

Abstract
When humans eat more and exercise less, they tend to become obese and unhealthy. The molecular pathways that link obesity to serious diseases like Type 2 diabetes and cardiovascular disease have become a subject of intensive scientific investigation because the exploding prevalence of obesity worldwide represents a grave new threat to the health of hundreds of millions of people. However, obesity is not always destiny. Two important clinical populations have been valuable to understand the mechanisms behind this conundrum: individuals who exhibit metabolic dysfunction, diabetes and elevated cardiovascular disease risk despite a lean body type, and individuals who are relatively protected from these dangers despite significant obesity. Study of this second group of ‘metabolically healthy obese’ people in particular has been revealing because such individuals exhibit specific, identifiable, anatomic, cellular and molecular features that set them apart from the rest of us who suffer declining health with increasing weight. Here, we examine some of these features, including some mouse models that are informative of mechanism, and suggest hypotheses for further study, including the possibility that genes and pathways of the immune system might offer new diagnostic or therapeutic targets.
1. Introduction
1.1. The problem of obesity
Human metabolism promotes a central tendency in response to high calorie diets and inactivity: obesity. Many serious diseases, such as metabolic syndrome (MS) and Type 2 diabetes (T2D) are directly attributable to elevated body mass index (BMI), so that the increased prevalence of overweight (BMI 25–29.9), obesity (BMI≥30) and morbid obesity (BMI≥40) has generated widespread alarm in the United States and internationally. Obesity also promotes cardiovascular disease (CVD), hypertension, stroke and cancer (Van Gaal et al., 2006; Guh et al., 2009). Worldwide, 1.7 billion people are currently classified as overweight (Haslam and James, 2009). Some years ago, the World Health Organization estimated that by 2030, 370 million people worldwide would be expected to be suffering from T2D, driven primarily by obesity, compared to 177 million in 2000 (Wild et al., 2004). However, this estimate appears to have been unduly conservative; midway through 2011 the number of diabetic adults worldwide reached 347 million. The same report indicates that the US now has the highest BMI of the high-income countries (Finucane et al., 2011). All US states report that the prevalence of adult obesity is at least 20%; one in three American adults are now obese. The health care costs associated with these diseases will be ruinous; the annual costs for care now account for nine percent of all medical spending: about $147 billion. No American political party, governmental or nongovernmental organization has adequately planned for the expected impact on the nation’s resources. Furthermore, demand for medical intervention is expected to grow: bariatric surgeries in America increased from 13,000 in 1990 to 100,000 in 2003 (Santry et al., 2005) and 200,000 in 2006 (Pear, 2006). In the face of this catastrophe, community-, school-, and Federally-based intervention programs to encourage better eating patterns and increased physical activity are attempting to blunt the expected collision of disease prevalence with budget and taxation realities, but concern remains grave.
2. Obesity and metabolic dysfunction
In pre-T2D obesity, specific anatomical, cellular, immunological and molecular changes are associated with progressive deterioration in metabolic function, including elevated blood glucose and reduced glucose clearance, measured in peripheral blood after oral bolus; hyperinsulinemia, insulin resistance (IR) and dyslipidemia, which contribute to MS; and increasing dysfunction of pancreatic islet β-cells that culminates in β-cell failure and insulin dependence. MS has been defined as any three or more of: fasting plasma glucose of 5.6–6.9 mmol/liter; waist circumference >102 cm (in men) or >88 cm (in women); fasting triglycerides ≥1.7 mmol/liter; high-density lipoprotein-cholesterol <1.0 mmol/liter (in men) or <1.3 mmol/liter (in women); and blood pressure≥130/85 mmHg or current treatment for hypertension (Grundy et al., 2005; Janiszewski and Ross, 2010). Insulin sensitivity has been defined by the homeostasis model [(fasting glucose × fasting insulin)/22.5)]. IR has been defined in the revised Adults Treatment Panel III (2001) as a homeostasis model assessment (HOMA) IR level in the top quartile of the distribution among subjects without diabetes (Matthews et al., 1985). Some investigators adopt a stringent measure of IR, with a value of HOMA >2 as a criterion. Nevertheless, there is no universally accepted definition of MS.
3. Features of unhealthy adipose tissue
3.1. Central obesity
It has been appreciated since 1947 that subcutaneous, or lower body obesity, typified by a ‘pear-shaped’ body distribution of adipose tissue common in women, is associated with metabolic protection (Vague, 1947), whereas central obesity, an ‘apple-shaped’ distribution common in men, is associated with MS and CVD (Lapidus et al., 1984; Donahue et al., 1987). The size of the intra-abdominal fat depot is strongly correlated with MS (Fujioka et al., 1987; Després et al., 1989) and reduced levels of an insulin-sensitizing adipokine, adiponectin (Côté et al., 2005). Visceral white adipose tissue (WAT) is correlated with hepatosteatosis and hepatic IR (Bergman et al., 2006) and is a risk factor for glucose intolerance, independent of BMI and subcutaneous depot size (Després et al., 1989; Pouliot et al., 1992). Visceral storage capacity is relatively low compared to subcutaneous. Without available storage, lipid is distributed to hepatic, cardiac, skeletal muscle and other highly undesirable locations, called ectopic fat deposition (Dubois et al., 2006), contributing to metabolic damage. Ectopic fat arises upon insufficient adipogenesis in the obese, pre-diabetic individual (Trayhurn et al., 2008). However, pro-adipogenic drugs of the thiazolidinedione (TZD) family, rosiglitazone (Avandia™), pioglitazone (Actos™) and troglitazone (Rezulin™), nicknamed ‘glitazones’, can induce WAT to expand (Tan and Vidal-Puig, 2008; Tontonoz and Spiegelman, 2008). TZDs have clinical efficacy; they stimulate peroxisome proliferator-activated receptor (PPAR)γ (Rosen et al., 1999, 2000) transcriptional programs that promote adipogenesis. However, unfavorable side effect profiles prompted the US Food and Drug Administration in 2011 to issue new restrictions on glitazones, leaving the future of this drug class uncertain.
3.2. Adipocyte stress and fibrosis
Lean and obese humans are born with a limited number of adipocytes, thus obese individuals have been thought to expand WAT through hypertrophy, not hyperplasia (Spalding et al., 2008). Adipocytes are individually ‘wrapped’ in a supporting sheath of extracellular matrix (ECM), in particular collagens. Remodeling of the ECM, and cycles of collagen breakdown/ re-deposition in particular, are essential for adipocyte and adipose tissue expansion (Chun et al., 2006). However, in the obese state, excessive, dysregulated deposition of collagens and other ECM components (i.e., fibrosis) eventually constrains adipocyte expansion, thereby promoting adipocyte stress, inflammatory/stress kinase activation and resulting AT and systemic metabolic dysfunction (Henegar et al., 2008; Halberg et al., 2009; Khan et al., 2009; Divoux et al., 2010). Collagen VI is specifically implicated in the pathogenesis of obesity-associated AT fibrosis and metabolic dysfunction, as collagen VI deposition is increased in WAT of obese, insulin resistant humans (Spencer et al., 2010), and the absence of this ECM component in obese (knockout) mice permits greater adipocyte hypertrophy while normalizing key metabolic parameters (Khan et al., 2009; see Section 5). Collagen VI deposition and AT fibrosis in human WAT is coincident with the presence of ‘alternatively activated’ (CD150+) adipose tissue macrophages (ATMs) that are known to promote ECM remodeling and wound healing (Spencer et al., 2010; see also Shaul et al., 2010).
