

Abstract
The identification of elevated circulating levels of the osteocytic protein fibroblast growth factor 23 (FGF23) in patients with chronic kidney disease (CKD), along with recent data linking these values to the pathogenesis of secondary hyperparathyroidism and to systemic complications, has changed the approach to the pathophysiology and treatment of disordered bone and mineral metabolism in renal failure. It now appears that osteocyte biology is altered very early in the course of CKD and these changes have implications for bone biology, as well as for progressive cardiovascular and renal disease. Since circulating FGF23 values are influenced by therapies used to treat secondary hyperparathyroidism, the effects of different therapeutic paradigms on FGF23 have important implications for mineral metabolism as well as for morbidity and mortality. Further studies are critically needed to identify the initial trigger for abnormalities of skeletal mineralization and turnover as well as the potential effects that current therapeutic options may have on osteocyte biology.
Keywords: osteocytes, FGF23, chronic kidney disease, secondary hyperparathyroidism
Introduction
Historically, the evaluation of abnormal mineral metabolism in chronic kidney disease (CKD) has focused on the development of secondary hyperparathyroidism. While increases in circulating PTH, and the consequences of this on the skeleton, remain important complications of CKD, recent advances in the field of bone and mineral metabolism suggest that the parathyroid gland is not the only endocrine organ whose physiology is altered by kidney dysfunction. Indeed, osteocytes in bone produce a circulating hormone, fibroblast growth factor 23 (FGF23), whose endocrine effects of increasing renal phosphate excretion and suppressing renal 1α-hydroxylase were initially characterized in patients with hypophosphatemic rickets [1]. In patients with early CKD, increased levels of FGF23 may be the initial indicator of altered mineral metabolism [2] and play a key role in the development of secondary hyperparathyroidism. Elevated values of FGF23 also have systemic effects, however, including the promotion of left ventricular hypertrophy [3], increased mortality rates [4], and faster deterioration of renal function [5] and, since circulating FGF23 values are influenced by therapies used to treat secondary hyperparathyroidism [6-8], the effects of different therapeutic paradigms on FGF23 have important implications for mineral metabolism as well as for morbidity and mortality.
Chronic Kidney Disease Mineral and Bone Disorder: Changing Concepts
Over the past decade, two major events have triggered a change in our concepts as to the pathogenesis of mineral metabolism and its consequences in CKD. The first major shift came from the recognition that a then newly discovered hormone, fibroblast growth factor 23 (FGF23), which is elevated in CKD [9], has a critical role in the development of abnormal mineral ion homeostasis [10, 11] and is associated with deleterious systemic consequences in the general population as well as in patients with CKD [3, 4, 12, 13]. This hormone is made in osteocytes in bone and the identification of its role in CKD-MBD has changed the way in which the regulation of mineral metabolism in CKD is viewed. The second major shift came as the result of an increasing awareness that altered mineral metabolism in CKD is not a linear process culminating in the ultimate abnormality of elevated PTH values but instead that multiple factors involved in the regulation of mineral ion homeostasis, including abnormal mineral ion and hormone levels, are linked not only to bone disease but also to cardiovascular disease, the leading cause of death in this patient population [14]. These concepts culminated in the reworked definition of renal osteodystrophy by the KDIGO working group in 2006 to reflect its nature as a systemic disorder, involving abnormalities in calcium, phosphorus, PTH and vitamin D, abnormalities in bone structure, strength, linear growth, and histology, and abnormalities in cardiovascular and soft tissue calcium deposition. Together, these alterations were re-named the “Chronic Kidney Disease Mineral and Bone Disorder” (CKD-MBD) in order to more accurately reflect their complex, intertwined, systemic nature [14]. The more complex nature of bone disease itself was also emphasized, with the recognition that changes in bone turnover are not the sole abnormality in CKD, but that poor skeletal mineralization and altered bone volume and structure also plague many patients [14]; thus, new recommendations suggested that three areas of bone histology be examined in CKD: bone turnover, mineralization and volume. Since bone biopsies are not routinely use in clinical practice, suitable biomarkers are needed for non-invasive prediction of these skeletal parameters [14, 15].
The Role of FGF23 in CKD-MBD
The decline in circulating levels of 1,25(OH)2vitamin D that occurs in early CKD has traditionally been attributed to a decrease in functional renal tissue. The concept of declining renal mass as the primary determinant of reduced 1,25(OH)2vitamin D levels in early CKD changed in 2005 when Gutierrez et al. described a significantly higher prevalence of 1,25(OH)2vitamin D deficiency than of anemia (i.e. erythropoietin deficiency) in adult patients with early CKD [11]. This finding, combined with earlier studies demonstrating that levels of FGF23 were increased in patients with various degrees of renal dysfunction [9], suggested that suppression of renal 1-α hydroxylase, rather than loss of renal mass (which would be expected to affect renal 1α-hydroxylase and erythropoietin to a similar degree), results in decreased values of 1,25(OH)2vitamin D, particularly in early stages of CKD, and that this suppression may be mediated by increasing circulating FGF23 levels.
FGF23 is a skeletal hormone which binds to one or several FGF receptors [16-18] and their co-receptor Klotho to inhibit the expression of the types IIa and IIc sodium-phosphate co-transporters on the apical membrane of renal proximal tubular cells, thus inducing phosphaturia [19, 20]. In contrast to PTH, which also induces phosphaturia while increasing renal 1α-hydroxylase activity, FGF23 both suppresses renal 1α-hydroxylase and stimulates renal 24-hydroxylase activity [19, 20], thus suppressing the production of calcitriol and increasing degradation of both 25(OH)vitamin D and 1,25(OH)2 vitamin D. In addition to regulating phosphate and vitamin D levels, FGF23 is itself regulated by both phosphorus and 1,25(OH)2vitamin D [8, 21-23], and probably also by PTH [10, 24]. Sustained increases in dietary phosphorus and the administration of 1,25(OH)2vitamin D increase circulating FGF23 levels [8, 21-23] [25], while dietary phosphorus restriction reverses this trend [21].
