

Abstract
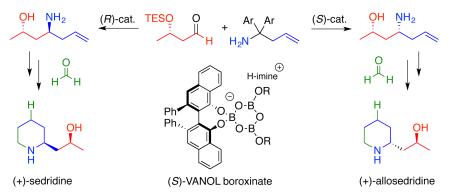
The catalytic asymmetric aminoallylation of chiral aldehydes is developed as a new method for the catalyst controlled synthesis of syn- and anti-1,3-aminoalcohols. This methodology is highlighted in the synthesis of the sedum alkaloids (+)-sedridine and (+)-allosedridine both of which have their final carbon incorporated during closure of the piperidine ring via a hydroformylation with formaldehyde.
The sedum alkaloids exist widely in nature and these types of alkaloids have memory-enhancing properties and are effective in the treatment of cognitive disorders.1 The most commonly occurring members of this alkaloid family are 2-substituted piperidines with various combinations of hydroxyl functionalities in the side chains, featuring the 1,3-amino alcohol moiety and a select set are shown in Figure 1.2 A review of the syntheses of sedium alkaloids has appeared2 and the field has remained very active.3
Figure 1.

Sedum and Related Alkaloid Natural Products
Our efforts in this field began with an interest in developing an approach to the sedum alkaloids that has a hydroformylation with formaldehyde and a catalyst controlled asymmetric aza-Cope rearrangement (or aminoallylation) as the key steps (Scheme 1). The catalyst controlled aza-Cope rearrangement was envisioned to be featured in ordered stereoselective syntheses of (+)-sedridine and (+)-allosedridine which constitute one of the diastereomeric pairs of natural products in the sedum alkaloid family.4
Scheme 1.

Retrosynthesis of (+)-Sedridine and (+)-Allosedridine
Conventionally, hydroformylation utilizes syngas (CO/H2) in the presence of a transition metal catalyst to give homologous linear and/or branched aldehydes. A recent innovation in hydroformylation chemistry features an experimentally convenient alternative using formaldehyde as a syngas substitute. This synthetically attractive method was recently described by Morimoto and coworkers when they smartly applied two types of insitu generated catalysts to separately direct the two cooperative catalytic processes involved: 1) decarbonylation of an aldehyde, and 2) hydroformylation of an olefin (Scheme 2).5 This dual catalyst system provides homologated aldehydes in excellent yields and with very high linear to branched ratios. The intention for the application of this hydroformylation in the synthesis of sedum alkaloids will be to incorporate it into an intramolecular amidocarbonylation6 that takes the alkenyl amino alcohol directly to a piperidine ring.
Scheme 2.

Hydroformylation with Formaldehyde
The key transformation in the synthesis of sedridine and allosedridine involves an aza-Cope rearrangement of an in-situ generated imine 3 to give imine 4 which upon hydrolysis provides the homoallyic amine 5 in high asymmetric inductions over a broad range of aromatic, alkenyl and aliphatic substrates.7 The successful catalyst system results from the incorporation of a molecule of benzoic acid into the VANOL boroxinate catalyst 6.7,8
The pertinent issue to address here is whether the presence of a chiral center in the aldehyde 1 (Scheme 3) will interfere with the normal operations of the boroxinate/benzoate catalyst in the aza-Cope rearrangement? If the answer is no then this reaction would represent a new method for the conrolled synthesis of syn- and anti-1,3-aminoalcohols. The controlled synthesis of both syn- and anti-1,3-aminoalcohols from a single substrate have been reported from β-aminoketones9 and β-borylamines.10 We only know of a single example where a catalyst contolled process can be used to access syn- or anti-1,3-aminoalcohols from a single substrate.11,12
Scheme 3.