3.3. Chronic inflammation
In diet-induced obesity, adipose tissue is often in a chronic, subclinical state of inflammation (Shoelson et al., 2006) caused by cytokines like tumor necrosis factor (TNF)-α that directly promote IR (Hotamisligil et al., 1993; Bastard et al., 2006). In both humans and rodent models of obesity, these cytokines are produced by infiltrating ATMs (Weisberg et al., 2003; Xu et al., 2003), which communicate with T cells (Kintscher et al., 2008) and B cells (discussed in this volume in detail by Nikolajczyk et al.). Obese IR humans also exhibit an elevated inflammatory profile (Kahn et al., 2006; Kim et al., 2006) that correlates well with cardiometabolic risk (Blüher, 2009, 2012) and central obesity (Lemieux et al., 2001). Immunohistochemistry of insulin-resistant adipocytes in diet-induced obesity has revealed that ATMs surround dead or dying adipocytes in “crown-like structures” (CLS) (Cinti et al., 2005; discussed in this volume in detail by Carey et al.) and promote fibrosis (Keophiphath et al., 2009) that is virtually diagnostic of metabolically unhealthy WAT. Consistent with this pattern, the larger adipocytes of IR obese individuals also express an elevated pro-inflammatory adipokine signature (Skurk et al., 2007). Interestingly, women with a common reproductive disorder, called Polycystic Ovary Syndrome (PCOS) that features anovulatory infertility, exhibit chronic, systemic low-grade inflammation that is more commonly associated with IR obesity (Tarkun et al., 2004). Aspirin was one of the first anti-inflammatory drugs recognized to have efficacy for T2D (Colwell, 1997). Indeed, sodium salicylate was used as long ago as the late 19th century to reduce blood glucose in these patients (Ebstein and Muller, 1876). Although it is well established that reduction of pro-inflammatory factors in human obesity potently mitigates insulin resistance, there is clearly an urgent need for new immunological agents to treat insulin-resistant obesity in the clinic (Nikolajczyk et al., 2012). Despite its efficacy in many cases, aspirin is far from innovative.
4. Informative exceptions to the pattern
A lay observer might be tempted to paraphrase Tolstoy and suggest that lean individuals seem to be mostly alike, whereas obese individuals are different, each in their own way. Some are apple shaped, some pear shaped, some morbidly obese, some mildly so, whereas some, like sumo wrestlers, are impressively obese but remain robustly athletic. However, not all individuals with metabolic dysfunction are obese. For example, PCOS women show impaired glucose tolerance and insulin hypersecretion, although about half of such women are lean and some are underweight (Vrbíková et al., 2004).
4.1. MONW
The phenotype of the ‘metabolically obese but normal weight’ (MONW) individual (Fig. 1), first identified by Ruderman and colleagues (Ruderman et al., 1981; Ruderman et al., 1998) and characterized by hyperinsulinemia, hyperglycemia, IR, impaired glucose tolerance, hypercholesterolemia and hypertriglyceridemia, but normal adipocyte volume and BMI, suggests that there must be genes and signal transduction pathways that ordinarily couple obesity to IR, and that these are deficient in certain individuals. These characteristics in a lean individual mark a departure from common human patterns, in which metabolic disease is a consequence of weight gain. Yet, these phenotypes are very prevalent. For example, PCOS has been diagnosed in up to 10% of one study of women of reproductive age (Goodarzi et al., 2011) and MONW in 12.7% of normal-weight subjects in a Korean study (Lee, 2009). Elevated risk for CVD is seen among both MONW (Ruderman et al., 1982) and PCOS individuals (Goodarzi et al., 2011), as well as elevated risk for hypertension, T2D and other metabolic complications. Thus, metabolic dysfunction and CVD risk come in many sizes and shapes (Fig. 1).
Fig 1.
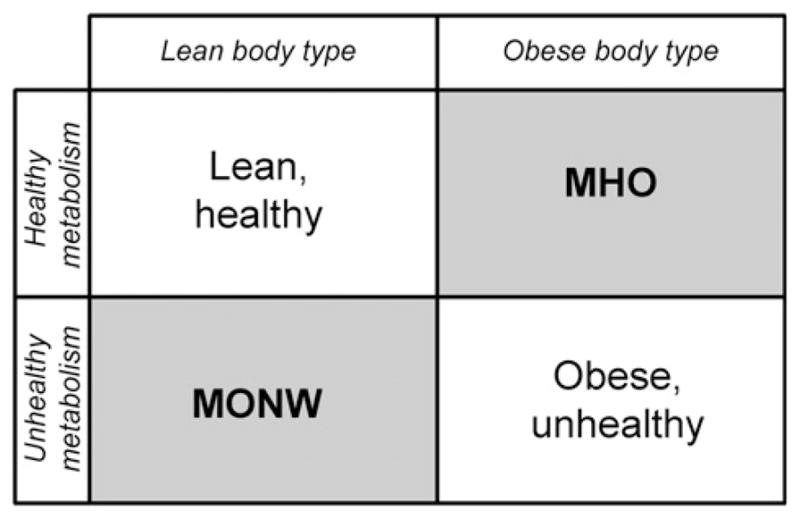
A classification model for obese and metabolic phenotypes. A ‘two-by-two’ map of the effects of obesity on health, with two off-diagonal entries.
4.2. Mouse models of the MONW individual
Recent study of Goto-Kakizaki rats (Xue et al., 2011) has shown that the Major Histocompatibility Complex (MHC) genes RT1-Ba, Bb and Db1, which are homologous to human HLA-DQA1, HLA-DQ beta 1 chain-like and HLA-DRB1, respectively, are differentially expressed in a phenotype associated with IR and increased inflammation, but a defect in pre-adipocyte differentiation and lack of obesity, which taken together resembles MONW individuals. The field would benefit from additional models of this phenotype.
4.3. MHO
Another important ‘off-diagonal’ population is characterized by certain obese individuals that exhibit a normal CVD risk profile, without increased risk for atherosclerosis, hypertension or T2D. This group, which also displays an absence of dyslipidemia, hyperuricemia and hypertension (Bonora et al., 1991; Wildman et al., 2008) has been thought of as ‘metabolically healthy obese’ (MHO, Fig. 1) (Sims, 2001). MHO adults have been defined as abdominally obese (BMI≥30 kg/m2) and lacking MS (Meigs et al., 2006). In hyperinsulinemic/euglycemic clamp studies, they demonstrate higher rates of glucose infusion than IR obese subjects, indicative of preserved whole-body glucose disposal (Klöting et al., 2010). However, there is no universally accepted definition of the MHO phenotype.
MHO individuals are also prevalent; by general agreement they comprise up to 25%, but not more than 30%, of the adult obese population. There is variance in the reported prevalence of MHO individuals: they comprised 15.2% of total subjects and 47.9% of obese subjects in a Korean study (Lee, 2009), and 11% of obese subjects in an Italian study (Calori et al., 2011). Others have concluded that the MHO phenotype does not define a discrete subpopulation of obese individuals, but is more precisely thought of as continuous distribution of preserved insulin sensitivity as a function of increasing BMI (Blüher, 2010, 2012; Klöting et al., 2010). Furthermore, it is not fully understood whether MHO individuals are genetically or epigenetically predisposed to maintain their insulin sensitivity as they age, or whether the phenotype is inherently unstable and can evolve rapidly to an IR state (Kim and Reaven, 2008). Sumo wrestlers, for example, often convert from a metabolically healthy to an unhealthy state upon retirement from the ring (Matsuzawa, 1997). At this stage, it seems wise to think of MHO as a syndrome or a cluster of traits that have some interpretive value, rather than as a clear category with specific prognostic implications.