In patients with CKD, circulating levels of FGF23 rise progressively as renal function declines (Figure 1). Controversy exists as to the etiology of increased FGF23 values in early CKD with some studies suggesting a role for early, possibly intermittent increases in enteral phosphate burden and others suggesting that PTH levels increase first and result in elevated FGF23 concentrations [10, 24]. Regardless of etiology, current data demonstrate that increased FGF23 expression in osteocytes occurs early in the course of CKD and that circulating FGF23 levels are increased in 30-40% of adults with an estimated GFR of 70-90 ml/min/1.73 m2, while PTH concentrations are above the normal range in only 10% of these same individuals [26]. Thus, it appears that an early increase in FGF23 is the first detectable biochemical evidence for an abnormal regulation of mineral ion homeostasis in early CKD, leading to the early decline in 1,25(OH)2vitamin D levels.
Fig. 1.



Serum values of phosphate (a), PTH (b), and FGF23 (c) in a cross-section of pediatric patients with pre-dialysis CKD. *P<0.01 CKD 1–2 vs. CKD 3, ***P<0.001 CKD 1–2 vs. CKD 4, ***P<0.001 CKD 1–2 vs. CKD 5. Boxes and bars represent the interquartile range and the median value, respectively; whiskers represent the distance to the smallest and largest unbooked sample value.
Following increases in FGF23, a decline in 1,25(OH)2vitamin D levels leads both to impaired intestinal calcium absorption as well as to increased serum parathyroid hormone (PTH) levels (2). Increased PTH levels maintain normocalcemia in the face of impaired intestinal calcium absorption by increasing calcium release from bone. Bone resorption, however, leads to an increase in the amount of phosphate which must be excreted by a declining number of functional nephrons. When renal function becomes severely impaired, phosphate levels rise, further suppressing renal 1α-hydroxylase activity and increasing PTH levels (3). Thus, in late stages of CKD, hypocalcemia, hyperphosphatemia, and low circulating 1,25(OH)2vitamin D values all contribute to the development of secondary hyperparathyroidism (4).
Renal Osteodystrophy Redefined and the Role of the Osteocyte
Turnover
In addition to the recognition that altered mineral metabolism in CKD is not fully reflected in the term “secondary hyperparathyroidism”, previous definitions of renal osteodystrophy which described abnormalities in bone turnover alone were proven to be too simplistic and applied primarily to patients treated with maintenance dialysis. In fact, abnormalities in bone turnover, driven by circulating PTH concentrations, are prevalent in both adults [27, 28] and pediatric patients with advanced stages of CKD [29]. PTH activates the PTH/PTHrP receptor on osteocytes and osteoblasts, thereby directly increasing the cellular activity of osteoblasts and, indirectly, the activity of osteoclasts [30-32]. Elevated bone turnover is evident in patients with moderate to advanced stages of CKD [33] and increases in prevalence as GFR declines [33]. Skeletal resistance to PTH develops as CKD progresses, due to multiple factors including, although likely not limited to the accumulation of circulating PTH fragments [34] and downregulation of the PTH receptor [35]; however, excessive levels of PTH result in increased resorption of bone matrix and release of mineral into the circulation. Prolonged exposure to elevated PTH levels and increases in bone turnover may lead to peri-trabecular fibrous changes in bones; thus, the renal osteodystrophy associated with prolonged secondary hyperparathyroidism is often termed “osteitis fibrosa cystica” [36]. A state of low-turnover bone disease (adynamic renal osteodystrophy), although most common in adult dialysis patients [28], also occurs in children treated with maintenance dialysis as a result of excess treatment with vitamin D and calcium salts. In addition to the increased risk for fractures and vascular calcifications that is observed in adults with adyamic bone, this form of bone disease in children treated with dialysis is associated with a further decline in growth [37, 38].
Although bone turnover is undoubtedly affected by changes in circulating PTH in advanced CKD, recent data from animals and humans have suggested that changes in bone turnover occur before alterations in circulating mineral, PTH, or vitamin D concentrations are apparent [32, 39] and that these changes are mediated in large part by alterations in osteocyte biology [32, 40]. Indeed, in 2004 Lund et al. demonstrated that renal ablation in mice resulted in low bone turnover if circulating levels of calcium, phosphate, 1,25(OH)2vitamin D and PTH were kept constant, suggesting that some changes in bone turnover, particularly in early CKD, are independent of measurable changes in mineral ion homeostasis [39]. Subsequently, Sabbagh et al. demonstrated similar findings in Jck mice [32], in whom increased osteocytic sclerostin expression was related to changes in bone turnover with progressive CKD. Indeed, sclerostin is an inhibitor of Wnt signaling-mediated bone turnover; thus, increases in its expression result in suppression of bone formation and the development of adynamic bone in early stages of CKD [32]. PTH, however, suppresses sclerostin expression [41] and, as moderate to severe CKD develops, rising PTH levels inhibit skeletal sclerostin expression, allowing bone formation rates to rise [32].