Direct Aminoallylation of Non-Chiral Aldehydes
The initial screen of chiral aldehydes was carried out with the TBS protected aldehyde (R)-8a derived from the commercially available methyl (R)-3-hydroxybutyrate (Table 1). The diastereoselectivity is nearly equal and opposite with the (R)- and (S)- ligands of VANOL (33:1 vs 1:23) and thus this is a case of catalyst control (entries 1 & 2). The total yield of 10a and 11a was low and the elimination product 12 was observed as a by-product. The reaction of the benzyl protected aldehyde 8b with amine 2 gave a 4:1 mixture of aza-Cope product (10b+11b) and by-product 12 (entry 3). Incorporation of a larger protecting group (TBDPS) lead to a mixture largely consisting of the eliminated imine 12 (entry 4). However, when the sterically less hindered triethylsilyl protecting group (TES) was installed, the formation of 12 was completely shut down (Table 1, entry 5) giving an 87% yield and a 20:1 diastereoselectivity in favor of 11 with (R)-VANOL and a 74% yield and a 26:1 diastereoselectivity in favor of 10 with (S)-VANOL (these two reactions were with (S)-8)
Table 1.
Direct Aminoallylation of Chiral β-Alkoxy Aldehydes
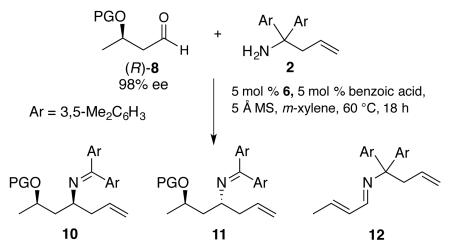
| entrya | series | ligand | PG | conv (%)b |
(10+11) : 12c |
10:11 d | % yield (10+11 )e |
|---|---|---|---|---|---|---|---|
| 1 | a | (R)-VANOL | TBS | 73 (70) | 3:1 | 33:1 | 48 |
| 2 | a | (S)-VANOL | TBS | 72 (67) | 4:1 | 1:23 | 44 |
| 3 | b | (R)-VANOL | Bn | (80) | 4:1 | nd | nd |
| 4 | c | (R)-VANOL | TBDPS | (52) | 1:10 | nd | nd |
| 5 f | d | (R)-VANOL | TES | 100 | 100:0 | 1:20 | 87 |
| 6f | d | (S)-VANOL | TES | 100 | 100:0 | 26:1 | 74 |
Unless otherwise specified all reactions were run at 0.2 M in amine 2 with 1.1 equiv 8. The catalyst was prepared from 1 equiv of the ligand, 2 equiv of 2,4,6-trimethylphenol, 3 equiv of H2O and 3 equiv BH3·SMe2. nd = not determined.
Calculated from the 1H NMR spectrum of the crude reaction mixture from the ratio (10+11 ):2 (or 2 + imine 9) and the isolated yield of 10+11. Value in parentheses based on the ratios of 2 (or 2 + imine 9) 10, 11 and 12 and assuming no other products are formed. In most cases, the unreacted material is in the form of amine 2 but in some cases a small amount of imine 9 formed from 8 and 2 is present.
Determined from the 1H NMR spectrum of the crude reaction mixture.
Isolated ratio.
Isolated yield of a mixture of 10 + 11 after chromatography on silica gel.
Reaction performed on (S)-8 also of 98% ee. This reaction gives the enantiomer of 10 and 11.
The interplay of the catalyst with a pre-existing α - chiral center was also investigated. As shown in Scheme 4, the reaction of the chiral aldehyde (S)-13 is not under catalyst control but rather displays a match and mis-matched relationship. A 12:1 selectivity was observed for the matched case with the (S)-VANOL catalyst resulting in a 71% isolated yield. The same diasteromer predominated in the mis-matched case with the (R)-VANOL catalyst but the selectivity dropped to 2.5:1. The stereochemistry of 14 was assigned as anti since the matched case would be expected to be with the Re-face addition to the imine with the (S)-VANOL catalyst since this is the preference with non-chiral aldehydes.7
Scheme 4.

