

Abstract
Recent studies using both dissociated and organotypic cell cultures have shown that heterozygous Lurcher (Lc/+) Purkinje cells (PCs) grown in vitro share many of the same survival and morphological characteristics as Lc/+ PCs in vivo. We have used this established tissue culture system as a valuable model for studying cell death mechanisms in a relatively simple system where neurodegeneration is induced by a constitutive cation leak mediated by the Lurcher mutation in theδ2 glutamate receptor (GluRδ2). In this study, Ca++ imaging and immunocytochemistry studies indicate that intracellular levels of Ca++ are chronically increased in Lc/+ PCs and the concentration and/or distribution of the conventional PKCγ isoform is altered in degenerating Lc/+ PCs. To begin to characterize the molecular mechanisms that regulate Lc/+ PC death, the contributions of conventional PKC pathways and of two MAP kinase family members, JNK and p38, were examined in slice cultures from wild type and Lc/+ mutant mouse cerebellum. Cerebellar slice cultures from P0 pups were treated with either a conventional PKC inhibitor, a JNK inhibitor, or a p38 inhibitor either from 0 to 14 or 7 to 14 DIV. Treatment with either of the three inhibitors from 0 DIV significantly increased wild type and Lc/+ PC survival through 14 DIV, but only Lc/+ PC survival was significantly increased following treatments from 7 to 14 DIV. The results suggest that multiple PC death pathways are induced by the physical trauma of making organotypic slice cultures, naturally-occurring postnatal cell death, and the GluRδ2Lc mutation.
Keywords: Cell death, Lurcher mutant mouse, JNK, PKC, MAP Kinase, Organotypic culture, Purkinje cells, JNK
Introduction
Neurons actively enter cell death pathways both during development and in pathological processes. Identifying the precise mechanisms that regulate neuronal cell death is one of the fundamental goals in neurobiology. However, our understanding of cell death is hampered by the fact that multiple complex biochemical pathways are involved in the various modes of cell death. Thus, there is a need for well-defined and relatively simple models. This study is part of an overall analysis of the molecular mechanisms of cell death in Purkinje cells (PCs) in the Lc/+ mutant as a relatively simple model of neuronal death caused by a well-defined lesion in a single, well-characterized cell type.
Lurcher (gene symbol, Lc) is a gain-of-function point mutation in the δ2 glutamate receptor gene that turns the receptor (GluRδ2) into a constitutively open cation channel (1, 2). GluRδ2 receptors are predominantly expressed in cerebellar PCs, and the leak current mediated by the GluRδ2Lc receptor chronically depolarizes cerebellar PCs starting during the first postnatal week of development (3). In the heterozygous Lc/+ mutant almost all PCs degenerate after the first week of postnatal life via pathways that have been described as either apoptotic, autophagic, or necrotic (2–7). Homozygous Lc mutants die around birth subsequent to massive neuronal cell loss in the hindbrain during embryonic development (8).
A general hypothesis guiding our studies of PC death in the GluRδ2Lc/+ mutant is that chronic depolarization of PCs mediated by the GluRδ2Lc leak current affects a wide range of cellular homeostatic systems, including key signaling pathways that are important in the regulation of PC dendritic development and cell death. Genetic studies have indicated that there are likely to be multiple apoptotic molecular pathways that can contribute to GluRδ2Lc/+ PC death. For example, one hallmark of apoptotic GluRδ2Lc/+ PC death is the up-regulation of procaspase-3 expression in many Lc/+ PCs, and the expression of activated caspase-3 in PCs that appear to be degenerating. Deletion of the pro-apoptotic gene Bax can transiently delay GluRδ2Lc/+ PC death, but in Lc/+:Bax−/− double mutants, activated caspase-3 can no longer be detected, suggesting that an alternative cell death pathway has been invoked in the absence of Bax expression (9, 10). Other recent studies have shown evidence for an increase in oxidative stress in GluRδ2Lc/+ PCs (11), which may lead to the deleterious activation or suppression of a number of cellular developmental or homeostatic pathways.
The purpose of this study is to investigate the role of three critical protein kinase pathways associated with cellular responses to stress on the survival and differentiation of chronically depolarized and stressed GluRδ2Lc/+ PCs. The three pathways investigated are the conventional, Ca2+–activated, phospholipid-dependent protein kinases (cPKC) and the stress related MAPK pathways represented by the downstream effectors, c-Jun N-terminal kinase (JNK) and p38. The family of conventional PKC isoforms are involved in signal transduction systems associated with cell proliferation, differentiation, and apoptosis, and they are highly sensitive to the redox status of their environment (reviewed in (12)). JNK and p38 are downstream kinases in a sequence of MAPK signaling cascades that are associated with a variety of stressors, including inflammation, activation of death receptors, apoptosis, and oxidative stress (reviewed in (13, 14)). Activation of the stress activated JNK pathway, for example, is thought to induce apoptosis by transcription-dependent or – independent mechanisms. With the aid of selective inhibitors for the conventional PKCs, JNK, and p38, we show that treatment of WT and Lc/+ cerebellar organotypic slice cultures with Gö6976 (cPKC inhibitor), SP600125 (JNK inhibitor) or SB 203580 (p38 inhibitor) significantly increases both GluRδ2+/+ and GluRδ2Lc/+ PC survival in vitro. These results suggest that activation of all three pathways is associated with PC death processes in vitro related to the stress of slicing and culturing tissue, normal developmental neuronal cell death, and the homeostatic stress of chronic depolarization mediated by the GluRδ2Lc mutant receptor.
Materials and Methods
Animals
GluRδ2Lc/+ mutant and wild type (GluRδ2+/+) pups were generated by mating B6CBACa Aw-J/A-Grid2Lc/J males with wild type B6CBA females from Janvier Laboratories or Jackson Laboratories (NB: Grid2 is the official name of the Lc gene). Males were harem mated with one male to two or three females and the females were checked for copulatory plugs every day the mice remained together. The day of finding the copulatory plug was considered embryonic day 0.5 (E0.5) and the day of birth is counted as postnatal day 0 (P0). All animals were housed in standard conditions (14 hours light, 10 hours dark) in animal facilities either at the Université Pierre et Marie Curie (UPMC) or the Maryland Psychiatric Research Center (MPRC) and provided with food and water ad libitum. The animal facility at the UPMC is fully accredited by the French Research and Higher Education ministry, and studies were conducted in accordance with the Guide for Care and Use of Laboratory Animals provided by the guidelines established by “le comité national d’éthique pour les sciences de la vie et de la santé.” The animal facilities at the MPRC are fully accredited by the American Association for the Accreditation of Laboratory Animal Care (AAALAC) and the studies were conducted in accordance with the Guide for Care and Use of Laboratory Animals provided by the NIH.
The Lc/+ or +/+ genotype of P0 pups was identified by PCR and single-stranded conformation polymorphism (SSCP) as described previously (3). GluRδ2 S1 and AS primers were used to amplify a stretch of DNA that spans the single base pair change in the Grid2Lc mutation (15).