Some commentators dislike the term ‘metabolically healthy obese’ because of the implication that these individuals are somehow normal. In fact, ultrasound studies reveal worrisome subclinical changes in certain subjects in this population, such as thickening of the intima-media of the common carotid artery and decreased flow-mediated dilation of the brachial artery, which suggests deterioration in endothelial function and nascent atherosclerosis (Oflaz et al., 2003). MHO biology is still associated with gall bladder disease, osteoarthritis and other co-morbidities (Guh et al., 2009) including cardiomyopathy. A more precise term might be ‘metabolically protected obese’ because not only are a number of deleterious factors reduced in these individuals, but other protective factors are relatively undiminished, such as serum adiponectin (Berg and Scherer, 2005; Matsuzawa, 2006). In some MHO subjects, adiponectin concentrations even exceed the values seen in normal BMI individuals (Aguilar-Salinas et al., 2008). Interestingly, a meta-analysis of PCOS women showed that both lean and obese subjects have significantly lower adiponectin than normal controls (Toulis et al., 2009), consistent with the contention that increased metabolic risk need not be a result of elevated BMI.
5. Features of the MHO individual
5.1. Reduced central obesity
The MHO phenotype is strongly associated with a smaller visceral depot, although not necessarily with expanded subcutaneous; the clamped glucose infusion rate strongly correlates with visceral WAT area (Klöting et al., 2010). Population studies suggest that propensity to accumulate fat in central or peripheral depots has a strong genetic component (Bouchard et al., 1990; Wajchenberg, 2000), thus certain features of the MHO phenotype are likely to be heritable (Després et al., 1992), including body fat distribution.
5.2. Reduced adipocyte stress
In humans, WAT fibrosis as measured by collagen VI expression is positively correlated with IR and inflammatory markers, such as the number of ATMs (Spencer et al., 2010). The relationship between stress/fibrosis and unhealthy WAT (Pasarica et al., 2009) supports a hypothesis that alleles of genes that encode different forms of collagen or enzymes that modify collagen, such as lysyl oxidase (Huang et al., 2010), correlate with the ability of WAT to expand and remodel in obesity while avoiding stress and remaining metabolically healthy. Adipocyte size per se in both omental and subcutaneous depots is also strongly negatively correlated with metabolic health (Klöting et al., 2010; Spencer et al., 2010), with smaller adipocytes and preserved insulin-sensitive glucose transport characteristic of MHO individuals. A hypothesis follows that some MHO humans will show increased adipogenesis, based on functional allelic variants of PPARγ, PGC-1α, PRDM16 or other components of the adipogenic transcriptional program that expand subcutaneous WAT. Not much clinical evidence has yet been marshaled to support this idea, although Patti et al. (2003) noted in a study of Mexican–American subjects that expression of PGC-1α and PPARγ-directed transcriptional networks are decreased in pre-diabetic and T2D obese subjects. Their metabolic dysfunction, including reduced oxidative metabolism and attenuated mitochondrial electron transport, is consistent with defective PGC-1α and PPARγ function, although primary adipogenesis was not studied in this cohort. Well matched MHO and IR obese human subjects should be evaluated for these hypothesized variants.
5.3. Reduced inflammation
MHO individuals display a reduced inflammatory profile (Karelis et al., 2005), with reduced hepatosteatosis (O’Connell et al., 2011), lower numbers of infiltrating ATMs and CLS in WAT (Klöting et al., 2010) and reduced serum levels of TNF-α, monocyte chemotactic protein-1 (O’Connell et al., 2011), interleukin (IL)-6 and C-reactive protein (Klöting et al., 2010). Elevated serum adiponectin and reduced ATM infiltration are the strongest predictors of preserved ability to clear glucose (Combs et al., 2004; Klöting et al., 2010); in men, preserved adiponectin levels are associated with a reduced rate of myocardial infarction (Pischon et al., 2004). Adiponectin also promotes protective (M2) macrophage differentiation (Ohashi et al., 2010). The mechanistic details of the ways in which macrophages, T cells, B cells and adipocytes respond to dietary interventions, bariatric surgery or drug treatment for IR, or can be mobilized for therapeutic benefit, are very poorly understood which has provoked active inquiry (Nikolajczyk et al., 2012). Study of MHO individuals is likely to reveal critical new principles for how their specific anatomical, cellular, immunological and molecular features (Succurro et al., 2008) protect them from T2D and CVD.
6. Murine models of the MHO individual: fatter but fitter mice
Mouse models of obesity have provided robust insights into human obesity and its metabolic complications, including metabolically protected obesity. In this section we highlight genetic models in which murine obesity is associated with a salutary profile of glucose–insulin homeostasis and adipocyte/AT function. The models selected are representative of five (overlapping) categories of proximate physiological effect: (1) altered expression of adipokines or inflammatory mediators, (2) disrupted inflammatory signal transduction, (3) reduction or enhancement of AT immune cell populations, (4) attenuated adipocyte stress, and (5) enhanced adipogenesis and/or adipocyte lipogenesis. Intriguingly, in several of these models, the MHO phenotype is associated with greater obesity, including greater hypertrophic obesity. Because weight loss per se is metabolically beneficial, we do not discuss the myriad examples in which metabolic protection is associated with reduced adiposity or body weight, including those in which metabolic improvements in obese mice are obtained following ablation or supplementation of specific innate or adaptive immune cell populations (see articles by Lumeng and by Snyder–Cappione and Nikolajczyk, this volume).
6.1. Adiponectin transgenic mouse
Adiponectin is an anti-inflammatory, insulin-sensitizing adipokine expressed exclusively by adipocytes that exerts beneficial effects on lipid and glucose homeostasis via multiple mechanisms, most notably via ceramidase activity and AMPK/ SIRT1-dependent activation of PGC-1α (Iwabu et al., 2010; Holland et al., 2011). Elevated (~2-fold) circulating adiponectin is a hallmark and predictor of the MHO phenotype in humans (Klöting et al., 2010). Adiponectin transgenic mice on an ob/ob background (AdTg) have 2- to 3-fold higher levels of circulating adiponectin than ob/ob controls (Kim et al., 2007). These AdTg mice become extraordinarily obese on a normal ‘chow’ diet, weighing on average 40 g more than ob/ob mice. This extreme obesity largely reflects preferential hyperplastic expansion of subcutaneous AT depots, although visceral depots and adipocytes are also reduced in size to wild-type (ob/+) levels. Remarkably, despite morbid obesity, AdTg mice remain insulin sensitive and glucose tolerant, have fewer AT macrophages and inflammatory markers, less liver steatosis and improved circulating insulin, glucose and triacylglycerol. Thus, adiponectin overexpression results in a hyper-obese mouse with predominantly subcutaneous fat, smaller adipocytes, attenuated inflammation and metabolic protection – i.e., all hallmark features of human MHO.
