

Abstract
Nanoparticle-based therapeutics are emerging technologies that have the potential to greatly impact the treatment of many human diseases. However, drug instability and premature release from the nanoparticles during circulation currently preclude clinical translation. Herein, we use a lipase-labile (Sn 2) fumagillin prodrug platform coupled with a unique lipid surface-to-surface targeted delivery mechanism, termed contact-facilitated drug delivery, to counter the premature drug release and overcome the inherent photo-instability of fumagillin, an established anti-angiogenic agent. We show that αvβ3-integrin targeted fumagillin prodrug nanoparticles, administered at 0.3 mg of fumagillin prodrug/kg of body weight suppress the clinical disease indices of KRN serum-mediated arthritis in a dose-dependent manner when compared to treatment with the control nanoparticles with no drug. This study demonstrates the effectiveness of this lipase-labile prodrug nanocarrier in a relevant preclinical model that approximates human rheumatoid arthritis. The lipase-labile prodrug paradigm offers a translatable approach that is broadly applicable to many targeted nanosystems and increases the translational potential of this platform for many diseases.
Keywords: Arthritis, Angiogenesis, Inflammation, Nanoparticles, Prodrug, Drug delivery
1. Introduction
Angiogenesis is the formation of new blood vessels from preexisting vessels and represents one of the earliest findings in the inflamed rheumatoid arthritis (RA) synovium. RA is a chronic inflammatory disease characterizedbyproliferative synovitis leading to progressive joint destruction. In RA a known imbalance between pro-angiogenic and anti-angiogenic factors favors the proliferation of endothelial cells and perpetuates angiogenesis [1-3]. These new blood vessels are not bystander products of inflammation but actively participate in the inflammatory process by supplying nutrients and oxygen to the proliferating RA synovium. Thus inhibition of angiogenesis may halt the perpetuating cycle of inflammation and represent an attractive approach to the treatment of RA [4,5].
Fumagillin is a mycotoxin produced by Aspergillus fumagatus that inhibits methionine aminopeptidase 2 (MetAP-2). MetAP-2 is one of two methionine aminopeptidases in eukaryotes that are responsible for cleavage of the N-terminal methionine residue from nascent proteins [6-8]. Although both MetAP-1 and MetAP-2 have common activity and substrates, MetAP-2 is upregulated during cellular proliferation [9] and has greater efficiency (1000-fold) in catalyzing methionine removal from certain proteins such as glyceraldehydes-3-phosphate dehydrogenase, a key glycolytic enzyme involved in the initiation of apoptosis [10]. Recent evidence suggests that MetAP-2 inhibition by fumagillin perturbed angiogenesis in zebrafish embryos, likely through the modulation of the noncanonical Wnt pathway signaling and erk1/2 phosphorylation, whose level is strictly regulated during angiogenesis [11,12]. However the exact mechanism by which fumagillin exerts its antiangiogenic effect remains undefined.
Our laboratory has previously explored the delivery of native fumagillin using a nanocarrier system targeted to angiogenic endothelium expressing αvβ3-integrin [13-16]. Because native fumagillin is highly hydrophobic, it was originally dissolved into the outer phospholipid monolayer membrane of perfluorooctylbromide (PFOB) nanoparticles and delivered by a mechanism referred to as “contact-facilitated drug delivery” (CFDD), a process in which ligand-tethering of the phospholipid encapsulated nanoparticles to the target cell surface (i.e. via αvβ3-integrin) facilitates the interaction and hemifusion of the two lipid membranes [17], promoting the passive transfer of the drug and phospholipids from the nanoparticle surface to the inner leaflet of the target cell membrane through an ATP-dependent mechanism [18,19]. These αvβ3-targeted PFOB nanoparticles home specifically to angiogenic blood vessels in arthritic paws and ameliorate the progression of disease in a mouse model of KRN serum-induced inflammatory arthritis [20,21]. Although the fumagillin appeared to be stably incorporated into the nanoparticles surfactant during in vitro dissolution testing, the compound was found to be largely released from the lipid membrane during transit to the target in vivo [22]. Moreover, fumagillin is chemically unstable in blood and inherently has significant photo-instability during storage and processing [23-25] thus requiring the compensatory administration of higher nanoparticle doses, which collectively detract from the beneficial intent of site-specific drug delivery and increase the likelihood of off-target effects.
We have recently developed a translatable lipase-labile (Sn 2) fumagillin prodrug (Fum-PD) that eliminates the photo-instability of the compound and is anticipated to improve drug retention in the circulation, enhancing net drug delivery to angiogenic endothelial cells via CFDD (Fig. 1) [22]. While this prodrug concept is broadly applicable to many compounds, lipid nanocarriers, and disease applications, the focus and objectives of this study were: 1) to evaluate the efficacy of this αvβ3-targeted fumagillin prodrug nanoparticle (αvβ3-Fum-PD NP) platform in a relevant model of inflammatory arthritis, 2) to demonstrate effectiveness of the platform at decreased drug doses, 3) to correlate the therapeutic effect of Fum-PD NP to changes in joint histopathology, and 4) to assess toxicity profile in vivo.
Fig. 1.
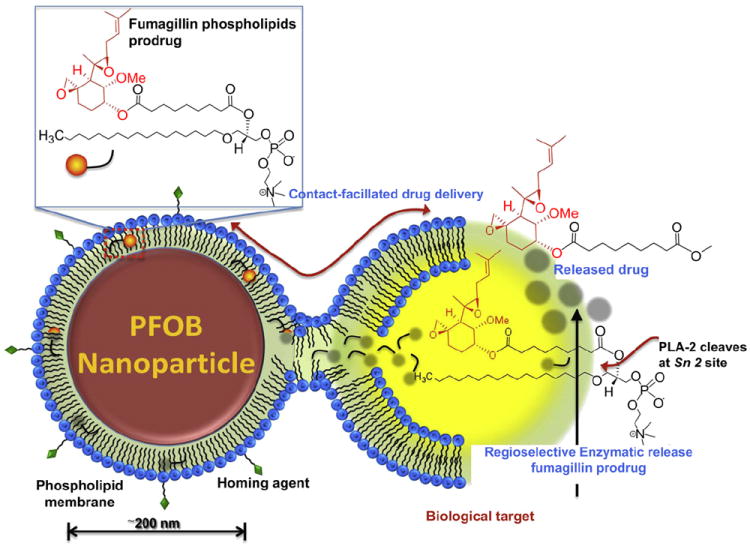
Schematic representation of the contact-facilitated drug delivery mechanism. Phospholipid encapsulated PFOB nanoparticle with incorporated homing ligand and fumagillin prodrug intereacts with a target cell via ligand-tethering, leading to the hemifusion of the two lipid membranes. The continuity of the nanoparticle monolayer and the outer leaflet of the target cell permit the transfer of the fumagillin prodrug from the nanoparticle to the inner leaflet of the target cell membrane. Once inside the cell, the prodrug undergoes a regioselective enzymatic cleavage at the Sn 2 site by local phospholipase (such as phospholipase A2, PLA-2) to release the active form of the drug.
2. Materials and methods
2.1. Chemicals and reagents
Unless otherwise listed, all solvents and reagents were purchased from Aldrich Chemical Co. (St. Louis, MO, USA) and used as received. Anhydrous chloroform was purchased from Aldrich Chemical Co. and distilled over calcium hydride prior to use. Biotinylated dipalmitoyl-phosphatidylethanolamine (DPPE), C16–09:0 (COOH) PC 1-hexadecyl-2-azelaoyl-sn-glycero-3-phosphocholine (PAzPC) and high purity egg yolk phosphatidylcholine (PC) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, USA) Perfluorooctylbromide (PFOB) was purchased and used as received from (Exfluor, Inc., Round Rock, TX, USA). Fumagillin was obtained as a dicyclohexylamine salt from NCI (Bethesda, MD, USA) as a gift (http://dtp.nci.nih.gov) and was used as received. Argon and nitrogen (UHP, 99.99%) were used for storage of materials. The Spectra/Por membrane (Cellulose MWCO: 20 kDa) used for dialysis was obtained from Spectrum Medical Industries, Inc. (Laguna Hills, CA, USA).