Organotypic tissue slice cultures
Cerebellar slices were prepared from GluRδ2Lc/+ mutant and GluRδ2+/+ pups at P0. Pups were decapitated and the brain removed in ice cold Gey’s balanced salt solution with 5 mg/mL glucose. The cerebellum was separated from the rest of the brain with forceps after removing the choroid plexus and dura. The entire cerebellum was then sliced into 350 μm sagittal sections, and the slices were placed on the membrane of Millicell CM inserts (Millipore, MA). All of the sections from one cerebellum were arranged on one culture insert. Slices were maintained at the interface between the air and the culture media consisting of 50% Basal Medium Eagle (BME), 25% Hank’s Balanced Salt Solution (HBSS), 25% heat inactivated horse serum, 1 mM L-glutamine, and 5 mg/ml D-glucose in a humidified chamber with 5% CO2 (pH 7.3) at 35° C. The media was changed every 2–3 days and cultures were fixed at 14 days in vitro. JNK inhibitor SP600125, P38 inhibitor SB 203580 or PKC inhibitor Gö6976 were added in the culture medium either from DIV 1 or DIV 7. All of the in vitro organotypic experiments with cerebellar slices were conducted at the Université Pierre et Marie Curie, including the Ca++ imaging and electrophysiology studies.
Immunohistochemistry
Slice cultures were fixed in 4% paraformaldehyde in 0.1M phosphate buffer (pH 7.4) for 30 minutes at room temperature followed by multiple washes with 10 mM phosphate buffered saline (0.9% NaCl, PBS). Slices were then incubated for 1 hour in PBS containing 0.2 % Triton X-100, 0.2% gelatin, 0.1M lysine before immunostaining with mouse monoclonal antibodies against calbindin (dilution 1:5000, Swant, Bellinzona, Switzerland). Antibodies were revealed with CY3- conjugated Donkey anti-mouse antibody (1:500 dilutions, Jackson ImmunoResearch Laboratories, Inc). After 2 h incubation in buffer containing the secondary antibody, the slices were washed several times with PBS and counterstained with DNA fluochrome Hoechst 33258 (diluted 1/50000, Sigma) and mounted in Mowiol (Calbiochem, La Jolla, CA, USA).
For immunocytochemistry in fixed cerebellar sections, mouse pups were euthanized by cardiac perfusion with 0.9% saline followed by 4% paraformaldehyde (while deeply anesthetized with Euthasol, >100 μg/g). Following the perfusion, brains were removed from the skull, postfixed for 2 hours in fresh fixative, and then cryoprotected with 20% sucrose in 10 mM phosphate buffed saline (PBS). At least 48 h later, the fixed brains were embedded in OCT and frozen in isopentane. Fixed, frozen brains were cut at 12 μm on a Leica cryostat, collected directly on slides, and stored at −70°C until stained. To prepare slides for immunofluoresence studies, slides were rinsed in 10mM PBS, followed by incubation in two changes of 0.1M glycine for 5 min each. Endogenous fluorescence was reduced by incubating the sections in 50 mM ammonium chloride for 1 hour. The sections were then rinsed three times in 10mM PBS and incubated for an hour in blocking solution containing 3% normal goat serum and 0.3%Triton X-100. Sections were then incubated in the following primary antibodies overnight at 4°C: mouse monoclonal anti-PKCγ(Zymed: 1/100) and rabbit polyclonal anti-calbindin (Sigma: 1/1000) or rabbit polyclonal anti-activated caspase-3 (R&D: 1/500). The following day, sections were rinsed 3 times in PBS and then incubated for 2 hours with fluorescent-labeled secondary antibodies (anti-mouse or anti-rabbit Alexa 594 and Alexa 488: Molecular Probes: 1/200). After incubation, they were rinsed once in 10mM PBS and incubated with 300nM DAPI, then rinsed 3 times in 10 mM PBS, once in distilled water, and coverslipped with gelmount. The finished slides were then photographed using a Zeiss Axioplan fluorescence microscope with an Olympus DP70 CCD camera. All immunofluorescence experiments included slides (wild type and Lc/+) with no primary antibody incubation as a control for non-specific immunolabeling. Digital images were cropped and adjusted using Adobe Photoshop for color balance and intensity. All of the immunohistochemistry studies on cerebellar sections from brains fixed in vivo were performed at the Maryland Psychiatric Research Center.
PC counts
In most cases, the total number of PCs per culture was directly determined by systematically scanning the entire culture area and counting all fluorescent calbindin-labeled PCs. The cell counts were conducted with a 40x objective and an eyepiece graticule was used to systematically sweep across the culture and to keep track of counted and uncounted areas. For cerebellar slice cultures treated with Gö6976 it was not possible to count all of the PCs since the density and total number was too high. Instead, the total number of PCs per slice was estimated from sampling the density of PCs and measuring the total area of each slice. The total area of the slices per culture was calculated by photographing each cultured slice and then measuring the area of the slice with Image-Pro 4.5 (Media Cybernetics, Maryland). The PC density was estimated in each slice by counting the number of PCs in 180 × 180 μm grids systematically randomly selected throughout each slice. The total number of PCs per slice was then calculated as the product of the PC density and slice area. The total number of PCs was estimated as the sum of PCs in all of the slices cultured from each whole cerebellum. The average number and density of PCs is reported as the mean ± standard error.
Calcium imaging and electrophysiology
Standard patch-clamp recordings were used to determine the size of leak currents in GluRδ2+/+ and GluRδ2Lc/+ PCs either from slice cultures from 8–16 DIV, or from acute slices prepared from pups aged P8–P11 using standard procedures (16). Cerebellar slices were placed in artificial CSF (aCSF) containing (in mM) 125 NaCl, 25 NaHCO3, 10 glucose, 3.5 KCl, 1.25 NaH2PO4, 2 CaCl2, and 1 MgCl2 and oxygenated with 95%:5% O2:CO2. Slices are incubated at 32–34°C for at least 1 h until ready for recording. For the recordings, slices were placed in a chamber mounted on an Olympus BX51-WI microscope, and continuously perfused with aCSF at 2 ml/min. Whole-cell configuration was acquired with glass pipettes containing a 0.125% Neurobiotin solution containing (in mM) 115 K gluconate, 10 HEPES, 2 MgCl2, 20 KCl, 2 MgATP, 2 Na2-ATP, and 0.3 GTP (pH 7.3, 280±5 mOsm). Electrophysiological recordings were acquired with an Axopatch 200B amplifier (Axon Instruments), digitized, and analyzed using Acquis (Biologic, France).
Leak current was measured for each PC. For measurements of Ca++ levels, individual PCs in acute slice preparations were patch clamped and filled with Fura (100 μM). Fluorescence levels were measured using a CCD camera (Cascade 1K, Photometrics) piloted by the Metamorph image program at 340 and 380 nm to calculate 340nm/380nm ratios as a measure of the basal intracellular Ca++ concentration. In some experiments, a brief depolarizing voltage step was applied to the PC to evaluate the time course and amplitude of increased Ca++ concentration.
Statistical analysis
The number of surviving PCs in drug-treated and control GluRδ2Lc/+ and GluRδ2+/+ slice cultures was initially analyzed with two-way analysis of variance (ANOVA) including genotype and drug treatment as co-variants (StatView 5.0, Cary, North Carolina). A log transformation of the number of PCs per culture was used to stabilize the variance for the ANOVA analysis. Post-hoc comparisons between groups were made using the Bonferroni/Dunn test.
For the statistical analysis of Ca++ levels using 340n/380nm fluorescence ratios, the non-parametric Mann-Whitney U test was used to compare the differences between Ca++ levels. ANOVA was used to compare mean leak currents in GluRδ2Lc/+ and GluRδ2+/+ PCs.