Significant effort is currently directed toward the development of novel therapeutics that elevate production and secretion of the metabolically effective, high molecular weight (HMW) form of adiponectin. Some success has been reported with natriuretic peptides (Tsukamoto et al., 2009) and biflavonoids (Liu et al., 2007). More recently overexpression of the disulfide bond A oxidoreductase-like protein in fat (fDsbA-L) was reported to increase levels of total and HMW adiponectin and to confer resistance to obesity-associated IR through these effects on adiponectin expression (Liu et al., 2012).
6.2. Brd2 hypomorph
The Brd2 gene is located near the TNF-α locus in the class II MHC region of human chromosome 6. Recent work has shown that a leaky knockout of this gene in mice produces a hypomorphic phenotype characterized by extreme obesity coincident with elevated adiponectin (nearing levels of male AdTg mice) and protection from IR (Wang et al., 2009). These mice show hepatosteatosis and hyperinsulinemia, but hypoglycemia and better clamped glucose infusion rates than wild type (Wang et al., 2012; Jornayvaz et al., forthcoming). Brd2 ordinarily co-represses PPARγ-dependent transcription, thus reduced Brd2 in the animals resembles TZD treatment and adipogenesis is potentiated (Denis et al., 2010). Reduced expression of Brd2 also creates a low-inflammatory environment (Belkina and Denis, 2010, 2012; Belkina et al., 2010; Belkina et al., forthcoming) that protects against cytokines that would ordinarily cause IR in adipocytes. Endotoxin-stimulated production of pro-inflammatory cytokines such as TNF-α, IL-1β and IL-6 from bone marrow-derived macrophages is reduced by about half, compared to wild type controls, establishing a ‘protective deficiency’. In MHO humans, it is not yet clear if certain alleles in inflammatory loci create such mild hypo-responsiveness to inflammation signal transduction to confer protection. Consideration of the association among MHC alleles and complex human diseases with an inflammatory component provokes the hypothesis that certain MHO individuals are protected from adipose tissue inflammation and metabolic complications because of specific alleles in their MHC. Several genes in the MHC are likely candidates, including LTA, which encodes lymphotoxin-α and is important for the inflammatory component of coronary artery disease (Ozaki et al., 2002); TNF, BRD2 and HLA-DRB1. These alleles should be evaluated for correlation with the MHO phenotype.
6.3. COL6 KO mice
As discussed above, recent studies suggest that excessive deposition of collagen(s) or other ECM components (fibrosis) promotes inflammatory and metabolic pathology in individuals with hypertrophic obesity. In a seminal study, collagen VI (COL6) KO mice crossed onto the genetically obese ob/ob background developed dramatically-hypertrophic adipocytes and greater AT mass in both the gonadal and mesenteric depots, but had lower fasting glucose, enhanced glucose tolerance and reduced circulating triacylglycerol following lipid challenge as compared with control ob/ob mice (Khan et al., 2009). This metabolic protection was associated with reductions in ER stress markers, stress kinase (JNK, ERK) activation and adipocyte death, as well as with lower systemic IL-6 and TNF-α following LPS challenge. Thus, reducing AT collagen promotes the development of metabolically ‘protective’ adipocyte hypertrophy and an MHO phenotype. A plausible interpretation is that the (hyper)obese COL6 KO mouse uncouples hypertrophy per se from obesity complications and suggests that therapeutic approaches that reduce adipocyte stress can maintain metabolically healthy fat storage in enlarged adipocytes.
6.4. TBP-2/Txnip/VDUP1 KO mice
Thioredoxin binding protein (TBP)-2 is a member of the α-arrestin protein family with demonstrated roles in the regulation of cell fate, immune responses and energy metabolism. TBP-2 is a negative regulator of adipogenesis, in part through its inhibitory actions on PPARγ expression and activity (Chutkow et al., 2010; Yoshihara et al., 2010; Chutkow and Lee, 2011; see also Chen et al., 2008). Consistent with these observations, TBP-2 KO mice fed either a normal or HFD diet or TBP-2 KO mice on an ob/ob background consumed more energy and gained significantly more weight (up to 100%) and adiposity (up to 50%) than similarly-fed WT or ob/ob mice (Chutkow et al., 2010; Yoshihara et al., 2010). Notably, greater adipose mass reflected adipocyte hyperplasia in multiple AT depots (Chutkow et al., 2010). Despite greater adiposity, both models of obesity in TBP-2 KO mice were more glucose tolerant and insulin sensitive than obese controls, reflecting augmented glucose transport in AT and skeletal muscle and improved glucose-stimulated insulin secretion (GSIS). Thus, as in AdTg mice (see above) adipose accretion by adipocyte hyperplasia is associated with an MHO phenotype in obese TBP-2 KO mice. HBC-19 mice that express a naturally-occurring TBP-2 truncation mutation also become more obese than WT mice but retain glucose tolerance and insulin responsiveness (Chen et al., 2008). Moreover, HcB-19 mice crossed with the ob/ob mice become even more obese than ob/ob mice, but are protected against peripheral insulin resistance, β-cell apoptosis and subsequent T2DM. The protection from β-cell apoptosis despite morbid obesity in mice lacking functional TBP-2 was demonstrated in β cell-specific TBP-2 KO mice to reflect enhanced Akt/Bcl-xL signaling and associated mitochondrial protection (Chen et al., 2008).
6.5. Tumor-progression locus 2 (TPL2) KO mice
TPL2 (MAP3K8) is a serine/threonine kinase that is activated by TNFα, TLR4-signaling and inflammatory mediators that activate the NFκB and MAPK pathways (Vougioukalaki et al., 2011). Thus, TPL2 is uniquely situated to integrate inflammatory signaling pathways that promote obesity-associated inflammation and IR. Perfield et al. (2011) placed TPL2 KO mice on a HFD for 16 weeks and reported increases in body weight, adipose depot weights and gonadal adipocyte size comparable to those observed in HFD-fed WT mice. However, HFD-fed KO mice had decreased fasting glucose and insulin, and improved glucose dynamics in euglycemic/hyperinsulinemic clamp studies. This protected metabolic phenotype was associated with reduced MAPK (ERK, JNK) activation in peripheral tissues, as well as with reduced frequency of CLS and attenuated inflammatory gene expression in the epididymal fat depot. Thus, the absence of TPL2-dependent inflammatory signaling confers an MHO phenotype of reduced AT inflammation and enhanced insulin sensitivity despite hypertrophic obesity.
6.6. Apoptosis inhibitor of macrophage (AIM) KO mice
AIM (also known as Spa, Api6, and CD5L) is a macrophage-secreted member of the scavenger receptor cysteine-rich superfamily (SRCR-SF). AIM expression and serum levels are upregulated in murine obesity (reviewed in Miyazaki et al., 2011). Macrophage-derived AIM is taken up by adipocytes via CD36-mediated endocytosis. AIM inhibits fatty acid synthase, thereby stimulating lipolysis and the release of fatty acids (Kurokawa et al., 2010). This release of fatty acids activates adipocyte chemokine production via TLR4 (Kurokawa et al., 2011). The pathophysiologic impact of AIM on inflammation and obesity complications was assessed in AIM-deficient (AIM KO) mice (Kurokawa et al., 2011). AIM KO mice fed a HFD for 12 weeks became more obese than WT mice. However, greater adiposity was associated with fewer M1 macrophages, reflecting reductions in AT chemokine expression. Systemic protection was also evident, including reduced circulating TNFα, IL-6 and IL-1β, enhanced insulin-dependent phosphorylation of Akt and GSK3β in peripheral tissues and improved GTT and ITT performance (Kurokawa et al., 2011). Thus, despite greater adiposity, impaired recruitment of M1 macrophages into AT protects AIM KO mice from the inflammatory and metabolic complications of obesity.
