

Abstract
Many Gram-negative bacteria contain specific systems for uptake of foreign DNA, which play a critical role in the acquisition of antibiotic resistance. The TtPilF (PilF ATPase from Thermus thermophilus) is required for high transformation efficiency, but its mechanism of action is unknown. In the present study, we show that TtPilF is able to bind to both DNA and RNA. The structure of TtPilF was determined by cryoelectron microscopy in the presence and absence of the ATP analogue p[NH]ppA (adenosine 5′-[β,γ-imido]triphosphate), at 10 and 12 Å (1 Å=0.1 nm) resolutions respectively. It consists of two distinct N- and C-terminal regions, separated by a short stem-like structure. Binding of p[NH]ppA induces structural changes in the C-terminal domains, which are transmitted via the stem to the N-terminal domains. Molecular models were generated for the apoenzyme and p[NH]ppA-bound states in the C-terminal regions by docking of a model based on a crystal structure from a closely related enzyme. Analysis of DNA binding by electron microscopy, using gold labelling, localized the binding site to the N-terminal domains. The results suggest a model in which DNA uptake by TtPilF is powered by ATP hydrolysis, causing conformational changes in the C-terminal domains, which are transmitted via the stem to take up DNA into the cell.
Keywords: ATPase, cryoelectron microscopy, DNA transformation, pilus biogenesis
Abbreviations: AAA+, ATPase associated with diverse cellular activities; CCD, charge-coupled device; EMDB, Electron Microscopy Data Bank; p[NH]ppA, adenosine 5′-[β,γ-imido]triphosphate; T2SS, type II secretion system; TEM, transmission electron microscopy; TFP, type IV pilus; TtPilF, PilF ATPase from Thermus thermophilus
INTRODUCTION
TFP (type IV pili) are hair-like filaments that are found on the surface of Gram-negative and Gram-positive bacteria. They play a major role in several processes in Gram-negative pathogens including host cell adherence, biofilm formation, twitching motility and DNA transformation. DNA transformation, also known as competence, is a process by which bacteria can take up DNA from the environment. The thermophile Thermus thermophilus is known to be highly transformable and the protein components of its natural transformation machinery have been identified [1,2]. T. thermophilus can take up DNA with high efficiency and has been shown to have a broad DNA specificity [3]. DNA translocation and TFP biogenesis are known to be functionally linked in Gram-negative bacteria [4], and TFP are essential for high levels of DNA transformation in most organisms [5], although the mechanistic details of both processes remain to be elucidated.
Genome-wide analyses have identified 16 proteins that are necessary for DNA transport in T. thermophilus HB27, including the competence-specific proteins ComEA, ComEC, ComZ and DprA [6,7]. Proteins specific to TFP biogenesis include the pilins PilA1–4, the outer membrane secretin PilQ, the FtsA-like protein PilM [8], PilW and inner membrane proteins PilC, PilD, PilN and PilO [9]. The PilF protein in T. thermophilus belongs to the AAA+ (ATPase associated with diverse cellular activities) family [10] (referred to as TtPilF in the present paper, to distinguish it from ATPases in other bacteria that are also called PilF). TtPilF shares sequence similarity with TFP biogenesis ATPases such as PilB in Pseudomonas aeruginosa and PilF in Neisseria meningitidis [11,12]. Unlike its counterparts in P. aeruginosa and N. meningitidis, however, where mutation of PilB or PilF abolishes TFP assembly, a T. thermophilus pilF mutant is reported as still being covered in pili [6]. This observation led to the suggestion that TtPilF is a vital component in the uptake of DNA and may supply the energy required for this process by ATP hydrolysis. Other AAA+ ATPases have been identified in T. thermophilus, including PilT-like proteins, but they have no apparent role in DNA transformation [13]. It has also been suggested that the inner membrane TFP biogenesis proteins PilC, PilM, PilN and PilO are responsible for DNA transport across the cytoplasmic membrane, as mutations to these proteins promote accumulation of DNA in the periplasm [11].
Many TFP biogenesis proteins share sequence and structural similarities with the T2SS (type II secretion system) proteins [14–16]. The ATPases associated with the T2SS, which provide the energy for secretion, belong to a larger family of ‘traffic NTPases’, which also includes the PilB/PilF subfamily associated with TFP biogenesis and DNA competence [17]. Structural studies of the T2SS ATPase Archaeoglobus fulgidus GspE differentiated closed and open states of the hexamer associated with ATP binding, thus linking ATP hydrolysis with changes in conformation that could power the secretion process [18]. Similar observations have been made on the PilT ATPases from Aquifex aeolicus [19] and P. aeruginosa [20]. PilT has been shown to be responsible for retraction of TFP in Neisseria gonorrhoeae [21], a process associated with twitching motility [17].
On the basis of analogy with the ATPases from the T2SS, the PilB/PilF subfamily is thought to provide the energy for TFP assembly, although the process whereby ATP hydrolysis in the cytoplasm is able to power pilus assembly in the periplasm is unclear. Proteins in the PilB/PilF subfamily share some characteristics with the T2SS ATPases, including Walker A and Walker B motifs, histidine and aspartic acid boxes and, notably, a tetracysteine motif, which has been structurally characterized in the T2SS ATPase EpsE [22]. Analysis of the PilF sequence in T. thermophilus shows that it has an extended N-terminal region (>300 amino acids longer) compared with other ATPases in the PilB/PilF subfamily. This larger sequence is predicted to contain three copies of GSPII, a structural fold that is found in proteins associated with type II secretion, as well as TFP biogenesis (Figure 1). GSPII consists of a small N-terminal helical domain followed by an α/β sandwich domain [23]. It was shown that, in the T2SS, the N-terminal helical region of XpsE can undergo large structural rearrangements that are crucial for binding to its partner, XpsL. Typically, only one GSPII region is found in the members of the PilB/PilF subfamily, and its precise function in relation to TFP formation or DNA uptake is unclear. The C-terminus of TtPilF is predicted to have a similar structure to Vibrio cholerae EpsE, comprising three subdomains: C1, CM and C2 (VcEpsE in Figure 1). The C1 subdomain contains the Walker A and B motifs, histidine and aspartic acid boxes, and the CM subdomain contains the tetracysteine motif that binds Zn2+ [22]. TtPilF has been expressed and purified; unlike EpsE, it forms a hexamer in solution and was also shown to bind Zn2+, although this was not essential for ATPase activity [24].