Results
Intracellular Ca++ levels in Lc/+ PCs
To test whether intracellular Ca++ levels are affected by the GluRδ2Lc leak current, individual PCs in acute slice preparations were patch clamped and filled with Fura-2 (100 μM). Fluorescence levels were measured at 340 and 380 nm to calculate the basal [Ca++] as 340nm/380nm ratios. Pseudo-color is used to show fluorescence intensity in a filled WT PC at 340 nm (Fig. 1A) and for the 340nm/380nm ratio (Fig. 1B). The labeling is much less intense than in an equivalent Lc/+ PC (Fig. 1C, D) where the filled Lc/+ PC body and dendrites can be seen in the 340nm/380nm ratio image. Comparison of Lc/+ and wild type PCs at P8–12 shows that intracellular Ca++ levels are increased in resting Lc/+ PCs compared with wild type PCs (Fig. 1E; Mann-Whitney test, p < 0.0001).
Figure 1.

Elevated calcium concentrations in Lc/+ PCs. Fura-2 imaging of WT (A–B) and Lc/+ (C–D) PCs, measured by the ratio of fluorescence intensity at 340/380nm. The patch clamp electrode used to fill the cells is shown touching the cells from the left. Scale bar 20 μm. E. Mean fluorescence ratio for WT and Lc/+ PCs. F. Representative ratiometric recordings of voltage clamped PCs show that resting basal Ca++ levels in Lc/+ PCs are higher than in WT PCs and the Lc/+ PCs do not respond to a step depolarizing pulse (arrow) with an increase in Ca++ levels as in WT PCs.
In addition to increased basal levels of Ca++, voltage clamp studies indicate that Lc/+ PCs do not have a normal Ca++ response to depolarization (Fig. 1F). WT and Lc/+ PCs were voltage clamped at −60 mV in acute cerebellar slices at P8–11 and then given a test depolarizing pulse (to −10 mV for 50 msec) and changes in the 340nm/380nm fluorescence ratio measured. Wild type PCs respond with a robust Ca++ increase (n=13; representative response shown in red) but Lc/+ PCs do not show any response to the depolarizing pulse (n=6, representative response shown in blue). The lack of a Ca++ response in Lc/+ PCs is likely due to the abnormally high basal levels of Ca++.
Thus our Ca++ imaging results show that resting levels of Ca++ are constitutively increased in Lc/+ PCs, which could cause activation of the conventional PKC isoforms.
Altered expression of PKCγ in Lc/+ PCs
To test the hypothesis that chronic increases in resting Ca++ levels may affect the expression patterns of conventional PKC isoforms, we have focused our studies on the expression of PKCγ, the PKC isoform with the highest and most uniform expression levels in cerebellar PCs. Cerebellar PCs express a wide range of PKC isoforms, although the exact complement is disputed (17). There is some agreement, though, that PCs express the conventional PKCs (cPKC), alpha (α) and gamma (γ), and the novel PKCs (nPKC), delta (δ) and epsilon (ε). The developmental distribution of PKCγ was qualitatively analyzed in PCs in WT and Lc/+ PCs in cerebellar sections from pups at P10, P15, and P25 using immunocytochemistry (ICC) with an anti-PKCγ antibody from Invitrogen. The pattern and level of ICC labeling for PKCγ is similar in WT and Lc/+ cerebellar slices at P10 (data not shown), but by P15 the intensity of PKCγ immunolabeling is reduced in most Lc/+ PCs compared with WT PCs (Fig. 2) and by P25 PKCγ immunolabeling has virtually disappeared in Lc/+ PCs (data not shown). However, at P15 there are distinct heterogeneities in PKCγ staining among the Lc/+ PCs. Figure 2 shows epifluoresence double-labeling for PKCγ (green) and calbindin (red: a marker for PCs) in P15 WT and Lc/+ cerebellar sections at the same antibody concentration and at the same photographic exposure times. The overall intensity of immunolabeling for PKCγ is reduced in the Lc/+ PCs (Fig. 2: D–F) compared with WT PCs (Fig. 2: A–C), which may indicate that PKCγ levels decline in the Lc/+ PCs. There are some exceptions, however, where the intensity of PKCγ immunolabeling is increased compared with other Lc/+ PCs (arrows and arrowheads in Fig. 2: D–F). In some cases, the morphology of these intensely-labelled Lc/+ PCs does not differ from the other Lc/+ PCs (arrowheads in Fig. 2 D–F), but in other cases they have the characteristic appearance of Lc/+ PCs during degeneration (arrows in Fig. 2: D–F). These observations suggest that the overall levels of PKCγ may be reduced in depolarized Lc/+ PCs, but they also raise the possibility that the expression or distribution of PKCγ may undergo a change in Lc/+ PCs as they actually degenerate.
Figure 2.

Double immunolabeling for PKCγ (green; A, D) and calbindin (red; B. E) in WT (AC) and Lc/+ (D–F) in P15 cerebellar slices indicates that PKCγ is expressed in WT and Lc/+ PCs, although the intensity of PKCγ immunolabeling is highly variable in Lc/+ PCs. The images were taken at the same exposure settings. The merged images (C, F) were created by overlaying the original images in Adobe Photoshop, and no other editing was performed. Scale bar 20 μm.
To explore the possibility that Lc/+ PCs with more intense PKCg immunolabeling are degenerating, a small sample of P15 Lc/+ cerebellar slices were double labeled with antibodies to PKCγ (Fig. 3 A; green) and activated caspase-3 (Fig. 3 B; red). In general, Lc/+ PCs that express activated caspase-3 have more rounded and stumpy dendrites that are often associated with calbindin-positive blebs that look like dendrites that have broken off the rest of the PC ((6) and unpublished observations). As shown in Fig. 3, many of the degenerating Lc/+ PCs expressing activated caspase-3 also stained more intensely for PKCγ (lower arrow), but this was not always the case (upper arrow). While a quantitative analysis of the distribution and intensity of PKCg immunolabeling in Lc/+ PCs that express activated caspase-3 was outside the scope of this project, these qualitative descriptions raise the possibility that the expression or distribution of PKCγ may be altered in Lc/+ PCs that are actively degenerating and that PKCγ activation could play a role in the Lc/+ cell death pathway.
Figure 3.
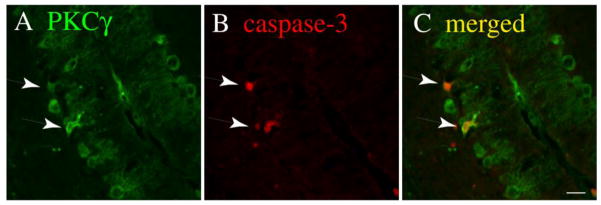
Double immunolabeling for PKCγ (green; A) and activated caspase-3 (red; B) in P15 Lc/+ cerebellar slices indicates that PKCγ is expressed in degenerating Lc/+ PCs. The merged image (C) was created by overlaying the original images in Adobe Photoshop. Scale bar 20 μm.
Lc/+ and WT PC survival in vitro is enhanced by treatment with a conventional PKC inhibitor, Gö6976
The family of PKC kinases plays a critical role in signaling pathways in stressed cells (12), and conventional PKC (cPKC) isoforms are stimulated by Ca++. Since Ca++ signaling is disrupted in GluRδ2Lc/+ PCs and the expression and/or distribution of PKCγ appears to be altered in Lc/+ PCs in vivo, we tested the hypothesis that activation of cPKC isoforms contributes to GluRδ2Lc/+ PC death by treating GluRδ2+/+ and GluRδ2Lc/+ cerebellar slice cultures with the general conventional PKC inhibitor, Gö6976 (1μM), from 0 to 14 DIV. The total number of calbindin-labeled PCs was then counted or calculated at 14 DIV. These experiments were conducted in vitro since it was not feasible to consistently inhibit cPKC activity selectively in the in vivo cerebellum with pharmacological tools and Lc/+ PC differentiation and survival in vitro appears to recapitulate their in vivo development in key aspects (18).