7. Future directions
7.1. MHC genes and metabolic risk
It is well known that alleles in the class I and II MHC, which is located in humans on the short arm of Chromosome 6 at p21.3, are linked to Type 1 diabetes, specifically through the mobilization of autoimmune processes during childhood (Grant and Hakonarson, 2009). Here, the β-cells of pancreatic islets are destroyed by autoreactive T cells, leading directly to irreversible, insulin-dependent diabetes, but the details of this process are beyond the scope of the present discussion of obesity and T2D. Many autoimmune and inflammatory diseases are associated with the class II MHC (de Bakker et al., 2006), particularly inflammatory bowel disease (IBD), rheumatoid arthritis (RA), lupus and ankylosing spondylitis (Handunnetthi et al., 2010). Reports continue to emerge that emphasize new significant associations, such as most recently, between multiple sclerosis and HLA-DRB1 alleles (Sawcer et al., 2011). Study of this region for correlation of diseases with specific genes has been particularly difficult because it is the most polymorphic region of the human genome; numerous inflammation-relevant alleles exhibit non-random association (linkage disequilibrium). Certain class II MHC genes that are implicated in IBD and RA, such as TNF (also at 6p21.3), are also clearly important for IR and T2D.
Genome-wide association studies of Pima Indians (Malhotra et al., 2011), an aboriginal American people who have the highest prevalence of T2D in the world, have shown a significant association between the BRD2 SNP rs12216336, with a risk allele frequency of 0.87 and BMI. Individuals homozygous for the risk allele have a mean BMI that is 4 kg/m2 higher than individuals homozygous for the non-risk allele, supporting the hypothesis that BRD2 is an obesity susceptibility gene in this population (Muller et al., 2011). Recent results link Brd2 levels directly to regulation of transcriptional activity of the Ins1 promoter in mouse embryonic stem cells (Wang et al., 2012), suggesting that Brd2 regulates insulin production, insulin sensitivity and chronic inflammation in insulin-resistant obesity. Reinforcing this concept, meta-analyses of the literature have concluded that there is likely an association among inflammatory markers, obesity/IR and certain inflammatory diseases, such as RA. Patients with RA exhibit elevated risk for IR, T2D and CVD (Wasko et al., 2011), likely mediated through elevated TNF-α. Interestingly, BRD2 polymorphism has also been independently linked to RA (Mahdi et al., 2009). Elevated serum inflammatory profiles, reduced adiponectin (Ozgen et al., 2010) and IR likely co-vary and contribute to common comorbidities, such as CVD and atherosclerosis that is prevalent among obese IR patients (Mangge et al., 2010; Westlake et al., 2011).
Certain individuals with this ‘pro-inflammatory phenotype’, who are prone to a cluster of diseases of chronic inflammation (e.g. CVD, IBD, RA), would be predicted upon the development of overweight or obesity to exhibit higher relative risk for T2D and obesity-associated cancers, for example, than the mean risk of the obese population (Denis, 2010; Belkina and Denis, 2012; Denis and Bowen, 2013). If the population distribution of inflammatory responses is symmetrical and two-tailed, as many as 18 million Americans could be included in this ‘at-risk’ cohort, but the relevant biomarkers are unknown. Three hypothetical relationships are summarized in Fig. 2. To address such a question, it would be crucial therefore to stratify obese populations by inflammatory markers and IR, and compare their co-morbidity incidence. Thus, there is clear need for deeper analysis of the MHC, with the translational goal of developing an intervention component to address patients at the greatest risk for multiple serious conditions that build on their underlying inflammatory status.
Fig 2.
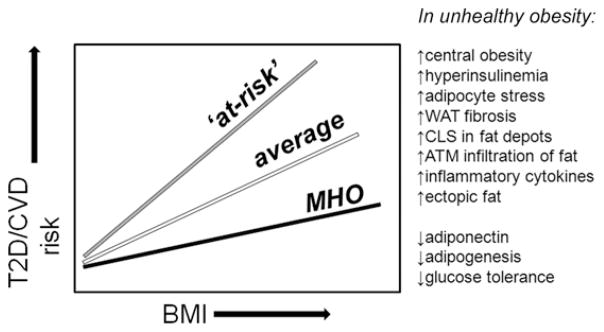
Three populations of humans or mouse models define risk profiles for obesity-driven metabolic dysfunction. As BMI increases, the risks of T2D and CVD increase, but less steeply for MHO individuals than for the general population. MHO individuals are characterized by less serious increases in central obesity, adipocyte stress and other factors. On the other hand, this model predicts that some individuals will experience greater risk than the general population; these individuals (‘at-risk’) may have predisposition to chronic inflammatory disease, hyper-reactive T cell function, or unresolved co-morbidity such as rheumatoid arthritis, lupus or other autoimmune disorders. ATM; adipose tissue macrophage; BMI, body mass index; CLS, ‘crown-like’ structures; CVD, cardiovascular disease; MHO, metabolically healthy obese; T2D, Type 2 diabetes; WAT, white adipose tissue.
7.2. Shared chromatin complexes integrate apparently unrelated diseases
Biochemical crosstalk likely links certain diseases discussed above through a limited, shared subset of chromatin-directed co-regulators, such as the double bromodomain protein family (Denis, 2010; Belkina and Denis, 2012). An overall hypothesis that integrates many of the phenomena discussed is presented in Fig. 3. Manipulation of this single chromatin-directed co-regulator Brd2 reveals that transcriptional networks are related through shared molecular complexes, much the same way that SWI/SNF chromatin remodeling complexes both activate and repress related sets of genes. Reduced action of Brd2 alleviates of Brd2 co-repression of PPARγ and insulin gene transcription, which is pro-adipogenic and shifts energy balance towards storage (Fig. 3B), while simultaneously ablating both inflammation and cancer through reduced transcription of inflammatory cytokine genes and cell cycle genes (Belkina and Denis, 2012; Denis and Bowen, 2013). Thus, this hypothesis predicts that the MHO phenotype will be associated with reduced occurrence or progression of cancer for the obesity-associated malignancies (Chadid et al., forthcoming).
Fig 3.
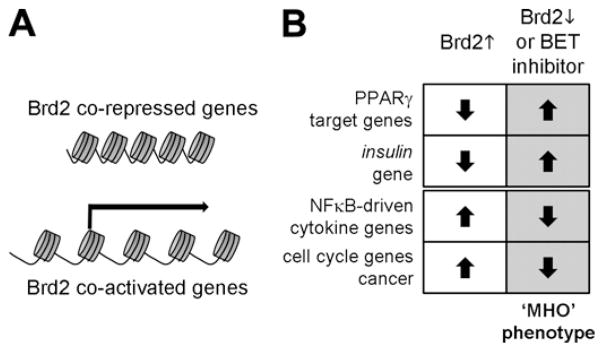
Hypothesis for crosstalk among Brd2-regulated genes. (A) Certain genes are transcriptionally co-repressed by Brd2, such as PPARγ target genes (Wang et al., 2009) and the insulin-1 gene (Wang et al., 2012), whereas other genes are transcriptionally co-activated by Brd2, such as cyclin A (Sinha et al., 2005), which causes cell cycle acceleration and cancer (Greenwald et al., 2004) and pro-inflammatory cytokine genes (Belkina et al., 2010; Belkina et al., forthcoming). (B) Brd2 action permits the MHO phenotype to be interpreted as a coherent transcriptional phenotype of increased adipogenesis (Wang et al., 2009; Belkina and Denis, 2010), reduced inflammation and reduced obesity-associated cancer (Belkina and Denis, 2012; Chadid et al., forthcoming; Denis and Bowen, 2013).
