, J = 124.6 Hz, C-1), 48.2 (t, J = 3.9 Hz, C-2), 51.6 (C-4); 31P NMR (202.46 MHz, D2O) δ 18.03 mAB. HRMS (ESI) calcd for C9H22O7P2SNa [M+Na]+ 359.0459; found 359.0441.
1-[(n-Octylsulfinyl)ethyl]-1,1-bisphosphonic Acid (44)
Amorphous solid; 1H NMR (500.13 MHz, D2O) δ 0.79 (t, J = 7.0 Hz, 3H, H-11), 1.22 (m, 6H, -CH2-), 1.29 (p, J = 7.0 Hz, 2H, H-7), 1.41 (m, 2H, H-6), 1.70 (m, 2H, H-5), 2.34 (ddt, J = 21.0, 9.0, 5.9 Hz, 1H, H-1), 2.77 (ddd, J = 13.4, 8.5, 5.9 Hz, 1H, H-4a), 2.94 (ddd, J = 13.3, 9.0, 7.4 Hz, 1H, H-4b), 3.15 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.5 (C-11), 22.1 (C-10), 22.2 (C-5), 28.0 (C-6), 28.4 (C-7), 28.5 (C-8), 31.3 (C-9), 33.1 (t, J = 124.8 Hz, C-1), 48.3 (C-2), 51.7 (C-4); 31P NMR (202.46 MHz, D2O) δ 16.36 (d, J = 99.4 Hz). HRMS (ESI) calcd for C10H24O7P2SNa [M+Na]+ 373.0616; found 373.0595.
1-[(n-Nonylsulfinyl)ethyl]-1,1-bisphosphonic Acid (45)
Amorphous solid; 1H NMR (500.13 MHz, D2O) δ 0.77 (t, J = 7.0 Hz, 3H, H-12), 1.19 (m, 8H, -CH2-), 1.27 (p, J = 6.8 Hz, 2H, H-7), 1.38 (m, 2H, H-6), 1.96 (m, 2H, H-5), 2.42 (m, 1H, H-1), 2.81 (ddd, J = 13.4, 8.5, 5.8 Hz, 1H, Hs4a), 2.92 (ddd, J = 13.3, 9.0, 7.4 Hz, 1H, H-4b), 3.15 (m, 2H, H-2); 31P NMR (202.46 MHz, D2O) δ 16.99 mAB. HRMS (ESI) Calcd. for C11H27O7P2S [M+H]+ Calcd 365.0953. Found 365.0936.
1-[(n-Decylsulfinyl)ethyl]-1,1-bisphosphonic Acid (46)
Amorphous solid; 1H NMR (500.13 MHz, D2O) δ 0.82 (t, J = 7.0 Hz, 3H, H-13), 1.24 (m, 10H, -CH2-), 1.33 (p, J = 7.0 Hz, 2H, H-7), 1.44 (m, 2H, H-6), 1.74 (m, 2H, H-5), 2.38 (m, 1H, H-1), 2.81 (ddd, J = 13.6, 8.5, 5.5 Hz, 1H, H-4a), 2.98 (ddd, J = 13.2, 8.9, 7.4 Hz, 1H, H-4b), 3.20 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.8 (C-13), 22.6 (C-12), 22.7 (C-5), 28.7 (C-6, C-7), 29.4 (C-10), 29.6 (C-8), 29.7 (C-9), 31.9 (C-11), 33.0 (t, J = 128.1 Hz, C-1), 48.4 (C-2), 51.9 (C-4); 31P NMR (202.46 MHz, D2O) δ 14.15. HRMS (ESI) Calcd. for C12H28O7P2SNa [M+Na]+ Calcd 401.0929. Found 401.0904.
1-[(n-Pentylsulfonyl)ethyl]-1,1-biphosphonic Acid (47)
Amorphous solid; 1H NMR (500.13 MHz, D2O) δ 0.82 (t, J = 7.2 Hz, 3H, H-8) 1.28 (m, 2H, H-7), 1.36 (m, 2H, H-6), 1.78 (p, J = 7.7 Hz, 2H, H-5) 2.63 (tt, J = 22.8, 5.1 Hz, 2H, H-1), 3.27 (m, 2H, H-4), 3.59 (dt, J = 23.8, 5.2 Hz, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 12.9 (C-8), 21.3 (C-7), 21.4 (C-6), 29.8 (C-5), 33.2 (t, J = 122.9 Hz, C-1), 48.3 (t, J = 3.7 Hz, C-2), 51.3 (C-4); 31P NMR (202.46 MHz, D2O) δ 17.49. HRMS (ESI) Calcd. for C7H18O8P2SNa [M+Na]+ Calcd 347.0095. Found 347.0100.