Previous studies have shown that after 14 DIV in control slice cultures, GluRδ2Lc/+ Purkinje survival is reduced by over 70% compared to control values (18). However, treating cultures with the PKC inhibitor Gö6976 from 0 DIV dramatically increased PC survival in both GluRδ2+/+ and GluRδ2Lc/+ slice cultures at 14 DIV (Fig. 4A–D). Cultures from 54 P0 pups were treated with Gö6976 or vehicle, and a random sample was selected for PC counts (Fig. 4E). ANOVA analysis of the resulting counts showed that there were significant effects of genotype and treatment (Genotype: ANOVA, F2,51= 46.0, p < 0.0001; Treatment: ANOVA, F2,51=93.3, p < 0.0001). The results of the PC counts indicate that treatment with 1μM Gö6976 resulted in a 6 fold increase in the number of surviving WT PCs by 14 DIV (Treated: 14,490 ± 585, n=7 vs. control: 2216 ± 266, n=10; p < 0.0001) and an over 20 fold increase in the number of surviving GluRδ2Lc/+ PCs per cerebellar slice culture (Treated: 14,780± 1054, n=7 vs. control: 625±65, n=13, p < 0.0001). There is no significant difference between the numbers of surviving PCs in treated control or GluRδ2Lc/+ cultures (p>0.5), suggesting that treatment with Gö6976 restores GluRδ2Lc/+ PC survival to the level of wild type cerebellar slice cultures also treated with Gö6976.
Figure 4.

Treatment of both WT and Lc/+ cerebellar slice cultures with the conventional PKC inhibitor Gö6976 (1μM) from 0 to 14 DIV dramatically increases PC survival. A–D) The images of calbindin labeled control (A and B) and Gö6976-treated (C and D) WT (A, C) and Lc/+ (B, D) cerebellar slices demonstrates the dramatic increase in the number of surviving PCs after 14 DIV due to ongoing treatment with Gö6976 (Scale bar 200μm). E) Treatment with Gö6976 dramatically increases both WT and Lc/+ PC survival when cultures are treated from the first day in vitro. However, delaying Gö6976 treatment until 7 DIV to 14 DIV still significantly increases WT and Lc/+ PC survival, although the effects are not as dramatic. F) Measurement of the leak current in WT and Lc/+ PCs did not show any evidence that treatment with Gö6976 has long term effects on the leak current induced by the mutant GluRδ2Lc channel. Lc/+ PCs had larger leak currents whether or not they had been treated with Gö6976.
While treatment with Gö6976 from 0 DIV dramatically increases both GluRδ2+/+ and GluRδ2Lc/+ PC survival, the mechanism is unknown so it was not clear that Gö6976 treatment was affecting PC death due to the GluRδ2Lc leak current. In vivo, PC death due to the GluRδ2Lc mutation in the GluRδ2 receptor is not apparent until approximately 1 week after birth, and a similar delay in GluRδ2Lc/+ PC death is also observed in slice and isolated cell cultures (18). To test the hypothesis that cPKC activation contributes specifically to GluRδ2Lc/+ PC death in vitro, in a separate set of experiments GluRδ2+/+ and GluRδ2Lc/+ PC slice cultures were treated with Gö6976 (1 μM) from 7 to 14 DIV (Fig. 4E). Delayed treatment of cerebellar slice cultures from 7 DIV only increased the number of surviving wild type PCs 1.5 fold (3,706±495; n=9), while GluRδ2Lc/+ PC survival increased by about three fold (1,858±170; n=8) compared with untreated cerebellar slice cultures. However, the increase in PC survival following Gö6976 treatment from 7 to 14 DIV is still significant for both GluRδ2+/+ and GluRδ2Lc/+ PCs (p < 0.002).
Previous studies have shown that blocking the GluRδ2Lc/+ leak current with 1-naphtylacetylspermine (NASP), a channel blocker for Ca++ permeable iGluRs, also restores GluRδ2Lc/+ PC survival to in vitro wild type levels (although WT PC survival is not affected by NASP treatment: (18)). To determine whether Gö6976 treatment increases GluRδ2Lc/+ PC survival by eliminating the leak current, a series of cerebellar slice cultures were treated with Gö6976 (1 μM) until 14 DIV. The GluRδ2Lc – mediated leak current was then measured in PCs from Gö6976 treated and untreated GluRδ2+/+ and GluRδ2Lc/+ PC slice cultures slice cultures. For technical reasons Gö6976 could not be included in the electrophysiological bath solution. Since cerebellar slices were rinsed in the electrophysiological aCSF for at least an hour before recordings were made, the drug is likely to have washed out at the time of the experiments. However, as shown in Figure 4F there are no significant differences in the magnitude of the leak current between treated and untreated Lc/+ PCs. There is a significant effect of genotype, with the Lc/+ PCs having significantly higher leak currents compared to WT controls (ANOVA F3, 84 = 13.187, p < 0.0001). The results indicate that treatment with Gö6976 does not permanently eliminate the leak current mediated by the GluRδ2Lc channel. While we cannot rule out the possibility that the continuous presence of Gö6976 might have had an effect on the leak current, there is no evidence that Gö6976 acts on glutamate channels. Instead we hypothesize that Gö6976 is acting downstream of the leak current to block activation of one or more cell death pathways following the activation of cPKC isoforms.
PC survival following treatments with inhibitors for the stress-related MAP kinases JNK and p38
The MAP kinases JNK and p38 are critical downstream effector kinases for signaling pathways involved in the cellular response to various types of oxidative stress. To compare the effects of a cPKC inhibitor on GluRδ2+/+ and GluRδ2Lc/+ PC survival with the effects of JNK and p38 inhibitors, wild type and Lc/+ cerebellar slice cultures were treated from 0 to 14 DIV with either the JNK II inhibitor SP600125 or the p38 inhibitor SB 203580 (Fig. 5). Counts of calbindin-labeled GluRδ2+/+ and GluRδ2Lc/+ PCs at 14 DIV treated with the JNK inhibitor SP600125 (10 μM) from 0 DIV indicate there were significant effects of genotype (ANOVA, F1, 41 = 65.21; p < 0.0001) and treatment (ANOVA, F2, 41=79.42, p < 0.0001) on PC survival, but no significant genotype-by-treatment interactions (p > 0.1; Fig. 5G). In wild type cultures, SP600125 treatment significantly increased GluRδ2+/+ PC survival by almost 500% (Treated: 10,855±774, n=5 vs. control: 2216±266, n=10; p<0.0001), while in Lc/+ slice cultures, GluRδ2Lc/+ PC survival increased by about 675% (Treated: 4,205±407; n=7 vs. control: 625±65; n=13; p < 0.0001).
Figure 5.

A) Low magnification images of calbindin immunolabeled WT (A, C, E) and Lc/+ (B, D, F) cerebellar slice cultures treated with the JNK inhibitor, SP600125, and the p38 inhibitor, SB 203580, demonstrate that both of these inhibitors increase WT and Lc/+ PC survival when cultures are treated from 0 to 14 DIV. However, the increase in PC survival is not as dramatic as treatment with the cPKC inhibitor Gö6976. G, H) Estimates of total PC numbers in control and drug treated WT and Lc/+ cerebellar slice cultures from 0 to 14 and 7 to 14 DIV (G: SP600125; H: SB 203580). Scale bar 200μm.