Figure 1. Arrangement of domains within selected secretory ATPases.
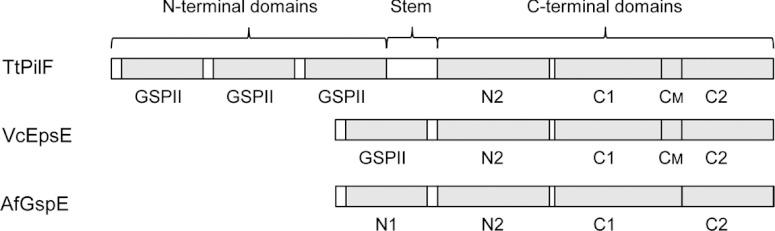
The figure compares the proposed domain arrangement in TtPilF (top) with EpsE from Vibrio cholerae (VcEpsE) [22] and GspE from A. fulgidus (AfGspE) [18].
In the present paper, we report the structure of the entire TtPilF hexamer by cryoelectron microscopy, revealing an unusual dumbbell-like structure. We show that the C-terminal regions undergo structural changes on binding to p[NH]ppA (adenosine 5′-[β,γ-imido]triphosphate), which are transmitted to the N-terminal half of the molecule through the stem-like structure that links them. We also show that TtPilF binds nucleic acid and that the binding site is located within the N-terminal half of the macromolecule. Together, these observations suggest a model for the mechanism of TtPilF, whereby ATP hydrolysis in the C-terminal domains is linked to structural changes within the GSPII domains which, through binding to DNA, are linked to mechanical force generation and DNA uptake into the cell.
EXPERIMENTAL
Protein expression and purification
The full-length pilF gene was amplified from T. thermophilus HB8 genomic DNA using primers TtPilF forward (5′-CCTCGAGGGGGTCCATGGAGATGAGCGTGCTGAC-3′) and TtPilF reverse (5′-GCTTTGGCCATCGCTTTCCGCGGCCGCCTCAATGGTAC-3′). The amplified gene and the pET52b vector (Novagen) were treated with restriction enzymes NcoI and NotI, purified and ligated. The final construct, pilF–52b, codes for the TtPilF protein followed by a thrombin cleavage site and 6×histidine tag at the C-terminus. The pilF–52b plasmid was transformed into T7 express cells (New England Biolabs) and three to four colonies were inoculated in 50 ml of 2YT medium [1.6% (w/v) tryptone/1% (w/v) yeast extract/0.5% NaCl] containing 100 μg/ml ampicillin and grown for 3–4 h at 37°C. This startup culture was then diluted into 2 litres of 2YT medium and the cells were allowed to grow at 37°C until the absorbance at 600 nm reached 0.8–1.0. At this point, the temperature was reduced to 16°C and IPTG (isopropyl β-D-thiogalactopyranoside) was added to a final concentration of 0.1 mM. The cells were harvested after 16 h by centrifugation at 6000 g for 20 min. Cells from 2 litres of culture were resuspended in 40 ml of buffer A [25 mM Tris (pH 8.0), 100 mM NaCl and 10 mM MgCl2] containing 20 mg of lysozyme, 1×EDTA-free protease inhibitor cocktail (Roche) and 0.35 mg of DNAse (Sigma). The cell suspension was lysed using a sonication probe (TT13/FZ, Bandelin Sonopuls HD3200) at 30% amplitude for 5 min, pulsed on for 10 s and pulsed off for 10 s. The debris was removed by centrifugation at 16000 g for 30 min and the supernatant passed through a 0.45 μm filter. The filtered lysate was then pumped into a 5 ml HisTrap column (GE Healthcare) and washed with ten column volumes of buffer A containing 40 mM imidazole. Increasing the imidazole concentration to 500 mM eluted the bound protein. The TtPilF preparation was concentrated using a 100 kDa cut-off concentrator (Sartorius) and treated with RNAse (100 units) for 15 min at 37°C before further purification using a Hiload 16/600 Superdex 200 column (GE Healthcare) equilibrated with buffer A at a flow rate of 1 ml/min. The TtPilF peak eluted at ~54 ml, close to the void volume of the column. The final sample was found to be devoid of DNA/RNA as assessed by agarose gel electrophoresis.
DNA binding
An EMSA (electrophoretic mobility shift assay) was used to examine the ability of TtPilF to bind single- and double-stranded DNA. PolyA (3.8 μg) or polyAT (0.2 μg) were incubated with different amounts of TtPilF (25–100 μg) at 22°C for 5 min. The samples were then run on 2% agarose gels containing 0.42 μg/ml ethidium bromide, at 100 V for 15 min. p[NH]ppA, if required in the experiment, was added to TtPilF to a concentration of 4 mM. The ability of TtPilF to protect DNA from DNAse degradation was examined by incubating polyAT (0.2 μg) with TtPilF (50 μg) at 22°C for 5 min. The TtPilF–polyAT samples were then treated with different amounts of DNase (2–150 units) at 37°C for 15 min and subject to electrophoresis on an agarose gel as described above. A control gel for the DNA protection experiment that has no TtPilF was also run for comparison.
ATP hydrolysis assay
The ATPase activity of TtPilF was measured at different temperatures (37, 50, 55, 60, 65, 70, 75 and 80°C) by incubating TtPilF (100 μg, in buffer A) with 1 mM ATP for 30 min at the specified temperature and then placing the sample on ice to cool. A separate control experiment without TtPilF was run at each temperature and used to apply the correction for non-enzymatic hydrolysis. The total phosphate released during the incubation time was measured using the EnzChek® phosphate assay kit (Invitrogen) and calibrated using phosphate solutions of known concentration. To examine the effect of DNA binding on TtPilF ATPase activity, 0.2 μg of either polyA or polyAT was incubated with TtPilF (100 μg) at 22°C for 5 min before the assay. All ATPase assay experiments were carried out in triplicate.