2.2. Synthesis of the αvβ3-targeted fumagillin prodrug PFOB nanoparticles
Synthesis of the fumagillin prodrug was accomplished in two simple steps. Briefly, fumagillin dicyclohexylamine salt (NCI) was suspended in 1:1 methanol:- water and treated with 35% NaOH solution, stirred in an ice bath for 2 h, then warmed to room temperature and treated with another equivalent of NaOH solution. The mixture was monitored by TLC (~4 h); the methanol was evaporated and the residue extracted into ethyl acetate and washed with 5% citric acid, brine, bicarbonate, and brine again, then dried with MgSO4 and concentrated in vacuo. The crude product of fumagillol ((3R,4S,5S,6R)-5-Methoxy-4-((2R,3R)-2-methyl-3-(3-methylbut-2-enyl)oxiran-2-yl)-1oxaspiro[2.5]octan-6-ol) was further purified with activated charcoal in acetonitrile then filtered through a celite pad. (Yield: a colorless solid, 5960 mg (71%). HRMS found: MH+ (283.5)).
A solution of C16–09:0 (COOH) PC 1-hexadecyl-2-azelaoyl-sn-glycero-3-phosphocholine followed by DMAP and DCC was added to a solution of fumagillol in dry dichloromethane. The reaction mixture was stirred for 12 h at ambient temperature then purified (silica gel, EtOAc/n-hexane). The filtered solvent was removed in vacuo, leaving an oil residue that was purified by column chromatography on SiO2 with EtOAc/n-hexane as the elution solvent to give the fumagillin prodrug 1 compound as a pale yellowish solid (53%). 1H NMR (CDCl3): δ 0.88 (t, 3H), 1.22–1.37 (m, 37H), 1.58–1.96 (m, 20H), 2.26–2.60 (m, 7H), 3.40 (m, 12H), 3.48 (m, 3H), 3.86–4.00 (m, 4H), 4.10–4.37 (m, 5H), 5.21 (m, 1H). HRMS found: 930.6 (MH+).
All nanoparticles comprised 20% (v/v) perfluorooctylbromide (Exfluor), 1.3–2% (w/v) of a surfactant co-mixture, 1.7% (w/v) glycerin and water for the balance. The surfactant co-mixture for the nanoparticles included 98.7 mol% lecithin (Avanti Polar Lipids, Inc.), 0.1 mol% peptidomimetic αvβ3–integrin antagonist (US Patent 6,322,770) conjugated to PEG2000-phosphatidylethanolamine (Avanti Polar Lipids, Inc.). The fumagillin prodrug was included at the proportionate expense of lecithin in the surfactant at 2.28 mol% of the surfactant mixture. The αvβ3–integrin antagonist used is a quinalone nonpeptide developed by Bristol-Myers Squibb Medical Imaging (US patent 6,511,648) [26].
2.3. Dynamic light scattering measurements
Hydrodynamic diameter distribution and distribution averages for the PFC nanoparticles and controls in aqueous solutions were determined by dynamic light scattering. Hydrodynamic diameters were determined using a Brookhaven Instrument Co. (Holtsville, NY) Model Zeta Plus particle size analyzer. Measurements were made following dialysis (MWCO 10 kDa dialysis tubing, Spectrum Laboratories, Rancho Dominguez, CA, USA) of nanoparticle suspensions into deionized water (0.2 μm). Nanoparticles were dialyzed into water prior to analysis. Scattered light was collected at a fixed angle of 90°. A photomultiplier aperture of 400 mm was used, and the incident laser power was adjusted to obtain a photon counting rate between 200 and 300 kcps. Only measurements for which the measured and calculated baselines of the intensity autocorrelation function agreed to within +0.1% were use to calculate nanoparticle hydrodynamic diameter values. All determinations were made in multiples of five consecutive measurements.
2.4. Electrophoretic potential measurements
Zeta potential (ζ) values for the PFC nanoparticles were determined with a Brookhaven Instrument Co. (Holtsville, NY) model Zeta Plus zeta potential analyzer. Measurements were made following dialysis (MWCO 10 kDa dialysis tubing, Spectrum Laboratories) of the nanoparticle suspensions into water. Data were acquired in the phase analysis light scattering (PALS) mode following solution equilibration at 25 °C. Calculation of ζ from the measured nanoparticle electrophoretic mobility (μ) employed the Smoluchowski equation: μ = εζ/η, where ε and η are the dielectric constant and the absolute viscosity of the medium, respectively. Measurements of ζ were reproducible to within ±4 mV of the mean value given by 16 determinations of 10 data accumulations.
2.5. Immunofluorescence
Mice were injected i.v. with 200 μl of rhodamine-conjugated non-targeted or αvβ3-targeted nanoparticles along with FITC-conjugated tomato lectin (Lycopersicon Esculentum, Vector Laboratories, Burlingame, CA, USA) to visualize the blood vessels. Mice were sacrificed 60 min after injections, flushed extensively through a transcardial approach with PBS to remove circulating blood, and the paws embedded in OCT compound and sectioned. All images were visualized on a Nikon Eclipse microscope and acquired with QCapture software.
2.6. Arthritis induction and treatment
All animal experiments were performed in strict accordance with the guidelines established by the Division of Comparative Medicine at Washington University. Arthritis was induced using the KRN mouse model of inflammatory arthritis as previously described [20]. Six to eight week-old male C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME, USA) were injected i.p. with 150 μl of KRN serum on day 0 to induce arthritis. Clinical manifestation of arthritis was assessed daily on a scale of 0–3 (0 = no swelling or erythema, 1 = slight swelling or erythema, 2 = moderate erythema and swelling in multiple digits or entire paw, 3 = pronounced erythema and swelling of entire paw, maximum score of 12 per mouse). Change from baseline in paw thickness was determined daily by dial calipers and an average change in ankle thickness was determined for each mouse from the two hind paw measurements. On the days indicated, mice were injected i.v. at 2 μl of nanoparticles/g body weight with αvβ3-Ctrl NP without drug or αvβ3-Fum-PD NP at 0.3 μg of drug/g body weight.
2.7. Histological analysis
Paws were harvested on day 7 or day 9 after serum transfer, fixed in 10% buffered formalin for 48 h, decalcified in EDTA solution, embedded in paraffin, and sectioned at 5 μm. The sections were stained with hematoxylin and eosin (H&E) or toluidine blue. Inflammatory cell influx, bone erosions and cartilage integrity were assessed as previously described [20,21]. Digital images of five random areas per H&E-stained paw section were acquired at 400× and the number of exuded inflammatory cells enumerated. The number of bone erosions was enumerated per mm of bone surface using ImageJ program (http://rsb.info.nih.gov/ij). Proteoglycan content, an indication of cartilage integrity, was graded on toluidine blue-stained sections on a scale of 0–4 as previously described [27] (0 = fully stained cartilage; 1 = less than 25% unstained; 2 = 25–50% unstained; 3 = 50–75% unstained; and 4 = greater than 75% unstained cartilage). Quantitative scoring was performed by an observer blinded to the treatments. Each value represents the average per animal derived from the cumulative scoring of two hind paws.
2.8. Hematologic parameters and serum chemistry analysis
Animals were injected i.v. with the indicated doses of NP and blood samples were drawn at the indicated time from the inferior vena cava after euthanasia. WBC, differentials, and serum chemistry were analyzed by the Washington University Department of Comparative Medicine. Values were compared to those obtained in unmanipulated C57BL/6 male mice.