As in the study of PKC inhibition, to address the concern that that treatment of cerebellar slice cultures with SP600125 (10μM) from the first day of culture through 14 DIV is non-specifically reducing PC loss from the stress of being placed in slice cultures or rescuing PCs from developmental cell death, wild type and Lc/+ cerebellar slice cultures were also treated with SP600125 (10 μM) from 7 to 14 DIV (Fig. 5G). This delayed treatment schedule did not significantly affect the number of surviving wild type PCs at 14 DIV (2,851±563 (SP600125-treated), n=6 vs. 2216±266 (non-treated), n=10, p>0.05), but GluRδ2Lc/+ PC survival increased by approximately 230% in the treated cultures (1,460±318, n=6 vs. 625±65, n=13; p < 0.0005). A similar treatment of wild type and Lc/+ cerebellar slices from 7 DIV with the highly selective peptide JNK inhibitor, DJNKII, produced similar findings of increased GluRδ2Lc/+ PC survival (19), but no effect on wild type PC survival. In the current study, among the Lc/+ slice cultures, treatment with SP600125 (10 μM) starting from 0 DIV significantly increased GluRδ2Lc/+ PC survival compared with treatment starting at 7 DIV (p < 0.001). This could mean that some GluRδ2Lc/+ PCs start dying before P7 or that early treatment with the JNK inhibitor increases the overall initial survival of PCs in slice culture, leading to relatively higher PC survival at 14 DIV, assuming that the rate of cell death is the same.
Treatment of wild type and Lc/+ cerebellar slice cultures with the p38 inhibitor SB 203580 (10 μM) from 0 to 14 DIV also had significant treatment and genotype effects on PC survival (Fig. 5H: Genotype: ANOVA, F1,75=77, p < 0.001; Treatment: ANOVA, F2,75=30.85, p < 0.0001) with no significant interactions (p > 0.1). The number of surviving WT PCs was increased by about 2-fold following SB 203580 treatments (4359±470, n=4 vs. 2005±215, n=10; p < 0.0005) and Lc/+ PC survival was increased by over 3-fold (2192±290, n=8 vs. 593±54, n=13; p < 0.0001). When cerebellar slice cultures were treated with SB 203580 from 7 to 14 DIV, there was no significant change in the number of GluRδ2+/+ PCs (2719±355) compared with untreated WT cultures (2005±215 PCs, p > 0.1). However, there was still an almost 2-fold increase in the number of GluRδ2Lc/+ PCs (1081 ± 188) compared with untreated Lc/+ cerebellar cultures (593±54 PCs, p<0.005).
Low-power magnification photomicrographs of wild type and Lc/+ untreated cultures and those treated with the JNK II inhibitor (SP600125) or the p38 inhibitor (SB 203580) confirm the quantitative results (Fig. 5A–F). The density of calbindin-labeled PCs is greater in the Lc/+ cultures treated with the JNK II and p38 inhibitors compared with controls and many of the pharmacologically treated Lc/+ cerebellar slices look more like normal cerebellar slices with PCs arranged in distinct lobules sending axons to what looks like the deep cerebellar nuclei.
GluRδ2Lc/+ PC dendritic differentiation is not affected by treatment with inhibitors for PKC, JNK, or p38
The treatment of cerebellar slice cultures with the pharmacological inhibitors for cPKCs and the MAP kinases JNK and p38 all resulted in significant increases in both GluRδ2+/+ and GluRδ2Lc/+ PC survival. However, the images of the dendritic trees of GluRδ2Lc/+ PCs in the treated slice cultures indicates that exposure to either the cPKC, JNK, or p38 inhibitors does not reverse the phenotype of deficient dendritic growth in GluRδ2Lc/+ PCs (Fig. 6). GluRδ2Lc/+ PC dendrites in all of the treated Lc/+ slice cultures showed decreased dendritic growth and complexity compared with treated and control GluRδ2+/+ PC dendrites. In their general appearance, the dendrites of Lc/+ PCs treated with Gö6976, SP600125, or SB 203580 may even appear less well differentiated and stunted compared with untreated Lc/+ PCS (compare Fig. 6B with 6D, F. and H). PC morphology was not quantified in this study, however, because of concerns it would not be possible to differentiate the effects of increased PC packing density in the treated cultures with the direct effects of the inhibitors on individual PCs.
Figure 6.

High magnification, confocal images of WT (A, C, E, G) and Lc/+ (B, D, F, H) PCs in cerebellar slice cultures treated with vehicle (Control) or cPKC, JNK, or p38 inhibitors. The images of the PC dendrites indicate that although the treatment with the three different inhibitors prevented PC death to varying extents, the inhibitors did not affect the stunted dendritic differentiation of Lc/+ PCs. Scale bar 20 μm.
Discussion
The results of this study demonstrate that treating cerebellar slice cultures with the kinase inhibitors Gö6976, SP600125, or SB 203580 from 0 to 14 DIV dramatically increases both GluRδ2+/+ and GluRδ2Lc/+ PC survival, although the inhibitors do not appear to stimulate the differentiation of GluRδ2Lc/+ PC dendrites. Treatment of the cultures with the inhibitors starting at 7 DIV also rescues significant numbers of GluRδ2Lc/+ PCs, with a much less dramatic affect on the survival of wild type PCs. The results suggest that the activation of conventional PKC isoforms, JNK, and p38 are all involved in the mechanisms for PC death in cerebellar slice cultures in general, and in the mechanisms of GluRδ2Lc/+ PC in particular. Inhibiting the activation of cPKCs, JNK, or p38 starting at the time when the slice cultures are established may reduce the overall PC death that is due to the stress of sectioning the cerebellum and placing the slices in culture, and may even have an effect on naturally occurring PC death in the slice cultures. However, we hypothesize that the decrease in GluRδ2Lc/+ PC death following treatment with any of the three kinase inhibitors starting from 7 DIV is due to the inhibition of cell death pathways involved in excitotoxic Lc/+ PC death.
Previous studies of wild type and Lc/+ PC death in vivo and in vitro have presented immunohistochemical evidence for the expression of phosphorylated c-Jun in wild type and Lc/+ PCs as a marker for activation of the JNK kinases during periods of PC death (19, 20). In this study, we present qualitative evidence for potential changes in the density or distribution of PKCγ in Lc/+ PCs, as reflected in changes in the intensity of immunolabeling for PKCγ at P15 and P25. In most Lc/+ PCs at P15, the intensity of PKCγ immunolabeling is reduced compared with controls, but in a few cases the intensity is increased. While we recognize that the intensity of immunofluorescence should be interpreted cautiously as an indicator for protein expression or concentration changes (there could be many factors that influence the intensity of labeling beyond changes in the concentration of the antigen, e.g. changes in antigen accessibility as a cell degenerates), one possible interpretation of this observation is that there is an increase in PKCγ protein or a change in its distribution (indicating activation) in a small subset of Lc/+ PCs at a given time at P15. This raises the intriguing possibility that PKCγ expression or activation is dynamic in Lc/+ PCs and at some point in the life/death history of Lc/+ PCs, cPKC isoforms are activated and this leads to their rapid death. Since the period of PC death in Lc/+ mutants in vivo (and in vitro) extends over a fairly long period it is statistically likely that only a few Lc/+ PCs will be in the process of dying at a given time point (the time of perfusion). This would account for the observation that only a few Lc/+ PCs immunolabel for activated caspase-3 in P15 cerebella in vivo (9, 10). Gö6976 may dramatically increase Lc/+ PC survival in vitro because it prevents Lc/+ PC death when cPKC isoforms are activated at some point as they begin to degenerate. However, it is not at all clear what triggers the death of individual Lc/+ PCs over the course of the period of PC death in the Lc/+ cerebellum, especially since the available electrophysiological evidence suggests that all Lc/+ PCs are chronically depolarized by the end of the first week of postnatal development and blocking the GluRδ2Lc leak current rescues Lc/+ PCs, at least in vitro (3, 18). There is some evidence that heterogeneity in the expression of neuroprotective factors in PC compartments may influence the timing of Lc/+ PC death (see (21, 22)).