Electron microscopy sample preparation
Cryoelectron microscopy experiments were carried out at a TtPilF concentration of 170 μg/ml in 25 mM Tris (pH 8.0), 100 mM NaCl and 10 mM MgCl2. The complex with p[NH]ppA bound was formed by the addition of p[NH]ppA to a final concentration of 1.5 mM and incubation for 30 min at 20°C, before application to the electron microscopy grid. For polyA-gold labelling, 1 μl of TtPilF sample (17 mg/ml) in buffer A was diluted to 68 μl of Mes buffer [50 mM Mes (pH 6.5), 200 mM NaCl and 10 mM MgCl2] and 1 μl (2 μg/μl) of polyA–biotin (25 bases, biotin conjugated at the 3′ end). Nanogold-streptavidin (nanogold diameter: 1.4 nm, nanoprobes) (30 μl) was then added to achieve a final TtPilF concentration of 170 μg/ml.
Electron microscopy data processing and analysis
Negatively stained samples of TtPilF were prepared as previously described [25]. In brief, 10 μl of PilF (25 μg/ml) was absorbed to glow-discharged 400 mesh carbon-coated grids (Agar Scientific) for 30 s, washed in distilled water and placed on a 10 μl droplet of 2% (w/v) uranyl acetate for 30 s before air drying. Data were recorded on a Tecnai Biotwin operating at 120 kV and a nominal magnification of 23000×. Cryoelectron microscopy TEM (transmission electron microscopy) grids of TtPilF were prepared in a FEI Vitrobot using 3 μl of sample (170 μg/ml) absorbed to freshly glow-discharged 2×2 Quantifoil grids. Grids were continuously blotted for 4–5 s in a 90% humidity chamber before plunge-freezing into liquid ethane. Data were then recorded on a Polara FEG operating at 200 kV on a 4K Gatan Ultrascan CCD (charge-coupled device) in low-dose mode. CCD images were recorded between 0.5 and 5.0 μm defocus at 3 Å/pixel (1 Å=0.1 nm) and had a maximum electron dose of 20–40 electrons/Å2. Single-particle averaging was performed using EMAN2 [26]. Particles were selected using semi-automated picking and, following CTF (contrast transfer function) correction, were combined into particle datasets and two-dimensional classification was performed. Approximately 20–30 projection averages of unique and different classes representing different particle orientations were selected and used to generate an initial three-dimensional model for refinement. Examination of the Eigenimages and rotational symmetry analysis revealed a six-fold symmetry (results not shown); subsequently, six rounds of iterative refinement were then performed using FRC (Fourier ring correlation) as the main alignment comparator to produce the final three-dimensional structures with six-fold symmetry applied. Resolution was estimated using the same method as applied previously [27]: each dataset is split into two halves, and the estimated resolution is set at the point at which the FSC (Fourier shell correlation) of one half with the other reaches 0.5. Both density maps were deposited at the EMDB (Electron Microscopy Data Bank) [28] with accession numbers 2222 (apoprotein form) and 2223 (p[NH]ppA-bound form).
Construction of a model for the TtPilF C-terminal region and fitting into electron density
A homology model was constructed using the automated Swissmodel server [29] and EpsE apoprotein co-ordinates ([22]; PDB accession 1P9R) as a template. The sequence identity over the modelled residue range was 45%. The TtPilF hexamer was constructed by secondary structure alignment in COOT [30] on to the co-ordinates of AfGspE from the thermophilic bacterium A. fulgidus ([18]; PDB accession 2OAP). This model was initially docked manually into the electron density map for TtPilF (p[NH]ppA-bound form), and subjected to an initial round of refinement using Situs (version 2.7) [31], treating each separate chain as a rigid body. For a second round of refinement in Situs, each chain was separated into two regions, one spanning the N2 domain, and the other comprising the C1, CM and C2 domains, as defined by Robien et al. [22]. The process was then repeated, starting with the electron density map for the apoprotein form of TtPilF. For both models, the distance between the C-terminus of the N2 domain and the N-terminus of the C1, CM and C2 domains could plausibly be spanned by a linker in a fully extended state, although the gap in the p[NH]ppA model (28 Å) might require some localized unfolding of the respective N- and C-termini.
RESULTS
The association of TtPilF with DNA transformation [32] led us to investigate the possibility that it binds nucleic acid. We noted that the purified recombinant TtPilF had an absorbance peak at 260 nm, rather than 280 nm (Figure 2A), and was able to bind and elute from a heparin chromatography column (results not shown). Gel electrophoresis indicated a tightly bound nucleic acid contaminant of approximately 30 bp, which was identified as RNA, rather than DNA, as it was susceptible to degradation by RNase (Figure 2B). We subsequently introduced an RNaase treatment step into the purification protocol to remove residual contaminant RNA, which we presume binds to TtPilF during the cellular expression and extraction process and is carried through the purification.
Figure 2. Nucleic acid contamination of purified TtPilF.

(A) UV absorption spectrum of TtPilF (continuous line) and BSA (broken line), both at 0.1 mg/ml, recorded in 25 mM Tris/HCl (pH 8.0), 100 mM NaCl and 10 mM MgCl2. (B) Agarose gel (1.5%) electrophoresis of TtPilF. A total of 50 μg of purified TtPilF was loaded in each lane. Lane 1 has TtPilF without pretreatment, lane 2 has TtPilF after treatment with 2 units of DNase (RNase-free) and lane 3 has TtPilF after treatment with 2 units of RNase (DNase-free). DNA markers are indicated to the left of the gel.