(2,2-Diphosphonoethyl)(methyl)(octyl)sulfonium Tetrafluoroborate (48)
To a mixture of compound 37 (295 mg, 0.88 mmol), iodomethane (0.6 mL) in acetonitrile (20 mL) was added silver tetrafluoroborate (150 mg, 0.88 mmol) under argon atmosphere. The reaction mixture was stirred at room temperature for 2 h. The solvent was evaporated and the product was purified by column chromatography (C-18 silica gel) eluting with methanol to afford 131 mg (36% yield) of pure 48 as a white solid: mp = 96–97 °C; 1H NMR (500.13 MHz, D2O) δ 0.78 (t, J = 6.6 Hz, 3H, H-11), 1.20 (m, 8H, -CH2-), 1.28 (p, J = 6.8 Hz, 2H, H-7), 1.41 (p, J = 7.2 Hz, 2H, H-6), 1.76 (m, 2H, H-5), 2.51 (m, 1H, H-1), 2.86 (s, 3H, S(+)CH3), 3.19 (ddd, J = 12.3, 8.9, 6.3 Hz, 1H, H-4a), 3.35 (ddd, J = 12.3, 9.1, 7.0 Hz, 1H, H-4b), 3.50 (m, 1H, H-2a), 3.59 (m, 1H, H-2b); 13C NMR (125.77 MHz, D2O) δ 13.3 (C-11), 21.93 (C-10), 23.12 (C-5), 23.17 (S(+)CH3), 27.5 (C-6), 27.9 (C-7), 28.0 (C-8), 30.9 (C-9), 35.6 (t, J = 118.5 Hz, C-1), 40.8 (t, J = 3.2 Hz, C-2), 42.7 (C-4); 31P NMR (202.46 MHz, D2O) δ 14.59. HRMS (ESI) calcd for C11H27O6P2S [M]+ 349.1004; found 349.1008.
Drug Screening
T. cruzi amastigote assays
Gamma-irradiated (2,000 Rads) Vero cells (3.4 × 104 cells/well) were seeded in 96 well plates (black, clear bottom plates from Greiner Bio-One) in 100 μL RPMI media (Sigma) with 10% FBS. Plates were incubated overnight at 35 °C and 7% CO2. After overnight incubation, Vero cells were challenged with 3.4 × 105 trypomastigotes/well (CL strain overexpressing a tdTomato red fluorescent protein) in 50 μL volume and incubated for 5 h at 35 °C and 7% CO2. After infection, cells were washed once with Hanks solution (150 μL/well) to eliminate any extracellular parasites and compounds were added in serial dilutions in RPMI media in 150 μL volumes. Each dilution was tested in quadruplicate. Each plate also contained controls with host cells and no parasites (for background check), and controls with parasites and no drugs (positive control). Drugs were tested on T. cruzi at 1.56 μM, 3.125 μM, 6.25 μM, 12.5 μM, 25 μM. For each set of experiments, benznidazole was also used as a positive control 0.39 μM, 0.78 μM, 1.56 μM, 3.125 μM, and 6.25 μM. After drug addition, plates were incubated at 35 °C and 7% CO2. At day 3 post-infection, plates were assayed for fluorescence [69] IC50 values were determined by non-linear regression analysis using SigmaPlot. There was no evident cytotoxicity on the host cells (visual assay) with any of the drugs tested at concentrations as high as 25 μM.
T. gondii tachyzoites assays
Experiments on T. gondii tachyzoites were carried out as described previously [70] using T.gondii tachyzoites expressing red fluorescent protein [71]. Cells were routinely maintained in hTerT cells grown in High Glucose Dulbecco’s modified Eagle’s medium (DMEM-HG) supplemented with 1% fetal bovine serum, 2 mM glutamine, 1 mM pyruvate, at 37 °C in a humid 5% CO2 atmosphere. Confluent monolayers grown in 96-well black plates with optical bottoms (black, clear bottom plates from Greiner Bio-One) were used and drugs dissolved in the same medium and serially diluted in the plates. Freshly isolated tachyzoites were filtered through a 3 μm filter and passed through a 22 gauge needle, before use. The cultures were inoculated with 104 tachyzoites/well in the same media. The plates were incubated at 37 °C and read daily in a Molecular Devices fluorescence plate reader. To preserve sterility the plates were read with covered lids, and both excitation (510 nm) and emission (540 nm) were read from the bottom [72]. For the calculation of the EC50, the percent of growth inhibition was plotted as a function of drug concentration by fitting the values to the function: I = Imax C/(EC50 + C), where I is the percent inhibition, Imax = 100% inhibition, C is the concentration of the inhibitor, and EC50 is the concentration for 50% growth inhibition. There was no evident cytotoxicity on the host cells with any of the drugs tested (visual assay).