In cerebellar slice cultures taken from P1 to P5 pups, Dusart and colleagues have shown that there is enhanced PC death at these ages that may correspond to a postnatal period of developmental PC death (19, 23–27). As shown by Western blots, the JNK and p38 kinases are also activated in wild type cerebellar slice cultures, though p38 appears to be activated early in response to the physical disruption of the slices being put in culture while JNK activity appears to be associated with programmed cell death in the slice culture (26). In results similar to the current study, treatment of wild type P3 cerebellar slice cultures with the PKC inhibitor Gö6976, the JNK inhibitor D-JNKII, or the p38 inhibitor SB 203580 for 5 DIV significantly increased the survival of PCs (24, 26). In a separate study (19), treatment of wild type and Lc/+ cerebellar slice cultures from P0 pups with the selective JNK II inhibitor, D-JNKII, from 7 to 14 DIV significantly increased Lc/+ PC survival, but not wild type PC survival.
In this study, we have not directly analyzed the activation of any of the conventional PKCs, JNK, or p38, in wild type or Lc/+ PCs so our interpretation of the data depends on the specificity of the drug treatments. In the slice culture experiments, we deliberately used low concentrations of both SP600125 and SB 203580 (10μM), that have been reported to be selective for the JNK 1–3 and p38 α and β isoforms, respectively (28, 29). Gö6976 is reported to be a potent and selective inhibitor for conventional PKC isoforms (30, 31), but there is also evidence that Gö6976 has an equal affinity for inhibition of TrkA and TrkB neurotrophin receptors (32). In their study of the effects of Gö6976 on PC survival and axonal regeneration, Ghoumari et al. (24) demonstrated, using L7-PKCI transgenic mice (33), that the increased survival of PCs following treatment with Gö6976 was likely to be due to direct inhibition of cPKC isoforms and not due to the inhibition of Trk receptors. Since we observed similar results with Gö6976 with respect to PC survival, we hypothesize that Gö6976 is acting through inhibition of one or more of the conventional PKC isoforms. There is agreement in the literature that rodent PCs express the conventional α and γ PKC isoforms, while the I and II isoforms may only be expressed by granule cells (although there are some disagreements in the literature because of potential cross-reactivity of antibodies: see (17, 34, 35)). Qualitative evidence is presented here for changes in the pattern of immunolabeling for PKCγ in Lc/+ PCs in vivo, but our results do not indicate which cPKC isoforms are activated in Lc/+ PCs and whether one or all are primarily responsible for activating cell death pathways.
PKC activity in PCs is necessary for the induction of long-term depression at PC-parallel fiber synapses, the elimination of multiple climbing fiber innervation, and the differentiation of PC dendritic trees (17, 36–38). The elimination of supernumerary climbing fibers from PCs also involves activation of the GluRδ2 and mGluR1 receptors and Ca++-dependent activation of PKC (17). In this case, deletion of PKCγ interferes with climbing fiber elimination so this isoform may play a key role in activity dependent mechanisms (38). Similarly, PKCγ may play a key role in the activity-dependent regulation of PC dendritic differentiation; stimulation of PKC activity reduces PC growth in vitro while PKC inhibition promotes dendritic differentiation (17). PC dendritic growth in vitro is enhanced in PKCγ knock-out mice and PCs in slice cultures from PKCγ deficient mice do not respond to PKC inhibitors with enhanced dendritic growth (39). Based on these results, the activation of cPKC isoforms by increased intracellular Ca++ levels in Lc/+ PCs may play a role in the failure of Lc/+ PC dendrites to differentiate normally. However, treatment with the cPKC inhbitor, Gö6976, at levels that prevents significant amounts of Lc/+ PC cell death is not sufficient to restore Lc/+ dendritic development, suggesting that there may be additional factors that control dendritic differentiation.
Activation of PKC isoforms is also associated with the induction of apoptosis in a variety of neurodegenerative diseases, from cerebral ischemia and reperfusion injury to Alzheimer’s disease (31, 40–42). The interactions between PKC isoforms and apoptosis pathways are complex and, to a certain degree, cell type and stimulus specific. Yet in general PKCα,β,ε and ζ are often associated with suppression of apoptosis, while PKCδ is a critical pro-apoptotic signal in many cell types. PKCδ is activated by a variety of apoptotic stimuli including oxidative stress, death receptors, and by cleavage with activated caspase-3 (41). Cleavage of PKCδ by caspase-3 creates a constitutively active enzyme that serves both as a marker and effector of apoptosis. Oxidative stress can have differential effects on PKC activity, with low levels of reactive oxygen or reactive nitrogen stimulating PKC activity while higher concentrations can inhibit PKC activity, although high ONOO− concentrations can also increase intracellular protease activity which leads to proteolytic activation of PKC isoforms (42–45).
A recent study (46) presented evidence that the key stimulus for cell death mediated by the GluRδ2Lc channel is the influx of Na+ ions and a subsequent decrease in ATP levels. Ionic stress caused by the increase in intracellular Na+ and Ca++ levels in Lc/+ PCs can lead to a variety of changes in intracellular homeostasis, and one of the most important of these could be the increased production of reactive oxygen species (ROS) that can interact with nitric oxide (NO) to form reactive nitrogen species (RNS). We hypothesize that the initiation of cell death processes in Lc/+ PCs is strongly dependent on oxidative stress and the production of RNS caused by the ionic imbalance from the constitutive GluRδ2Lc Na+ leak. Among many possibilities, protein oxidation or nitration by RNS can cause the activation of various PKC isoforms and the p38 and JNK/c-Jun pathways (14, 42–45, 47). Activation of PKC isoforms can trigger the production of even more ROS and direct activation of cell death pathways (12). Phosphorylation of c-Jun by JNK may activate a number of target genes, including the cell death promoters Fas-L and Bim, leading to stimulation of apoptotic cell death pathways. Furthermore, activation of the p38 and JNK pathways can also suppress activity in the ERK pathway that promotes cell survival (13). It is important to note that there are multiple complex biochemical interactions that are responsible for regulating a cell’s response to stress and many of the interactions may be cell-type specific, depending on the matrix of protein expression within a particular cell. This may be particularly true for cerebellar PCs which are known to have heterogeneous patterns of protein expression in cerebellar compartments that may affect cell death processes (21, 22). The timing of Lc/+ PC death may ultimately depend on a complex interaction among pro- and anti-death factors with individual PCs.