We then examined the ability of TtPilF to bind DNA oligonucleotides: the addition of TtPilF to either single-stranded (25-mer polyA) or double-stranded (25-mer polyAT) DNA demonstrated a band shift for both, although affinity appeared to be higher for polyAT than polyA (Figure 3A). DNA binding was retained in the presence of p[NH]ppA (Figure 3B), an observation that has implications for the mechanism of TtPilF in mediating DNA uptake. We also investigated DNA binding by examining the ability of TtPilF to protect polyAT from DNAse degradation: TtPilF was added to polyAT and incubated with increasing concentrations of DNase (Figure 3C). The ‘shifted’ TtPilF–DNA complex retained DNA (top panel, Figure 3C), compared with polyAT DNA alone (bottom panel, Figure 3C). The ATPase activity of TtPilF increased at temperatures up to 75°C, as would be expected for an enzyme from a thermophilic organism, but the activity was not affected by the presence of DNA (Figure 4).
Figure 3. Binding of DNA to TtPilF.

(A) Agarose gel (2%) band-shift assay: top panel, 3.85 μg of polyA; bottom panel, 0.2 μg of polyAT. TtPilF quantities used were: lane 1, 50 μg; lane 2, 0 μg; lane 3, 25 μg; lane 4, 50 μg; lane 5, 75 μg and lane 6, 100 μg. (B) Band-shift assay for TtPilF with 4 mM p[NH]ppA. TtPilF and polyA/polyAT quantities used were the same as in (A). (C) Comparison of DNase digestion of polyAT in the presence (top panel) and absence (bottom panel) of TtPilF. Lane 1 has 50 μg of TtPilF, lane 2 has 200 ng of polyAT, lanes 3–7 have both 50 μg of TtPilF and 0.2 μg of polyAT with DNaase (Sigma) added as follows: lane 3, 2 units; lane 4, 6 units; lane 5, 18 units; lane 6, 54 units; and lane 7, 150 units. AMPPNP, p[NH]ppA.
Figure 4. TtPilF ATPase activity.

Results shown are means±S.D. (n=3).
The quality of the TtPilF sample was then examined using TEM. Our initial experiments used negative stain to enhance contrast and enable rapid screening; the TEM data showed that the sample was of excellent quality, with the particles forming an oligomeric, well dispersed and minimally aggregated complex. By eye, the data presented a variety of different particle views (Supplementary Figure S1A at http://www.biochemj.org/bj/450/bj4500417add.htm) and this impression was confirmed by multistatistical analysis of individually selected particles (Supplementary Figure S1B). Side views were characterized by a distinct double-banded or four-lobed feature (~130 Å×150 Å), whereas top views had a ring-like appearance with a diameter of ~130 Å (Supplementary Figure S1B). A preliminary three-dimensional reconstruction was generated from 6500 unique single particles, at a resolution of ~28 Å. Supplementary Figure S1(C) shows that the TtPilF hexamer is made up of two stacked disks of approximately equal size (130 Å×60 Å) and volume. Each domain has distinctive features, with one of the disks appearing essentially solid whereas the other is much more ring-like with a hollow centre. This significant invagination readily distinguished it from the other disk domain and allowed for an accurate orientation of the complex. In negative stain, the central sections of the complex appeared to be void of any protein density.
Given the high sample quality, we proceeded to collect cryoelectron microscopy data on the TtPilF sample. To examine any potential structural changes that might be induced by ATP binding, parallel datasets of the TtPilF apoenzyme and the TtPilF–p[NH]ppA binary complex were collected. An example of data obtained from the latter is shown in Figure 5(A). As was the case with the negatively stained sample, the TtPilF particles were well dispersed, readily identifiable and positioned in the thin ice layer in many orientations. The inset to Figure 5(A) shows a selection of projection class averages produced from singular value decomposition and multistatistical analysis of the particles; these data show a good correspondence with the negatively stained dataset, in terms of size and distribution of orientations. Using data from 44000 particles of the TtPilF–p[NH]ppA binary complex, a final three-dimensional structure was obtained with a resolution of ~10 Å (Figure 5B). The parallel experiment carried out on data obtained from the TtPilF apoenzyme used 37000 particles and produced a broadly similar structure (the two volumes are compared below), but at a slightly lower resolution of 12 Å. In both structures, the two ring/disk features identified in the negative stain study are clearly identified but, in addition, a central connector stem region is discernible which links the two. The most likely reason for the failure to identify this connector stem in the negative stain-derived structure is because the stain pools and accumulates throughout the crevice between the ring and the disk, obscuring the protein density.
Figure 5. Determination of TtPilF structure by single particle averaging and cryoelectron microscopy.

(A) A sample field of ‘raw’ TtPilF particles embedded in thin vitreous ice. The inset shows examples of projection averages determined from the raw data (note that contrast is inverted for analysis so that protein appears white). The sample data shown are from the TtPilF–p[NH]ppA complex. Scale bar=1000 Å. (B) Surface-rendered three-dimensional volume of the six-fold symmetric TtPilF–p[NH]ppA complex (orthogonal views) displayed at a sigma value to accommodate 600 kDa of protein mass. The slab view has the foremost 50% of volume removed to display protein density through the complex. The figure was prepared using Chimera [37].
The ring structure, which is at the top of the side view shown at the bottom of Figure 5(B), forms a cavity that is sealed at one end by the stem. A slab view (bottom right, Figure 5B) shows continuous density from the ring to the disk at the bottom, which forms a narrow cavity at its base. A comparison of the structural changes induced by binding of p[NH]ppA is shown in Figure 6(A). The most pronounced difference is in the stem connector region, which undergoes a significant narrowing in the transition from the apoprotein to p[NH]ppA-bound state (arrowed in Figure 6A). This is accompanied by a downward shift in mass within the disk, and also by changes in the ring structure at the top. These structural perturbations are best illustrated by an animation that shows the transition between the two states (Supplementary Movie S1 at http://www.biochemj.org/bj/450/bj4500417add.htm).
Figure 6. Structural changes in TtPilF on binding of p[NH]ppA and modelling of the C-domains into cryoelectron density maps.