Mineralization
Despite the traditional emphasis on bone turnover, abnormalities in skeletal mineralization have been observed in both adults [28] and children with CKD [33, 42]. Until recently, bone histology data in early CKD from humans were lacking; however, current bone biopsy data have filled some of this information gap and have identified a skeletal mineralization defect in as many as 30-40% of children with early CKD (a glomerular filtration rate of 60-90 ml/min/1.73m2). Although the prevalence of the mineralization defect in adults remains controversial [27], data from Brazil suggest that it may also be common in the adult pre-dialysis CKD population, particularly in those individuals with low bone mineral density [28]. The prevalence of defective mineralization increases with progressive renal dysfunction and a startlingly high (50-80%) incidence of mineralization has been described in pediatric dialysis patients both before and during therapy with active vitamin D sterols for secondary hyperparathyroidism [8]. Defective mineralization that is associated with high turnover bone disease is termed “mixed lesion”; when associated with low to normal bone turnover, it is referred to as “osteomalacia” [14, 15]. While the clinical implications of defective mineralization in patients with CKD remain to be established, increased fracture rates, bone deformities, and growth retardation—common features of children with rickets [1, 43]—are common in the pediatric CKD population [44] and these lesions persist despite adequate suppression of secondary hyperparathyroidism [8].
Recent advances in bone preparation techniques have improved the ability to assess skeletal protein expression in un-decalcified samples, thus allowing for the simultaneous comparison of bone protein expression and bone histomorphometry in human bone [45]. Through these new techniques, in 2009, Pereira et al. identified that osteocytic protein expression is altered early in the course of CKD and that expression of some of these proteins may be linked to abnormalities in skeletal mineralization that are apparent in early pediatric CKD. Indeed, skeletal expression FGF23 is already markedly upregulated in osteocytes in very early CKD [40], occurring as early as stage 2 CKD (Figure 2), at a time when serum PTH, vitamin D, calcium, and phosphorus concentrations are still within the normal range [46], but when circulating values of FGF23 are increased [26]. Increased plasma FGF23 levels may be secondary to defects in regulatory proteins, including the osteocytic proteins dentin matrix protein 1 (DMP1) and ectonucleotide pyrophosphatase/phosphodiesterase (ENPP1), as documented in patients with normal renal function, who develop different forms of hypophosphatemic rickets [40, 47, 48]. These findings, which are consistent with those in animals lacking these two proteins [47, 49], have suggested that DMP1 and ENPP1 are negative regulators of FGF23 synthesis and secretion. However, the early increases in FGF23 in pediatric patients with CKD are associated with an increase, rather than a decrease, in osteocytic DMP1 expression [40] (Figure 3). In the context of CKD, osteocytic expression of both FGF23 and DMP1 correlates with histomorphometric parameters of skeletal mineralization [40]. It appears that osteocytes, which are presumably the source of increased circulating FGF23 values early in the course of CKD, are unable to generate sufficiently high levels of a biologically active DMP1 to suppress osteocytic FGF23 production; abnormalities in osteocyte function thus occur even with only mild decreases in kidney function. Moreover, the osteocyte, previously thought to be a relatively quiescent cell with a role limited to skeletal mechanosensation, is an endocrine cell and that disruption of its normal biology triggers alterations in parathyroid gland biology, mineral metabolism, and skeletal mineralization in patients with all stages of CKD.
Fig. 2.
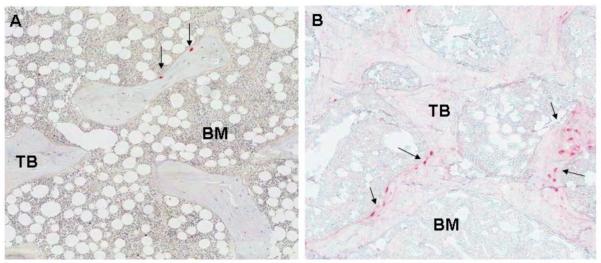
FGF23 expression is markedly increased in patients with early (stage 2) CKD (B) as compared to control (A).
Fig. 3.
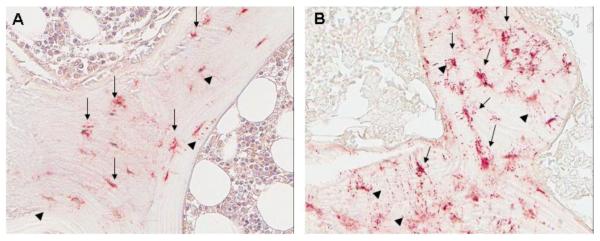
DMP1 expression is markedly increased in patients with early (stage 2) CKD (B) as compared to control (A).
Volume
Although children with CKD—except those treated with corticosteroids who may display loss of bone volume, termed “osteoporosis”—typically have normal or high bone volume as assessed by bone histomorphometry, adults with CKD may display low bone mineral density which, a significant proportion of the time, may be associated with low bone volume [28, 50]. Although expression of one osteocytic protein, matrix extracellular phosphoglycoprotein (MEPE), has been associated with bone volume—indeed a complete absence of MEPE results in an increase in bone volume [51]—changes in osteocytic MEPE expression have not been observed in individuals with CKD [40].
Systemic Implications of Osteocyte Dysfunction in CKD-MBD
Cardiovascular Disease
Cardiovascular disease is the most common cause of mortality in patients with all stages of CKD, with young (i.e. 20 and 30 year old) dialysis patients displaying the same rates of mortality from cardiovascular disease as 80 year old individuals in the general population [52]. Cardiovascular disease is not limited to the adults, but is also common in children with CKD [53, 54]. In addition to traditional risk factors for cardiovascular disease (i.e. hypertension and smoking) found in the general population, risk factors for vascular disease in pre-dialysis CKD and dialysis patients include hypercalcemia, hyperphosphatemia, elevated levels of the calcium x phosphorus product and treatment with high doses of calcium salts and vitamin D sterols [53-59]. However, 40% of adult patients with stage 3 CKD, without these risk factors, show evidence of calcification [60] and changes in carotid artery wall thickness are apparent in children as early as CKD stage 2 [56, 61]. The prevalence of vascular changes in children with early CKD—patients who not only lack the mineral metabolism-associated risk factors common in the dialysis population but who also lack traditional adult risk factors such as diabetes, hypertension and smoking [56, 61]—suggest that factors unique to CKD and independent of circulating mineral content contribute to vascular disease.