2.9. C3a ELISA
Mice were left uninjected or injected with 2 μl of αvβ3-Fum-PD NP/g body weight (once or three times) and their plasma obtained at 30 min after the last injection for C3a ELISA. Cobra venom factor (CVF), a strong complement activator [28], was injected i.v. at 2 U/mouse and used as a positive control for complement activation. For the ELISA, plates were coated overnight at 4 °C with anti-mouse C3a (4 μg/mL) monoclonal antibody (BD Pharmingen, San Jose, CA, USA). After being blocked with 1% BSA, the plates were washed and incubated with samples (100 μL of fresh plasma diluted 1:100 in reagent diluent) for 2 h at room temperature, followed by biotinylated anti-mouse C3a (250 ng/mL) monoclonal antibody (BD Pharmingen). After incubation with streptavidin-peroxidase (400 ng/mL; Sigma), 100 μL of peroxide-chromogen solution (R&D Systems, Minneapolis, MN, USA) was added to each well, and color development was read at 450 nm with a SpectraMax Plus reader (Molecular Devices, Sunnyvale, CA, USA). Mouse recombinant C3a (BD Pharmingen) was used to establish the standard curve.
2.10. Statistical analysis
In all experiments, statistical comparisons between study groups were performed using Student’s t-test. Differences between means with P < 0.05 were considered significant. Numerical values are reported as mean ± SEM.
3. Results
3.1. Synthesis and characterization of the fumagillin prodrug nanoparticles
As stated above, a rapid loss of the membrane dissolved fumagillin relative to the perfluorocarbon core and lipid-gadolinium conjugates in vivo [22], combined with the well-known chemical and photo-instability of fumagillin [23-25], prompted us to explore an alternate synthetic route to photostable fumagillin prodrug (Fum-PD) in this work. The synthesis of Fum-PD followed two straightforward steps starting from the parent fumagillin compound, as illustrated in Fig. 2. In the preliminary step, fumagillin underwent saponification in the presence of 35% NaOH and methanol/water (1:1) to produce fumagillol. The decatetraene photosensitive moiety was effectively substituted with the seven-carbon Sn 2 acyl group of the phosphatidylcholine backbone of PAzPC by a carbodiimide-mediated esterification. The fumagillol was then incorporated as a component of the lipid surfactant mixture and microfluidized with PFOB under high pressure (20,000 psi) for 4 min (Fig. 2). Synthesis was completed with the addition of 0.1 mol% αvβ3-integrin antagonist conjugated to PEG2000-phosphatidylethanolamine to the lipid surfactant mixture. The αvβ3-integrin antagonist was a quinalone nonpeptide developed by Bristol-Myers Squibb Medical Imaging (US patent 6,511,648 and related patents) and initially reported and characterized as the 111In-DOTA conjugate RP478 and cyan 5.5 homolog TA145 [29]. The targeted nanoparticles presented ~300 ligands/particle with an IC50 of 50 pm for the Mn2+-activated αvβ3-integrin (unpublished data from Kereos, Inc., St. Louis, MO, USA and [14]). Homing specificity of the vascular constrained PFOB nanoparticle analog has been extensively demonstrated through in vivo competition studies in preclinical cancer [14,16,30-34], atherosclerosis [13,35,36], rheumatoid arthritis [20,21], and Matrigel™ plug [37] models. Pharmacokinetic and biodistribution studies of 111In αvβ3-targeted nanoparticles were previously reported [32].
Fig. 2.
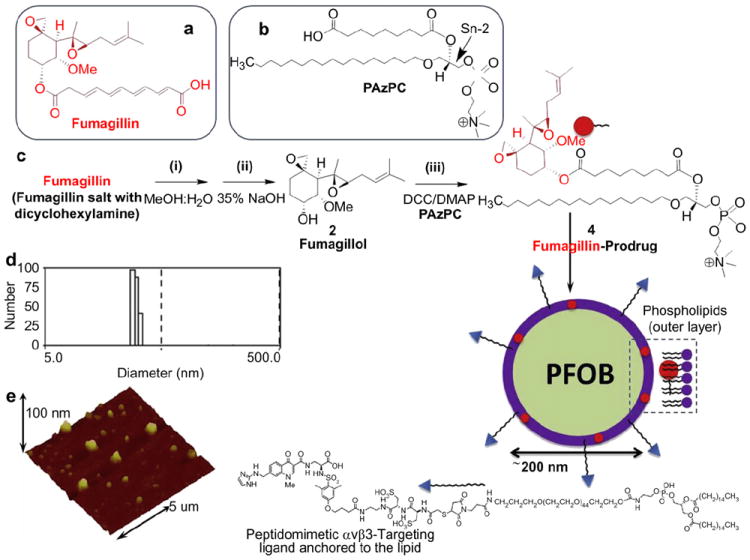
Preparation of fumagillin prodrug and its incorporation into perfluorooctylbromide (PFOB) nanoparticle. Chemical structure of fumagillin (a) and PAzPC (b); (c) Saponification of fumagillin: (i) MeOH:water (1:1), 35% NaOH; (ii) PAzPC, DCC/DMAP; (iii) synthesis of the nanoparticle: a lipid thin film is prepared from a phospholipids mixture of 98.7 mol% lecithin PC, 0.15 mol% of αvβ3-ligand conjugated lipid and 2.28 mol% of fumagillin prodrug; the nanoparticle self-assembles upon sonification and microfluidization of PFOB and glycerin pH 6.5, at 20,000 psi for 4 min; (d) dynamic light scattering shows the number and average particle size distribution; (e) Anhydrous state atomic force microscopy image of αvβ3-targeted PFOB fumagillin prodrug nanoparticle.
These particles were extensively characterized with multiple characterization methods. Hydrodynamic diameters (Dh) of the αvβ3-targeted fumagillin prodrug PFOB nanoparticle and control samples in aqueous solution were within 230–260 nm as measured from dynamic light scattering (DLS) (Fig. 2d). Negligible changes in particle sizes were noticed after the encapsulation of prodrug at 2.28 mol% (~0.5 μm) within the surfactant co-mixture. Successful phospholipid encapsulation was confirmed by the presence of negative Zeta potential values (ζ, ca. −25 mV). Anhydrous state morphologies of these particles were observed by atomic force microscopy, which confirmed the spherical nature of these particles on a glass substrate. (Fig. 2e).
3.2. αvβ3-targeted nanoparticles localize to inflamed joints
The KRN serum-mediated inflammatory arthritis is a widely used mouse model that recapitulates many key features of human RA. It is a highly reproducible disease model of RA with robust joint inflammation induced by the passive transfer of serum from the K/BxN mice [38]. We induced arthritis by injecting 150 μl of KRN serum i.p. on day 0 and on day 7, at the peak of disease, mice were administered 200 μl of rhodamine-conjugated αvβ3-targeted or non-targeted NPs. Targeted NPs were detected prominently, lining small blood vessel wall in the arthritic paw (Fig. 3a). In contrast, we saw no accumulation in the arthritic paw of animals injected with the non-targeted NPs (Fig. 3a). These results confirm that the αvβ3-targeted NPs home specifically to inflamed paws, as previously reported [20].
Fig. 3.
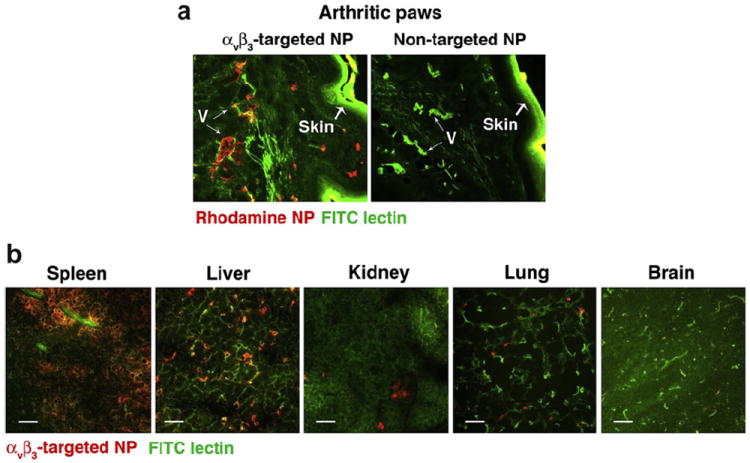
αvβ3-targeted fumagillin prodrug nanoparticles (αvβ3-Fum-PD NP) localize specifically to the inflamed paws. Arthritis was induced by the i.p. administration of KRN serum. On day 7, at the peak of disease, mice were injected with rhodamine-conjugated αvβ3-Fum-PD NP or non-targeted NP. (a) Sections of paws were examined for NP accumulation (red). FITC lectin (green) was used to label blood vessels (V). The skin also autofluoresces green. (b) Sections of different organs were examined for NP accumulation 1 h after injection. Scale bar = 0.05 mm.