TcFPPS and TgFPPS Assays and Product Analysis
Drugs were tested on the enzymes first at 1 and 20 μM (T. cruzi) or 1 and 10 μM (T. gondii). If no activity was detected at 20 or 10 μM, respectively, then they were not further tested. For TcFPPS [73–75] 100 μL of assay buffer (10 mM Hepes, pH 7.4, 5 mM MgCl2, 2 mM dithiothreitol, 4.7 μM [4-14C]IPP (10 μCi/μmol)), and 55 μM DMAPP were prewarmed to 37 °C. The assay was initiated by the addition of recombinant protein (10–20 ng). The assay was allowed to proceed for 30 min at 37 °C and was quenched by the addition of 6 M HCl (10 μL). The reactions were made alkaline with 6.0 M NaOH (15 μL), diluted in water (0.7 mL), and extracted with hexane (1 mL). The hexane solution was washed with water and transferred to a scintillation vial for counting. One unit of enzyme activity was defined as the activity required to incorporate 1 nmol of [4-14C]IPP into [14-14C]FPP in 1 min. For TgFPPS the reaction conditions were the same except that 1 mM MgCl2 was used.
General method for measuring cytotoxicity or proliferation using AlamarBlue™ by spectrophotometry
Confluent monolayers of hTERT cells were seeded in 96 well plates (black, clear bottom from Greiner Bio-One Cat#655090) in 150 μL DMEM high glucose no phenol red (Gibco Cat# 21063) with 10% Cosmic Calf Serum. Plates were incubated overnight at 35 °C and 7% CO2. After overnight incubation, wells were washed once with Hanks (150 μL/well) to eliminate any detached host cells, and drug compounds were added in serial dilutions in DMEM media in 150 μL volumes. Each dilution was tested in quadruplicate. Each plate also contained controls with host cells and no drug added. Plates containing drug dilutions were incubated at 35°C and 7% CO2 for 3–4 days. After 3–4 days, Alamar Blue indicator (AbD serotec cat# BUF012B) was aseptically added in an amount equal to 10% of the culture volume. Cultures were incubated at 35°C for 6 hours. After incubation, absorbance was measured at 570 and 600 nm. To calculate the percent difference in reduction (of Alamar Blue) between treated and control cells the following formula was used:
Where:
εOX = molar extinction coefficient of Alamar Blue TM oxidized form (BLUE)
A = absorbance of test wells
A0 =absorbance of positive growth control well (cells plus Alamar BlueTM but no test agent)
λ1 = 570nm
-
λ2 = 600nm
Wavelength (λ) εOX 570nm 80,586 600 nm 117,216
The Percentage difference obtained is then subtracted from 100 to obtain the percent of growth inhibition in the test well compared to that of the control.
Example calculation: Percent difference between treatment and control cells = 62%. This would indicate that the amount of reduction in the test well is 62% of that in the control well, or put another way, that growth in the test well is inhibited by 38% when compared to that of the control. As this assay was performed using non-irradiated hTERT cells, cell confluency was also checked in control wells during the 4 days of the assay to evaluate cell death as a consequence of overgrowth. At day 4, there was a minimum amount of cells detached in control wells. Different concentrations of DMSO were tested as positive control of toxicity.
Supplementary Material
Sulfur-containing bisphosphonates were active against amastigotes of T. cruzi
These compounds were also active against tachyzoites of T. gondii
This effect was associated with farnesyl pyrophosphate synthase (FPPS) blockage
Some compounds inhibited the TgFPPS with IC50 values at the low nanomolar range
Acknowledgments
We thank Leena Malayil for initial enzymatic determinations. This work was supported by grants from the National Research Council of Argentina (PIP 1888), ANPCyT (PICT 2008 #1690), and the Universidad de Buenos Aires (200201001003801) to J.B.R., the Bunge & Born Foundation to S.H.S, and the U.S. National Institutes of Health to R.D. (AI-082542) and S.N.J.M. (AI-102254).