The pathophysiology of calcification in patients with CKD clearly differs from that observed in the general population, although the mechanisms by which vascular calcification develop remain to be fully elucidated. Indeed, calcium deposition is observed primarily in intimal atherosclerotic plaques in individuals with preserved kidney function, while deposition of mineral occurs primarily in the tunica media in the presence of CKD [62]. In patients with compromised renal function, the entire smooth muscle layer surrounding arteries may be replaced not only by calcium deposits, but by tissue that resembles bone. Indeed, osteoblasts and vascular smooth muscle cells have a common mesenchymal origin and, in patients with CKD, core binding factor-1 (Cbfa1) is thought to trigger mesenchymal cell to osteoblast transformation. Mice deficient in Cbfa1 fail to mineralize bone [63] and arteries obtained from patients undergoing renal transplantation show increased levels of the protein [64]. Expression of the sodium-dependent phosphate transporters PIT-1 and PIT-2 likely contributes to the increased calcification [65, 66] and upregulation of pro-mineralization factors such as osteopontin, bone sialoprotein, osteonectin, alkaline phosphatase, type I collagen, and bone morphogenic protein-2 (BMP-2) is potentiated by the uremic milieu [67-70], while expression of calcification inhibitors, such as fetuin A and matrix Gla protein, is suppressed [71-73].
Throughout the past several years, elevated circulating values of FGF23 have been linked to cardiovascular disease in patients with all stages of CKD and recent experimental data have defined a direct role for FGF23, independent of changes in mineral metabolism, on pathological changes in cardiac myocytes [74]. Although the effects of circulating FGF23 on mineral metabolism require the presence of Klotho, as the co-receptor of FGF receptors [75], some of the effects on the cardiovascular system appear to be Klotho-independent. FGF23 levels are often hundreds- to thousands-fold higher in patients with renal failure as compared to those detected in individuals with normal kidney function (78), and these values have been associated with an increased prevalence of left ventricular hypertrophy (87), with an increased burden of cardiovascular calcification in adults and pediatric patients (89;99)[76], and with premature mortality in adults with all stages of CKD (Figure 4). Recent experimental data suggest that these associations may reflect a direct effect of FGF23 on the myocardium that induces myocyte hypertrophy in a Klotho-independent manner [74]. However, it is important to note that Klotho is expressed in arteries, although not in myocardium, and decreases in vascular Klotho, as occur with progressive CKD [77], have been linked to loss of vascular elasticity [78].
Fig. 4.
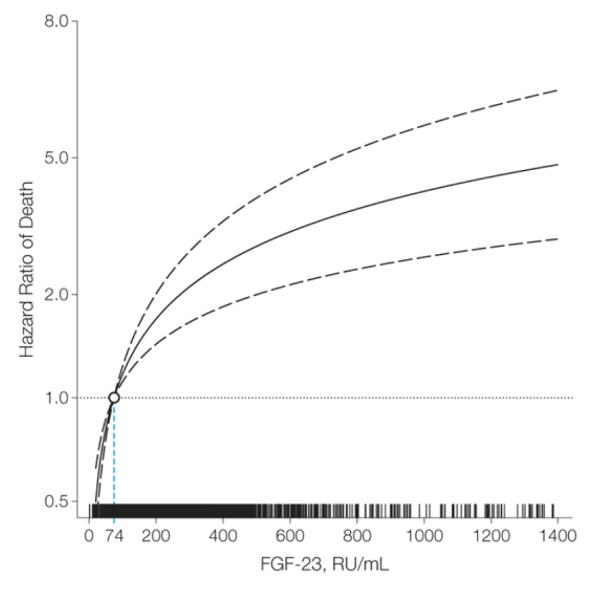
Hazard ratio for death associated with increasing FGF23 concentrations in patients with pre-dialysis CKD. The median fibroblast growth factor 23 (FGF-23) level within the lowest FGF-23 quartile (74 RU/mL) served as the referent value (hazard = 1.0). The model was stratified by center and adjusted for age; sex; race; ethnicity; estimated glomerular filtration rate; natural log-transformed urine albumin-to-creatinine ratio; hemoglobin; serum albumin; systolic blood pressure; body mass index; diabetes; smoking status; low-density lipoprotein; history of coronary artery disease, congestive heart failure, stroke, and peripheral vascular disease; use of aspirin, β-blockers, statins, and angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers; and serum calcium, phosphate, and natural log-transformed parathyroid hormone. Tick marks on the x-axis indicate individual observations at corresponding levels of FGF-23. The solid black line represents the multivariable-adjusted hazard of mortality as a function of the measured (nontransformed) FGF-23 level. The dashed lines indicate the 95% confidence intervals.
Link to Progressive Renal Dysfunction
In the early 1980s, a link between dietary phosphate intake in CKD patients and the rate of progression to renal failure was established. More recent evidence suggests increased levels of FGF23—potentially reflecting chronically increased dietary phosphate loads—may also predict a more rapid deterioration of renal function. In a longitudinal analysis of adult patients with CKD stages 1-4, older age, higher protein excretion rates, lower glomerular filtration rates, higher serum phosphorus, PTH, and FGF23 levels, were all associated with an increased rate of CKD progression, as defined by a doubling of serum creatinine or the need for renal replacement therapy during the follow-up period. However, in multivariable analysis, only baseline GFR and FGF23 were independent predictors of progression [5, 12]. The etiology of the association between increased FGF23 and renal failure progression is incompletely understood but may reflect some deleterious effect of phosphate itself, such as progressive tissue calcification, or some as yet uncharacterized toxic effects of FGF23 on the renal parenchyma.
