

Abstract
The blood glucose-lowering hormone glucagon-like peptide-1 (GLP-1) stimulates cAMP production, promotes Ca2+ influx, and mobilizes an intracellular source of Ca2+ in pancreatic β cells. Here we provide evidence that these actions of GLP-1 are functionally related: they reflect a process of Ca2+-induced Ca2+ release (CICR) that requires activation of protein kinase A (PKA) and the Epac family of cAMP-regulated guanine nucleotide exchange factors (cAMPGEFs). In rat insulin-secreting INS-1 cells or mouse β cells loaded with caged Ca2+ (NP-EGTA), a GLP-1 receptor agonist (exendin-4) is demonstrated to sensitize intracellular Ca2+ release channels to stimulatory effects of cytosolic Ca2+, thereby allowing CICR to be generated by the uncaging of Ca2+ (UV flash photolysis). This sensitizing action of exendin-4 is diminished by an inhibitor of PKA (H-89) or by overexpression of dominant negative Epac. It is reproduced by cell-permeant cAMP analogues that activate PKA (6-Bnz-cAMP) or Epac (8-pCPT-2′-O-Me-cAMP) selectively. Depletion of Ca2+ stores with thapsigargin abolishes CICR, while inhibitors of Ca2+ release channels (ryanodine and heparin) attenuate CICR in an additive manner. Because the uncaging of Ca2+ fails to stimulate CICR in the absence of cAMP-elevating agents, it is concluded that there exists in β cells a process of second messenger coincidence detection, whereby intracellular Ca2+ release channels (ryanodine receptors, inositol 1,4,5-trisphosphate (IP3) receptors) monitor a simultaneous increase of cAMP and Ca2+ concentrations. We propose that second messenger coincidence detection of this type may explain how GLP-1 interacts with β cell glucose metabolism to stimulate insulin secretion.
Pancreatic β cells express GTP-binding protein-coupled receptors that mediate stimulatory actions of the blood glucose-lowering hormone glucagon-like peptide-1-(7–36)-amide (GLP-1) on insulin biosynthesis and secretion (Kieffer & Habener, 1999). Insulinotropic actions of GLP-1 are mimicked by exendin-4 (Ex-4), a peptide related in structure to GLP-1. Ex-4 acts as a high-affinity agonist at the GLP-1 receptor (GLP-1-R) and, as is the case for GLP-1, it is currently under investigation for use in the treatment of diabetes mellitus (Holz & Chepurny, 2003). Activation of the GLP-1-R on β cells initiates a complex series of signalling events that include cAMP production, membrane depolarization, an increase of intracellular calcium concentration ([Ca2+]i), and exocytosis (Thorens, 1992; Holz et al. 1993, 1995, 1999; Gromada et al. 1995, 1998a,b; Bode et al. 1999; Nakazaki et al. 2002; Eliasson et al. 2003). Because multiple processes within the β cell are regulated in a cAMP- and Ca2+-dependent manner, it is of interest to ascertain how these two second messengers interact. In particular, the potential importance of second messenger coincidence detection as a determinant of GLP-1-R signal transduction has yet to be explored fully (Holz & Habener, 1992).
Coincidence detection in biological systems is a phenomenon in which two or more complementary signals interact synergistically to generate a cellular response. Neither signal is an adequate stimulus in the absence of its complement. The type VIII isoform of adenylyl cyclase expressed in β cells acts as a molecular coincidence detector because it is stimulated not only by GS GTP-binding proteins, but also by Ca2+/calmodulin (Pipeleers et al. 1985; Schuit & Pipeleers, 1985; Delmeire et al. 2003). Coincidence detection also exists when the activity of an effector molecule is governed by multiple second messengers. This may be the case for intracellular Ca2+ release channels (ryanodine receptors, RYR; inositol 1,4,5-trisphosphate (IP3) receptors, IP3-R), the opening of which is reported to be facilitated by Ca2+ and cAMP (Marx et al. 2000; Bruce et al. 2003). Because GLP-1 stimulates cAMP production, and because β cell glucose metabolism stimulates influx of Ca2+ through voltage-dependent Ca2+ channels (VDCCs), it is predicted that GLP-1 and glucose should interact synergistically to gate Ca2+ release channels from a closed to open state. Indeed, second messenger coincidence detection of this type might explain the unusual interaction of GLP-1 and glucose to mobilize an intracellular source of Ca2+ in the β cell (Gromada et al. 1995; Bode et al. 1999; Holz et al. 1999; Kang et al. 2001, 2003; Kang & Holz, 2003; Sasaki et al. 2002; Tsuboi et al. 2003; Dyachok & Gylfe, 2004).
Here we demonstrate that Ex-4 acts via cAMP, protein kinase A (PKA), and the Epac family of cAMP-regulated guanine nucleotide exchange factors (cAMPGEFs; also known as Epac1 and Epac2; Holz, 2004a) to sensitize Ca2+-induced Ca2+ release (CICR) mediated by the RYR and IP3-R. Sensitization allows CICR to be triggered by the uncaging of Ca2+ in INS-1 cells or mouse β cells loaded with a photolabile Ca2+ chelator (NP-EGTA; Ellis-Davies et al. 1994). Because the uncaging of Ca2+ fails to stimulate CICR in the absence of cAMP-elevating agents, it is concluded that there exists in β cells a process of second messenger coincidence detection whereby intracellular Ca2+ release channels monitor a simultaneous increase of cAMP and Ca2+ concentrations. Some of these findings have been published in preliminary form (Kang et al. 2005).
Methods
Islet isolation and cell culture
Islets were isolated from male C57BL/6 mice fed ad libitum (20–25 g body weight; Charles River Laboratories, Inc., Wilmington, MA, USA). The mice were anaesthetized by inhalation of CO2 (100%; 2–3 min exposure), and were killed by cervical dislocation. Surgical procedures for removal of the pancreas were performed in accordance with NYU School of Medicine policies governing the ethical use of mice for experimentation (IACUC Protocol no. 040602-01). After digestion of the pancreas with collagenase P (Roche Applied Science, Indianapolis, IN, USA; 2 mg ml−1 dissolved in RPMI 1640 medium), batches of 150–200 islets were subjected to mild trypsinization and were dispersed by trituration in a Ca2+-free saline in order to generate a single cell suspension. Isolated cells were then allowed to adhere to glass coverslips (25CIR-1; Fisher Sci.) coated with concanavalin A (type V; Sigma-Aldrich, St Louis, MO, USA). Primary cultures were maintained in a humidified incubator (95% air, 5% CO2) at 37°C in RPMI 1640 supplemented with 10% FBS, 100 units ml−1 penicillin G, and 100 μg ml−1 streptomycin. β cells were identified on the basis of their large diameter and granular appearance. INS-1 cells (passage numbers 70–90) were maintained in RPMI 1640 containing 10 mm Hepes, 11.1 mm glucose, 10% FBS, 100 units ml−1 penicillin G, 100 μg ml−1 streptomycin, 2.0 mml-glutamine, 1.0 mm sodium pyruvate, and 50 μm 2-mercaptoethanol (Asfari et al. 1992). INS-1 cells were passaged by trypsinization and subcultured once a week. All reagents for cell culture were obtained from Invitrogen-Life Technologies (Rockville, MD).
Measurement of [Ca2+]i
The fura−2 loading solution consisted of standard extracellular saline (SES) containing (mm): 138 NaCl, 5.6 KCl, 2.6 CaCl2, 1.2 MgCl2, 10 Hepes, 11.1 d–glucose and supplemented with 1 μm fura−2 AM (Molecular Probes Inc., Eugene, OR, USA), 2% FBS, and 0.02% Pluronic F-127 (w/v; Molecular Probes Inc.). Cells were exposed to the fura−2 loading solution for 20–30 min at 22°C. Experiments were performed in SES at 32°C using a TE300 inverted microscope (Nikon, Melville, NY, USA) equipped with a temperature-controlled stage (Medical Systems Corp., Greenvale, NY, USA) and a 100× Nikon UVF oil immersion objective (NA 1.3). Microfluorimetry was performed ratiometrically at 0.5 s intervals using a video imaging system outfitted with an intensified CCD camera (IonOptix Corp., Milton, MA, USA). A rotating mirror delivered excitation light at 340 or 380 nm. The emitted light was measured at 510 nm, and the average of 29 frames of imaging data was used to calculate numerator and denominator values for determination of 340/380 ratios after background subtraction. [Ca2+]i was calculated according to established methods (Grynkiewicz et al. 1985). Calibration of raw fura−2 fluorescence values was performed as described (Kang & Holz, 2003) using fura−2 [K+]5 salt dissolved in calibration buffers from Molecular Probes Inc. (Calcium Calibration Kit 1 with Mg2+). Values of Rmin and Rmax were 0.20 and 7.70. Components of the imaging system are illustrated (Supplementary Fig. 1).
Figure 1.