Footnotes
Supporting Information: Copies of the 1H NMR, 13C NMR and 31P NMR spectra Time-dependent 1H and 31P NMR spectra of tetraethyl esters and free bisphosphonic acids under oxidizing conditions are included as supporting information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roelofs AJ, Thompson K, Ebetino FH, Rogers MJ, Coxon FP. Bisphosphonates: Molecular mechanisms of action and effects on bone cells, monocytes and macrophages. Curr Pharm Des. 2010;16:2950–2960. doi: 10.2174/138161210793563635. [DOI] [PubMed] [Google Scholar]
- 2.Fleisch H, Russell RGG, Straumann F. Effect of pyrophosphate on hydroxyapatite and its implications in calcium homeostasis. Nature. 1966;212:901–903. doi: 10.1038/212901a0. [DOI] [PubMed] [Google Scholar]
- 3.Fleisch H, Russell RGG, Francis MD. Diphosphonates inhibit hydroxyapatite dissolution in vitro and bone resorption in tissue culture and in vivo. Science. 1969;165:1262–1264. doi: 10.1126/science.165.3899.1262. [DOI] [PubMed] [Google Scholar]
- 4.Francis MD, Russell RGG, Fleisch H. Diphosphonates inhibit formation of calcium phosphate crystals in vitro and pathological calcification in vivo. Science. 1969;165:1264–1266. doi: 10.1126/science.165.3899.1264. [DOI] [PubMed] [Google Scholar]
- 5.Russell RGG. Bisphosphonates: The first 40 years. Bone. 2011;49:2–19. doi: 10.1016/j.bone.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 6.Reszka AA, Rodan GA. Nitrogen-containing bisphosphonate mechanism of action. Mini-Rev Med Chem. 2004;4:711–717. [PubMed] [Google Scholar]
- 7.Rogers MJ. Bisphosphonates: From the laboratory to the clinic and back again. Bone. 1999;25:97–106. doi: 10.1016/s8756-3282(99)00116-7. [DOI] [PubMed] [Google Scholar]
- 8.Reszka AA, Rodan GA. Mechanism of action of bisphosphonates. Curr Osteoporos Rep. 2003;1:45–52. doi: 10.1007/s11914-003-0008-5. [DOI] [PubMed] [Google Scholar]
- 9.Reddy R, Dietrich E, Lafontaine Y, Houghton TJ, Belanger O, Dubois A, Arhin FF, Sarmiento I, Fadhil I, Laquerre K, Ostiguy V, Lehoux D, Moeck G, Parr TR, Jr, Rafai Far A. Bisphosphonated Benzoxazinorifamycin Prodrugs for the Prevention and Treatment of Osteomyelitis. Chem Med Chem. 2008;3:1863–1868. doi: 10.1002/cmdc.200800255. [DOI] [PubMed] [Google Scholar]
- 10.Miller K, Erez R, Segal E, Shabat D, Satchi-Fainaro R. Targeting Bone Metastases with a Bispecific Anticancer and Antiangiogenic Polymer–Alendronate–Taxane Conjugate. Angew Chem Int Ed. 2009;48:2949–2954. doi: 10.1002/anie.200805133. [DOI] [PubMed] [Google Scholar]
- 11.Clézardin P, Massaia M. Nitrogen-containing bisphosphonates and cancer immunotherapy. Curr Pharm Des. 2010;16:3007–3014. doi: 10.2174/138161210793563545. [DOI] [PubMed] [Google Scholar]
- 12.Coleman RE. Risks and benefits of bisphosphonates. British J Cancer. 2008;98:1736–1740. doi: 10.1038/sj.bjc.6604382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Cao R, Yin F, Hudock MP, Guo R–T, Krysiak K, Mukherjee S, Gao Y–G, Robinson H, Song Y, No JH, Bergan K, Leon A, Cass L, Goddard A, Chang T–K, Lin F–Y, Van Beek E, Papapoulos S, Wang AH–J, Kubo T, Ochi M, Mukkamala D, Oldfield E. Lipophilic Bisphosphonates as Dual Farnesyl/Geranylgeranyl Diphosphate Synthase Inhibitors: An X-ray and NMR Investigation. J Am Chem Soc. 2009;131:5153–5162. doi: 10.1021/ja808285e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roth AG, Drescher D, Yang Y, Redmer S, Uhlig S, Arenz C. Potent and selective inhibition of acid sphingomyelinase by bisphosphonates. Angew Chem Int Ed. 2009;48:7560–7563. doi: 10.1002/anie.200903288. [DOI] [PubMed] [Google Scholar]