To examine the biodistribution of αvβ3-targeted NPs, we administered rhodamine-conjugated αvβ3-targeted to animals and surveyed for NP accumulation in various organs 60 min following the NP injection. αvβ3-targeted NPs accumulate mainly in the spleen, followed by liver, while levels in the kidney, lung and brain were significantly lower (Fig. 3b). These results were consistent with previous studies [20,32].
3.3. αvβ3-Fum-PD nanoparticles suppress inflammation in the KRN model of arthritis
Arthritis was induced as above and disease manifestation, assessed as increase in ankle thickness and arthritis score, was monitored daily. On day 2 the majority of mice had established arthritis with an average arthritis score of 3 (maximum score of 12). Mice with established arthritis received one, two, or three serial i.v. injections of αvβ3-targeted control (no drug) nanoparticles (αvβ3-Ctrl NPs) or αvβ3-Fum-PD NPs, starting on day 2 after KRN serum transfer. Animals treated with one dose (X1) of αvβ3-Fum-PD NPs on day 2 exhibited a rapid decrease in ankle thickness observed within 24 h of NP administration (P < 0.05) that was unsustained and recrudesced to the severity of disease observed in the αvβ3-Ctrl NP group by the end of the 7-day study (Fig. 4a). On the other hand, we did not see a significant difference in the arthritis score between the αvβ3-Ctrl NP- and the αvβ3-Fum-PD NP treated groups (Fig. 4d). Administration of αvβ3-Fum-PD NPs on days 2 and 4 (X2) achieved a modest decrease in disease indices (20–25%, Fig. 4a and e P < 0.05 starting on day 4). Mice treated serially with αvβ3-Fum-PD NPs (X3 on days 2, 3, and 4) also showed rapid stabilization in ankle thickness and improvement in clinical scores compared to the αvβ3-Ctrl NP treated animals (P < 0.05, Fig. 4c and f). These improved clinical features continued for the αvβ3-Fum-PD NP treated mice while inflammation progressed unabated in the αvβ3-Ctrl NP group. Following the last NP treatment on day 4, changes in ankle thickness and arthritis score began to slowly increase in the αvβ3-Fum-PD NP group but the rate of disease progression was significantly slower compared to the αvβ3-Ctrl NP treated animals. With time, the absolute difference between the Fum-PD and no drug groups became more evident (a 45–50% decrease in disease indices in the group treated with Fum-PD NPs, Fig. 4c and f). On day 7, mice receiving αvβ3-Fum-PD NPs had significantly less joint disease than the animals that received control NPs [maximum increase in ankle thickness of 0.90 ± 0.04 mm in the αvβ3-Ctrl NP group compared with 0.45 ± 0.08 in the αvβ3-Fum-PD NP group, P < 0.01; maximum clinical score of 10.6 ± 0.8 in the αvβ3-Ctrl NP group compared with 6.0 ± 0.7 in the αvβ3-Fum-PD NP group, P < 0.01].
Fig. 4.
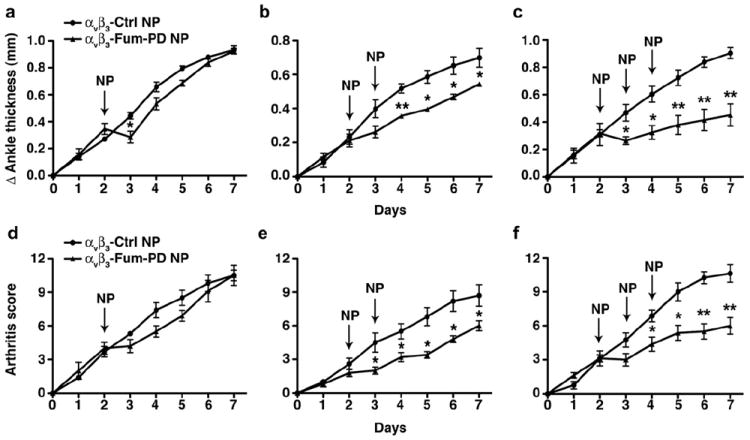
Serial doses of αvβ3-Fum-PD NPs suppress inflammatory arthritis. Arthritis was induced with 150 μl KRN serum on day 0. αvβ3-targeted nanoparticles without drug (αvβ3-Ctrl NP) or αvβ3-targeted fumagillin prodrug nanoparticles (αvβ3-Fum-PD NP) were administered once, twice, or three times (arrows). Changes (Δ) in ankle thickness (a–c) and arthritis score (d–f) were monitored daily. Values represent mean ± SEM, n = 4–6 animals per treatment group. *P < 0.05, **P < 0.01.
Histologic examination of arthritic paws on day 7 revealed that treatment with αvβ3-Fum-PD NPs (X3) limited the number of inflammatory leukocytes recruited to the inflamed paws [12.5 ± 2.1 cells per high power field (HPF) versus 54.6 ± 4.1 cells per HPF in the αvβ3-Ctrl NP group, P < 0.0001] (Fig. 5a–b and c). Similarly, the number of bone erosions in the αvβ3-Ctrl NP group (2.3 ± 0.2 erosions/mm of bone surface) were 10-fold higher than that measured in mice treated with αvβ3-Fum-PD NPs (0.2 ± 0.1 erosions/mm of bone surface, P < 0.0001) (Fig. 5a and F). Moreover, cartilage damage, as reflected by proteoglycan depletion score, was nearly 50% less in the αvβ3-Fum-PD NP group (1.4 ± 0.1) compared with the αvβ3-Ctrl NP group (2.4 ± 0.2, P < 0.0001) (Fig. 5c–d and g). These results suggest that Fum-PD NPs effectively suppress inflammation in a relevant model of robust inflammatory arthritis.
Fig. 5.
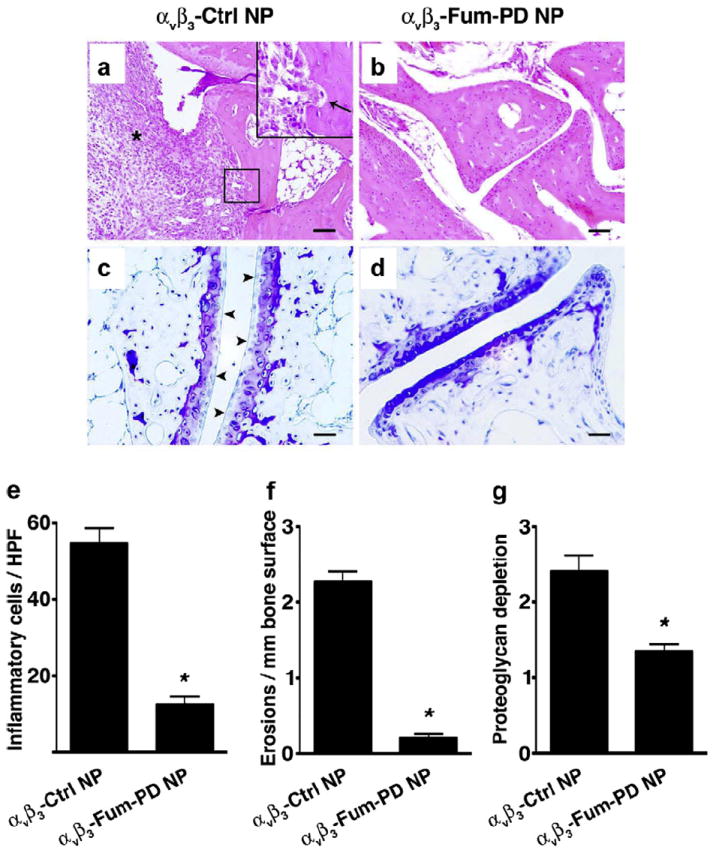
Serial doses of αvβ3-Fum-PD NPs suppress inflammation and preserve bone and cartilage integrity. On day 7 after KRN serum transfer and Fum-PD or Ctrl NP treatment (3 doses), mice were sacrificed and their paws analyzed histologically for the number of inflammatory cells (asterisk) per high power field (HPF) by H&E (a, b & e), erosions (arrow in a, b & f), and degree of proteoglycan depletion by toluidine blue staining (arrowheads in e, f & g). Values represent mean ± SEM (n = 4–6 animals per treatment group). Scale bar = 0.1 mm (a, b); 0.05 mm (c, d). Inset in A represents higher magnification of boxed area. *P < 0.0001.