Sensitization of CICR by Ex-4 and forskolinA, an INS-1 cell loaded with NP-EGTA was bathed in SES containing 11.1 mm glucose. The uncaging of Ca2+ by UV flash photolysis (UV, arrows) produced a small increase of [Ca2+]i but did not trigger CICR. B, a large transient increase of [Ca2+]i (CICR) was observed when UV flash photolysis was performed in the presence but not the absence of Ex-4 (compare y-axis scale bars of A and B). C, the action of Ex-4, as illustrated on an expanded time scale. CICR amplitude was determined by subtracting the baseline Ca2+ concentration from the Ca2+ concentration measured at the peak of the Ca2+ transient. Also illustrated are the 50% and 10% threshold values of [Ca2+]i that were used when determining the percentage of cells exhibiting CICR (see Methods). D, sensitization of CICR by the cAMP-elevating agent forskolin (2 μm). A and D are from two different cells. B and C are from the same cell.
UV flash photolysis for liberation of caged Ca2+ and caged IP3
Cells were bathed for 60 min at 23°C in SES containing cell-permeable caged Ca2+ (NP-EGTA-AM, 5 μm, Molecular Probes, Inc.) or caged IP3 (ci-IP3/PM, 10 μm). The loading solution also contained 1 μm fura-2-AM, 2% FBS, and 0.02% Pluronic F-127. ci-IP3/PM is a photolabile caged IP3 related in structure to cm-IP3/PM (Li et al. 1998). It was synthesized in nine steps from myo-inositol in the laboratory of W-H Li, using a procedure similar to that used for making cm-IP3/PM (W-H Li, manuscript in preparation). In ci-IP3/PM, 2- and 3-hydroxyl groups of myo-inositol are protected by an isopropylidene group; whereas in cm-IP3/PM, 2- and 3-hydroxyl groups are protected by a methoxymethylene group. Like cm-IP3/PM, ci-IP3/PM is cell permeable, and once inside cells, induces Ca2+ release from intracellular stores upon UV flash photolysis (W-H Li, unpublished results; Wagner et al. 2004). Uncaging of caged compounds was achieved using a flash photolysis system (Model JML-C2, Rapp OptoElectronic, Hamburg, Germany). The excitation light of 80 J intensity and 600 μs duration was filtered using a short-pass filter (cut-off 390 nm), and was delivered to the specimen by way of the microscope's objective (Supplementary Fig. 1). The intensity and duration of the flash were minimized so that no measurable photo-bleaching of fura−2 was observed. The intensity of 340 and 380 nm excitation light for detection of fura−2 was also reduced so as to be so low as to produce no measurable uncaging of caged compounds.
Measurement of endoplasmic reticulum [Ca2+] using YC3.3-er
INS-1 cells were transiently transfected with a plasmid encoding yellow cameleon 3.3-er (YC3.3-er) (Griesbeck et al. 2001) using Lipofectamine 2000 (Invitrogen-Life Tech.). Cells on glass coverslips were placed within a microperfusion chamber mounted on an inverted microscope (TE-2000, Nikon) and were visualized with a 40× objective (Hara et al. 2004). The excitation light (440 nm) was attenuated 50–90% using neutral density filters. Changes in fluorescence emission intensities at 535 nm (citrine; FRET acceptor) and 485 nm (enhanced cyan fluorescent protein, ECFP; FRET donor) were monitored using emission filters mounted on a computer-controlled filter wheel (Lambda 10–2 Optical Filter Changer, Sutter Instruments, Novato, CA, USA). Images (50–100 ms exposure time) were captured with a 16-bit Cascade 650 digital camera (Photometrics, Tucson, AZ, USA) at 2–10 s intervals, and were analysed using MetaMorph/MetaFluor software (Universal Imaging Corp., Downington, PA, USA). Data were expressed as the background subtracted ratios of the FRET acceptor and FRET donor emission intensities monitored at 535 and 485 nm, respectively. Cells were superfused at 32°C in saline of high pH-buffering capacity consisting of (mm): 138 NaCl, 5.6 KCl, 2.6 CaCl2, 1.2 MgCl2, 25 NaHCO3, 10 Hepes-NaOH (pH 7.40), and 11.1 glucose (Varadi & Rutter, 2004).
Confocal microscopy for measurement of [Ca2+]i
Cells plated on 0.15 mm glass coverslips were incubated for 20 min at room temperature in SES containing 5 μm fluo-4 AM (Molecular Probes). The [Ca2+]i was imaged using a laser scanning confocal microscope (Leica DM IRE2; Leica Microsystems Heidelberg GmbH) equipped with a 63× water immersion objective (NA 1.2). Fluo-4 was excited at 488 nm using an argon laser. The emitted light passed through a 500 nm dichroic filter for detection using a fluo-4 emission filter (Leica TCS SP2; Leica Microsystems Heidelberg GmbH). Images of the x–y optical sections were recorded with a resolution of 512 pixels line−1 at 400 Hz. Raster point size was 0.1 μm, with an overall lateral resolution of 0.16 μm. For each data set, 20 x–y scans of a small cluster of cells were acquired. After the third section was scanned, a test solution was applied via a micropipette. Image analysis was performed using Leica Confocal Software (Version 2.5; Leica Microsystems).
Microinjection of heparin
Low molecular weight heparin (MW 5000 kDa; Calbiochem) was dissolved in buffer containing (mm): 110 KCl, 10 NaCl, 2 MgCl2, 20 Hepes, 5 KH2PO (297 mOsmol, pH 7.2). The heparin was injected from Femtotip II needles into individual β cells using a Transjector 5246 microinjection system (Eppendorf, Hamburg, Germany) mounted on a Nikon TE200 inverted microscope. The injection pressure was 100 hPa, the compensation pressure was 33 hPa, and the duration of injection was 0.2 s. In order to identify β cells injected with heparin, the injection solution also contained fluorescein (1 mg ml−1; Sigma-Aldrich). The injected cells were then loaded with fura 2 AM and caged compounds. Prior to imaging of Ca2+, cells injected with heparin and fluorescein were visualized using an EYFP filter set.
Epac constructs and transfection protocol
Human wild-type Epac1 (GenBank Accession No. AAF103905) (de Rooij et al. 1998) and dominant negative Epac1 (R279E) in pcDNA3.1 were obtained from Dr X. Cheng (Galveston, TX) (Qiao et al. 2002). Mouse wild-type Epac2 (cAMPGEFII; Accession No. AB021132) and dominant negative Epac2 (G114E, G422D) in pSRα were provided by Dr S. Seino (Kobe, Japan) (Ozaki et al. 2000; Kashima et al. 2001). Epac1 and Epac2 cDNAs were subcloned into pCMV2-FLAG (Sigma-Aldrich, USA) to insert the FLAG epitope at the exchange factor's N-terminus. Epac constructs were introduced into INS-1 cells using LipofectAMINE Plus (Invitrogen-Life Tech.). Cells transfected with wild-type or dominant negative Epac were identified by cotransfection with pEYFP-N1 (Clonetech, Palo Alto, CA, USA). A 1: 4 molar ratio of pEYFP-N1 relative to Epac plasmid was used in all transfections (Kang et al. 2001). EYFP fluorescence was monitored in fura-2-loaded cells 2 days post-transfection, using 513 nm excitation and 527 nm emission filters. Once an EYFP-positive cell was identified as having been transfected, the filter set was manually switched to a fura-2 filter set, allowing ratiometric determinations of [Ca2+]i. Control experiments demonstrated that less than 1% crossover existed between fura-2 and EYFP when using filter sets selective for each reporter (see Supplementary Figs 2–4).
Detection of recombinant Epac immunoreactivity
Whole cell lysates of transfected INS-1 cells expressing recombinant Epac were dissolved in 1× Laemmli's sample buffer, boiled for 5 min, centrifuged to remove unsolubilized material, and resolved by SDS-PAGE using 4% stacking and 12% resolving gels. The resolved Epac proteins were transferred to Immobilon-P PVDF membrane (Millipore, Bedford, MA, USA) by electrophoretic transfer (120 V, 1 h). Western immunoblot analyses were performed using mouse anti-FLAG monoclonal primary antiserum (Sigma-Aldrich; 1: 1000 dilution) in combination with goat antimouse polyclonal secondary antiserum (1: 5000 dilution) conjugated to horseradish peroxidase (Sigma-Aldrich).
CRE-Luc reporter assay
Luciferase activity was measured in lysates of INS-1 cells transfected with CRE-Luc (Stratagene, La Jolla, CA, USA) as described (Chepurny et al. 2002,Chepurny & Holz, 2002). This construct allows expression of luciferase to be regulated by cAMP response elements (CREs) located within a minimal promoter. Monolayers of INS–1 cells were exposed to test substances for 4 h, lysed and assayed for luciferase-catalysed photoemissions using a luciferase assay kit (Promega, Madison, WI, USA) and a luminometer allowing automated application of solutions containing ATP and luciferin (Model TR-717, Perkin Elmer Applied Biosystems, Foster City, CA, USA). Experiments were carried out in triplicate. Statistical analyses were performed using an ANOVA test combined with Fisher's PLSD test.