- 15.Sanders JM, Ghosh S, Chan JMW, Meints G, Wang H, Raker AM, Song Y, Colantino A, Burzynska A, Kafarski P, Morita CT, Oldfield E. Quantitative structure-activity relationships for γδ T cell activation by bisphosphonates. J Med Chem. 2004;47:375–384. doi: 10.1021/jm0303709. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh S, Chan JMW, Lea CR, Meints GA, Lewis JC, Tovian ZS, Flessner RM, Loftus TC, Bruchhaus I, Kendrick H, Croft SL, Kemp RG, Kobayashi S, Nozaki T, Oldfield E. Effects of bisphosphonates on the growth of Entamoeba histolytica and plasmodium species in vitro and in vivo. J Med Chem. 2004;47:175–187. doi: 10.1021/jm030084x. [DOI] [PubMed] [Google Scholar]
- 17.Martin MB, Grimley JS, Lewis JC, Heath HT, III, Bailey BN, Kendrick H, Yardley V, Caldera A, Lira R, Urbina JA, Moreno SN, Docampo R, Croft SL, Oldfield E. Bisphosphonates inhibit the growth of Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondii, and Plasmodium falciparum: a potential route to chemotherapy. J Med Chem. 2001;44:909–916. doi: 10.1021/jm0002578. [DOI] [PubMed] [Google Scholar]
- 18.Yardley V, Khan AA, Martin MB, Slifer TR, Araujo FG, Moreno SNJ, Docampo R, Croft SL, Oldfield E. In vivo activities of farnesyl pyrophosphate synthase inhibitors against Leishmania donovani and Toxoplasma gondii. Antimicrob, Agents Chemother. 2002;46:929–931. doi: 10.1128/AAC.46.3.929-931.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin MB, Sanders JM, Kendrick H, de Luca-Fradley K, Lewis JC, Grimley JS, Van Brussel EM, Olsen JR, Meints GA, Burzynska A, Kafarski P, Croft SL, Oldfield E. Activity of Bisphosphonates against Trypanosoma brucei rhodesiense. J Med Chem. 2002;45:2904–2914. doi: 10.1021/jm0102809. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez JB, Szajnman SH. New antibacterials for the treatment of toxoplasmosis; a patent review. Expert Opinion Ther Patents. 2012;22:311–334. doi: 10.1517/13543776.2012.668886. [DOI] [PubMed] [Google Scholar]
- 21.García Liñares G, Ravaschino EL, Rodriguez JB. Progresses in the Field of Drug Design to Combat Tropical Protozoan Parasitic Diseases. Curr Med Chem. 2006;13:335–360. doi: 10.2174/092986706775476043. [DOI] [PubMed] [Google Scholar]
- 22.Urbina JA, Moreno B, Vierkotter S, Oldfield E, Payares G, Sanoja C, Bailey BN, Yan W, Scott DA, Moreno SN, Docampo R. Trypanosoma cruzi contains major pyrophosphate stores, and its growth in vitro and in vivo is blocked by pyrophosphate analogs. J Biol Chem. 1999;274:33609–33615. doi: 10.1074/jbc.274.47.33609. [DOI] [PubMed] [Google Scholar]
- 23.Docampo R, Moreno SNJ. The Acidocalcisome as a Target for Chemotherapeutic Agents in Protozoan Parasites. Curr Pharm Des. 2008;14:882–888. doi: 10.2174/138161208784041079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosso VS, Szajnman SH, Malayil L, Galizzi M, Moreno SNJ, Docampo R, Rodriguez JB. Synthesis and Biological Evaluation of New 2-alkylaminoethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii targeting Farnesyl Diphosphate Synthase. Bioorg Med Chem. 2011;19:2211–2217. doi: 10.1016/j.bmc.2011.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szajnman SH, García Liñares GE, Li Z–H, Galizzi M, Jiang C, Bontempi E, Ferella M, Moreno SNJ, Docampo R, Rodriguez JB. Synthesis and Biological Evaluation of 2-alkylaminoethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii targeting Farnesyl Diphosphate Synthase. Bioorg, Med Chem. 2008;16:3283–3290. doi: 10.1016/j.bmc.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szajnman SH, Bailey BN, Docampo R, Rodriguez JB. Bisphosphonates Derived from Fatty Acids are Potent Growth Inhibitors of Trypanosoma cruzi. Bioorg Med Chem Lett. 2001;11:789–792. doi: 10.1016/s0960-894x(01)00057-9. [DOI] [PubMed] [Google Scholar]
- 27.Szajnman SH, Montalvetti A, Wang Y, Docampo R, Rodriguez JB. Bisphosphonates Derived from Fatty Acids are Potent Inhibitors of Trypanosoma cruzi Farnesyl Pyrophosphate Synthase. Bioorg Med Chem Lett. 2003;13:3231–3235. doi: 10.1016/s0960-894x(03)00663-2. [DOI] [PubMed] [Google Scholar]