Acknowledgments
GVD is the 2011–2012 Chair of Basic Science Section of The Obesity Society and gratefully acknowledges the Society for its strong support and promotion of scientific interactions and collaborations. Drs. C. Apovian, B. Corkey, S. Fried, and B. Nikolajczyk are particularly acknowledged for valuable discussion related to this work, which is supported by the National Institutes of Health (R56 DK090455), and the Boston University Clinical and Translational Science Institute (UL1-TR000157).
Abbreviations
- AIM
apoptosis inhibitor of macrophage
- ATM
adipose tissue macrophage
- BMI
body mass index
- CLS
crown-like structure
- CVD
cardiovascular disease
- ECM
extracellular matrix
- HOMA
homeostasis model assessment
- IBD
inflammatory bowel disease
- IL
interleukin
- IR
insulin resistance
- MHC
major histocompatibility complex
- MHO
metabolically healthy obese
- MONW
metabolically obese normal weight
- MS
metabolic syndrome
- PCOS
polycystic ovary syndrome
- PPAR
peroxisome proliferator-activated receptor
- RA
rheumatoid arthritis
- TBP
thioredoxin binding protein
- TPL2
tumor-progression locus 2
- T2D
Type 2 diabetes
- TNF
tumor necrosis factor
- TZD
thiazolidinedione
- WAT
white adipose tissue
Footnotes
Disclosure
The authors report no conflicts of interest.
Contributor Information
Gerald V. Denis, Email: gdenis@bu.edu.
Martin S. Obin, Email: Martin.obin@tufts.edu.
References
- Aguilar-Salinas CA, García EG, Robles L, Riaño D, Ruiz-Gomez DG, García-Ulloa AC, Melgarejo MA, Zamora M, Guillen-Pineda LE, Mehta R, Canizales-Quinteros S, Tusie Luna MT, Gómez-Pérez FJ. High adiponectin concentrations are associated with the metabolically healthy obese phenotype. J Clin Endocrinol Metab. 2008;93:4075–4079. doi: 10.1210/jc.2007-2724. [DOI] [PubMed] [Google Scholar]
- Bastard JP, Maachi M, Lagathu C, Kim MJ, Caron M, et al. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. 2006;17:4–12. [PubMed] [Google Scholar]
- Belkina AC, Blanton W, Wang F, Liu H, Denis GV. Whole body Brd2 deficiency protects obese mice from insulin resistance by creating a low inflammatory environment. Obesity. 2010;18:S58. [Google Scholar]
- Belkina AC, Denis GV. Obesity genes and insulin resistance. Curr Opin Endocrinol Diabetes Obes. 2010;17:472–477. doi: 10.1097/MED.0b013e32833c5c48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12:465–477. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96:939–949. doi: 10.1161/01.RES.0000163635.62927.34. [DOI] [PubMed] [Google Scholar]
- Bergman RN, Kim SP, Catalano KJ, Hsu IR, Chiu JD, Kabir M, Hucking K, Ader M. Why visceral fat is bad: mechanisms of the metabolic syndrome. Obesity (Silver Spring) 2006;14 (Suppl 1):16S–19S. doi: 10.1038/oby.2006.277. [DOI] [PubMed] [Google Scholar]
- Blüher M. Adipose tissue dysfunction in obesity. Exp Clin Endocrinol Diabetes. 2009;117:241–250. doi: 10.1055/s-0029-1192044. [DOI] [PubMed] [Google Scholar]
- Blüher M. The distinction of metabolically ‘healthy’ from ‘unhealthy’ obese individuals. Curr Opin Lipidol. 2010;21:38–43. doi: 10.1097/MOL.0b013e3283346ccc. [DOI] [PubMed] [Google Scholar]
- Blüher M. Are there still healthy obese patients? Curr Opin Endocrinol Diabetes Obes. 2012;19:341–346. doi: 10.1097/MED.0b013e328357f0a3. [DOI] [PubMed] [Google Scholar]
- Bonora E, Willeit J, Kiechl S, Oberhollenzer F, Egger G, Bonadonna R, Muggeo M. U-shaped and J-shaped relationships between serum insulin and coronary heart disease in the general population. The Bruneck study. Diabetes Care. 1991;21:221–230. doi: 10.2337/diacare.21.2.221. [DOI] [PubMed] [Google Scholar]
- Bouchard C, Tremblay A, Després JP, Nadeau A, Lupien PJ, Thériault G, Dussault J, Moorjani S, Pinault S, Fournier G. The response to long-term overfeeding in identical twins. N Engl J Med. 1990;322:1477–1482. doi: 10.1056/NEJM199005243222101. [DOI] [PubMed] [Google Scholar]
- Calori G, Lattuada G, Piemonti L, Garancini MP, Ragogna F, Villa M, Mannino S, Crosignani P, Bosi E, Luzi L, Ruotolo G, Perseghin G. Prevalence, metabolic features, and prognosis of metabolically healthy obese Italian individuals: the Cremona study. Diabetes Care. 2011;34:210–215. doi: 10.2337/dc10-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Hui ST, Couto FM, Mungrue IN, Davis DB, Attie AD, Lusis AJ, Davis RA, Shalev A. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB J. 2008;22:3581–3594. doi: 10.1096/fj.08-111690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TH, Hotary KB, Sabeh F, Saltiel AR, Allen ED, Weiss SJ. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell. 2006;125:577–591. doi: 10.1016/j.cell.2006.02.050. [DOI] [PubMed] [Google Scholar]
- Chutkow WA, Birkenfeld AL, Brown JD, Lee HY, Frederick DW, Yoshioka J, Patwari P, Kursawe R, Cushman SW, Plutzky J, Shulman GI, Samuel VT, Lee RT. Deletion of the alpha-arrestin protein Txnip in mice promotes adiposity and adipogenesis while preserving insulin sensitivity. Diabetes. 2010;59 (6):1424–1434. doi: 10.2337/db09-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chutkow WA, Lee RT. Thioredoxin regulates adipogenesis through thioredoxin-interacting protein (Txnip) protein stability. J Biol Chem. 2011;286:29139–29145. doi: 10.1074/jbc.M111.267666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, Wang S, Greenberg AS, Obin MS. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- Colwell JA. Multifactorial aspects of the treatment of the type II diabetic patient. Metabolism. 1997;46 (12 Suppl 1):1–4. doi: 10.1016/s0026-0495(97)90308-5. [DOI] [PubMed] [Google Scholar]
- Combs TP, Pajvani UB, Berg AH, Lin Y, Jelicks LA, Laplante M, Nawrocki AR, Rajala MW, Parlow AF, Cheeseboro L, Ding YY, Russell RG, Lindemann D, Hartley A, Baker GR, Obici S, Deshaies Y, Ludgate M, Rossetti L, Scherer PE. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology. 2004;145:367–383. doi: 10.1210/en.2003-1068. [DOI] [PubMed] [Google Scholar]
- Côté M, Mauriège P, Bergeron J, Alméras N, Tremblay A, Lemieux I, Després JP. Adiponectinemia in visceral obesity: impact on glucose tolerance and plasma lipoprotein and lipid levels in men. J Clin Endocrinol Metab. 2005;90:1434–1439. doi: 10.1210/jc.2004-1711. [DOI] [PubMed] [Google Scholar]