Sources of reagents and application of test substances
Exendin-4, forskolin, caffeine, ryanodine, and thapsigargin were from Sigma-Aldrich. H-89 was from Calbiochem (San Diego, CA, USA). 8-pCPT-cAMP, 6-Bnz-cAMP, and 8-pCPT-2′-O-Me-cAMP were from BioLog Life Science (Bremen, Germany). Test solutions dissolved in SES were added to the bath solution or were applied to individual cells from glass ‘puffer’ micropipettes (type 1B150-6; World Precision Institute Inc., Sarasota, FL, USA) using a pressure ejection system (PicoSpritzer II, General Valve Corp., NJ, USA) as described (Holz et al. 1993).
Statistical analyses of CICR
Population studies were performed at the single-cell level in order to determine the percentage of cells exhibiting CICR under conditions in which cells were treated with pharmacological agents added directly to the bath and ‘puffer’ pipette solutions. A coverslip with adherent cells served as a control, while a ‘sister’ culture served as the test. At least 10 cells (coverslip)−1 were selected one at a time in random order to determine basal [Ca2+]i and CICR amplitude. CICR in INS-1 cells was defined as a transient increase of [Ca2+]i, the duration of which did not exceed 30 s when measured at the 10% amplitude cut-off (Fig. 1C). An additional requirement was that the increase of [Ca2+]i for INS-1 cells must have exceeded 200 nm when measured at the 50% amplitude cut-off (Fig. 1C). Because CICR in mouse β cells tended to be of smaller amplitude, these criteria were modified so that the increase of [Ca2+]i must have exceeded 125 nm when measured at the 50% amplitude cut-off. Each experiment was performed in triplicate, and statistical analyses were performed using the ANOVA test combined with Fisher's PLSD test.
Results
Sensitization of CICR by exendin-4 and forskolin
To assess the properties of CICR in INS-1 cells, we performed UV flash photolysis to uncage Ca2+ from NP-EGTA, a photolabile Ca2+ chelator. Prior to the loading of cells with NP-EGTA, the resting [Ca2+]i was 135 ± 65 nm (mean ±s.d.; n= 20 cells). After loading with NP-EGTA, there was a statistically significant reduction of [Ca2+]i to 91 ± 5 nm (P < 0.001; n= 20 cells). A 600 μsec flash of UV light produced a small increase of [Ca2+]i in these cells loaded with NP-EGTA (122 ± 26 nm increase; n= 30 cells) (Fig. 1A). This increase of [Ca2+]i recovered to its original baseline and was fully repeatable (Fig. 1A). No such increase of [Ca2+]i was measured in cells not loaded with NP-EGTA (n= 20 cells; data not shown). It may be concluded that the increase of [Ca2+]i depicted in Fig. 1A resulted from the uncaging of Ca2+.
When the GLP-1-R agonist exendin-4 (Ex-4, 1 nm) was applied to single INS-1 cells loaded with NP-EGTA, no increase of [Ca2+]i was measured (Fig. 1B). In contrast, a large and transient increase of [Ca2+]i (CICR) was observed when Ex-4 was applied simultaneous with the uncaging of Ca2+ (Fig. 1B). As summarized in Table 1A, this sensitizing action of Ex-4 to promote CICR was observed in 17 of 24 cells tested. The mean amplitude of the Ca2+ spike measured under these conditions was 1110 ± 176 nm, and the mean duration measured at the 50% amplitude cut-off was 5.1 ± 1.3 s (n= 15 cells). Sensitization of CICR was also observed when UV flash photolysis was performed in the presence of cAMP-elevating agent forskolin (2 μm) (Fig. 1D). This action of forskolin was repeatable and was observed in 23 of 30 cells tested.
Table 1.
Ryanodine sensitivity of CICR
| Treatment | % of cells (n) exhibiting CICR (no ryanodine) | Mean ±s.d. increase of [Ca2+]iwith UV flash (nm) | % of cells (n) exhibiting CICR (with ryanodine) |
|---|---|---|---|
| A INS-1 cells | |||
| Ex-4 | 71 (24) | 1119 ± 176 | 22 (18)* |
| 8-pCPT-cAMP | 69 (13) | 1046 ± 156 | 24 (17)* |
| Caffeine | 74 (27) | 1252 ± 216 | 18 (17)* |
| B Mouse β cells | |||
| Forskolin | 73 (11) | 422 ± 63 | 33 (12)* |
| Caffeine | 67 (12) | 487 ± 79 | 27 (11)* |
Values of n shown in parentheses. The concentrations of test substances were: Ex-4 (1 nm), 8-pCPT-cAMP (100 μm), caffeine (1 mm), and forskolin (2 μm). Each test substance was applied for 30 s to individual cells while performing the uncaging of Ca2+. The mean increase of [Ca2+]i refers to cells exhibiting CICR. Ryanodine (10 μm) was added directly to the bath solution and cells were allowed to equilibrate with ryanodine for 10–15 min at 34°C. *The percentage of cells exhibiting CICR in the presence of ryanodine was significantly different from that in the absence of ryanodine (P < 0.001).
Sensitization of CICR by cAMP analogues
cAMP and Ca2+ may interact to generate CICR in INS-1 cells. To evaluate this possibility, we examined whether cell-permeant cAMP analogues sensitize the CICR mechanism of INS-1 cells in a manner analogous to that described for Ex-4 and forskolin. When NP-EGTA-loaded INS-1 cells were exposed to 8-pCPT-cAMP (100 μm), an activator of both PKA and Epac, the uncaging of Ca2+-triggered CICR (Fig. 2A, Table 1A). This action of 8-pCPT-cAMP was reduced but not blocked by treatment with H-89 (10 μm), a PKA inhibitor (Fig. 2A; inset). CICR was also observed when cells were exposed to 8-pCPT-2′-O-Me-cAMP (100 μm), a cAMP analogue active at Epac only (Fig. 2B), or 6-Bnz-cAMP (100 μm), an analogue active at PKA only (Fig. 2C). As predicted, 8-pCPT-2′-O-Me-cAMP remained effective in cells treated with H-89, whereas the action of 6-Bnz-cAMP was nearly abrogated (cf. Fig. 2B and C; insets). Given that INS-1 cells express mRNA corresponding to Epac1 and Epac2 (Leech et al. 2000), such findings are expected if the sensitizing action of cAMP is mediated not only by PKA but also by Epac. To confirm the efficacy of H-89 as an inhibitor of PKA, the activity of a PKA-regulated luciferase reporter (CRE-Luc) was assessed in transfected INS-1 cells. Under conditions of 8-pCPT-cAMP treatment, both the basal and stimulated activities of CRE-Luc were inhibited by H-89 (Fig. 2D).
Figure 2.

Sensitization of CICR by cAMP analogues The uncaging of Ca2+ produced an increase of [Ca2+]i in INS-1 cells treated with 8-pCPT-cAMP (A), 8-pCPT-2′-O-Me-cAMP (B), or 6-Bnz-cAMP (C). Horizontal bars indicate the time at which 100 μm of each cAMP analogue was administered. Insets of A–C illustrate the percentage of cells (n= 20 cells per cAMP analogue) exhibiting CICR under control conditions or under conditions in which cells were treated with the H-89 (10 μm). *P < 0.01 compared to control. D, the activity of CRE-Luc in INS-1 cells was stimulated by 8-pCPT-cAMP and the action of 8-pCPT-cAMP was inhibited by H-89. Luciferase activity is expressed as relative light units (RLUs).
Exendin-4 exerts its action via PKA and Epac
We next sought to ascertain if it is PKA or Epac that mediates the cAMP-dependent mobilization of Ca2+ by Ex-4. To this end, the action of Ex-4 was evaluated after treatment of INS-1 cells with H-89 or after transfection with dominant negative isoforms of Epac that do not bind cAMP (Holz, 2004a). Population studies demonstrated that H-89 exerted partial inhibitory effects when evaluating its ability to suppress CICR. Whereas a low concentration of H-89 (1 μm) was without effect, a higher concentration (10 μm) reduced the percentage of cells responding to Ex-4 by 60% (Fig. 3A). The ineffectiveness of 1 μm H-89 in this assay of CICR is notable, because this concentration of H-89 reduced CRE-Luc activity by 33% (Fig. 2D). Therefore, there may exist a PKA-independent signalling pathway by which Ex-4 exerts its sensitizing action. This is likely to be the case because the action of Ex-4 was inhibited by overexpression (see immunoblot, Fig. 3B) of dominant negative (DN) FLAG epitope-tagged Epac1 (Fig. 3C) or Epac2 (Fig. 3D). These DN Epacs incorporate inactivating amino acid substitutions within their cAMP-binding domains (Holz, 2004a). Importantly, overexpression of wild type (WT) FLAG-Epac1 or FLAG-Epac2 failed to confer such an inhibitory effect (Figs 3C and D).
Figure 3.