Implications for Renal Osteodystrophy Therapy
Current treatment paradigms advocate the use of 1,25(OH)2vitamin D and its derivatives for the control of secondary hyperparathyroidism and current data support the concept that active vitamin D sterols have a positive effect on renal disease, cardiovascular disease, and mortality [79, 80]. At the same time, however, active vitamin D sterol therapy also significantly increases FGF23 levels (Figure 5) [8] and high levels of FGF23 are independently associated with progressive renal disease, cardiac disease, and an increased mortality risk in the CKD population [3-5, 12, 13]. Thus, the interplay between PTH, FGF23, Klotho, phosphorus, and vitamin D appears to be critical for advancing the still limited understanding of the pathogenesis and future treatment of CKD-MBD. Since FGF23 levels increase in response to oral phosphate loading in healthy volunteers [21] as well as in wild-type and CKD rats [81], an increased, possibly intermittent, phosphate burden in the context of even mildly decreased renal function may contribute to enhanced FGF23 secretion. Phosphate-independent mechanisms, such as subtle, sub-clinical elevations in PTH [82, 83] or alterations in function of osteocytic regulators of FGF23 expression, may also contribute to the increase in FGF23 levels in CKD. Since circulating levels of FGF23 have been identified as an independent risk factor for cardiovascular morbidity and mortality in CKD patients [4, 12, 84], identification of the etiology for these increased circulating concentrations is critical for the management of the complications of CKD-MBD. Current efforts have focused on pharmacologic reduction in enteral phosphate burden and short-term trials have demonstrated that treatment with either sevelamer-HCl or lanthanum carbonate may reduce elevated FGF23 levels, resulting in increased circulating 1,25(OH)2vitamin D and decreased PTH values without altering serum phosphorus levels in normophosphatemic patients with CKD stages 3 and 4 [6, 85] (Figure 6). However, newer data also suggest that an allosteric activator of the calcium-sensing receptor, cinacalcet, also reduces circulating FGF23 values [86], potentially by means of reducing both circulating PTH and circulating phosphate concentrations, and thus may provide an effective approach for simultaneously reducing both FGF23 and PTH in late stages of CKD.
Fig. 5.

Percentage change in serum FGF23 as determined by the C-terminal (a) and Intact (b) Immutopics assays in pediatric dialysis patients treated for 8 months with 1αD2+CaCO3 (closed diamonds), 1αD2+sevelamer (open diamonds), calcitriol+CaCO3 (closed squares), and calcitriol+sevelamer (open squares). Asterisk indicates p<0.01 from baseline. 1αD2, doxercalciferol; CaCO3, calcium carbonate.
Fig. 6.
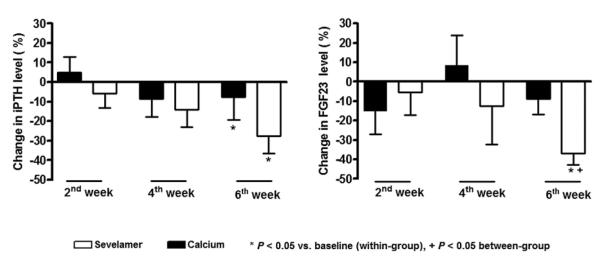
Percentage change in serum FGF23 and PTH levels in patients treated with sevelamer hydrochloride (□) or calcium acetate (■) over a 6-wk period.
Although much excitement surrounds the potential for new treatment paradigms to control FGF23, similar to the excitement and emphasis surrounding the pharmacologic control of PTH a decade ago, further studies are required to assess not only the long-term systemic consequences of targeting FGF23 levels, but the effects of different treatment strategies on bone. Indeed, the association between FGF23 with height standard deviation score in the pediatric population [61] and previous observations that FGF23 levels correlate with age, BMI, and IGF1 levels in children with CKD [87] suggest that this hormone may be a predictor of growth and that therapies aimed at lowering FGF23 levels in children may have additional consequences in the pediatric age group. Moreover, it is now clear that alterations in osteocyte biology, manifested by changes in osteocytic FGF23, DMP1, and sclerostin expression very early in the course of CKD, occur before abnormalities in traditional measures of mineral metabolism. Recent data, while identifying the central role of the osteocyte in CKD bone and mineral metabolism, have also highlighted just how little is known regarding the pathogenesis of renal osteodystrophy. Thus, further studies are critically needed to identify the initial trigger for abnormalities of skeletal mineralization and turnover as well as the potential effects that current therapeutic options may have on osteocyte biology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.ADHR Consortium Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat.Genet. 2000;26(3):345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 2.Isakova T, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney international. 2011;79(12):1370–8. doi: 10.1038/ki.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gutierrez OM, et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009;119(19):2545–2552. doi: 10.1161/CIRCULATIONAHA.108.844506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gutierrez OM, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N.Engl.J.Med. 2008;359(6):584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fliser D, et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J.Am.Soc.Nephrol. 2007;18(9):2600–2608. doi: 10.1681/ASN.2006080936. [DOI] [PubMed] [Google Scholar]