- de Bakker PI, McVean G, Sabeti PC, Miretti MM, Green T, Marchini J, Ke X, Monsuur AJ, Whittaker P, Delgado M, Morrison J, Richardson A, Walsh EC, Gao X, Galver L, Hart J, Hafler DA, Pericak-Vance M, Todd JA, Daly MJ, et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet. 2006;38:1166–1172. doi: 10.1038/ng1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis GV. Bromodomain coactivators in cancer, obesity, Type 2 diabetes, and inflammation. Discovery Med. 2010;10:489–499. [PMC free article] [PubMed] [Google Scholar]
- Denis GV, Nikolajczyk BS, Schnitzler GR. An emerging role for bromodomain-containing proteins in chromatin regulation and transcriptional control of adipogenesis. FEBS Lett. 2010;584:3260–3268. doi: 10.1016/j.febslet.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis GV, Bowen DJ. Uncoupling obesity from cancer: bromodomain co-regulators that control networks of inflammatory genes. In: Dannenberg AJ, Berger NA, editors. Energy Balance and Cancer, Obesity, Inflammation and Cancer. Vol. 7. Springer; New York: 2013. (Expected 2013) [Google Scholar]
- Després JP, Nadeau A, Tremblay A, Ferland M, Moorjani S, Lupien PJ, Thériault G, Pinault S, Bouchard C. Role of deep abdominal fat in the association between regional adipose tissue distribution and glucose tolerance in obese women. Diabetes. 1989;38:304–309. doi: 10.2337/diab.38.3.304. [DOI] [PubMed] [Google Scholar]
- Després JP, Moorjani S, Lupien PJ, Tremblay A, Nadeau A, Bouchard C. Genetic aspects of susceptibility to obesity and related dyslipidemias. Mol Cell Biochem. 1992;113:151–169. doi: 10.1007/BF00231535. [DOI] [PubMed] [Google Scholar]
- Divoux A, Tordjman J, Lacasa D, Veyrie N, Hugol D, Aissat A, et al. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes. 2010;59:2817–2825. doi: 10.2337/db10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue RP, Abbott RD, Bloom E, Reed DM, Yano K. Central obesity and coronary heart disease in men. Lancet. 1987;1:821–824. doi: 10.1016/s0140-6736(87)91605-9. [DOI] [PubMed] [Google Scholar]
- Dubois SG, Heilbronn LK, Smith SR, Albu JB, Kelley DE, Ravussin E. Look AHEAD Adipose Research Group, Decreased expression of adipogenic genes in obese subjects with type 2 diabetes. Obesity (Silver Spring) 2006;(1):1543–1552. doi: 10.1038/oby.2006.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebstein W, Muller J. Weitere mitteillungen uber die behandlung des diabetes mellitus mot carbisaure nebst bemerkunger uber die anwendung der salicylsaure bie dieser krankheit. Berl Klin Wochenschr. 1876;13:53–56. [Google Scholar]
- Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ, Singh GM, Gutierrez HR, Lu Y, Bahalim AN, Farzadfar F, Riley LM, Ezzati M. Global burden of metabolic risk factors of chronic diseases collaborating group (body mass index). National, regional, and global trends in body-s index since 1980 systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9·1 million participants. Lancet. 2011;377:557–567. doi: 10.1016/S0140-6736(10)62037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka S, Matsuzawa Y, Tokunaga K, Tarui S. Contribution of intra-abdominal fat accumulation to the impairment of glucose and lipid metabolism in human obesity. Metabolism. 1987;36:54–59. doi: 10.1016/0026-0495(87)90063-1. [DOI] [PubMed] [Google Scholar]
- Goodarzi MO, Dumesic DA, Chazenbalk G, Azziz R. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nat Rev Endocrinol. 2011;7:219–231. doi: 10.1038/nrendo.2010.217. [DOI] [PubMed] [Google Scholar]
- Grant SF, Hakonarson H. Genome-wide association studies in type 1 diabetes. Curr Diabetes Rep. 2009;9:157–163. doi: 10.1007/s11892-009-0026-5. [DOI] [PubMed] [Google Scholar]
- Greenwald RJ, Tumang JR, Sinha A, Currier N, Cardiff RD, Rothstein TL, Faller DV, Denis GV. Eμ-BRD2 transgenic mice develop B cell lymphoma and leukemia. Blood. 2004;103:1475–1484. doi: 10.1182/blood-2003-06-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC, Jr, Spertus JA, Costa F. American Heart Association, diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–2752. doi: 10.1161/CIRCULATIONAHA.105.169404. Erratum in: Circulation 112, e297, e298. [DOI] [PubMed] [Google Scholar]
- Guh DP, Zhang W, Bansback N, Amarsi Z, Birmingham CL, Anis AH. The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health. 2009;9:88. doi: 10.1186/1471-2458-9-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, Brekken RA, Scherer PE. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol. 2009;29:4467–4483. doi: 10.1128/MCB.00192-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handunnetthi L, Ramagopalan SV, Ebers GC, Knight JC. Regulation of major histocompatibility complex class II gene expression, genetic variation and disease. Genes Immun. 2010;11:99–112. doi: 10.1038/gene.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslam DW, James WP. Obesity. Lancet. 2009;366:1197–1209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- Henegar C, Tordjman J, Achard V, Lacasa D, Cremer I, Guerre-Millo M, Poitou C, Basdevant A, Stich V, Viguerie N, Langin D, Bedossa P, Zucker JD, Clement K. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol. 2008;9:R14. doi: 10.1186/gb-2008-9-1-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, Wade MR, Tenorio VM, Kuo MS, Brozinick JT, Zhang BB, Birnbaum MJ, Summers SA, Scherer PE. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011;17 (1):55–63. doi: 10.1038/nm.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Huang Y, Yan X, Zhu MJ, McCormick RJ, Ford SP, Nathanielsz PW, Du M. Enhanced transforming growth factor-beta signaling and fibrogenesis in ovine fetal skeletal muscle of obese dams at late gestation. Am J Physiol Endocrinol Metab. 2010;298:E1254–E1260. doi: 10.1152/ajpendo.00015.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata M, Ogata H, Kubota N, Takamoto I, Hayashi YK, Yamauchi N, Waki H, Fukayama M, Nishino I, Tokuyama K, Ueki K, Oike Y, Ishii S, Hirose K, Shimizu T, Touhara K, Kadowaki T. Adiponectin and adipor1 regulate PGC-1a and mitochondria by Ca2+ and AMPK/SIRT1. Nature. 2010;464:1313–1319. doi: 10.1038/nature08991. [DOI] [PubMed] [Google Scholar]
- Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. International multiple sclerosis genetics consortium; wellcome trust case control consortium 2. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiszewski PM, Ross R. Effects of weight loss among metabolically healthy obese men and women. Diabetes Care. 2010;33:1957–1959. doi: 10.2337/dc10-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn SE, Zinman B, Haffner SM, O’Neill MC, Kravitz BG, Yu D, Freed MI, Herman WH, Holman RR, Jones NP, Lachin JM, Viberti GC. Diabetes. Vol. 55. ADOPT Study Group; 2006. Obesity is a major determinant of the association of C-reactive protein levels and the metabolic syndrome in type 2 diabetes; pp. 2357–2364. [DOI] [PubMed] [Google Scholar]