The action of Ex-4 is mediated by PKA and Epac A, the percentage of INS-1 cells exhibiting CICR was determined under conditions of UV flash photolysis in which cells were treated with Ex-4 (1 nm) in the absence or presence of H-89. B, western blot analyses confirmed that wild-type (WT) and dominant negative (DN) FLAG-Epac1 and FLAG-Epac2 were expressed in transiently transfected INS-1 cells. C and D, transient transfection of INS-1 cells with DN FLAG-Epac1 or FLAG-Epac2 reduced the percentage of cells exhibiting CICR under conditions of UV flash photolysis in which cells were treated with Ex-4 (1 nm). No such inhibitory effect was observed after transfection of cells with empty vector (EV) or wild-type (WT) FLAG-Epac constructs. For each histogram bar a minimum of 20 cells was tested. *P < 0.005).
Ryanodine and thapsigargin-sensitive Ca2+ stores contribute to CICR
A 10–15 min pretreatment of INS-1 cells with ryanodine (10 μm) reduced the percentage of INS-1 cells exhibiting CICR under conditions of UV flash photolysis. This was the case when the uncaging of Ca2+ was performed in the presence of Ex-4 (1 nm) or 8-pCPT-cAMP (100 μm) (Fig. 4A and B; Table 1A). Caffeine (1 mm) mimicked the action of Ex-4 (Fig. 4C), and the action of caffeine was also inhibited by ryanodine (Fig. 4D; Table 1A). To validate that an intracellular source of Ca2+ was mobilized as a consequence of CICR, the actions of caffeine and forskolin were examined under conditions in which the SES was nominally Ca2+ free. Under such conditions, caffeine (1 mm) and forskolin (2 μm) allowed for the appearance of CICR (Fig. 5A and B). The source of Ca2+ mobilized by caffeine and forskolin included thapsigargin-sensitive Ca2+ stores, because the actions of both agents were abrogated by pretreatment with thapsigargin (1 μm) (Fig. 5C and D).
Figure 4.

Exendin-4 and caffeine mobilize Ca2+ from ryanodine-sensitive Ca2+ stores. A and B, the uncaging of Ca2+-stimulated CICR under conditions in which INS-1 cells were treated with Ex-4 (1 nm) in the absence (A) but not the presence (B) of ryanodine. C and D, sensitization of CICR by caffeine (1 mm, Caff.) was blocked by treatment of INS-1 cells with ryanodine. In B and D, ryanodine (10 μm) was added to the bath solution for 10–20 min prior to the uncaging of Ca2+. Each panel illustrates representative findings obtained for a minimum of 10 cells studied.
Figure 5.
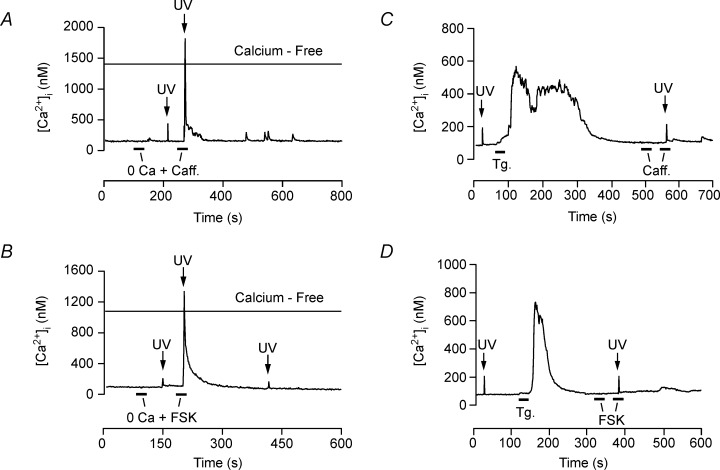
Extracellular Ca2+ independence and thapsigargin sensitivity of CICR. A and B, treatment of INS-1 cells with caffeine (1 mm, Caff.) or forskolin (2 μm; FSK) allowed for the appearance of CICR under conditions in which cells were bathed in a nominally Ca2+-free solution (0 Ca). C and D, treatment of INS-1 cells with thapsigargin (1 μm, Tg.) stimulated an increase of [Ca2+]i and abolished CICR measured under conditions in which cells were exposed to caffeine or forskolin. Each panel illustrates representative findings obtained for a minimum of 10 cells studied.
Caffeine mobilizes endoplasmic reticulum Ca2+
A high concentration of caffeine (10 mm) produced a transient increase of [Ca2+]i in INS-1 cells not loaded with NP-EGTA. This action of caffeine was fast in onset and recovered to baseline within 10–20 s (Fig. 6A). The increase of [Ca2+]i was measured in the cytoplasm and also the nucleus (Fig. 6B). The source of Ca2+ mobilized included the endoplasmic reticulum (ER), as demonstrated using INS-1 cells expressing a cameleon Ca2+ reporter (YC3.3-er) targeted to the ER. When 10 mm caffeine was administered to these cells, a transient decrease of 535/485 nm emission ratio was detected (Fig. 6C). This signifies a decrease of ER calcium concentration ([Ca2+]ER) (Griesbeck et al. 2001). The [Ca2+]ER of INS-1 cells was also lowered when these cells were exposed to thapsigargin (Fig. 6D).
Figure 6.

Mobilization of endoplasmic reticulum Ca2+. A, a high concentration of caffeine (10 mm) stimulated an increase of [Ca2+]i in an INS-1 cell not loaded with NP-EGTA. B, this action of caffeine appeared as an increase of [Ca2+]i in the cytosol and nucleus. The photomontage illustrates the time course of the response to caffeine (10 mm) applied at time = 0 s. The gain on the imaging system's detector was increased to allow measurement of cytosolic fluorescence (orange) under conditions in which an apparently large increase of nuclear Ca2+ concentration saturated the detector (blue). Scale bar, 10 μm. C, FRET-based detection of YC3.3er in INS-1 cells demonstrated that caffeine (10 mm) reduced ER Ca2+ concentration. D, a decrease of ER Ca2+ concentration was also observed during treatment of an INS-1 cell with thapsigargin (5 μm).
Sensitization of CICR by cAMP-elevating agents in mouse β cells
To examine how cAMP-elevating agents influence CICR in mouse β cells, UV flash photolysis was performed under conditions of NP-EGTA loading identical to that described for INS-1 cells. Prior to loading, the resting [Ca2+]i was 104 ± 29 nm (n= 25 cells). This value decreased to 87 ± 14 nm (n= 22 cells) in NP-EGTA-loaded cells (P < 0.001; n= 20 cells). The uncaging of Ca2+ in β cells generated a small increase of [Ca2+]i but did not initiate CICR (Fig. 7A; 55 ± 11 nm increase; n= 17 cells). In these same cells, application of Ex-4 (1 nm), forskolin (2 μm), or the Epac-selective cAMP analogue 8-pCPT-2′-O-Me-cAMP (100 μm) failed to alter resting [Ca2+]i (Fig. 7A–C). However, all three cAMP-elevating agents allowed for the appearance of CICR under conditions of UV flash photolysis (Fig. 7A–C; Table 1B). 8-pCPT-2′-O-Me-cAMP (100 μm) was highly effective in this assay. It allowed for the appearance of CICR in 8 of 15 cells tested. Similar to findings obtained with INS-1 cells, no sensitization of CICR by forskolin was observed when mouse β cells were treated with thapsigargin (Fig. 7D).
Figure 7.

cAMP-elevating agents sensitize CICR in mouse β cells. A, uncaging of Ca2+ in a mouse β cell produced a small increase of [Ca2+]i in the absence of Ex-4, whereas CICR was observed in the presence of Ex-4 (1 nm). B and C, CICR was also observed when Ca2+ was uncaged in the presence of forskolin (2 μm) or 8-pCPT-2′-O-Me-cAMP (100 μm). D, the action of forskolin was abolished when a mouse β cell was pretreated with thapsigargin (1 μm). Note that thapsigargin, alone, stimulated a fast transient increase of [Ca2+]i (*). Each panel illustrates representative findings obtained for a minimum of 10 cells studied.
Characterization of Ca2+ release channels targeted by cAMP in mouse β cells
To assess whether CICR sensitized by forskolin resulted from activation of RYR, the action of ryanodine was examined. A role for RYR is indicated because pretreatment with ryanodine (10 μm) rendered forskolin less effective in the assay of β cell CICR reported here (Table 1B). Furthermore, caffeine (1 mm), a sensitizer of RYR, allowed for the appearance of CICR under conditions of UV flash photolysis. This action of caffeine was also diminished by ryanodine (Table 1B).
Because ryanodine-resistant CICR was sometimes observed (Table 1B), a sensitizing action of cAMP at the IP3-R may also exist in β cells. Therefore, we assessed the efficacy of an IP3-R inhibitor (low molecular weight heparin, 100 mg ml−1; Ehrlich et al. 1994) administered intracellularly under conditions of NP-EGTA loading and UV flash photolysis. Administration of heparin led to a reduction in the percentage of forskolin-treated β cells exhibiting CICR (Fig. 8A). Moreover, CICR was nearly abolished after combined treatment with heparin and ryanodine, thereby demonstrating that these two inhibitors of Ca2+ release channels acted in an additive manner (Fig. 8B).