Direct Aminoallylation of a Chiral α-Alkoxy Aldehyde
When Morimoto’s protocol was applied to an intramolecular amidocarbonylation with a homo-allylic amine, some branched-hydroformylation and other side-products were obtained. When formalin, which contains methanol as a stabilizer, was utilized as the formaldehyde source, a significant amount of the 2-alkoxypiperidine 18 was observed in the 1H NMR spectrum of the crude reaction mixture (Table 2, entry 1). para-Formaldehyde gave didehydropiperidine 17 and its five-membered analog 19 along with some of the olefin isomerization product 20. Even with less than complete regio- and chemoselection, the didehydropiperidine 17 could be obtained in 73% isolated yield (Table 2, entry 3).13
Table 2.
Optimization of the Intramolecular Amidocarbonylation with Formaldehyde
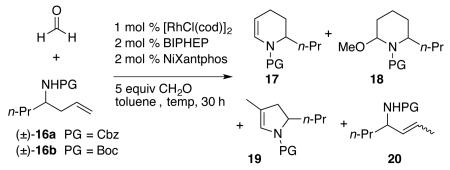
| entrya | PG | formaldehyde source |
temp (°C) |
yield 17 (%) b |
17:19:20 c |
|---|---|---|---|---|---|
| 1 | Cbz | formalin | 90 | 46 | _ d |
| 2 | Cbz | paraformaldehyde | 90 | 69 | 6:1:1 |
| 3 | Boc | paraformaldehyde | 90 | 73 | 6:1:1 |
| 4e | Boc | paraformaldehyde | 90 | nd | 2:3:0 |
Unless otherwise specified all reactions were run at 0.15 M in 16 with 5.0 equiv formaldehyde. nd = not determined.
Isolated yield after chromatography on silica gel.
Calculated from the 1H NMR spectrum of the crude reaction mixture.
A 38% yield of 18 also formed in this reaction.
PPTS (5 mol %) was added to this reaction.
The intramolecular amidocarbonylation was then applied to the synthesis of (−)-coniine 22. The chiral center is installed in acyclic amine 21 in 83% yield and 95% ee with catalytic asymmetric direct aminoallylation of n-butanal as shown in Scheme 5.7 The piperidine ring is closed using the intramolecular amidocarbonylation in 71% yield. Subsequent reduction of the double bond and cleavage of Boc give the target compound (−)-coniine 22 in 91% yield over two steps.
Scheme 5.

Synthesis of (−)-Coniine
Finally, we coupled the catalyst controlled aza-Cope rearrangement and intramolecular amidocarbonylation in the total synthesis of (+)-sedridine and (+)-allosedridine (Scheme 6). Chiral aldehyde (S)-8d was subjected to a diastereoselective aza-Cope rearrangement catalyzed by the boroxinate catalyst 6 derived from (R)-VANOL (Scheme 6). Following hydrolysis and protection with a Boc group, purification gave 26 as a single diastereomer in 72% yield over three steps in a one-pot fashion. After protection an intramolecular amidocarbonylation reaction afforded 28 in 78% yield. Reduction and deprotection gave (+)-sedridine in 83% yield. (+)-Allosedridine was obtained in a similar manner utilizing the boroxinate catalyst 6 derived from (S)-VANOL which allowed for the isolation of 23 in 61% yield as a single diastereomer. Protection with TBS and hydroformylation with formaldehyde gave 25 in 72% yield and to a final conversion to (+)-allosedridine in 74% yield in two steps.
Scheme 6.