- 28.Szajnman SH, Ravaschino EL, Docampo R, Rodriguez JB. Synthesis and Biological Evaluation of 1-Amino-1,1-Bisphosphonates Derived from Fatty Acids against Trypanosoma cruzi targeting Farnesyl Pyrophosphate Synthase. Bioorg Med Chem Lett. 2005;15:4685–4690. doi: 10.1016/j.bmcl.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 29.Ling Y, Sahota G, Odeh S, Chan JM, Araujo FG, Moreno SN, Oldfield E. Bisphosphonate inhibitors of Toxoplasma gondii growth: In vitro, QSAR, and in vivo investigations. J Med Chem. 2005;48:3130–3140. doi: 10.1021/jm040132t. [DOI] [PubMed] [Google Scholar]
- 30.Ling Y, Li Z–H, Miranda K, Oldfield E, Moreno SN. The Farnesyl-diphosphate/Geranylgeranyl-diphosphate Synthase of Toxoplasma gondii is a Bifunctional Enzyme and a Molecular Target of Bisphosphonates. J Biol Chem. 2007;282:30804–30816. doi: 10.1074/jbc.M703178200. [DOI] [PubMed] [Google Scholar]
- 31.Szajnman SH, Rosso VS, Malayil L, Smith A, Moreno SN, Docampo R, Rodriguez JB. Design Synthesis and Biological Evaluation of 1-(Fluoroalkylidene)-1,1-bisphosphonic Acids against Toxoplasma gondii targeting Farnesyl Diphosphate Synthase. Org Biomol Chem. 2012;10:1424–1433. doi: 10.1039/c1ob06602a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Urbina JA, Docampo R. Specific chemotherapy of Chagas’ disease: controversies and advances. Trends Parasitol. 2003;19:495–501. doi: 10.1016/j.pt.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 33.Urbina JA. Specific chemotherapy of Chagas disease: relevance, current limitations and new approaches. Acta Tropica. 2010;115:55–68. doi: 10.1016/j.actatropica.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 34.Urbina JA. New insights in Chagas’ disease treatment. Drugs Fut. 2010;35:409–420. [Google Scholar]
- 35.Brener Z. Biology of Trypanosoma cruzi. Annu Rev Microbiol. 1973;27:347–382. doi: 10.1146/annurev.mi.27.100173.002023. [DOI] [PubMed] [Google Scholar]
- 36.Kirchhoff VL. Epidemiology of American trypanosomiasis (Chagas disease) Adv Parasitol. 2011;75:1–18. doi: 10.1016/B978-0-12-385863-4.00001-0. [DOI] [PubMed] [Google Scholar]
- 37.Innes EA. A brief history and overview of Toxoplasma gondii. Zoonoses Public Health. 2010;57:1–7. doi: 10.1111/j.1863-2378.2009.01276.x. [DOI] [PubMed] [Google Scholar]
- 38.Tenter AM, Heckerost AR, Weiss LM. Toxoplasma gondii: from animals to humans. Int J Parasitol. 2000;30:1217–1258. doi: 10.1016/s0020-7519(00)00124-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dubey JP, Linsday DS, Speer CA. Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin Microbiol Rev. 1998;11:267–299. doi: 10.1128/cmr.11.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.C–Huang H, Gabelli SB, Oldfield E, Amzel LM. Binding of nitrogen-containing bisphosphonates (N-BPs) to the Trypanosoma cruzi farnesyl diphosphate synthase homodimer. Proteins. 2010;78:888–899. doi: 10.1002/prot.22614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao R, Chen CK–M, Guo R–T, Wang AH–J, Oldfield E. Structures of a potent phenylalkyl bisphosphonate inhibitor bound to farnesyl and geranylgeranyl diphosphate synthases. Proteins. 2008;73:431–439. doi: 10.1002/prot.22066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gabelli SB, McLellan JS, Montalvetti A, Oldfield E, Docampo R, Amzel LM. Structure and Mechanism of the Farnesyl Diphosphate Synthase from Trypanosoma cruzi: Implications for Drug Design. Proteins. 2006;62:80–88. doi: 10.1002/prot.20754. [DOI] [PubMed] [Google Scholar]
- 43.Aripirala S, Szajnman SH, Jakoncic J, Rodriguez JB, Docampo R, Gabelli SB, Amzel LM. Design, Synthesis, Calorimetry and Crystallographic analysis of 2-Alkylaminoethyl-1,1-Bisphosphonates as inhibitors of Trypanosoma cruzi Farnesyl Diphosphate Synthase. J Med Chem. 2012;55:6445–6454. doi: 10.1021/jm300425y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laskovics FM, Poulter CD. Prenyltransferase; determination of the binding mechanism and individual kinetic constants for farnesylpyrophosphate synthetase by rapid quench and isotope partitioning experiments. Biochemistry. 1981;20:1893–1901. doi: 10.1021/bi00510a027. [DOI] [PubMed] [Google Scholar]