(A) Comparison of TtPilF apoprotein and p[NH]ppA-bound structures. Left-hand panel, TtPilF apoenzyme, contoured at 4.9σ, in red; right-hand panel, as left-hand panel, but with the TtPilF–p[NH]ppA complex superimposed, contoured at 4.8σ, in green. The resolution of the TtPilF–p[NH]ppA complex was truncated at 12 Å resolution; sigma levels of both maps were selected such that the volumes were equal. The arrow in the right-hand panel shows the region of the connector stem that contracts on binding of p[NH]ppA. (B) Modelling of TtPilF C-domains into cryoelectron density maps. Top and side views of apoprotein (left-hand panel) and p[NH]ppA-bound (right-hand panel) models for the C-domains, superimposed on their respective electron density maps. One protomer in each hexamer is coloured differently to indicate subunit contacts. (C) Comparison of TtPilF C-domains in the apoprotein and p[NH]ppA-bound states. Superinposition of ribbon plots of TtPilF C-domain models for the apoprotein (red) and p[NH]ppA-bound (green) forms. Left-hand panel: a single subunit, viewed from the side; the N2 domain in the apoprotein is circled, with an arrow to indicate the lateral movement that occurs on binding of p[NH]ppA. Right-hand panel: top view of the assembled hexamers; the dimensions of the interior cavity are unchanged, but the apoprotein structure has a wider diameter, as a result of the movement of the N2 domain. AMPPNP, p[NH]ppA.
The predicted domain structure of TtPilF suggests a clear division between the C-terminal domains, which constitute the ATP-binding regions and are well conserved, and the N-terminal domains (Figure 1). A number of crystal structures of AAA+ family members are available; as outlined above, the closest orthologue for TtPilF is the T2SS ATPase EpsE [22]. Unfortunately, EpsE in these structures does not crystallize as an assembled hexamer. There is, however, another structure of a T2SS ATPase available from the thermophilic bacterium A. fulgidus, AfGspE, where the hexamer is intact [18]. We therefore constructed a homology molecular model for the C-terminal region of TtPilF using EpsE as a template, and assembled the hexamer by structural alignment on to the AfGspE ATPase oligomer. The hexameric model for the C-terminal region retains a central channel which is a major feature of AAA+ family hexamers [10]. A side-by-side comparison of the model with the ring and disk features demonstrates that it is the ring that is most likely to correspond to the C-terminal region of TtPilF (Supplementary Figure S2 at http://www.biochemj.org/bj/450/bj4500417add.htm). There is no clear channel through the disk structure, this is most readily seen from the slab view in the bottom right of Figure 5(B). In addition, the individual subunits in the disk structure are closer together than is the case for the ring, and fitting of the model for the C-terminal region would not be possible without introducing unacceptable steric clashes. We then used the software package Situs [31] to optimize separately the fit of the generated model to the cryoelectron microscopy density maps for the apoprotein and p[NH]ppA-bound forms. For the purposes of fitting into the density maps, we split each chain into two, with one region spanning the N2 domain and the second comprising the C1, CM and C2 domains, as originally defined by Robien et al. [22]. The connecting loop between the N2 and C1, CM and C2 domains was not included in the model, owing to poor homology with TtPilF. The results, shown in Figure 6(B), show an excellent agreement between the model and electron density map in each case.
A comparison of the modelled structures of the C-domains in the apoprotein and p[NH]ppA-bound states revealed domain motions induced by p[NH]ppA binding. In particular, the N2 domain moves laterally, accompanied by a pivoted motion of the C1, CM and C2 domains about a point at the top of the TtPilF oligomer (Figure 6C, left-hand panel). This movement is most easily seen from animations (Supplementary Movies S2 and S3 at http://www.biochemj.org/bj/450/bj4500417add.htm). The view from the top (Figure 6C, right-hand panel and Supplementary Movie S3) also shows how the N2 domain swings inwards on the transition from the apoprotein to the p[NH]ppA-bound state. These observations are in accordance with the changes observed in the electron density maps of the two states, this is most readily apparent from a comparison of the two, where the bulge from the protruding N2 domain is seen in the apoprotein state (left-hand panel of Figure 6A).
In principle, it should be possible to construct homology models for each GSPII fold in TtPilF (Figure 1), on the basis of the structure of the XpsE N-terminal domain from Xanthomonas campestris [23], and model these into the cryoelectron microscopy density maps. In practice, however, the resolution of the maps was not sufficient to distinguish density for individual α-helices, without which it was not possible to fix the relative orientation of each GSPII fold relative to the others.
Our observations on the ability of TtPilF to bind nucleic acid prompted us to examine how DNA recognition related to the TtPilF structure. Initial experiments incubated TtPilF with the 25-mer polyAT duplex DNA used for the experiment shown in Figure 3; however, when studied by cryoelectron microscopy, the particles were generally less well dispersed and showed a tendency to self-adhere. This behaviour led to a lower resolution in the resulting three-dimensional reconstruction (16 Å) and a map that was indistinguishable from the apoprotein form (results not shown). The small additional mass contributed from the DNA duplex would be difficult to resolve at this resolution. We therefore adopted a different approach, by incubating biotinylated DNA with a conjugate consisting of avidin linked to gold particles, we were able to generate a DNA–gold particle ligand. This reagent was incubated with TtPilF and particles observed by negative stain; gold labelling of the TtPilF complex occurred at a low frequency, but gold particles were clearly observed associated with the oligomer. A montage of selected particles with the DNA–gold bound is shown in Figure 7(A). Class averages corresponding to different projection views of the complex showed specific electron dense scattering material associated with the periphery of the complex from the top view and at the interface of the lower ring and the ATPase ring in the side views (Figure 7B). These projection views were used to generate a new starting model and a three-dimensional structure of the TtPilF complex was generated from 2700 particles. Figure 7(C) shows that the location of the negative gold density is clustered around the edge of the disk/GSPII domains, close to the stem region, providing evidence that it is indeed this section of the TtPilF oligomer which is associated with nucleic acid binding. It also suggests a model in which the DNA might wrap around the disk and/or stem structures, and hence might be coupled with mechanical motion induced by ATP hydrolysis within the C-terminal ring domains.