Figure 8.

Antagonism of CICR by heparin and ryanodine in mouse β cells. A, antagonism of CICR by intracellularly administered heparin in mouse β cells loaded with NP-EGTA. The uncaging of Ca2+ by UV flash photolysis was performed under conditions in which β cells were exposed to forskolin (2 μm). Control conditions refer to cells injected with a solution containing fluorescein but not heparin (see Methods). B, the sensitization of CICR by forskolin was nearly abolished under conditions in which heparin (Hep.)-injected β cells were also treated with ryanodine (10 μm; Ryan.). Numbers above each histogram bar indicate the fraction of cells exhibiting CICR. **P < 0.01.
To confirm the specificity with which heparin exerted its inhibitory effect, β cells were loaded with a cell-permeant caged IP3 (ci-IP3/PM). The uncaging of IP3 generated a small increase of [Ca2+]i under conditions of low-intensity UV flash photolysis, but it did not trigger CICR (Fig. 9A). When β cells were exposed to forskolin (2 μm), the small increase of [Ca2+]i was converted into a large Ca2+ spike (Fig. 9A). This CICR sensitized by forskolin required the activity of IP3 receptors, because it was abolished after administration of heparin (Fig. 9B). It may be concluded that in β cells, IP3-R-mediated CICR complements RYR-mediated CICR, and that both processes are cAMP regulated.
Figure 9.

IP3-R-mediated CICR sensitized by forskolin in mouse β cells. A, low-intensity UV flash photolysis produced a small increase of [Ca2+]i (61 ± 17 nm; n= 15) in a mouse β cell loaded with membrane-permeant caged IP3 (ci-IP3/PM). When this cell was treated with forskolin (2 μm; FSK), the uncaging of IP3 generated a large Ca2+ spike (528 ± 96 nm; n= 14). B, sensitization of IP3-R-mediated CICR by forskolin was inhibited by intracellularly administered heparin. The uncaging of IP3 in forskolin-treated β cells generated CICR in 78% of cell tested, whereas this percentage was reduced to 17% under conditions of intracellular heparin administration. Numbers above each histogram bar indicate the fraction of cells exhibiting CICR. **P < 0.01.
Discussion
Physiological significance of CICR in β cells
Prior studies demonstrate that cAMP-elevating agents mobilize Ca2+ in β cells and insulin-secreting cell lines (Islam et al. 1998; Holz et al. 1999; Kang et al. 2001, 2003; Kang & Holz, 2003; Dyachok & Gylfe, 2004; Dyachok et al. 2004). Ca2+ mobilized in this manner acts as a direct stimulus for exocytosis of insulin (Kang & Holz, 2003; Kang et al. 2003; Dyachok & Gylfe, 2004; reviewed by Holz, 2004b). Thus, exocytosis in β cells is not simply dependent on influx of Ca2+ through VDCCs, but is also dependent on the interaction of Ca2+ and cAMP to promote CICR. In this regard, new studies provide evidence for the existence of a highly Ca2+-sensitive pool (HCSP) of secretory granules in β cells (Wan et al. 2004; Yang & Gillis, 2004). These granules are released under conditions in which the [Ca2+]i increases to low micromolar levels. Since the mechanism of cAMP-dependent Ca2+ mobilization reported here generates an increase of [Ca2+]i in the 0.5–2 μm range, it may serve as an adequate stimulus for exocytosis of the HCSP.
Second messenger coincidence detection in β cells
Here we describe an experimental strategy by which the existence of cAMP-regulated CICR in β cells may be validated. The strategy relies on the use of caged Ca2+ and UV flash photolysis to produce a small increase of [Ca2+]i. Under these conditions, the uncaging of Ca2+ triggers CICR when Ca2+ release channels are sensitized by cAMP-elevating agents. Of particular note is our demonstration that the uncaging of Ca2+ fails to generate CICR in the absence of cAMP-elevating agents. This key observation leads us to conclude that a simultaneous increase of intracellular Ca2+ and cAMP concentrations is a necessary prerequisite for the initiation of CICR in β cells. Fundamentally, this is a process of second messenger coincidence detection, and it results from dual stimulatory actions of Ca2+ and cAMP at Ca2+ release channels. These Ca2+ release channels correspond to RYR and the IP3-R, as demonstrated by antagonism of CICR following application of ryanodine or heparin.
IP3-R-mediated Ca2+ mobilization is also demonstrated through our use of ci-IP3/PM, a membrane-permeant caged IP3 that activates the IP3-R in a selective manner. Under conditions in which β cells are treated with forskolin, the uncaging of IP3 stimulates CICR. This action of forskolin might reflect cAMP-dependent sensitization of the IP3-R. Alternatively, cAMP may sensitize RYR to stimulatory effects of Ca2+ released from activated IP3 receptors. If so, heparin would act at the IP3-R to prevent the small rise of [Ca2+]i that acts as an initiator of CICR mediated by RYR. Indeed, the mobilization of Ca2+ by IP3 in pancreatic acinar cells is reported to trigger CICR mediated by ryanodine receptors (Straub et al. 2000; Ashby et al. 2002). Thus, it will be of interest to ascertain whether there also exist functional interactions between IP3 receptors and RYR in β cells.
Coincidence detection may explain context specificity
Prior studies of β cells demonstrate that CICR occurs in a context-specific manner (Lemmens et al. 2001; Bruton et al. 2003). CICR is only observed when β cells are equilibrated in elevated concentrations of extracellular glucose. Metabolism of glucose by β cells promotes the filling of ER Ca2+ stores (Maechler et al. 1999; Tengholm et al. 1999), and it also produces an increase of [Ca2+]i (Henquin, 2000). Therefore, under conditions in which β cells are exposed to glucose, two necessary conditions are met. ER Ca2+ stores are full and the cytosolic Ca2+ concentration is elevated sufficiently to allow CICR to be generated when Ca2+ release channels are sensitized by cAMP. Context specificity of this type is likely to be of physiological significance, because it may explain, at least in part, why the insulin secretagogue action of GLP-1 is strictly dependent on exposure of β cells to glucose (Kieffer & Habener, 1999).
RYR as a determinant of β cell function
Because insulin-secreting cells express RYR, albeit at low levels (Takasawa et al. 1998; Gamberucci et al. 1999; Holz et al. 1999; Islam, 2002; Lee et al. 2002; Mitchell et al. 2003; Beauvois et al. 2004; Johnson et al. 2004a,b), it is not surprising that caffeine, a sensitizer of RYR, recapitulates the Ca2+-mobilizing action of Ex-4 reported here. This action of caffeine is inhibited by ryanodine, thereby confirming the existence of CICR mediated by RYR. Such findings are consistent with prior studies demonstrating ryanodine and caffeine-sensitive mobilization of Ca2+ in β cells or β cell lines (Islam et al. 1992, 1998; Gromada et al. 1995; Gamberucci et al. 1999; Maechler et al. 1999; Holz et al. 1999; Kang et al. 2001, 2003; Kang & Holz, 2003; Mitchell et al. 2003; Sasaki et al. 2002; Varadi & Rutter, 2002; Bruton et al. 2003; Tsuboi et al. 2003; but see Dyachok & Gylfe, 2004). The source of Ca2+ mobilized may include the ER because we find that caffeine and forskolin fail to promote CICR in thapsigargin-treated cells. Moreover, caffeine releases ER Ca2+, as measured in INS-1 cells expressing YC3.3-er.
RYR is implicated in the regulation of [Ca2+]i, exocytosis, apoptosis, and endosome function in β cells (Islam et al. 1992, 1998; Takasawa et al. 1998; Holz et al. 1999; Lemmens et al. 2001; Quesada et al. 2002; Bruton et al. 2003; Kang & Holz, 2003; Johnson et al. 2004a,b). Because not all studies support these contentions (Tengholm et al. 1998, 1999, 2000), there may exist differences in the levels of expression of RYR when comparing strains of mice or when making comparisons across species lines. The failure of prior studies to detect RYR in β cells might also be explained by the use of an experimental strategy that relies on treatment of cells with diazoxide and verapamil (Dyachok & Gylfe, 2004; Dyachok et al. 2004). These agents disrupt CICR by virtue of their ability to inhibit Ca2+ influx (Meissner, 2002). In contrast, human β cells not treated with these agents exhibit an increase of [Ca2+]i in response to a low concentration of ryanodine, thereby demonstrating the presence of RYR in this cell type (Johnson et al. 2004a). Similarly, human β cells not treated with diazoxide or verapamil exhibit an increase of [Ca2+]i in response to GLP-1, an action inhibited by a high concentration of ryanodine (Holz et al. 1999).