- 45.Poulter CD, Argyle JC, Mash EA. Farnesyl pyrophosphate synthetase. Mechanistic studies of the 1′-4 coupling reaction with 2-fluorogeranyl pyrophosphate. J Biol Chem. 1978;253:7227–7233. [PubMed] [Google Scholar]
- 46.Degenhardt CR, Burdsall DC. Synthesis of Ethenylidenebis(phosphonic acid) and Its Tetraalkyl Esters. J Org Chem. 1986;51:3488–3490. [Google Scholar]
- 47.Lolli ML, Lazzarato L, Di Stilo A, Fruttero R, Gasco A. Michael addition of Grignard reagents to tetraethyl ethenylidenebisphosphonate. J Organomet Chem. 2002;650:77–83. [Google Scholar]
- 48.Szajnman SH, García Liñares GE, Moro P, Rodríguez JB. New Insights into the Chemistry of gem-Bis(phosphonates): Unexpected Rearrangement of Michael-Type Acceptors. Eur J Org Chem. 2005:3687–3696. [Google Scholar]
- 49.Lazzarato L, Rolando B, Lolli ML, Tron GC, Fruttero R, Gasco A, Deleide G, Guenther HL. Synthesis of NO-donor bisphosphonates and their in-vitro action on bone resorption. J Med Chem. 2005;48:1322–1329. doi: 10.1021/jm040830d. [DOI] [PubMed] [Google Scholar]
- 50.Pospíšil J, Potáček M. Microwave-assisted solvent-free intramolecular 1,3-dipolar cycloaddition reactions leading to hexahydrochromeno[4,3-b]pyrroles: scope and limitations. Tetrahedron. 2006;63:337–346. [Google Scholar]
- 51.Kieczykowski GR, Jobson RB, Melillo DG, Reinhold DF, Grenda VJ, Shinkai I. Preparation of (4-Amino-1-Hydroxybutylidene)bisphosphonic Acid Sodium Salt, MK-217 (Alendronate Sodium). An Improved Procedure for the Preparation of 1-Hydroxy-1,1-bisphosphonic Acids. J Org Chem. 1995;60:8310–8312. [Google Scholar]
- 52.Witczak ZJ, Lorchak D, Nguyen N. A click chemistry approach to glycomimetics: Michael addition of 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranose to 4-deoxy-1,2-O-isopropylidene-L-glycero-pent-4-enopyranos-3-ulose – a convenient route to novel 4-deoxy-(1→5)-5-C-thiodisaccharides. Carbohydr Res. 2007;352:1929–1933. doi: 10.1016/j.carres.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 53.Witczak ZJ, Kaplon P, Dey PM. Thio-sugars VII. Effect of 3-deoxy-4-S-(β-D-gluco- and β-D-galactopyranosyl)-4-thiodisaccharides and their sulfoxides and sulfones on the viability and growth of selected murine and human tumor cell lines. Carbohydr Res. 2003;338:11–18. doi: 10.1016/s0008-6215(02)00394-4. [DOI] [PubMed] [Google Scholar]
- 54.Uhrig ML, Szilágyi L, Kövér KE, Varela O. Synthesis of non-glycosidic 4,6′-thioether-linked disaccharides as hydrolytically stable glycomimetics. Carbohydr Res. 2007;352:1841–1849. doi: 10.1016/j.carres.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 55.Uhrig ML, Manzano VE, Varela O. Stereoselective Synthesis of 3-Deoxy-4-S-(1→4)-Thiodisaccharides and Their Inhibitory Activities Towards â-Glycoside Hydrolases. Eur J Org Chem. 2006:162–168. [Google Scholar]
- 56.Madesclaire M. Synthesis of sulfoxides by oxidation of thioethers. Tetrahedron. 1986;42:5459–5495. [Google Scholar]
- 57.Oldfield E, Song Y, Zhang Y, Sanders JM. WO2007/109585A2. Bisphosphonates compounds and methods. 2007
- 58.Mikołajczyk M, Zatorski A. α-Phosphorylsulfoxides. Synthesis. 1973:669–671. [Google Scholar]
- 59.Leonard NJ, Johnson CR. Periodate oxidation of sulfides to sulfoxides. Scope of the reaction. J Org Chem. 1962;27:282–284. [Google Scholar]
- 60.Mikołajczyk M, Grzejszczak S, Zatorski A. α-Phosphorylsulfoxides sulfoxides II. Synthesis of α,β-unsaturated sulfoxides and configurational assigments to geometrical isomers. J Org Chem. 1975;40:1979–1984. [Google Scholar]