Figure 7. DNA binding to TtPilF.
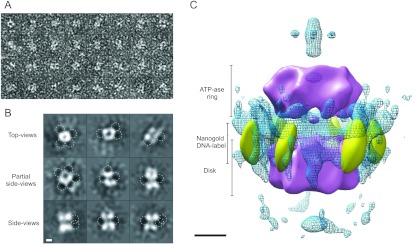
(A) A montage of TtPilF particles following incubation with biotin–DNA/avidin–gold. Box size=240 Å×240 Å. (B) Two-dimensional projection averages of negatively stained TtPilF incubated with biotin–DNA/avidin–gold. The locations of higher density peaks associated with gold particles are indicated by broken-lined circles. Scale bar=50 Å. (C) Three-dimensional alignment of the avidin–gold label within the TtPilF complex. The TtPilF volume is shown in purple surface render, contoured at a level to accommodate 600 kDa of mass. The positions of the gold densities at –4σ are indicated in gold surface render. The blue wireframe shows cross-correlated negative densities superimposed from a TtPilF volume with no gold label, contoured at –4σ and highlighting the specific electron density attributable to the gold particles. Scale bar=100 Å.
DISCUSSION
Previous work has established strong evidence which links TtPilF to a function associated with DNA uptake into T. thermophilus [6,11]. TtPilF was one of a group of proteins originally shown, through mutation and loss of competence, to be part of the natural transformation machinery in Thermus [6]. A previous study has demonstrated that mutation of TtPilF leads to an inhibition of DNA transport into the organism [11]. Interestingly, DNA translocation into T. thermophilus has been shown to be an energy-dependent process [3]. These observations all point to a role for TtPilF in providing the energy for DNA uptake. Our finding that TtPilF binds to nucleic acid suggests that this function could be mediated through direct interaction of the ATPase with DNA in the cytoplasm. Indeed, we would infer that the off-rate for dissociation of DNA or RNA from TtPilF must be low (in the region of per hour or slower), given that nucleic acid is persistently bound to the protein through several purification steps. Previous models for DNA translocation into T. thermophilus have placed TtPilF with other proteins associated with TFP biogenesis [13]: in particular, by analogy with work conducted on TFP assembly in other organisms, we would expect TtPilF to function alongside other inner membrane biogenesis components, such as PilC, PilM, PilN and PilO, to promote pilus formation. On the basis of this model, TtPilF would fill the equivalent of a role carried out by PilB in P. aeruginosa [33] or PilF in N. meningitidis [34]. This conclusion would appear to be justified by sequence comparison, which clearly associates TtPilF with members of the PilB/PilF/EpsE/PulE family of extension ATPases that promote assembly of TFP or pseudopili which drive type II secretion [24]. There are, however, some difficulties with this proposition. Mutation of TtPilF does not abolish piliation, although natural competence is much reduced [6]. Moreover, the sequence of TtPilF is considerably longer than its PilB/PilF/EpsE/PulE counterparts, as a result of additional repeats of the GSPII structural motif [24]. These observations raise doubts as to whether TtPilF indeed functions as the TFP assembly ATPase within T. thermophilus and suggest that a role in DNA uptake is more plausible.
How would TtPilF integrate into the current model for DNA uptake into T. thermophilus? A number of competence-associated proteins have been identified in T. thermophilus, including ComEA and ComEC [13]. The latter is a polytopic integral membrane protein, hypothesized to form a hydrophilic channel across the inner membrane for passage of DNA. One obvious possibility is that TtPilF interacts with ComEC in some way to promote DNA transport across the inner membrane. An obvious inference from our data is that because TtPilF binds DNA, and because p[NH]ppA causes structural change in the oligomer, ATP hydrolysis by TtPilF would be linked to active DNA transport across the inner membrane. This remains to be formally demonstrated, however, and may require reconstitution of a translocation complex, perhaps involving ComEC, for demonstration in vitro. As explained above, such a model would be attractive, in that it would explain a variety of observations concerning the provision of the energy source for DNA uptake. It is also interesting to note that, unlike the DNA uptake system in Neisseria for example, T. thermophilus does not exhibit any sequence specificity or distinguish between DNA of bacterial, eukaryal or archeal origins [13]. Our observations are that recognition of nucleic acid by TtPilF is apparently broad-based, extending to RNA as well as DNA, and would therefore be consistent with this conclusion.
Yamagata and Tainer [18] proposed a general model for the mechanism of secretion ATPases, on the basis of a crystal structure and SAXS data derived from the A. fulgidus secretion ATPase (AfGspE). The central N2 and C1 domains from AfGspE are similar in structure to the equivalent domains from V. cholerae EspE [22] and HP0525, a VirB11 homologue from the type IV secretion system [35]. The model proposed a transition between an open state, to which ATP binds, and a closed ATP-bound state. Transition from the open to the closed state is characterized by the closer approach of the N2 domain to the C1 domain. This structural change is transmitted to the N1 domain, which is thought to be the part of the ATPase that interacts with other components of the secretion machinery. This ‘piston-like’ mechanism, with an amplitude of approximately 10 Å, would be sufficient in principle to translate a pilin or pseudopilin into a nascent fibre. Our observations on TtPilF suggest that it functions in a similar way, producing a structural shift of comparable amplitude (Figure 6A). Our structures of TtPilF, corresponding to the open and closed states of the Yamagata and Tainer model, show how these structural changes within the C domains are transmitted extensively throughout the oligomer (Figure 6A and Supplementary Movie S1). Secondary structure predictions suggest that the stem region in the TtPilF structure is formed by a bundle of helices, which would act as a ‘connecting rod’ from the N2 domain to the GSPII domains in the bottom part of the structure, which is associated with DNA binding (Figure 7C). Our observation that DNA binding by TtPilF occurs in both the apoprotein and p[NH]ppA-bound forms (Figures 3A and 3B), suggests that DNA remains bound to the complex during the ATP hydrolysis cycle.