3.4. In vivo toxicity profile of αvβ3-Fum-PD nanoparticles
In previous studies, serial injections of larger volumes of nanoparticles (500% higher than those used in this study or for MRI imaging in rodent models) were used to elicit pharmacologic effect due to premature fumagillin loss in circulation [20,21]. These parenteral volumes induced a transient transaminitis that likely resulted from the physical sequestration of the PFOB NP in the reticulo-endothelial system (RES) and the resultant acute tense engorgement of the liver parenchyma within the less compliant tissue capsule. This transaminitis was independent of the drug load [20]. Despite the lower nanoparticle dosage used in this study, three daily injections of PFOB NP, independent of drugs, also induced a 2–3-fold increase in the serum level of aspartate aminotransferase (AST) in mice 3 days after the last injection of nanoparticles (Fig. 6a). Since the reported biological half-life of the chemically inert perfluorocarbon is about 4 days, it is clear that the three doses of particles spaced only two days apart resulted in significant liver accumulation. However, injection of 1 or 2 doses (2 days apart) of PFOB NPs (without or with drug) did not significantly affect liver transaminase levels (Fig. 6a–b).
Fig. 6.
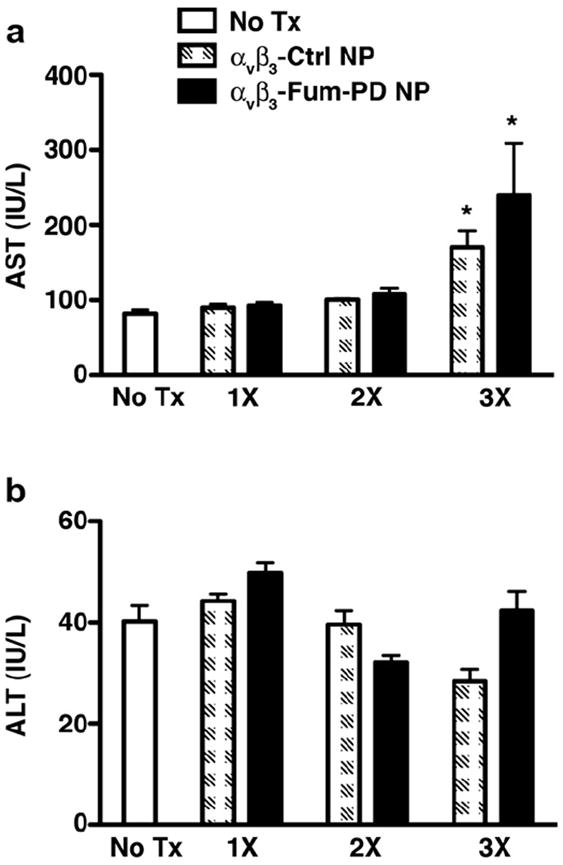
Transaminases in mice treated with nanoparticles. Mice were injected with one (1×), two (2×), or three (3×) doses of αvβ3-Ctrl NPs or αvβ3-Fum-PD NPs. Three days after the last injection, samples were drawn and serum levels of aspartate aminotransferase (AST, a) and alanine aminotransferase (ALT, b) were assessed. Values represent mean ± SEM, n = 9 animals in the no treatment (Tx) group and 4–6 animals per treatment group. *P < 0.01 compared with no Tx.
Studies using the soluble fumagillin derivative PPI-2458 in an animal model of arthritis suggest that repeated administration of this drug at higher doses significantly suppresses peripheral white blood cell counts (WBC) [39], specifically lymphocyte counts, prompting us to examine peripheral blood counts in our studies. Serial i.v. injections (×3 doses) of PFOB NPs (without or with Fum-PD) transiently decreased total peripheral WBC, specifically the lymphocyte subset (but not neutrophil number), which returned to normal levels within 24 h of the last injection. Of note, it is unlikely that the lower peripheral WBC accounts for the lower disease activity as 1) the neutrophil counts were relatively unaffected in all treatment groups (P > 0.05 compared with no treatment group) and 2) only the group that received αvβ3-Fum-PD NP had improved clinical scores and suppression of inflammation in the joints. On the other hand, injection of 1 dose of PFOB NP did not significantly affect total peripheral WBC or other hematologic parameters, including hemoglobin and platelet counts (Table 1).
Table 1.
Hematologic parameters in mice treated with nanoparticles.
| Treatment | Hgb (g/dl) | Platelets (103/mm3) | WBC (103/mm3) | Neutrophils (103/mm3) | Lymphocytes (103/mm3) |
|---|---|---|---|---|---|
| No treatment | 12.8 ± 0.6 | 747 ± 51 | 11.2 ± 0.4 | 1.3 ± 0.2 | 9.8 ± 0.3 |
| αvβ3-Ctrl NP (1×) | 13.5 ± 0.2 | 648 ± 14 | 8.8 ± 1.0 | 1.1 ± 0.1 | 7.8 ± 1.0 |
| αvβ3-Ctrl NP (3×) | 12.7 ± 0.3 | 698 ± 22 | 6.4 ± 0.4** | 0.7 ± 0.1 | 5.6 ± 0.4** |
| αvβ3-Fum-PD NP (1×) | 12.2 ± 0.1 | 738 ± 106 | 8.9 ± 0.5 | 1.7 ± 0.5 | 7.1 ± 0.8* |
| αvβ3-Fum-PD NP (3×) | 13.0 ± 0.4 | 874 ± 52 | 7.3 ± 0.4** | 0.9 ± 0.3 | 6.4 ± 0.4** |
| αvβ3-Fum-PD NP (3×)-24 h | 13.1 ± 0.2 | 637 ± 10 | 8.9 ± 0.1 | 0.8 ± 0.3 | 7.8 ± 0.3 |
P < 0.05,
P < 0.001 compared with no treatment. Values obtained 30 min after the last nanoparticle injection, unless otherwise indicated. n = 12 in the no treatment group; n = 3–4 in the treated groups.
Additionally we have recently begun to examine the innate immune response to nanoparticle administration and observed that injection of PFC nanoparticles can elicit swift complement activation, the intensity of which depends on nanoparticle surface functionalization [40]. To understand whether the interaction of nanoparticles with the host complement system may hamper clinical translation of this potential therapeutic platform, we looked for complement activation in mice injected with nanoparticles by C3a ELISA. We saw minimal, but not statistically significant, increase in C3a levels in mice treated with one dose of Fum-PD NPs. Moreover, we saw no cumulative effects in complement activation with serial injections (×3 doses) of Fum-PD NPs (Fig. 7). Collectively, these results suggest that one (or two) doses αvβ3-Fum-PD NP should be a well-tolerated therapeutic regimen in the clinical setting, especially if used as a combination therapy with other conventional drugs as previously shown [21].