- 6.Oliveira RB, et al. Early control of PTH and FGF23 in normophosphatemic CKD patients: a new target in CKD-MBD therapy? Clin.J.Am.Soc.Nephrol. 2010;5(2):286–291. doi: 10.2215/CJN.05420709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wetmore JB, et al. Effects of cinacalcet and concurrent low-dose vitamin D on FGF23 levels in ESRD. Clin.J.Am.Soc.Nephrol. 2010;5(1):110–116. doi: 10.2215/CJN.03630509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wesseling-Perry K, et al. Calcitriol and doxercalciferol are equivalent in controlling bone turnover, suppressing parathyroid hormone, and increasing fibroblast growth factor-23 in secondary hyperparathyroidism. Kidney international. 2011;79(1):112–9. doi: 10.1038/ki.2010.352. [DOI] [PubMed] [Google Scholar]
- 9.Larsson T, et al. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64(6):2272–2279. doi: 10.1046/j.1523-1755.2003.00328.x. [DOI] [PubMed] [Google Scholar]
- 10.Ben-Dov IZ, et al. The parathyroid is a target organ for FGF23 in rats. J.Clin.Invest. 2007;117(12):4003–4008. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gutierrez O, et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J.Am.Soc.Nephrol. 2005;16(7):2205–2215. doi: 10.1681/ASN.2005010052. [DOI] [PubMed] [Google Scholar]
- 12.Isakova T, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA : the journal of the American Medical Association. 2011;305(23):2432–9. doi: 10.1001/jama.2011.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolf M, et al. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. Journal of the American Society of Nephrology : JASN. 2011;22(5):956–66. doi: 10.1681/ASN.2010080894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moe S, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO) Kidney Int. 2006;69(11):1945–1953. doi: 10.1038/sj.ki.5000414. [DOI] [PubMed] [Google Scholar]
- 15.KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Kidney international. Supplement. 2009;(113):S1–130. doi: 10.1038/ki.2009.188. [DOI] [PubMed] [Google Scholar]
- 16.Gattineni J, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am.J.Physiol Renal Physiol. 2009;297(2):F282–F291. doi: 10.1152/ajprenal.90742.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gattineni J, et al. Regulation of serum 1,25(OH)2 vitamin D3 levels by fibroblast growth factor 23 is mediated by FGF receptors 3 and 4. American journal of physiology. Renal physiology. 2011;301(2):F371–7. doi: 10.1152/ajprenal.00740.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Urakawa I, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444(7120):770–4. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 19.Shimada T, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J.Bone Miner.Res. 2004;19(3):429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 20.Shimada T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J.Clin.Invest. 2004;113(4):561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J.Clin.Endocrinol.Metab. 2006;91(8):3144–3149. doi: 10.1210/jc.2006-0021. [DOI] [PubMed] [Google Scholar]
- 22.Perwad F, et al. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005;146(12):5358–5364. doi: 10.1210/en.2005-0777. [DOI] [PubMed] [Google Scholar]
- 23.Portale AA, et al. Effect of dietary phosphorus on circulating concentrations of 1,25-dihydroxyvitamin D and immunoreactive parathyroid hormone in children with moderate renal insufficiency. J.Clin.Invest. 1984;73(6):1580–1589. doi: 10.1172/JCI111365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krajisnik T, et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J.Endocrinol. 2007;195(1):125–131. doi: 10.1677/JOE-07-0267. [DOI] [PubMed] [Google Scholar]
- 25.Kolek OI, et al. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am.J.Physiol Gastrointest.Liver Physiol. 2005;289(6):G1036–G1042. doi: 10.1152/ajpgi.00243.2005. [DOI] [PubMed] [Google Scholar]
- 26.Isakova T, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79(12):1370–1378. doi: 10.1038/ki.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferreira A, et al. Effects of sevelamer hydrochloride and calcium carbonate on renal osteodystrophy in hemodialysis patients. J.Am.Soc.Nephrol. 2008;19(2):405–412. doi: 10.1681/ASN.2006101089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lobao R, et al. High prevalence of low bone mineral density in pre-dialysis chronic kidney disease patients: bone histomorphometric analysis. Clinical nephrology. 2004;62(6):432–9. doi: 10.5414/cnp62432. [DOI] [PubMed] [Google Scholar]
- 29.Bakkaloglu SA, et al. Value of the new bone classification system in pediatric renal osteodystrophy. Clin.J.Am.Soc.Nephrol. 2010;5(10):1860–1866. doi: 10.2215/CJN.01330210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atkins D, Peacock M. A comparison of the effects of the calcitonins, steroid hormones and thyroid hormones on the response of bone to parathyroid hormone in tissue culture. J.Endocrinol. 1975;64(3):573–583. [PubMed] [Google Scholar]
- 31.Lee K, et al. In situ localization of PTH/PTHrP receptor mRNA in the bone of fetal and young rats. Bone. 1993;14(3):341–345. doi: 10.1016/8756-3282(93)90162-4. [DOI] [PubMed] [Google Scholar]
- 32.Sabbagh Y, et al. Repression of osteocyte Wnt/beta-catenin signaling is an early event in the progression of renal osteodystrophy. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2012 doi: 10.1002/jbmr.1630. [DOI] [PubMed] [Google Scholar]
- 33.Wesseling-Perry K, et al. Early skeletal and biochemical alterations in pediatric chronic kidney disease. Clin J Am Soc Nephrol. 2012;7(1):146–52. doi: 10.2215/CJN.05940611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wesseling-Perry K, et al. The calcemic response to continuous parathyroid hormone (PTH)(1-34) infusion in end-stage kidney disease varies according to bone turnover: a potential role for PTH(7-84) The Journal of clinical endocrinology and metabolism. 2010;95(6):2772–80. doi: 10.1210/jc.2009-1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sebastian EM, Suva LJ, Friedman PA. Differential effects of intermittent PTH(1-34) and PTH(7-34) on bone microarchitecture and aortic calcification in experimental renal failure. Bone. 2008;43(6):1022–30. doi: 10.1016/j.bone.2008.07.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malluche H, Faugere MC. Renal bone disease 1990: an unmet challenge for the nephrologist. Kidney Int. 1990;38(2):193–211. doi: 10.1038/ki.1990.187. [DOI] [PubMed] [Google Scholar]