Observations on other DNA transport machines suggest that they work in a different way [5]. TrwB, for example, is a plasmid-encoded integral membrane protein that acts as a DNA transporter during bacterial conjugation [36]. The crystal structure of the soluble portion of TrwB revealed structural similarities with the F1 ATPase but, critically, the DNA passes through the centre of the hexamer where the coiled coil of the F1 ATPase γ-subunit is located [36]. It is proposed that structural changes that occur as part of the ATP hydrolysis cycle are transmitted into conformational changes in the subunits lining the DNA channel. Direct contacts between the DNA and the subunits lining the interior of channel would then be responsible for driving the DNA through in a pumping-type mechanism. The cut-away views of the density maps shown in Figure 6(A) effectively rule out a mechanism of this type for TtPilF: it is unlikely that there is a complete open channel through the central symmetry axis of sufficient dimensions to allow the passage of a DNA fibre, even once allowance has been made for the resolution of the cryoelectron microscopy reconstructions.
In summary, the results of the present study provide compelling evidence that TtPilF is a source of energy for DNA uptake into T. thermophilus, and that it does so by acting as a DNA translocation machine. The molecular details of this process remain to be elucidated and will probably require higher resolution structural data.
Online data
AUTHOR CONTRIBUTION
Darin Hassan, Vijaykumar Karuppiah and Angela Thistlethwaite expressed and purified TtPilF; Darin Hassan and Vijaykumar Karuppiah conducted the DNA-binding and biochemical experiments; Richard Collins collected the electron microscope data and determined the three-dimensional structures; Jeremy Derrick designed the research, performed the fitting of the C-terminal domains to the density maps and wrote the paper, with contributions from all authors.
ACKNOWLEDGEMENTS
We thank Jamie-Lee Berry for a critical reading of the paper before submission.
FUNDING
This work was funded by the Wellcome Trust [grant number 093388].
References
- 1.Averhoff B., Friedrich A. Type IV pili-related natural transformation systems: DNA transport in mesophilic and thermophilic bacteria. Arch. Microbiol. 2003;180:385–393. doi: 10.1007/s00203-003-0616-6. [DOI] [PubMed] [Google Scholar]
- 2.Averhoff B. DNA transport and natural transformation in mesophilic and thermophilic bacteria. J. Bioenerg. Biomembr. 2004;36:25–33. doi: 10.1023/b:jobb.0000019595.66733.fa. [DOI] [PubMed] [Google Scholar]
- 3.Schwarzenlander C., Averhoff B. Characterization of DNA transport in the thermophilic bacterium Thermus thermophilus HB27. FEBS J. 2006;273:4210–4218. doi: 10.1111/j.1742-4658.2006.05416.x. [DOI] [PubMed] [Google Scholar]
- 4.Chen I., Dubnau D. DNA uptake during bacterial transformation. Nat. Rev. Microbiol. 2004;2:241–249. doi: 10.1038/nrmicro844. [DOI] [PubMed] [Google Scholar]
- 5.Burton B., Dubnau D. Membrane-associated DNA transport machines. Cold Spring Harbour Perspect. Biol. 2010;2:a000406. doi: 10.1101/cshperspect.a000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedrich A., Prust C., Hartsch T., Henne A., Averhoff B. Molecular analyses of the natural transformation machinery and identification of pilus structures in the extremely thermophilic bacterium Thermus thermophilus strain HB27. Appl. Environ. Microbiol. 2002;68:745–755. doi: 10.1128/AEM.68.2.745-755.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedrich A., Rumszauer J., Henne A., Averhoff B. Pilin-like proteins in the extremely thermophilic bacterium Thermus thermophilus HB27: implication in competence for natural transformation and links to type IV pilus biogenesis. Appl. Environ. Microbiol. 2003;69:3695–3700. doi: 10.1128/AEM.69.7.3695-3700.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karuppiah V., Derrick J. P. Structure of the PilM–PilN inner membrane type IV pilus biogenesis complex from Thermus thermophilus. J. Biol. Chem. 2011;286:24434–24442. doi: 10.1074/jbc.M111.243535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rumszauer J., Schwarzenlander C., Averhoff B. Identification, subcellular localization and functional interactions of PilMNOWQ and PilA4 involved in transformation competency and pilus biogenesis in the thermophilic bacterium Thermus thermophilus HB27. FEBS J. 2006;273:3261–3272. doi: 10.1111/j.1742-4658.2006.05335.x. [DOI] [PubMed] [Google Scholar]
- 10.Wendler P., Ciniawsky S., Kock M., Kube S. Structure and function of the AAA plus nucleotide binding pocket. Biochim. Biophys. Acta. 2012;1823:2–14. doi: 10.1016/j.bbamcr.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 11.Schwarzenlander C., Haase W., Averhoff B. The role of single subunits of the DNA transport machinery of Thermus thermophilus HB27 in DNA binding and transport. Environ. Microbiol. 2009;11:801–808. doi: 10.1111/j.1462-2920.2008.01801.x. [DOI] [PubMed] [Google Scholar]
- 12.Pelicic V. Type IV pili: e pluribus unum? Mol. Microbiol. 2008;68:827–837. doi: 10.1111/j.1365-2958.2008.06197.x. [DOI] [PubMed] [Google Scholar]
- 13.Averhoff B. Shuffling genes around in hot environments: the unique DNA transporter of Thermus thermophilus. FEMS Microbiol. Rev. 2009;33:611–626. doi: 10.1111/j.1574-6976.2008.00160.x. [DOI] [PubMed] [Google Scholar]
- 14.Ayers M., Howell P. L., Burrows L. L. Architecture of the type II secretion and type IV pilus machineries. Future Microbiol. 2010;5:1203–1218. doi: 10.2217/fmb.10.76. [DOI] [PubMed] [Google Scholar]