In this study, treatment of wild type and Lc/+ cerebellar slices with Gö6976 had a much more dramatic effect on cell survival than did the treatments with the JNK and p38 inhibitors. One possible interpretation of this result is that activation of cPKC isoforms is earlier in the web of interactions that lead to Lc/+ PC death, so inhibiting cPKC activation is able to rescue more Lc/+ PCs from cell death. However, we do not yet know the extent to which the PKC, JNK, and p38 pathways were inhibited by the pharmacological treatments, since the drug doses were chosen to preserve specificity as much as possible, but not to maximize inhibition of a particular pathway. Yet the present results provide strong support for the hypothesis that there are multiple pathways for wild type and Lc/+ PC cell death. The magnitude of Lc/+ PC rescue following treatment with Gö6976 from 0 to 14 and 7 to 14 DIV is the largest that we have observed in vivo or in vitro (e.g. 9, 10, 19, 20, 48) and suggests that activation of PKC isoforms play a key role in Lc/+ PC death. Likewise, the dramatic increase in wild type PC survival following treatment with Gö6976 from 0 to 14 DIV indicates that activation of PKC isoforms also plays an important role in developmental PC death and in death due to the stress of being placed in tissue culture. The challenge is now to identify all of the molecular components of the web that regulates PC survival and death and to define the mechanisms by which they interact.
Acknowledgments
This work was supported by a NIH grant NS 34309 to M.W.V. and J.M. and by funds from “Centre National de la Recherche Scientifique” (CNRS) and Universite Pierre et Marie Curie; LIA (Laboratoire Internationaux Associé) grant from UPMC. MV was recipient of a visiting professorship grant from UPMC. We thank Fabrice Machulka for his assistance in animal breeding. We gratefully acknowledge the considerable contribution of Dr Pauline Cavelier in data acquisition. We thank the Cell Imaging and Flow Cytometry facility of the IFR83 (Paris, France) for access and technical support in microscopy.
Footnotes
Conflicts of Interest: The authors certify that there is no conflict of interest concerning the work presented in this manuscript.
This article was prepared while MWV was employed at the University of Maryland School of Medicine. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government.
References
- 1.Vogel MW, Caston J, Yuzaki M, Mariani J. The Lurcher mouse: Fresh insights from an old mutant. Brain Res. 2007;1140:4–18. doi: 10.1016/j.brainres.2005.11.086. [DOI] [PubMed] [Google Scholar]
- 2.Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N. A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35(5):921–33. doi: 10.1016/s0896-6273(02)00861-9. [DOI] [PubMed] [Google Scholar]
- 3.Selimi F, Lohof AM, Heitz S, Lalouette A, Jarvis CI, Bailly Y, et al. Lurcher GRID2-Induced Death and Depolarization Can Be Dissociated in Cerebellar Purkinje Cells. Neuron. 2003;37(5):813–9. doi: 10.1016/s0896-6273(03)00093-x. [DOI] [PubMed] [Google Scholar]
- 4.Dumesnil-Bousez N, Sotelo C. Early development of the Lurcher cerebellum: Purkinje cell alterations and impairment of synaptogenesis. Journal of Neurocytology. 1992;21:506–529. doi: 10.1007/BF01186954. [DOI] [PubMed] [Google Scholar]
- 5.Norman DJ, Feng L, Cheng SS, Gubbay J, Chan E, Heintz N. The lurcher gene induces apoptotic death in cerebellar Purkinje cells. Development. 1995;121(4):1183–93. doi: 10.1242/dev.121.4.1183. [DOI] [PubMed] [Google Scholar]
- 6.Selimi F, Doughty M, Delhaye-Bouchaud N, Mariani J. Target-related and intrinsic neuronal death in Lurcher mutant mice are both mediated by caspase-3 activation. J Neurosci. 2000;20:992–1000. doi: 10.1523/JNEUROSCI.20-03-00992.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wullner U, Loschmann P-A, Weller M, Klockgether T. Apoptotic cell death in the cerebellum of mutant weaver and lurcher mice. Neurosci. Lett. 1995;200:109–112. doi: 10.1016/0304-3940(95)12090-q. [DOI] [PubMed] [Google Scholar]
- 8.Cheng SS, Heintz N. Massive loss of mid- and hindbrain neurons during embryonic development of homozygous lurcher mice. J Neurosci. 1997;17(7):2400–7. doi: 10.1523/JNEUROSCI.17-07-02400.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selimi F, Vogel MW, Mariani J. Bax inactivation in Lurcher mutants rescues cerebellar granule cells but not Purkinje cells or inferior olivary neurons. J Neurosci. 2000;20(14):5339–45. doi: 10.1523/JNEUROSCI.20-14-05339.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doughty ML, De Jager PL, Korsmeyer SJ, Heintz N. Neurodegeneration in Lurcher mice occurs via multiple cell death pathways. J Neurosci. 2000;20(10):3687–94. doi: 10.1523/JNEUROSCI.20-10-03687.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McFarland R, Blokhin A, Sydnor J, Mariani J, Vogel MW. Oxidative stress, nitric oxide, and the mechanisms of cell death in Lurcher Purkinje cells. Devel Neurobio. 2007;67:1032–1046. doi: 10.1002/dneu.20391. [DOI] [PubMed] [Google Scholar]
- 12.Giorgi C, Agnoletto C, Baldini C, Bononi A, Bonora M, Marchi S, et al. Redox control of protein kinase C: cell- and disease-specific aspects. Antioxid Redox Signal. 2010;13(7):1051–85. doi: 10.1089/ars.2009.2825. [DOI] [PubMed] [Google Scholar]
- 13.Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22(4):954–65. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- 14.Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40(6):928–39. doi: 10.1016/j.freeradbiomed.2005.10.056. Epub 2005 Nov 21. [DOI] [PubMed] [Google Scholar]
- 15.Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N. Neurodegeneration in Lurcher mice caused by mutation in 2 glutamate receptor. Nature. 1997;388:769–773. doi: 10.1038/42009. [DOI] [PubMed] [Google Scholar]
- 16.Letellier M, Demais V, Bailly Y, Sherrard RM, Mariani J, Lohof AM. Reinnervation of late post-natal Purkinje cells by climbing fibres: neosynaptogenesis without transient multi-innervation. Journal of Neuroscience. 2007;27:5373–5383. doi: 10.1523/JNEUROSCI.0452-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metzger F, Kapfhammer JP. Protein kinase C: its role in activity-dependent Purkinje cell dendritic development and plasticity. Cerebellum. 2003;2(3):206–14. doi: 10.1080/14734220310016150. [DOI] [PubMed] [Google Scholar]
- 18.Zanjani HS, McFarland R, Cavelier P, Blokhin A, Gautheron V, Levenes C, et al. Death and survival of heterozygous Lurcher Purkinje cells in vitro. Dev Neurobiol. 2009;69(8):505–17. doi: 10.1002/dneu.20715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Repici M, Zanjani HS, Gautheron V, Borsello T, Dusart I, Mariani J. Specific JNK inhibition by D-JNKI1 protects Purkinje cells from cell death in Lurcher mutant mouse. Cerebellum. 2008;7(4):534–8. doi: 10.1007/s12311-008-0070-8. [DOI] [PubMed] [Google Scholar]