- 37.Kuizon BD, et al. Diminished linear growth during intermittent calcitriol therapy in children undergoing CCPD. Kidney Int. 1998;53(1):205–211. doi: 10.1046/j.1523-1755.1998.00724.x. [DOI] [PubMed] [Google Scholar]
- 38.Kuizon BD, Salusky IB. Intermittent calcitriol therapy and growth in children with chronic renal failure. Miner.Electrolyte Metab. 1998;24(4):290–295. doi: 10.1159/000057384. [DOI] [PubMed] [Google Scholar]
- 39.Lund RJ, et al. Successful treatment of an adynamic bone disorder with bone morphogenetic protein-7 in a renal ablation model. Journal of the American Society of Nephrology : JASN. 2004;15(2):359–69. doi: 10.1097/01.asn.0000109671.99498.08. [DOI] [PubMed] [Google Scholar]
- 40.Pereira RC, et al. Patterns of FGF-23, DMP1, and MEPE expression in patients with chronic kidney disease. Bone. 2009;45(6):1161–8. doi: 10.1016/j.bone.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rhee Y, et al. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone. 2011;49(4):636–43. doi: 10.1016/j.bone.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wesseling-Perry K, et al. Calcitriol and doxercalciferol are equivalent in controlling bone turnover, suppressing parathyroid hormone, and increasing fibroblast growth factor-23 in secondary hyperparathyroidism. Kidney Int. 2011;79(1):112–119. doi: 10.1038/ki.2010.352. [DOI] [PubMed] [Google Scholar]
- 43.Munns CF, et al. Three children with lower limb fractures and a mineralization defect: a novel bone fragility disorder? Bone. 2004;35(5):1023–8. doi: 10.1016/j.bone.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 44.Groothoff JW, et al. Severe bone disease and low bone mineral density after juvenile renal failure. Kidney Int. 2003;63(1):266–275. doi: 10.1046/j.1523-1755.2003.00727.x. [DOI] [PubMed] [Google Scholar]
- 45.Gomes SA, et al. Usefulness of a quick decalcification of bone sections embedded in methyl methacrylate[corrected]: an improved method for immunohistochemistry. J Bone Miner.Metab. 2008;26(1):110–113. doi: 10.1007/s00774-007-0788-2. [DOI] [PubMed] [Google Scholar]
- 46.Levin A, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int. 2007;71(1):31–38. doi: 10.1038/sj.ki.5002009. [DOI] [PubMed] [Google Scholar]
- 47.Liu S, et al. Pathogenic role of Fgf23 in Dmp1-null mice. Am J Physiol Endocrinol.Metab. 2008;295(2):E254–E261. doi: 10.1152/ajpendo.90201.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lorenz-Depiereux B, et al. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. American journal of human genetics. 2010;86(2):267–72. doi: 10.1016/j.ajhg.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mackenzie NC, et al. Altered bone development and an increase in FGF-23 expression in Enpp1(-/-) mice. PloS one. 2012;7(2):e32177. doi: 10.1371/journal.pone.0032177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sherrard DJ, et al. The spectrum of bone disease in end-stage renal failure--an evolving disorder. Kidney Int. 1993;43(2):436–442. doi: 10.1038/ki.1993.64. [DOI] [PubMed] [Google Scholar]
- 51.Gowen LC, et al. Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J.Biol.Chem. 2003;278(3):1998–2007. doi: 10.1074/jbc.M203250200. [DOI] [PubMed] [Google Scholar]
- 52.Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am.J.Kidney Dis. 1998;32(5 Suppl 3):S112–S119. doi: 10.1053/ajkd.1998.v32.pm9820470. [DOI] [PubMed] [Google Scholar]
- 53.Goodman WG, et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N.Engl.J.Med. 2000;342(20):1478–1483. doi: 10.1056/NEJM200005183422003. [DOI] [PubMed] [Google Scholar]
- 54.Oh J, et al. Advanced coronary and carotid arteriopathy in young adults with childhood-onset chronic renal failure. Circulation. 2002;106(1):100–105. doi: 10.1161/01.cir.0000020222.63035.c0. [DOI] [PubMed] [Google Scholar]
- 55.Milliner DS, et al. Soft tissue calcification in pediatric patients with end-stage renal disease. Kidney Int. 1990;38(5):931–936. doi: 10.1038/ki.1990.293. [DOI] [PubMed] [Google Scholar]
- 56.Litwin M, et al. Altered morphologic properties of large arteries in children with chronic renal failure and after renal transplantation. Journal of the American Society of Nephrology : JASN. 2005;16(5):1494–500. doi: 10.1681/ASN.2004110932. [DOI] [PubMed] [Google Scholar]
- 57.Chavers BM, et al. Cardiovascular disease in pediatric chronic dialysis patients. Kidney Int. 2002;62(2):648–653. doi: 10.1046/j.1523-1755.2002.00472.x. [DOI] [PubMed] [Google Scholar]
- 58.Shroff RC, et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008;118(17):1748–1757. doi: 10.1161/CIRCULATIONAHA.108.783738. [DOI] [PubMed] [Google Scholar]
- 59.Shroff RC, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J.Am.Soc.Nephrol. 2010;21(1):103–112. doi: 10.1681/ASN.2009060640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Russo D, et al. Coronary artery calcification in patients with CRF not undergoing dialysis. Am.J.Kidney Dis. 2004;44(6):1024–1030. doi: 10.1053/j.ajkd.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 61.Wesseling-Perry K, et al. Early skeletal and biochemical alterations in pediatric chronic kidney disease. Clinical journal of the American Society of Nephrology : CJASN. 2012;7(1):146–52. doi: 10.2215/CJN.05940611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shanahan CM. Mechanisms of vascular calcification in renal disease. Clin.Nephrol. 2005;63(2):146–157. doi: 10.5414/cnp63146. [DOI] [PubMed] [Google Scholar]