Fig. 7.
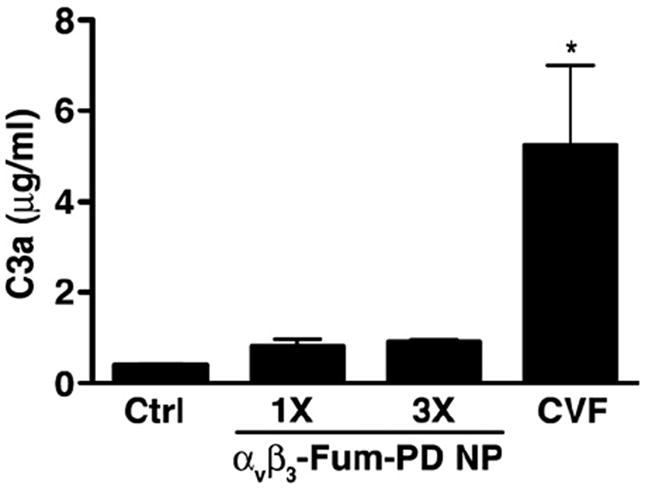
Complement activation by αvβ3-Fum-PD NPs. Mice were left uninjected (Ctrl) or injected with one dose (1×) or three serial daily doses of Pro-Fum NP (3×). Thirty min after the last injection, mice were sacrificed and plasma obtained for C3a ELISA. Values represent mean ± SEM (n = 3 animals per treatment group). CVF (2 U i.v.), a strong complement activator, was used as a positive control of complement activation. *P < 0.05 compared with Ctrl.
4. Discussion
These studies represent an in vivo evaluation of the effectiveness of an Sn 2 lipase-labile fumagillin prodrug nanocarrier platform in the treatment of KRN serum-induced inflammatory arthritis in mice, a pathological disease model that resembles human RA in many important aspects. We demonstrated that, by limiting premature drug loss in circulation and protecting the chemically unstable elements of the molecule, we were able to lower the effective drug dose in vivo (0.3 mg Fum-PD/kg of body weight) by ~8-fold compared to the dose we previously reported for native fumagillin (2.5 mg/kg of body weight) in the treatment of experimental arthritis [20,21], despite the extra requirement for intracellular lipase liberation of the active drug. We showed that serial αvβ3-Fum-PD NP treatment in mice with established arthritis slowed the progression of synovial inflammation, as evidenced by ~50% reduction in ankle thickness and arthritic score, a marked decrease in the number of inflammatory cells recruited into the subsynovial space, and preservation of bone and cartilage integrity. Moreover, we showed that the Fum-PD NP platform has a favorable toxicity profile.
Anti-angiogenesis has long been actively explored in cancer treatment, but these approaches have only begun to emerge in RA [2,4,5,41,42]. Indirect anti-angiogenesis effects have been observed with some of the current RA treatments, such as anti-TNF biologics, which reduce the circulating levels of several pro-angiogenic factors including VEGF, soluble intercellular adhesion molecule 1 (sICAM-1), and soluble vascular cell adhesion molecule 1 (sVCAM-1) in RA patients [43]. On the other hand, an attempt at inhibiting angiogenesis directly through immuno-blockade of the αvβ3-integrin, an adhesion molecule that is highly upregulated on angiogenic blood vessels, was unsuccessful and abandoned in a Phase II trial [5]. Water-soluble derivatives of fumagillin have been previously explored as anti-angiogenic therapy in animal models of inflammatory arthritis. The water-soluble fumagillin analog, TNP-470 (also known as AGM1470) ameliorates disease development only when administered at very high dose (90 mg/ kg s.c. every other day) prior to the onset of arthritis in rodents [44]. However, high-dose TNP-470 elicited moderate to severe neurotoxicity and greater than 50% mortality in treated animals [44]. PPI-2458, a more recent water-soluble fumagillin derivative, exhibits improved neurotoxicity profile and inhibits arthritis progression when given frequently (everyday to every other day), at high doses (15 mg/kg i.v., 30 mg/kg s.c., or 100 mg/kg p.o.) or in combination with MTX, to animals during the early phase of disease [39]. Unfortunately, cessation of treatment was followed by rapid recrudescence of the inflammatory process [39,45,46].
We previously showed that native fumagillin delivered via a αvβ3-integrin-targeted nanocarrier approach suppressed the progression of arthritis in mice when administered serially alone or as a single adjuvant dose in combination with MTX [20,21]. However, studies tracking the pharmacokinetics in whole blood of fumagillin (by mass spectroscopy) and the PFOB core (by gas chromatography) revealed a rapid, substantial loss of the active compound during circulation prior to reaching the neovascular targets, despite the high stability of the nanoparticle formulation during in vitro dissolution studies [16,22]. The premature loss of drugs from targeted nanoparticles during circulation, particularly lipid-based formulations (e.g., emulsions, micelles, liposomes), has been an intractable problem compensated for by increasing the drug load per particle and administering higher nanoparticle doses. In addition, fumagillin in its native form is highly unstable to light and hydrolysis, which severely complicates pharmaceutical production and precludes clinical translation [23-25]. To counter the instability of the compound and the premature release from lipid nanoparticles in circulation, a problem common to many lipid-based therapeutic agents, a fumagillin prodrug form was pursued wherein the active ingredient was chemically coupled to the Sn 2 acyl group of phosphatidylcholine. This chemical coupling eliminated the light-sensitive tail of the native compound and positioned the drug within the hydrophobic portion of the outer phospholipid membrane of the nanoparticle surfactant, which was anticipated to better protect the sensitive di-epoxide active site and the ester linkage to the prodrug acyl spacer from the aqueous medium [22].
The concept of Sn 2 phospholipid prodrugs has never been considered for targeted nanomedicine therapy as presented in this study. Jorgensen et al. developed untargeted liposomes containing a Sn 2 prodrug, anticipating that increased secretory phospholipase enzymes liberated by cancer cells would lead to the local release of high concentration of the active drug in the proximity of a tumor [47-51]. The effectiveness of this approach was modest but could be further enhanced by utilizing particles that were thermosensitive and disrupted at the tumor site with an exogenous heat source [47]. In general, activation of these liposomal Sn 2 prodrugs in the circulation was limited by the inaccessibility of the acyl bond to water [52]. For the targeted PFOB nanoparticles, the reduced water accessibility of the Sn 2 fumagillin prodrug is highly desirable as it prevents premature release or metabolism. Fumagillin prodrug retention in the particle during in vitro dissolution was excellent and stable in the presence of serum alone or serum-enriched with exogeneous phospholipase A2 [22]. Once the Sn 2 fumagillin prodrug enters the endothelial cell, the enzyme(s), mechanisms, and rate controlling elements affording the release of active fumagillin into the cytosol from the membrane remain to be elucidated. Regardless, our data herein clearly demonstrate that the use of the lipase-labile Sn 2 fumagillin prodrug in combination with ligand directed CFDD mechanism, which facilitated the intracellular delivery and enzymatic liberation of fumagillin without the need for particle internalization, was effective in an animal model that approximates a human disease. The same general approach could also be applied to a similar targeted liposomal, lipid micellar, or nanoemulsion system in which the surfaces of the particle and the cell membrane directly and freely interact, typically through a homing ligand, and the formation of a fusion complex.
5. Conclusions
The present study clearly demonstrates the efficacy of the targeted Sn 2 fumagillin prodrug nanocarrier platform in a pathological setting, the KRN serum-induced arthritis model. In this series of studies, the fumagillin prodrug formulation is not only bioactive but also effective at much lower total doses than previously achieved with the water-soluble derivatives. Combined with a favorable toxicity profile, these lipase-labile prodrugs used with lipid-based nanoparticles offer a scalable and clinically translatable approach to improve the efficacy and safety of fumagillin and many additional targeted nanomedicines.