- 20.Lu W, Tsirka SE. Partial rescue of neural apoptosis in the Lurcher mutant mouse through elimination of tissue plasminogen activator. Development. 2002;129(8):2043–50. doi: 10.1242/dev.129.8.2043. [DOI] [PubMed] [Google Scholar]
- 21.Armstrong CL, Duffin CA, McFarland R, Vogel MW. Mechanisms of compartmental purkinje cell death and survival in the lurcher mutant mouse. Cerebellum. 2010;10(3):504–14. doi: 10.1007/s12311-010-0231-4. [DOI] [PubMed] [Google Scholar]
- 22.Duffin CA, McFarland R, Sarna JR, Vogel MW, Armstrong CL. Heat shock protein 25 expression and preferential Purkinje cell survival in the Lurcher mutant mouse cerebellum. J Comp Neurol. 2010 doi: 10.1002/cne.22309. In press. [DOI] [PubMed] [Google Scholar]
- 23.Ghoumari AM, Wehrle R, Bernard O, Sotelo C, Dusart I. Implication of Bcl-2 and Caspase-3 in age-related Purkinje cell death in murine organotypic culture: an in vitro model to study apoptosis. Eur J Neurosci. 2000;12(8):2935–49. doi: 10.1046/j.1460-9568.2000.00186.x. [DOI] [PubMed] [Google Scholar]
- 24.Ghoumari AM, Wehrle R, De Zeeuw CI, Sotelo C, Dusart I. Inhibition of protein kinase C prevents Purkinje cell death but does not affect axonal regeneration. J Neurosci. 2002;22(9):3531–42. doi: 10.1523/JNEUROSCI.22-09-03531.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghoumari AM, Dusart I, El-Etr M, Tronche F, Sotelo C, Schumacher M, et al. Mifepristone (RU486) protects Purkinje cells from cell death in organotypic slice cultures of postnatal rat and mouse cerebellum. Proc Natl Acad Sci U S A. 2003;100(13):7953–8. doi: 10.1073/pnas.1332667100. Epub 2003 Jun 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Repici M, Wehrle R, Antoniou X, Borsello T, Dusart I. c-Jun N-terminal kinase (JNK) and p38 play different roles in age-related Purkinje cell death in murine organotypic culture. Cerebellum. 2010;10(2):281–90. doi: 10.1007/s12311-010-0244-z. [DOI] [PubMed] [Google Scholar]
- 27.Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41(4):535–47. doi: 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- 28.Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98(24):13681–6. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408(3):297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, et al. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem. 1993;268(13):9194–7. [PubMed] [Google Scholar]
- 31.Bright R, Mochly-Rosen D. The role of protein kinase C in cerebral ischemic and reperfusion injury. Stroke. 2005;36(12):2781–90. doi: 10.1161/01.STR.0000189996.71237.f7. Epub 2005 Oct 27. [DOI] [PubMed] [Google Scholar]
- 32.Behrens MM, Strasser U, Choi DW. Go 6976 is a potent inhibitor of neurotrophin-receptor intrinsic tyrosine kinase. J Neurochem. 1999;72(3):919–24. doi: 10.1046/j.1471-4159.1999.0720919.x. [DOI] [PubMed] [Google Scholar]
- 33.De Zeeuw CI, Hansel C, Bian F, Koekkoek SK, van Alphen AM, Linden DJ, et al. Expression of a protein kinase C inhibitor in Purkinje cells blocks cerebellar LTD and adaptation of the vestibulo-ocular reflex. Neuron. 1998;20(3):495–508. doi: 10.1016/s0896-6273(00)80990-3. [DOI] [PubMed] [Google Scholar]
- 34.Hirono M, Sugiyama T, Kishimoto Y, Sakai I, Miyazawa T, Kishio M, et al. Phospholipase Cbeta4 and protein kinase Calpha and/or protein kinase CbetaI are involved in the induction of long term depression in cerebellar Purkinje cells. J Biol Chem. 2001;276(48):45236–42. doi: 10.1074/jbc.M105413200. Epub 2001 Sep 10. [DOI] [PubMed] [Google Scholar]
- 35.Naik MU, Benedikz E, Hernandez I, Libien J, Hrabe J, Valsamis M, et al. Distribution of protein kinase Mzeta and the complete protein kinase C isoform family in rat brain. J Comp Neurol. 2000;426(2):243–58. doi: 10.1002/1096-9861(20001016)426:2<243::aid-cne6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 36.Leitges M, Kovac J, Plomann M, Linden DJ. A unique PDZ ligand in PKCalpha confers induction of cerebellar long-term synaptic depression. Neuron. 2004;44(4):585–94. doi: 10.1016/j.neuron.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 37.Metzger F, Kapfhammer JP. Protein kinase C activity modulates dendritic differentiation of rat Purkinje cells in cerebellar slice cultures. Eur J Neurosci. 2000;12(6):1993–2005. doi: 10.1046/j.1460-9568.2000.00086.x. [DOI] [PubMed] [Google Scholar]
- 38.Kano M, Hashimoto K, Chen C, Abeliovich A, Aiba A, Kurihara H, et al. Impaired synapse elimination during cerebellar development in PKC gamma mutant mice. Cell. 1995;83:1223–1231. doi: 10.1016/0092-8674(95)90147-7. [DOI] [PubMed] [Google Scholar]
- 39.Schrenk K, Kapfhammer JP, Metzger F. Altered dendritic development of cerebellar Purkinje cells in slice cultures from protein kinase Cgamma-deficient mice. Neuroscience. 2002;110(4):675–89. doi: 10.1016/s0306-4522(01)00559-0. [DOI] [PubMed] [Google Scholar]
- 40.Alkon DL, Sun MK, Nelson TJ. PKC signaling deficits: a mechanistic hypothesis for the origins of Alzheimer’s disease. Trends Pharmacol Sci. 2007;28(2):51–60. doi: 10.1016/j.tips.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Reyland ME. Protein kinase Cdelta and apoptosis. Biochem Soc Trans. 2007;35:1001–1004. doi: 10.1042/BST0351001. [DOI] [PubMed] [Google Scholar]
- 42.Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88(4):1341–78. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin D, Takemoto DJ. Oxidative activation of protein kinase Cgamma through the C1 domain. Effects on gap junctions. J Biol Chem. 2005;280(14):13682–93. doi: 10.1074/jbc.M407762200. [DOI] [PubMed] [Google Scholar]
- 44.Knapp LT, Kanterewicz BI, Hayes EL, Klann E. Peroxynitrite-induced tyrosine nitration and inhibition of protein kinase C. Biochem Biophys Res Commun. 2001;286(4):764–70. doi: 10.1006/bbrc.2001.5448. [DOI] [PubMed] [Google Scholar]
- 45.Chakraborti T, Das S, Chakraborti S. Proteolytic activation of protein kinase Calpha by peroxynitrite in stimulating cytosolic phospholipase A2 in pulmonary endothelium: involvement of a pertussis toxin sensitive protein. Biochemistry. 2005;44(13):5246–57. doi: 10.1021/bi0477889. [DOI] [PubMed] [Google Scholar]
- 46.Nishiyama J, Matsuda K, Kakegawa W, Yamada N, Motohashi J, Mizushima N, et al. Reevaluation of Neurodegeneration in lurcher Mice: Constitutive Ion Fluxes Cause Cell Death with, Not by, Autophagy. J Neurosci. 2010;30(6):2177–87. doi: 10.1523/JNEUROSCI.6030-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown GC. Nitric oxide and neuronal death. Nitric Oxide. 2010;23(3):153–65. doi: 10.1016/j.niox.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 48.Zanjani H, Rondi-Reig L, Vogel M, Martinou JC, Delhaye-Bouchaud N, Mariani J. Overexpression of a Hu-bcl-2 transgene in Lurcher mutant mice delays Purkinje cell death. C R Acad Sci III. 1998;321(8):633–40. doi: 10.1016/s0764-4469(98)80002-4. [DOI] [PubMed] [Google Scholar]