New studies demonstrate the presence of immunologically detectable RYR in human and mouse β cells (Johnson et al. 2004a,b). Although the molecular nature of β cell RYR remains to be determined, it may exhibit features somewhat different from the isoforms described to date. Indeed, splice variants of RYR1 and RYR2 mRNA are expressed in islets (Lee et al. 2002; Okamoto et al. 2004). Sequence variations of this type may explain why some have found it difficult or impossible to detect RYR mRNA by RT-PCR of purified preparations of mouse β cells (Beauvois et al. 2004).
Potential IP3-R-mediated signalling properties of the GLP-1-R
Actions of GLP-1 mediated by the IP3-R are also of interest. Although activation of the GLP-1-R fails to stimulate IP3 production in islets or INS-1 cells (Fridolf & Ahren, 1991; Zawalich et al. 1993; Kang et al. 2003), cAMP can, under certain conditions, facilitate IP3-R function. This action of cAMP is PKA mediated (Bruce et al. 2003), and is reported to be operational in mouse β cells (Liu et al. 1996; Dyachok et al. 2004). Mobilization of Ca2+ from IP3-R-regulated Ca2+ stores may explain, at least in part, how GLP-1 stimulates exocytosis in β cells (Dyachok et al. 2004). It may also play some role in the stimulation of β cell mitochondrial ATP production (Tsuboi et al. 2003) and mitogen-activated protein (MAP) kinase signalling (Arnette et al. 2003). Therefore, it is noteworthy that we demonstrate IP3-R-mediated CICR sensitized by a cAMP-elevating agent and which is blocked by treatment of β cells with heparin, an inhibitor of IP3-R function.
It is also important to note that Ca2+ exerts stimulatory effects on IP3 production in insulin-secreting cells (Roe et al. 1993; Gromada et al. 1996). This action of Ca2+ is most likely a consequence of its ability to activate phospholipase C (Berridge et al. 2003). If such an action of Ca2+ were to exist in β cells loaded with NP-EGTA, the uncaging of Ca2+ might raise levels of IP3, thereby stimulating the IP3-R. CICR triggered in this manner might be facilitated under conditions in which the IP3-R is sensitized by cAMP-elevating agents. Because evidence also exists for ‘atypical’ mechanisms of Ca2+ release in the β cell (Johnson & Misler, 2002; Masgrau et al. 2003; Mitchell et al. 2003; Beauvois et al. 2004), actions of cAMP-elevating agents described here may not be restricted to RYR or the IP3-R.
Epac-mediated sensitization of CICR
Prior studies of multiple cell types demonstrate that cAMP acts via PKA to sensitize RYR and the IP3-R (Marx et al. 2000; Bruce et al. 2003). Here we report that the action of cAMP may also be Epac-mediated. A cAMP analogue selective for Epac sensitizes the CICR mechanism of β cells, whereas transfection of INS-1 cells with dominant negative Epac diminishes CICR. Although prior studies implicate Epac2 in this process (Kang et al. 2001, 2003; Tsuboi et al. 2003), we are the first to demonstrate that Epac1 may also be a contributing factor. One established downstream effector of Epac is the small-molecular weight GTPase Rap. Although the signalling properties of Rap are not fully understood, evidence exists that it promotes protein kinase-mediated phosphorylation independently of PKA (Holz, 2004a). Therefore, cAMP-dependent sensitization of intracellular Ca2+ release channels might require formation of an activated Epac/Rap signalling complex. Such an action of Epac would complement its ability to interact with secretory granule-associated proteins (Kashima et al. 2001; Shibasaki et al. 2004) and to regulate fast Ca2+-dependent exocytosis in the β cell (Eliasson et al. 2003).
Conclusion
Findings presented here suggest that RYR and IP3-R Ca2+ release channels mediate the Ca2+ mobilizing action of GLP-1 in β cells. Both types of Ca2+ release channels act as cAMP and Ca2+ coincidence detectors, because their opening is facilitated by a simultaneous increase of intracellular cAMP and Ca2+ concentrations. PKA and Epac-mediated sensitization of CICR demonstrated here may provide a new explanation for how GLP-1 interacts with β cell glucose metabolism to stimulate insulin secretion.
Acknowledgments
The authors thank Dr Roger Tsien (HHMI/UCSD, San Diego, CA) for providing the YC3.3-er plasmid. G.G.H. acknowledges the support of the NIH (R01-DK45817) and the American Diabetes Association (Research Grant Award). W.-h. Li was supported by a research grant (I-1510) from the Welch Foundation and a Career Development Award from the American Diabetes Association. Special thanks to Dr C.A. Leech for critical reading of the manuscript and Dr Cheng Wang for technical assistance with mouse islet isolations.
Supplemental material
The online version of this paper can be accessed at: DOI: 10.1113/jphysiol.2005.087510
http://jp.physoc.org/cgi/content/full/jphysiol.2005.087510/DC1 and contains supplemental material consisting of four figures.
This material can also be found as part of the full-text HTML version available from http://www.blackwell-synergy.com
References
- Arnette D, Gibson TB, Lawrence MC, January B, Khoo S, McGlynn K, Vanderbilt CA, Cobb MH. Regulation of ERK1 and ERK2 by glucose and peptide hormones in pancreatic β cells. J Biol Chem. 2003;278:32517–32525. doi: 10.1074/jbc.M301174200. [DOI] [PubMed] [Google Scholar]
- Asfari M, Janjic D, Meda P, Li G, Halban PA, Wollheim CB. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology. 1992;130:167–178. doi: 10.1210/endo.130.1.1370150. [DOI] [PubMed] [Google Scholar]
- Ashby MC, Craske M, Park MK, Gerasimenko OV, Burgoyne RD, Petersen OH, Tepikin AV. Localized Ca2+ uncaging reveals polarized distribution of Ca2+-sensitive Ca2+ release sites: mechanism of unidirectional Ca2+ waves. J Cell Biol. 2002;158:283–292. doi: 10.1083/jcb.200112025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauvois MC, Arredouani A, Jonas JC, Rolland JF, Schuit F, Henquin JC, Gilon P. Atypical Ca2+-induced Ca2+ release from a SERCA3-dependent Ca2+ pool of the endoplasmic reticulum in mouse pancreatic β cells. J Physiol. 2004;559:141–156. doi: 10.1113/jphysiol.2004.067454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Bode HP, Moormann B, Dabew R, Goke B. Glucagon-like peptide-1 elevates cytosolic calcium in pancreatic β cells independently of protein kinase A. Endocrinology. 1999;140:3919–3927. doi: 10.1210/endo.140.9.6947. [DOI] [PubMed] [Google Scholar]
- Bruce JI, Straub SV, Yule DI. Crosstalk between cAMP and Ca2+ signaling in non-excitable cells. Cell Calcium. 2003;34:431–444. doi: 10.1016/s0143-4160(03)00150-7. [DOI] [PubMed] [Google Scholar]
- Bruton JD, Lemmens R, Shi CL, Persson-Sjogren S, Westerblad H, Ahmed M, Pyne NJ, Frame M, Furman BL, Islam MS. Ryanodine receptors of pancreatic β cells mediate a distinct context-dependent signal for insulin secretion. FASEB J. 2003;17:301–303. doi: 10.1096/fj.02-0481fje. [DOI] [PubMed] [Google Scholar]
- Chepurny OG, Holz GG. Over-expression of the glucagon-like peptide-1 receptor on INS-1 cells confers autocrine stimulation of insulin gene promoter activity: a strategy for production of pancreatic β-cell lines for use in transplantation. Cell Tissue Res. 2002;307:191–201. doi: 10.1007/s00441-001-0494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chepurny OG, Hussain MA, Holz GG. Exendin-4 as a stimulator of rat insulin I gene promoter activity via bZIP/CRE interactions sensitive to serine/threonine protein kinase inhibitor Ro 31–8220. Endocrinology. 2002;143:2303–2313. doi: 10.1210/endo.143.6.8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmeire D, Flamez D, Hinke SA, Cali JJ, Pipeleers D, Schuit F. Type VIII adenylyl cyclase in rat β cells: coincidence signal detector/generator for glucose and GLP-1. Diabetologia. 2003;46:1383–1393. doi: 10.1007/s00125-003-1203-8. [DOI] [PubMed] [Google Scholar]
- De Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- Dyachok O, Gylfe E. Ca2+-induced Ca2+ release via inositol 1,4,5-trisphosphate receptors is amplified by protein kinase A. J Biol Chem. 2004;279:45455–45461. doi: 10.1074/jbc.M407673200. [DOI] [PubMed] [Google Scholar]
- Dyachok O, Tufveson G, Gylfe E. Ca2+-induced Ca2+ release by activation of inositol 1,4,5-trisphosphate receptors in primary pancreatic β cells. Cell Calcium. 2004;36:1–9. doi: 10.1016/j.ceca.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Ehrlich BE, Kaftan E, Bezprozvannaya S, Bezprozvanny I. The pharmacology of intracellular Ca2+ release channels. Trends Pharmacol Sci. 1994;15:145–149. doi: 10.1016/0165-6147(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Eliasson L, Ma X, Renstrom E, Barg S, Berggren PO, Galvanovskis J, Gromada J, Jing X, Lundquist I, Salehi A, Sewing S, Rorsman P. SUR1 regulates PKA-independent cAMP-induced granule priming in mouse pancreatic β cells. J Gen Physiol. 2003;121:81–197. doi: 10.1085/jgp.20028707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis-Davies GCR, Kaplan JH. Nitrophenyl-EGTA, a photolabile chelator that selectively binds calcium with high affinity and releases it rapidly upon photolysis. Proc Natl Acad Sci U S A. 1994;91:187–191. doi: 10.1073/pnas.91.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridolf T, Ahren B. GLP-1 (7–36) amide stimulates insulin secretion in rat islets: studies on the mode of action. Diabetes Res. 1991;16:185–191. [PubMed] [Google Scholar]
- Gamberucci A, Fulceri R, Pralong W, Banhegyi G, Marcolongo P, Watkins SL, Benedetti A. Caffeine releases a glucose-primed endoplasmic reticulum Ca2+ pool in the insulin-secreting cell line INS-1. FEBS Lett. 1999;446:309–312. doi: 10.1016/s0014-5793(99)00220-3. [DOI] [PubMed] [Google Scholar]
- Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J Biol Chem. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- Gromada J, Bokvist K, Ding WG, Holst JJ, Nielsen JH, Rorsman P. Glucagon-like peptide 1 (7–36) amide stimulates exocytosis in human pancreatic β cells by both proximal and distal regulatory steps in stimulus-secretion coupling. Diabetes. 1998a;47:57–65. doi: 10.2337/diab.47.1.57. [DOI] [PubMed] [Google Scholar]
- Gromada J, Dissing S, Bokvist K, Renstrom E, Frokjaer-Jensen J, Wulff BS, Rorsman P. Glucagon-like peptide-1 increases cytoplasmic Ca2+ in insulin-secreting βTC3 cells by enhancement of intracellular Ca2+ mobilization. Diabetes. 1995;44:767–774. doi: 10.2337/diab.44.7.767. [DOI] [PubMed] [Google Scholar]
- Gromada J, Frokjaer-Jensen J, Dissing S. Glucose stimulates voltage- and calcium-dependent inositol trisphosphate production and intracellular Ca2+ mobilization in insulin-secreting βTC3 cells. Biochem J. 1996;314:339–345. doi: 10.1042/bj3140339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromada J, Holst JJ, Rorsman P. Cellular regulation of islet hormone secretion by the incretin hormone glucagon-like peptide-1. Pflügers Arch. 1998b;435:583–594. doi: 10.1007/s004240050558. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hara M, Bindokas V, Lopez JP, Kaihara K, Landa LR, Harbeck M, Roe MW. Imaging endoplasmic reticulum calcium with a fluorescent biosensor in transgenic mice. Am J Physiol Cell Physiol. 2004;287:C932–C938. doi: 10.1152/ajpcell.00151.2004. [DOI] [PubMed] [Google Scholar]
- Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- Holz GG. Epac–a new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic β-cell. Diabetes. 2004a;53:5–13. doi: 10.2337/diabetes.53.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GG. New insights concerning the glucose-dependent insulin secretagogue action of glucagon-like peptide-1 in pancreatic β cells. Horm Metabolic Res. 2004b;36:787–794. doi: 10.1055/s-2004-826165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GG, Chepurny OG. Glucagon-like peptide-1 synthetic analogs: New therapeutic agents for use in the treatment of diabetes mellitus. Current Med Chem. 2003;10:2471–2483. doi: 10.2174/0929867033456648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GG, Habener JF. Signal transduction crosstalk in the endocrine system: pancreatic β cells and the glucose competence concept. Trends Biochem Sci. 1992;17:388–393. doi: 10.1016/0968-0004(92)90006-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GG, Kuhtreiber WM, Habener JF. Pancreatic β cells are rendered glucose competent by the insulinotropic hormone glucagon-like peptide-1-(7–37) Nature. 1993;361:362–365. doi: 10.1038/361362a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GG, Leech CA, Habener JF. Activation of a cAMP-regulated Ca2+-signaling pathway in pancreatic β cells by the insulinotropic hormone glucagon-like peptide-1. J Biol Chem. 1995;270:17749–17757. [PMC free article] [PubMed] [Google Scholar]
- Holz GG, Leech CA, Heller RS, Castonguay M, Habener JF. cAMP-dependent mobilization of intracellular Ca2+ stores by activation of ryanodine receptors in pancreatic β cells. A Ca2+ signaling system stimulated by the insulinotropic hormone glucagon-like peptide-1-(7–37) J Biol Chem. 1999;274:14147–14156. doi: 10.1074/jbc.274.20.14147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MS. The ryanodine receptor calcium channel of β cells: molecular regulation and physiological significance. Diabetes. 2002;51:1299–1309. doi: 10.2337/diabetes.51.5.1299. [DOI] [PubMed] [Google Scholar]
- Islam MS, Leibiger I, Leibiger B, Rossi D, Sorrentino V, Ekstrom TJ, Westerblad H, Andrade FH, Berggren PO. In situ activation of the type 2 ryanodine receptor in pancreatic β cells requires cAMP-dependent phosphorylation. Proc Natl Acad Sci U S A. 1998;95:6145–6150. doi: 10.1073/pnas.95.11.6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MS, Rorsman P, Berggren PO. Ca2+-induced Ca2+ release in insulin-secreting cells. FEBS Lett. 1992;296:287–291. doi: 10.1016/0014-5793(92)80306-2. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Han Z, Otani K, Ye H, Zhang Y, Wu H, Horikawa Y, Misler S, Bell GI, Polonsky KS. RyR2 and calpain-10 delineate a novel apoptosis pathway in pancreatic islets. J Biol Chem. 2004b;279:24794–24802. doi: 10.1074/jbc.M401216200. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Kuang S, Misler S, Polonsky KS. Ryanodine receptors in human pancreatic β cells: localization and effects on insulin secretion. FASEB J. 2004a;18:878–880. doi: 10.1096/fj.03-1280fje. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Misler S. Nicotinic acid-adenine dinucleotide phosphate-sensitive calcium stores initiate insulin signaling in human β cells. Proc Natl Acad Sci U S A. 2002;99:14566–14571. doi: 10.1073/pnas.222099799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang G, Chepurny OG, Harbeck M, Roe MW, Holz GG. American Diabetes Association 65th Annual Scientific Sessions. San Diego, CA: 2005. Ca2+ mobilizing properties of the GLP-1 receptor: Role of a cAMP and Ca2+ coincidence detector in support of Ca2+-induced Ca2+ release in pancreatic β cells. [Google Scholar]
- Kang G, Chepurny OG, Holz GG. cAMP-regulated guanine nucleotide exchange factor-II (Epac2) mediates Ca2+- induced Ca2+ release in INS-1 pancreatic β cells. J Physiol. 2001;536:375–385. doi: 10.1111/j.1469-7793.2001.0375c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang G, Holz GG. Amplification of exocytosis by Ca2+-induced Ca2+ release in INS-1 pancreatic β cells. J Physiol. 2003;546:175–189. doi: 10.1113/jphysiol.2002.029959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang G, Joseph JW, Chepurny OG, Monaco M, Wheeler MB, Bos JL, Schwede F, Genieser HG, Holz GG. Epac-selective cAMP analog 8-pCPT-2′-O-Me-cAMP as a stimulus for Ca2+-induced Ca2+ release and exocytosis in pancreatic β cells. J Biol Chem. 2003;278:8279–8285. doi: 10.1074/jbc.M211682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima Y, Miki T, Shibasaki T, Ozaki N, Miyazaki M, Yano H, Seino S. Critical role of cAMP-GEFII – Rim2 complex in incretin-potentiated insulin secretion. J Biol Chem. 2001;276:46046–46053. doi: 10.1074/jbc.M108378200. [DOI] [PubMed] [Google Scholar]
- Kieffer TJ, Habener JF. The glucagon-like peptides. Endocrine Rev. 1999;20:876–913. doi: 10.1210/edrv.20.6.0385. [DOI] [PubMed] [Google Scholar]
- Lee BS, Sessanna S, Laychock SG, Rubin RP. Expression and cellular localization of a modified type 1 ryanodine receptor and L-type channel proteins in non-muscle cells. J Membr Biol. 2002;189:181–190. doi: 10.1007/s00232-002-1012-x. [DOI] [PubMed] [Google Scholar]
- Leech CA, Holz GG, Chepurny O, Habener JF. Expression of cAMP-regulated guanine nucleotide exchange factors in pancreatic β cells. Biochem Biophys Res Comm. 2000;278:44–47. doi: 10.1006/bbrc.2000.3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens R, Larsson O, Berggren PO, Islam MS. Ca2+- induced Ca2+ release from the endoplasmic reticulum amplifies the Ca2+ signal mediated by activation of voltagegated L-type Ca2+ channels in pancreatic β cells. J Biol Chem. 2001;276:9971–9977. doi: 10.1074/jbc.M009463200. [DOI] [PubMed] [Google Scholar]
- Li W, Llopis J, Whitney M, Zlokarnik G, Tsien RY. Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature. 1998;392:936–941. doi: 10.1038/31965. [DOI] [PubMed] [Google Scholar]
- Liu YJ, Grapengiesser E, Gylfe E, Hellman B. Crosstalk between the cAMP and inositol trisphosphate-signalling pathways in pancreatic β cells. Arch Biochem Biophys. 1996;334:295–302. doi: 10.1006/abbi.1996.0458. [DOI] [PubMed] [Google Scholar]
- Maechler P, Kennedy ED, Sebo E, Valeva A, Pozzan T, Wollheim CB. Secretagogues modulate the calcium concentration in the endoplasmic reticulum of insulin-secreting cells. Studies in aequorin-expressing intact and permeabilized INS-1 cells. J Biol Chem. 1999;274:12583–12592. doi: 10.1074/jbc.274.18.12583. [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Masgrau R, Churchill GC, Morgan AJ, Ashcroft SJH, Galione A. NAADP: a new second messenger for glucose-induced Ca2+ responses in clonal pancreatic β cells. Current Biol. 2003;13:247–251. doi: 10.1016/s0960-9822(03)00041-1. [DOI] [PubMed] [Google Scholar]
- Meissner G. Regulation of mammalian ryanodine receptors. Front Biosci. 2002;7:d2072–80. doi: 10.2741/A899. [DOI] [PubMed] [Google Scholar]
- Mitchell KJ, Lai FA, Rutter GA. Ryanodine receptor type I and nicotinic acid adenine dinucleotide phosphate receptors mediate Ca2+ release from insulin-containing vesicles in living pancreatic β cells (MIN6) J Biol Chem. 2003;278:11057–11064. doi: 10.1074/jbc.M210257200. [DOI] [PubMed] [Google Scholar]
- Nakazaki M, Crane A, Hu M, Seghers V, Ullrich S, Aguilar-Bryan L, Bryan J. cAMP-activated protein kinase-independent potentiation of insulin secretion by cAMP is impaired in SUR1 null islets. Diabetes. 2002;51:3440–3449. doi: 10.2337/diabetes.51.12.3440. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Nata K, Noguchi N, Kuroki M, Akiyama T, Ikeda T, Yamauchi A, Takahashi S, Takasawa S. A novel ryanodine receptor expressed in pancreatic islets and its possible roles in the cyclic ADP-ribose-induced intracellular calcium mobilization. Islet Study Post. 2004. EASD Symposium 2004, Prien/Chiemsse, Germany.
- Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, Takai Y, Seino S. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol. 2000;2:805–811. doi: 10.1038/35041046. [DOI] [PubMed] [Google Scholar]
- Pipeleers DG, Schuit FC, In't Veld PA, Maes E, Hooghe-Peters EL, Van de Winkel M, Gepts W. Interplay of nutrients and hormones in the regulation of insulin release. Endocrinology. 1985;117:824–833. doi: 10.1210/endo-117-3-824. [DOI] [PubMed] [Google Scholar]
- Qiao J, Mei FC, Popov VL, Vergara LA, Cheng X. Cell cycle-dependent subcellular localization of exchange factor directly activated by cAMP. J Biol Chem. 2002;277:26581–26586. doi: 10.1074/jbc.M203571200. [DOI] [PubMed] [Google Scholar]
- Quesada I, Rovira JM, Martin F, Roche E, Nadal A, Soria B. Nuclear KATP channels trigger nuclear Ca2+ transients that modulate nuclear function. Proc Natl Acad Sci USA. 2002;99:9544–9549. doi: 10.1073/pnas.142039299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe MW, Lancaster ME, Mertz RJ, Worley JF, 3rd, Dukes ID. Voltage-dependent intracellular calcium release from mouse islets stimulated by glucose. J Biol Chem. 1993;268:9953–9956. [PubMed] [Google Scholar]
- Sasaki S, Nakagaki I, Kondo H, Hori S. Involvement of the ryanodine-sensitive Ca2+ store in GLP-1-induced Ca2+ oscillations in insulin-secreting HIT cells. Pflugers Arch. 2002;445:342–351. doi: 10.1007/s00424-002-0965-z. [DOI] [PubMed] [Google Scholar]
- Schuit FC, Pipeleers DG. Regulation of adenosine 3,5-monophosphate levels in the pancreatic β cell. Endocrinology. 1985;117:834–840. doi: 10.1210/endo-117-3-834. [DOI] [PubMed] [Google Scholar]
- Shibasaki T, Sunaga Y, Fujimoto K, Kashima Y, Seino S. Interaction of ATP sensor, cAMP sensor, Ca2+ sensor, and voltage-dependent Ca2+ channel in insulin granule exocytosis. J Biol Chem. 2004;279:7956–7961. doi: 10.1074/jbc.M309068200. [DOI] [PubMed] [Google Scholar]
- Straub SV, Giovannucci DR, Yule DI. Calcium wave propagation in pancreatic acinar cells: functional interaction of inositol 1,4,5-trisphosphate receptors, ryanodine receptors, and mitochondria. J Gen Physiol. 2000;116:547–560. doi: 10.1085/jgp.116.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasawa S, Akiyama T, Nata K, Kuroki M, Tohgo A, Noguchi N, Kobayashi S, Kato I, Katada T, Okamoto H. Cyclic ADP-ribose and inositol 1,4,5-trisphosphate as alternate second messengers for intracellular Ca2+ mobilization in normal and diabetic β cells. J Biol Chem. 1998;273:2497–2500. doi: 10.1074/jbc.273.5.2497. [DOI] [PubMed] [Google Scholar]
- Tengholm A, Hagman C, Gylfe E, Hellman B. In situ characterization of non-mitochondrial Ca2+ stores in individual pancreatic β cells. Diabetes. 1998;47:1224–1230. doi: 10.2337/diab.47.8.1224. [DOI] [PubMed] [Google Scholar]
- Tengholm A, Hellman B, Gylfe E. Glucose regulation of free Ca2+ in the endoplasmic reticulum of mouse pancreatic β cells. J Biol Chem. 1999;274:36883–36890. doi: 10.1074/jbc.274.52.36883. [DOI] [PubMed] [Google Scholar]
- Tengholm A, Hellman B, Gylfe E. Mobilization of Ca2+ stores in individual pancreatic β cells permeabilized or not with digitonin or α-toxin. Cell Calcium. 2000;27:43–51. doi: 10.1054/ceca.1999.0087. [DOI] [PubMed] [Google Scholar]
- Thorens B. Expression cloning of the pancreatic β-cell receptor for the gluco-incretin hormone glucagon-like peptide-1. Proc Natl Acad Sci U S A. 1992;89:8641–8645. doi: 10.1073/pnas.89.18.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi T, Da Silva Xavier G, Holz GG, Jouaville LS, Thomas AP, Rutter GA. Glucagon-like peptide-1 mobilizes intracellular Ca2+ and stimulates mitochondrial ATP synthesis in pancreatic MIN6 β cells. Biochem J. 2003;369:287–299. doi: 10.1042/BJ20021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi A, Rutter GA. Dynamic imaging of endoplasmic reticulum Ca2+ concentration in insulin-secreting MIN6 cells using recombinant targeted cameleons: roles of sarco (endo) plasmic reticulum Ca2+-ATPase (SERCA)-2 and ryanodine receptors. Diabetes Suppl. 2002;51:S190–S201. doi: 10.2337/diabetes.51.2007.s190. [DOI] [PubMed] [Google Scholar]
- Varadi A, Rutter GA. Ca2+-induced Ca2+ release in pancreatic islet β cells: critical evaluation of the use of ER-targeted ‘Cameleons’. Endocrinology. 2004;45:4540–4549. doi: 10.1210/en.2004-0241. [DOI] [PubMed] [Google Scholar]
- Wagner LE, Li WH, Joseph SK, Yule DI. Functional consequences of phosphomimetic mutations at key cAMP-dependent protein kinase phosphorylation sites in the type 1 inositol 1,4,5-trisphosphate receptor. J Biol Chem. 2004;279:46242–46252. doi: 10.1074/jbc.M405849200. [DOI] [PubMed] [Google Scholar]
- Wan Q-F, Dong Y, Yang H, Lou X, Ding J, Xu T. Protein kinase activation increases insulin secretion by sensitizing the secretory machinery to Ca2+ J Gen Physiol. 2004;124:653–662. doi: 10.1085/jgp.200409082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gillis KD. A highly Ca2+-sensitive pool of granules is regulated by glucose and protein kinases in insulin-secreting INS-1 cells. J Gen Physiol. 2004;124:641–651. doi: 10.1085/jgp.200409081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawalich WS, Zawalich KC, Rasmussen H. Influence of glucagon-like peptide-1 on β-cell responsiveness. Regul Pept. 1993;44:277–283. doi: 10.1016/0167-0115(93)90137-w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.