Synthesis of (+)-Sedridine and (+)-Allosedridine
This work has demonstrated that chiral aldehydes bearing a β–alkoxy group will undergo a chiral Brønsted acid catalyzed 2-aza-Cope rearrangement with an in-situ generated imine to give either syn- or anti-1,3-homoallylic amino alcohols depending on the absolute configuration of the catalyst. This catalyst controlled 2-aza-Cope rearrangement is coupled with a syngas free hydroformylation in a highly stereoselective synthesis of (+)-sedridine and (+)-allosedridine from the same β-alkoxy aldehyde.
Supplementary Material
Acknowledgment
Support provided by the National Institute of General Medical Sciences (GM094478).
Footnotes
Supporting Information Available Procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Meth-Cohn O, Yu CY, Lestage P, Lebrun MC, Cagniard DH, Renard P. Eur. Pat. 2000:1050531. [Google Scholar]
- 2.For a review, see: Bates RW, Sa-Ei K. Tetrahedron. 2002;58:5957–5978.
- 3.For examples after 2008, see: Chen L-J, Hou D-R. Tetrahedron: Asymmetry. 2008;19:715–720. Chang MY, Lin CY, Wu TC, Sun PP. J. Chin. Chem. Soc. 2008;55:421–430. Rice GT, White MC. J. Am. Chem. Soc. 2009;131:11707–11711. doi: 10.1021/ja9054959. Wang G, Vedejs E. Org. Lett. 2009;11:1059–1061. doi: 10.1021/ol802781c. Davies SG, Fletcher AM, Roberts PM, Smith AD. Tetrahedron. 2009;65:10192–10213. Lebrun S, Couture A, Deniau E, Grandclaudon P. Chirality. 2010:212–216. doi: 10.1002/chir.20729. Shaikh TM, Sudalai A. Eur. J. Org. Chem. 2010:3437–3444. Coldham I, Leonori D. J. Org. Chem. 2010;75:4069–4077. doi: 10.1021/jo100415x. Bates RW, Lu Y. Org. Lett. 2010;12:3938–3941. doi: 10.1021/ol1016492. Boussonniere A, Ranaivondrambola T, Lebreton J, Mathe-Allainmat M. Synthesis. 2010:2456–2462. Liu J-D, Chen Y-C, Zhang G-B, Li Z-Q, Chem P, Du J-Y, Tu Y-Q, Fan C-A. Adv. Synth. Catal. 2011;353:2721–2730. Riva E, Rencurosi A, Gagliardi S, Passarella D, Martinelli Chem. Eur. J. 2011;17:6221–6226. doi: 10.1002/chem.201100300. Satyalakshmi G, Suneel K, Shinde DB, Das B. Tetrahedron: Asymmetry. 2011;22:1000–1005. Bhat C, Tilve S. Tetrahedron Lett. 2011;52:6566–6568.
- 4.For leading references, see reference 3s and: Butruille D, Fodor G, Huber CS, Letourneau F. Tetrahedron. 1971;27:2055–2067.
- 5.Makado G, Morimoto T, Sugimoto Y, Tsutsumi K, Kagawa N, Kiuchi K. Adv. Synth. Catal. 2010;352:299–304. [Google Scholar]
- 6.For a review on intramolecular amidocarbonylation, see: Chiou W, Lee S, Ojima I. Can. J. Chem. 2005;83:681–692.
- 7.Ren H, Wulff WD. J. Am. Chem. Soc. 2011;133:5656–5659. doi: 10.1021/ja1110865. [DOI] [PubMed] [Google Scholar]
- 8.(a) Hu G, Gupta AK, Huang RH, Mukherjee M, Wulff WD. J. Am. Chem. Soc. 2010;132:14669–14675. doi: 10.1021/ja1070224. [DOI] [PubMed] [Google Scholar]; (b) Vetticatt MJ, Desai AA, Wulff WD. J. Am. Chem. Soc. 2010;132:13104–13107. doi: 10.1021/ja103863j. [DOI] [PubMed] [Google Scholar]
- 9.Pillim RA, Russowsky D, Dias LC, Keck GE, Truong AP, Davis FA, Gaspari PM, Nolt BM, Xu P, Gonzalez-Gomez JC, Foubelo F, Yus M, De Lamo Marin S, Catala C, Kumar SR, Valleix A, Wagner A, Mioskowski C. J. Chem. Soc. Perkin Trans 1. Org. Lett. J. Org. Chem. Synthesis. Eur. J. Org. Chem. 1990;2002;2008;2009;2010;473 [Google Scholar]
- 10.Sole C, Whiting A, Gulyas H, Fernandez E. Adv. Synth. Catal. 2011;353:376–376. [Google Scholar]
- 11.Jha V, Kondekar NB, Kumar P. Org. Lett. 2010;12:2762–2765. doi: 10.1021/ol100856u. [DOI] [PubMed] [Google Scholar]
- 12.Another example would presumably be a catalyst controlled process if the enantiomer of the enzyme was available: Millet R, Traff AM, Petrus ML, Bäckvall J-E. J. Am. Chem. Soc. 2010;132:15182–15184. doi: 10.1021/ja107857v.
- 13.2-Alkoxypiperidines and didehydropiperidines are both useful intermediates for the synthesis of piperidine compounds6 and in the present case both could be reduced to provide access to the target alkaloids. This possibility was not pursued in the present work.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.