- 61.Drabowicz J, Mikołajczyk M. A facile and selective oxidation of organic sulphides to sulphoxides with hydrogen peroxide/selenium dioxide system. Synthesis. 1978:758–759. [Google Scholar]
- 62.Barthélémy P, Maurizis JC, Lacombe JM, Pucci B. A new class sulfoxide surfactant derived from Tris. Synthesis and preliminary assessments of their properties. Bioorg Med Chem Lett. 1998;8:1559–1562. doi: 10.1016/s0960-894x(98)00263-7. [DOI] [PubMed] [Google Scholar]
- 63.Aversa MC, Barattucci A, Bonaccorsi P, Giannetto P. L-Cysteine, a Versatile Source of Sulfenic Acids. Synthesis of Enantiopure Alliin Analogues. J Org Chem. 2005;70:1986–1992. doi: 10.1021/jo048662k. [DOI] [PubMed] [Google Scholar]
- 64.Aversa MC, Barattucci A, Bonaccorsi P, Marino-Merlo F, Mastino A, Sciortino MT. Synthesis and biological testing of thioalkane- and thioarene-spaced bis-β-D-glucopyranosides. Bioorg Med Chem. 2009;17:1456–1463. doi: 10.1016/j.bmc.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 65.Aversa MC, Barattucci A, Bilardo MC, Bonaccorsi P, Giannetto P, Rollin P, Tatibouët A. Sulfenic Acids in the Carbohydrate Field. An Example of Straightforward Access to Novel Multivalent Thiosaccharides. J Org Chem. 2005;70:7389–7396. doi: 10.1021/jo0510991. [DOI] [PubMed] [Google Scholar]
- 66.Horhant D, Le Lamer A–C, Boustie J, Uriac P, Gouault N. Separation of a mixture of paraconic acids from Cetraria islandica (L.) Ach. employing a fluorous tag—catch and release strategy. Tetrahedron Lett. 2007;48:6031–6033. [Google Scholar]
- 67.Hillis LR, Ronald RC. Total Synthesis of (−)-Grahamimycin A1. J Org Chem. 1985;50:470–473. [Google Scholar]
- 68.Demoro B, Caruso F, Rossi M, Benítez D, Gonzalez M, Cerecetto H, Parajón-Costa B, Castiglioni J, Galizzi M, Docampo R, Otero L, Gambino D. Risedronate metal complexes potentially active against Chagas disease. J Inorg Biochem. 2010;104:1252–1258. doi: 10.1016/j.jinorgbio.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Canavaci AM, Bustamante JM, Padilla AM, Pereza Brandan CM, Simpson LJ, Xu D, Boehlke CL, Tarleton RL. In vitro and in vivo high-throughput assays for the testing of anti-Trypanosoma cruzi compounds. PLOS Negl Trop Dis. 2010;4:e740. doi: 10.1371/journal.pntd.0000740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gubbels MJ, Li C, Striepen B. High-throughput growth assay for Toxoplasma gondii using yellow fluorescent protein. Antimicrob, Agents Chemother. 2003;43:309–316. doi: 10.1128/AAC.47.1.309-316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Agrawal S, van Dooren GG, Beatty WL, Striepen B. Genetic evidence that an endosymbiont-derived endoplasmic reticulum-associated protein degradation (ERAD) system functions in import of apicoplast proteins. J Biol Chem. 2009;284:33683–33691. doi: 10.1074/jbc.M109.044024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ravaschino EL, Docampo R, Rodriguez JB. Design, Synthesis and Biological Evaluation of Phosphinopeptides against Trypanosoma cruzi Targeting Trypanothione Biosynthesis. J Med Chem. 2006;49:426–435. doi: 10.1021/jm050922i. [DOI] [PubMed] [Google Scholar]
- 73.Montalvetti A, Bailey BN, Martin MB, Severin GW, Oldfield E, Docampo R. Bisphosphonates are potent inhibitors of Trypanosoma cruzi farnesyl pyrophosphate synthase. J Biol Chem. 2001;276:33930–33937. doi: 10.1074/jbc.M103950200. [DOI] [PubMed] [Google Scholar]
- 74.Montalvetti A, Fernandez A, Sanders JM, Ghosh S, Van Brussel E, Oldfield E, Docampo R. Farnesyl pyrophosphate synthase is an essential enzyme in Trypanosoma brucei. In vitro RNA interference and in vivo inhibition studies. J Biol Chem. 2003;278:17075–17083. doi: 10.1074/jbc.M210467200. [DOI] [PubMed] [Google Scholar]
- 75.Ogura K, Nishino T, Shinka T, Seto S. Prenyltransferases of pumpkin fruit. Methods Enzymol. 1985;110:167–171. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.