Acknowledgments
The authors extend sincere thanks to Ying Hu and Jennifer Huang for technical expertise, and Paul Allen for generous gift of KRN serum. This work was supported by grants from the NIH (HL113392, CA100623, CA154737, HL094470, AR056468, NS073457, CA136398) and The American Heart Association (0835426N and 11IRG5690011). Drs. Lanza and Pan are co-inventors on a Washington University Medical School pending patent application concerning the fumagillin prodrug technology. In addition, Drs. Lanza and Wickline are scientific cofounders of Kereos, Inc, St. Louis, which has licensed angiogenesis-targeted perfluorocarbon nanotechnology intellectual property from Washington University/Barnes-Jewish Hospital for clinical development.
References
- 1.Costa C, Incio J, Soares R. Angiogenesis and chronic inflammation: cause or consequence? Angiogenesis. 2007;10:149–66. doi: 10.1007/s10456-007-9074-0. [DOI] [PubMed] [Google Scholar]
- 2.Szekanecz Z, Koch AE. Angiogenesis and its targeting in rheumatoid arthritis. Vascul Pharmacol. 2009;51:1–7. doi: 10.1016/j.vph.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szekanecz Z, Besenyei T, Szentpetery A, Koch AE. Angiogenesis and vasculogenesis in rheumatoid arthritis. Curr Opin Rheumatol. 2010;22:299–306. doi: 10.1097/BOR.0b013e328337c95a. [DOI] [PubMed] [Google Scholar]
- 4.Lainer DT, Brahn E. New antiangiogenic strategies for the treatment of proliferative synovitis. Expert Opin Investig Drugs. 2005;14:1–17. doi: 10.1517/13543784.14.1.1. [DOI] [PubMed] [Google Scholar]
- 5.Lainer-Carr D, Brahn E. Angiogenesis inhibition as a therapeutic approach for inflammatory synovitis. Nat Clin Pract Rheumatol. 2007;3:434–42. doi: 10.1038/ncprheum0559. [DOI] [PubMed] [Google Scholar]
- 6.Arfin SM, Kendall RL, Hall L, Weaver LH, Stewart AE, Matthews BW, et al. Eukaryotic methionyl aminopeptidases: two classes of cobalt-dependent enzymes. Proc Natl Acad Sci USA. 1995;92:7714–8. doi: 10.1073/pnas.92.17.7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc Natl Acad Sci USA. 1997;94:6099–103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu S, Widom J, Kemp CW, Crews CM, Clardy J. Structure of human methionine aminopeptidase-2 complexed with fumagillin. Science. 1998;282:1324–7. doi: 10.1126/science.282.5392.1324. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Lou P, Henkin J. Selective inhibition of endothelial cell proliferation by fumagillin is not due to differential expression of methionine aminopeptidases. J Cell Biochem. 2000;77:465–73. [PubMed] [Google Scholar]
- 10.Tarze A, Deniaud A, Le Bras M, Maillier E, Molle D, Larochette N, et al. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane per-meabilization. Oncogene. 2007;26:2606–20. doi: 10.1038/sj.onc.1210074. [DOI] [PubMed] [Google Scholar]
- 11.Ma AC, Fung TK, Lin RH, Chung MI, Yang D, Ekker SC, et al. Methionine aminopeptidase 2 is required for HSC initiation and proliferation. Blood. 2011;118:5448–57. doi: 10.1182/blood-2011-04-350173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Yeh JR, Mara A, Ju R, Hines JF, Cirone P, et al. A chemical and genetic approach to the mode of action of fumagillin. Chem Biol. 2006;13:1001–9. doi: 10.1016/j.chembiol.2006.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winter PM, Neubauer AM, Caruthers SD, Harris TD, Robertson JD, Williams TA, et al. Endothelial alpha(v)beta3 integrin-targeted fumagillin nanoparticles inhibit angiogenesis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:2103–9. doi: 10.1161/01.ATV.0000235724.11299.76. [DOI] [PubMed] [Google Scholar]
- 14.Schmieder AH, Caruthers SD, Zhang H, Williams TA, Robertson JD, Wickline SA, et al. Three-dimensional MR mapping of angiogenesis with alpha5beta1(alpha nu beta3)-targeted theranostic nanoparticles in the MDA-MB-435 xenograft mouse model. FASEB J. 2008;22:4179–89. doi: 10.1096/fj.08-112060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winter PM, Caruthers SD, Zhang H, Williams TA, Wickline SA, Lanza GM. Antiangiogenic synergism of integrin-targeted fumagillin nanoparticles and atorvastatin in atherosclerosis. JACC Cardiovasc Imaging. 2008;1:624–34. doi: 10.1016/j.jcmg.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winter PM, Schmieder AH, Caruthers SD, Keene JL, Zhang H, Wickline SA, et al. Minute dosages of alpha(nu)beta3-targeted fumagillin nanoparticles impair Vx-2 tumor angiogenesis and development in rabbits. FASEB J. 2008;22:2758–67. doi: 10.1096/fj.07-103929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lanza GM, Yu X, Winter PM, Abendschein DR, Karukstis KK, Scott MJ, et al. Targeted antiproliferative drug delivery to vascular smooth muscle cells with a magnetic resonance imaging nanoparticle contrast agent: implications for rational therapy of restenosis. Circulation. 2002;106:2842–7. doi: 10.1161/01.cir.0000044020.27990.32. [DOI] [PubMed] [Google Scholar]
- 18.Partlow KC, Lanza GM, Wickline SA. Exploiting lipid raft transport with membrane targeted nanoparticles: a strategy for cytosolic drug delivery. Biomaterials. 2008;29:3367–75. doi: 10.1016/j.biomaterials.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soman NR, Baldwin SL, Hu G, Marsh JN, Lanza GM, Heuser JE, et al. Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J Clin Invest. 2009;119:2830–42. doi: 10.1172/JCI38842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou HF, Chan HW, Wickline SA, Lanza GM, Pham CT. Alphavbeta3-targeted nanotherapy suppresses inflammatory arthritis in mice. FASEB J. 2009;23:2978–85. doi: 10.1096/fj.09-129874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou HF, Hu G, Wickline SA, Lanza GM, Pham CT. Synergistic effect of anti-angiogenic nanotherapy combined with methotrexate in the treatment of experimental inflammatory arthritis. Nanomedicine (Lond) 2010;5:1065–74. doi: 10.2217/nnm.10.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan D, Sanyal N, Schmieder AH, Senpan A, Kim B, Yang X, et al. Anti-angiogenic nanotherapy with lipase-labile Sn-2 fumagillin prodrug. Nanomedicine (Lond) 2012 doi: 10.2217/nnm.12.27. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eble TE, Garrett ER. Studies on the stability of fumagillin. II. Photolytic degradation of crystalline fumagillin. J Am Pharm Assoc Am Pharm Assoc. 1954;43:536–8. doi: 10.1002/jps.3030430906. [DOI] [PubMed] [Google Scholar]
- 24.Garrett ER. Studies on the stability of fumagillin. III. Thermal degradation in the presence and absence of air. J Am Pharm Assoc Am Pharm Assoc. 1954;43:539–43. [PubMed] [Google Scholar]
- 25.Garrett ER, Eble TE. Studies on the stability of fumagillin. I. Photolytic degradation in alcohol solution. J Am Pharm Assoc Am Pharm Assoc. 1954;43:385–90. doi: 10.1002/jps.3030430702. [DOI] [PubMed] [Google Scholar]
- 26.Harris TD, Kalogeropoulos S, Nguyen T, Liu S, Bartis J, Ellars C, et al. Design, synthesis, and evaluation of radiolabeled integrin alpha v beta 3 receptor antagonists for tumor imaging and radiotherapy. Cancer Biother Radiopharm. 2003;18:627–41. doi: 10.1089/108497803322287727. [DOI] [PubMed] [Google Scholar]