- 63.Ducy P, et al. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89(5):747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 64.Moe SM, et al. Uremia induces the osteoblast differentiation factor Cbfa1 in human blood vessels. Kidney Int. 2003;63(3):1003–1011. doi: 10.1046/j.1523-1755.2003.00820.x. [DOI] [PubMed] [Google Scholar]
- 65.Jono S, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circ.Res. 2000;87(7):E10–E17. doi: 10.1161/01.res.87.7.e10. [DOI] [PubMed] [Google Scholar]
- 66.Villa-Bellosta R, et al. Characterization of phosphate transport in rat vascular smooth muscle cells: implications for vascular calcification. Arteriosclerosis, thrombosis, and vascular biology. 2007;27(5):1030–6. doi: 10.1161/ATVBAHA.106.132266. [DOI] [PubMed] [Google Scholar]
- 67.Ahmed S, et al. Calciphylaxis is associated with hyperphosphatemia and increased osteopontin expression by vascular smooth muscle cells. Am.J.Kidney Dis. 2001;37(6):1267–1276. doi: 10.1053/ajkd.2001.24533. [DOI] [PubMed] [Google Scholar]
- 68.Bostrom K. Insights into the mechanism of vascular calcification. Am.J.Cardiol. 2001;88(2A):20E–22E. doi: 10.1016/s0002-9149(01)01718-0. [DOI] [PubMed] [Google Scholar]
- 69.Moe SM, et al. Medial artery calcification in ESRD patients is associated with deposition of bone matrix proteins. Kidney Int. 2002;61(2):638–647. doi: 10.1046/j.1523-1755.2002.00170.x. [DOI] [PubMed] [Google Scholar]
- 70.Chen NX, et al. Phosphorus and uremic serum up-regulate osteopontin expression in vascular smooth muscle cells. Kidney Int. 2002;62(5):1724–1731. doi: 10.1046/j.1523-1755.2002.00625.x. [DOI] [PubMed] [Google Scholar]
- 71.Schinke T, et al. The serum protein alpha2-HS glycoprotein/fetuin inhibits apatite formation in vitro and in mineralizing calvaria cells. A possible role in mineralization and calcium homeostasis. J.Biol.Chem. 1996;271(34):20789–20796. doi: 10.1074/jbc.271.34.20789. [DOI] [PubMed] [Google Scholar]
- 72.Sweatt A, et al. Matrix Gla protein (MGP) and bone morphogenetic protein-2 in aortic calcified lesions of aging rats. J.Thromb.Haemost. 2003;1(1):178–185. doi: 10.1046/j.1538-7836.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- 73.Schafer C, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J.Clin.Invest. 2003;112(3):357–366. doi: 10.1172/JCI17202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Faul C, et al. FGF23 induces left ventricular hypertrophy. The Journal of clinical investigation. 2011;121(11):4393–408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Razzaque MS, et al. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J. 2006;20(6):720–722. doi: 10.1096/fj.05-5432fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Srivaths PR, et al. Elevated FGF 23 and phosphorus are associated with coronary calcification in hemodialysis patients. Pediatric nephrology. 2011;26(6):945–51. doi: 10.1007/s00467-011-1822-0. [DOI] [PubMed] [Google Scholar]
- 77.Lim K, et al. Vascular Klotho Deficiency Potentiates the Development of Human Artery Calcification and Mediates Resistance to FGF-23. Circulation. 2012 doi: 10.1161/CIRCULATIONAHA.111.053405. [DOI] [PubMed] [Google Scholar]
- 78.Hu MC, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. Journal of the American Society of Nephrology : JASN. 2011;22(1):124–36. doi: 10.1681/ASN.2009121311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Teng M, et al. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N.Engl.J.Med. 2003;349(5):446–456. doi: 10.1056/NEJMoa022536. [DOI] [PubMed] [Google Scholar]
- 80.Teng M, et al. Activated injectable vitamin D and hemodialysis survival: a historical cohort study. J.Am.Soc.Nephrol. 2005;16(4):1115–1125. doi: 10.1681/ASN.2004070573. [DOI] [PubMed] [Google Scholar]
- 81.Nagano N, et al. Effect of manipulating serum phosphorus with phosphate binder on circulating PTH and FGF23 in renal failure rats. Kidney Int. 2006;69(3):531–537. doi: 10.1038/sj.ki.5000020. [DOI] [PubMed] [Google Scholar]
- 82.Wesseling-Perry K, et al. The calcemic response to continous PTH(1-34) infusion in end-stage kidney disease varies according to bone turnover: a potential role for PTH(7-84) J.Clin.Endocrinol.Metab. 2010;95(6):2772–2780. doi: 10.1210/jc.2009-1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lavi-Moshayoff V, et al. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am.J.Physiol Renal Physiol. 2010;299(4):F882–F889. doi: 10.1152/ajprenal.00360.2010. [DOI] [PubMed] [Google Scholar]
- 84.Parker BD, et al. The associations of fibroblast growth factor 23 and uncarboxylated matrix Gla protein with mortality in coronary artery disease: the Heart and Soul Study. Ann.Intern.Med. 2010;152(10):640–648. doi: 10.1059/0003-4819-152-10-201005180-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gonzalez-Parra E, et al. Lanthanum carbonate reduces FGF23 in chronic kidney disease stage 3 patients. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2011 doi: 10.1093/ndt/gfr144. [DOI] [PubMed] [Google Scholar]
- 86.Koizumi M, et al. Cinacalcet treatment and serum FGF23 levels in haemodialysis patients with secondary hyperparathyroidism. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2012;27(2):784–90. doi: 10.1093/ndt/gfr384. [DOI] [PubMed] [Google Scholar]
- 87.Bacchetta J, et al. The influence of glomerular filtration rate and age on fibroblast growth factor 23 serum levels in pediatric chronic kidney disease. J.Clin.Endocrinol.Metab. 2010;95(4):1741–1748. doi: 10.1210/jc.2009-1576. [DOI] [PubMed] [Google Scholar]