- 15.Berry J.-L., Phelan M. M., Collins R. F., Adomavicius T., Tønjum T., Frye S., Bird L., Owens R., Ford R. C., Lian L.-Y., Derrick J. P. Structure and assembly of a trans-periplasmic channel for type IV pili in Neisseria meningitidis. PLoS Pathog. 2012;8:e1002923. doi: 10.1371/journal.ppat.1002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karuppiah V., Hassan D., Saleem M., Derrick J. P. Structure and oligomerization of the PilC type IV pilus biogenesis protein from Thermus thermophilus. Proteins. 2010;78:2049–2057. doi: 10.1002/prot.22720. [DOI] [PubMed] [Google Scholar]
- 17.Burrows L. L. Weapons of mass retraction. Mol. Microbiol. 2005;57:878–888. doi: 10.1111/j.1365-2958.2005.04703.x. [DOI] [PubMed] [Google Scholar]
- 18.Yamagata A., Tainer J. A. Hexameric structures of the archaeal secretion ATPase GspE and implications for a universal secretion mechanism. EMBO J. 2007;26:878–890. doi: 10.1038/sj.emboj.7601544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Satyshur K. A., Worzalla G. A., Meyer L. S., Heiniger E. K., Aukema K. G., Misic A. M., Forest K. T. Crystal structures of the pilus retraction motor PilT suggest large domain movements and subunit cooperation drive motility. Structure. 2007;15:363–376. doi: 10.1016/j.str.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Misic A. M., Satyshur K. A., Forest K. T. P. aeruginosa PilT structures with and without nucleotide reveal a dynamic type IV pilus retraction motor. J. Mol. Biol. 2010;400:1011–1021. doi: 10.1016/j.jmb.2010.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolfgang M., van Putten J. P. M., Hayes S. F., Dorward D., Koomey M. Components and dynamics of fiber formation define a ubiquitous biogenesis pathway for bacterial pili. EMBO J. 2000;19:6408–6418. doi: 10.1093/emboj/19.23.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robien M. A., Krumm B. E., Sandkvist M., Hol W. G. Crystal structure of the extracellular protein secretion NTPase EpsE of Vibrio cholerae. J. Mol. Biol. 2003;333:657–674. doi: 10.1016/j.jmb.2003.07.015. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y., Shiue S. J., Huang C. W., Chang J. L., Chien Y. L., Hu N. T., Chan N. L. Structure and function of the XpsE N-terminal domain, an essential component of the Xanthomonas campestris type II secretion system. J. Biol. Chem. 2005;280:42356–42363. doi: 10.1074/jbc.M506843200. [DOI] [PubMed] [Google Scholar]
- 24.Rose I., Biukovic G., Aderhold P., Muller V., Gruber G., Averhoff B. Identification and characterization of a unique, zinc-containing transport ATPase essential for natural transformation in Thermus thermophilus HB27. Extremophiles. 2011;15:191–202. doi: 10.1007/s00792-010-0343-2. [DOI] [PubMed] [Google Scholar]
- 25.Collins R. F., Davidsen L., Derrick J. P., Ford R. C., Tonjum T. Analysis of the PilQ secretin from Neisseria meningitidis by transmission electron microscopy reveals a dodecameric quaternary structure. J. Bacteriol. 2001;183:3825–3832. doi: 10.1128/JB.183.13.3825-3832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 27.Collins R. F., Frye S. A., Kitmitto A., Ford R. C., Tonjum T., Derrick J. P. Structure of the Neisseria meningitidis outer membrane PilQ secretin complex at 12 Å resolution. J. Biol. Chem. 2004;279:39750–39756. doi: 10.1074/jbc.M405971200. [DOI] [PubMed] [Google Scholar]
- 28.Tagari M., Newman R., Chagoyen M., Carazo J. M., Henrick K. New electron microscopy database and deposition system. Trends Biochem. Sci. 2002;27:589–589. doi: 10.1016/s0968-0004(02)02176-x. [DOI] [PubMed] [Google Scholar]
- 29.Schwede T., Kopp J., Guex N., Peitsch M. C. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Wriggers W. Using Situs for the integration of multi-resolution structures. Biophys. Rev. 2010;2:21–27. doi: 10.1007/s12551-009-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friedrich A., Hartsch T., Averhoff B. Natural transformation in mesophilic and thermophilic bacteria: identification and characterization of novel, closely related competence genes in Acinetobacter sp. strain BD413 and Thermus thermophilus HB27. Appl. Environ. Microbiol. 2001;67:3140–3148. doi: 10.1128/AEM.67.7.3140-3148.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiang P., Sampaleanu L. M., Ayers M., Pahuta M., Howel P. L., Burrows L. L. Functional role of conserved residues in the characteristic secretion NTPase motifs of the Pseudomonas aeruginosa type IV pilus motor proteins PilB, PilT and PilU. Microbiology. 2008;154:114–126. doi: 10.1099/mic.0.2007/011320-0. [DOI] [PubMed] [Google Scholar]
- 34.Carbonnelle E., Helaine S., Nassif X., Pelicic V. A systematic genetic analysis in Neisseria meningitidis defines the Pil proteins required for assembly, functionality, stabilization and export of type IV pili. Mol. Microbiol. 2006;61:1510–1522. doi: 10.1111/j.1365-2958.2006.05341.x. [DOI] [PubMed] [Google Scholar]
- 35.Savvides S. N., Yeo H. J., Beck M. R., Blaesing F., Lurz R., Lanka E., Buhrdorf R., Fischer W., Haas R., Waksman G. VirB11 ATPases are dynamic hexameric assemblies: new insights into bacterial type IV secretion. EMBO J. 2003;22:1969–1980. doi: 10.1093/emboj/cdg223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cabezon E., de la Cruz F. TrwB: an F-1-ATPase-like molecular motor involved in DNA transport during bacterial conjugation. Res. Microbiol. 2006;157:299–305. doi: 10.1016/j.resmic.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. UCSF chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.