- 27.Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363–71. doi: 10.1172/JCI13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alper CA, Balavitch D. Cobra venom factor: evidence for its being altered cobra C3 (the third component of complement) Science. 1976;191:1275–6. doi: 10.1126/science.56780. [DOI] [PubMed] [Google Scholar]
- 29.Meoli DF, Sadeghi MM, Krassilnikova S, Bourke BN, Giordano FJ, Dione DP, et al. Noninvasive imaging of myocardial angiogenesis following experimental myocardial infarction. J Clin Invest. 2004;113:1684–91. doi: 10.1172/JCI20352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winter PM, Caruthers SD, Kassner A, Harris TD, Chinen LK, Allen JS, et al. Molecular imaging of angiogenesis in nascent Vx-2 rabbit tumors using a novel alpha(nu)beta3-targeted nanoparticle and 1.5 tesla magnetic resonance imaging. Cancer Res. 2003;63:5838–43. [PubMed] [Google Scholar]
- 31.Schmieder AH, Winter PM, Caruthers SD, Harris TD, Williams TA, Allen JS, et al. Molecular MR imaging of melanoma angiogenesis with alphanubeta3-targeted paramagnetic nanoparticles. Magn Reson Med. 2005;53:621–7. doi: 10.1002/mrm.20391. [DOI] [PubMed] [Google Scholar]
- 32.Hu G, Lijowski M, Zhang H, Partlow KC, Caruthers SD, Kiefer G, et al. Imaging of Vx-2 rabbit tumors with alpha(nu)beta3-integrin-targeted 111In nanoparticles. Int J Cancer. 2007;120:1951–7. doi: 10.1002/ijc.22581. [DOI] [PubMed] [Google Scholar]
- 33.Boles KS, Schmieder AH, Koch AW, Carano RA, Wu Y, Caruthers SD, et al. MR angiogenesis imaging with Robo4- vs. alphaVbeta3-targeted nanoparticles in a B16/F10 mouse melanoma model. FASEB J. 2010;24:4262–70. doi: 10.1096/fj.10-157933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kassner A, Thornhill RE, Liu F, Winter PM, Caruthers SD, Wickline SA, et al. Assessment of tumor angiogenesis: dynamic contrast-enhanced MRI with paramagnetic nanoparticles compared with Gd-DTPA in a rabbit Vx-2 tumor model. Contrast Med Mol Imaging. 2010;5:155–61. doi: 10.1002/cmmi.380. [DOI] [PubMed] [Google Scholar]
- 35.Winter PM, Morawski AM, Caruthers SD, Fuhrhop RW, Zhang H, Williams TA, et al. Molecular imaging of angiogenesis in early-stage atherosclerosis with alpha(v)beta3-integrin-targeted nanoparticles. Circulation. 2003;108:2270–4. doi: 10.1161/01.CIR.0000093185.16083.95. [DOI] [PubMed] [Google Scholar]
- 36.Cai K, Caruthers SD, Huang W, Williams TA, Zhang H, Wickline SA, et al. MR molecular imaging of aortic angiogenesis. JACC Cardiovasc Imaging. 2010;3:824–32. doi: 10.1016/j.jcmg.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan D, Pramanik M, Senpan A, Allen JS, Zhang H, Wickline SA, et al. Molecular photoacoustic imaging of angiogenesis with integrin-targeted gold nanobeacons. FASEB J. 2011;25:875–82. doi: 10.1096/fj.10-171728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kyburz D, Corr M. The KRN mouse model of inflammatory arthritis. Springer Semin Immunopathol. 2003;25:79–90. doi: 10.1007/s00281-003-0131-5. [DOI] [PubMed] [Google Scholar]
- 39.Brahn E, Schoettler N, Lee S, Banquerigo ML. Involution of collagen-induced arthritis with an angiogenesis inhibitor, PPI-2458. J Pharmacol Exp Ther. 2009;329:615–24. doi: 10.1124/jpet.108.148478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pham CT, Mitchell LM, Huang JL, Lubniewski CM, Schall OF, Killgore JK, et al. Variable antibody-dependent activation of complement by functionalized phospholipid nanoparticle surfaces. J Biol Chem. 2011;286:123–30. doi: 10.1074/jbc.M110.180760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Firestein GS. Starving the synovium: angiogenesis and inflammation in rheumatoid arthritis. J Clin Invest. 1999;103:3–4. doi: 10.1172/JCI5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koch AE. Angiogenesis as a target in rheumatoid arthritis. Ann Rheum Dis. 2003;62(Suppl. 2):ii60–70. doi: 10.1136/ard.62.suppl_2.ii60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klimiuk PA, Sierakowski S, Domyslawska I, Fiedorczyk M, Chwiecko J. Reduction of soluble adhesion molecules (sICAM-1, sVCAM-1, and sE-selectin) and vascular endothelial growth factor levels in serum of rheumatoid arthritis patients following multiple intravenous infusions of infliximab. Arch Immunol Ther Exp. 2004;52:36–42. [PubMed] [Google Scholar]
- 44.de Bandt M, Grossin M, Weber AJ, Chopin M, Elbim C, Pla M, et al. Suppression of arthritis and protection from bone destruction by treatment with TNP-470/ AGM-1470 in a transgenic mouse model of rheumatoid arthritis. Arthritis Rheum. 2000;43:2056–63. doi: 10.1002/1529-0131(200009)43:9<2056::AID-ANR17>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 45.Bernier SG, Lazarus DD, Clark E, Doyle B, Labenski MT, Thompson CD, et al. A methionine aminopeptidase-2 inhibitor, PPI-2458, for the treatment of rheumatoid arthritis. Proc Natl Acad Sci USA. 2004;101:10768–73. doi: 10.1073/pnas.0404105101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hannig G, Bernier SG, Hoyt JG, Doyle B, Clark E, Karp RM, et al. Suppression of inflammation and structural damage in experimental arthritis through molecular targeted therapy with PPI-2458. Arthritis Rheum. 2007;56:850–60. doi: 10.1002/art.22402. [DOI] [PubMed] [Google Scholar]
- 47.Davidsen J, Jorgensen K, Andresen TL, Mouritsen OG. Secreted phospholipase A(2) as a new enzymatic trigger mechanism for localised liposomal drug release and absorption in diseased tissue. Biochim Biophys Acta. 2003;1609:95–101. doi: 10.1016/s0005-2736(02)00659-4. [DOI] [PubMed] [Google Scholar]
- 48.Andresen TL, Davidsen J, Begtrup M, Mouritsen OG, Jorgensen K. Enzymatic release of antitumor ether lipids by specific phospholipase A2 activation of liposome-forming prodrugs. J Med Chem. 2004;47:1694–703. doi: 10.1021/jm031029r. [DOI] [PubMed] [Google Scholar]
- 49.Jensen SS, Andresen TL, Davidsen J, Hoyrup P, Shnyder SD, Bibby MC, et al. Secretory phospholipase A2 as a tumor-specific trigger for targeted delivery of a novel class of liposomal prodrug anticancer etherlipids. Mol Cancer Ther. 2004;3:1451–8. [PubMed] [Google Scholar]
- 50.Andresen TL, Jensen SS, Kaasgaard T, Jorgensen K. Triggered activation and release of liposomal prodrugs and drugs in cancer tissue by secretory phospholipase A2. Curr Drug Deliv. 2005;2:353–62. doi: 10.2174/156720105774370203. [DOI] [PubMed] [Google Scholar]
- 51.Peters GH, Moller MS, Jorgensen K, Ronnholm P, Mikkelsen M, Andresen TL. Secretory phospholipase A2 hydrolysis of phospholipid analogues is dependent on water accessibility to the active site. J Am Chem Soc. 2007;129:5451–61. doi: 10.1021/ja067755b. [DOI] [PubMed] [Google Scholar]
- 52.Jorgensen K, Davidsen J, Mouritsen OG. Biophysical mechanisms of phospholipase A2 activation and their use in liposome-based drug delivery. FEBS Lett. 2002;531:23–7. doi: 10.1016/s0014-5793(02)03408-7. [DOI] [PubMed] [Google Scholar]