- Karelis AD, Faraj M, Bastard JP, St-Pierre DH, Brochu M, Prud’homme D, Rabasa-Lhoret R. The metabolically healthy but obese individual presents a favorable inflammation profile. J Clin Endocrinol Metab. 2005;90:4145–4150. doi: 10.1210/jc.2005-0482. [DOI] [PubMed] [Google Scholar]
- Keophiphath M, Achard V, Henegar C, Rouault C, Clément K, Lacasa D. Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Mol Endocrinol. 2009;23:11–24. doi: 10.1210/me.2008-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, Scherer PE. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;29:1575–1591. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CS, Park HS, Kawada T, Kim JH, Lim D, Hubbard NE, Kwon BS, Erickson KL, Yu R. Circulating levels of MCP-1 and IL-8 are elevated in human obese subjects and associated with obesity-related parameters. Int J Obes (London) 2006;30:1347–1355. doi: 10.1038/sj.ijo.0803259. [DOI] [PubMed] [Google Scholar]
- Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ, Scherer PE. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Investig. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Reaven GM. Insulin resistance and hyperinsulinemia: you can’t have one without the other. Diabetes Care. 2008;31:1433–1438. doi: 10.2337/dc08-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintscher U, Hartge M, Hess K, Foryst-Ludwig A, Clemenz M, Wabitsch M, Fischer-Posovszky P, Barth TF, Dragun D, Skurk T, Hauner H, Blüher M, Unger T, Wolf AM, Knippschild U, Hombach V, Marx N. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28:1304–1310. doi: 10.1161/ATVBAHA.108.165100. [DOI] [PubMed] [Google Scholar]
- Klöting N, Fasshauer M, Dietrich A, Kovacs P, Schön MR, Kern M, Stumvoll M, Blüher M. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab. 2010;299:E506–E515. doi: 10.1152/ajpendo.00586.2009. [DOI] [PubMed] [Google Scholar]
- Kurokawa J, Arai S, Nakashima K, Nagano H, Nishijima A, Miyata K, Ose R, Mori M, Kubota N, Kadowaki T, Oike Y, Koga H, Febbraio M, Iwanaga T, Miyazaki T. Macrophage-derived AIM is endocytosed into adipocytes and decreases lipid droplets via inhibition of fatty acid synthase activity. Cell Metab. 2010;11:479–492. doi: 10.1016/j.cmet.2010.04.013. [DOI] [PubMed] [Google Scholar]
- Kurokawa J, Nagano H, Ohara O, Kubota N, Kadowaki T, Arai S, Miyazaki T. Apoptosis inhibitor of macrophage (AIM) is required for obesity-associated recruitment of inflammatory macrophages into adipose tissue. Proc Natl Acad Sci USA. 2011;108:12072–12077. doi: 10.1073/pnas.1101841108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidus L, Bengtsson C, Larsson B, Pennert K, Rybo E, Sjöström L. Distribution of adipose tissue and risk of cardiovascular disease and death: a 12 year follow-up of participants in the population study of women in Gothenburg. Sweden Br Med J (Clin Res Ed) 1984;289:1257–1261. doi: 10.1136/bmj.289.6454.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. Metabolically obese but normal weight (MONW) and metabolically healthy but obese (MHO) phenotypes in Koreans: characteristics and health behaviors. Asia Pac J Clin Nutr. 2009;18:280–284. [PubMed] [Google Scholar]
- Lemieux I, Pascot A, Prud’homme D, Alméras N, Bogaty P, Nadeau A, Bergeron J, Després JP. Elevated C-reactive protein: another component of the atherothrombotic profile of abdominal obesity. Arterioscler Thromb Vasc Biol. 2001;21:961–967. doi: 10.1161/01.atv.21.6.961. [DOI] [PubMed] [Google Scholar]
- Liu G, Grifman M, Macdonald J, Moller P, Wong-Staal F, Li QX. Isoginkgetin enhances adiponectin secretion from differentiated adiposarcoma cells via a novel pathway involving AMP-activated protein kinase. J Endocrinol. 2007;194:569–578. doi: 10.1677/JOE-07-0200. [DOI] [PubMed] [Google Scholar]
- Liu M, Xiang R, Wilk SA, Zhang N, Sloane LB, Azarnoush K, Zhou L, Chen H, Xiang G, Walter CA, Austad SN, Musi N, Defronzo RA, Asmis R, Scherer PE, Dong LQ, Liu F. Fat-specific DsbA-L overexpression promotes adiponectin multimerization and protects mice from diet-induced obesity and insulin resistance. Diabetes. 2012 doi: 10.2337/db12-0169. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdi H, Fisher BA, Källberg H, Plant D, Malmström V, Rönnelid J, Charles P, Ding B, Alfredsson L, Padyukov L, Symmons DP, Venables PJ, Klareskog L, Lundberg K. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat Genet. 2009;41:1319–1324. doi: 10.1038/ng.480. [DOI] [PubMed] [Google Scholar]
- Malhotra A, Kobes S, Knowler WC, Baier LJ, Bogardus C, Hanson RL. A genome-wide association study of BMI in American Indians. Obesity (Silver Spring) 2011;19:2102–2106. doi: 10.1038/oby.2011.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangge H, Almer G, Truschnig-Wilders M, Schmidt A, Gasser R, Fuchs D. Inflammation, adiponectin, obesity and cardiovascular risk. Curr Med Chem. 2010;17:4511–4520. doi: 10.2174/092986710794183006. [DOI] [PubMed] [Google Scholar]
- Matsuzawa Y. Pathophysiology and molecular mechanisms of visceral fat syndrome: the Japanese experience. Diabetes Metab Rev. 1997;13:3–13. doi: 10.1002/(sici)1099-0895(199703)13:1<3::aid-dmr178>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Matsuzawa Y. Therapy insight: adipocytokines in metabolic syndrome and related cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2006;3:35–42. doi: 10.1038/ncpcardio0380. [DOI] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- Meigs JB, Wilson PW, Fox CS, Vasan RS, Nathan DM, Sullivan LM, D’Agostino RB. Body mass index, metabolic syndrome, and risk of Type 2 diabetes or cardiovascular disease. J Clin Endocrinol Metab. 2006;91:2906–2912. doi: 10.1210/jc.2006-0594. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Kurokawa J, Arai S. AIMing at metabolic syndrome. Towards the development of novel therapies for metabolic diseases via apoptosis inhibitor of macrophage (AIM) Circ J. 2011;75:2522–2531. doi: 10.1253/circj.cj-11-0891. [DOI] [PubMed] [Google Scholar]
- Muller Y, Abdussamad M, Hanson R, Knowler WC, Bogardus C, Baier L. Variants in/near BRD2 are associated with body mass index in Pima Indians. Diabetes. 2011;60 (Suppl 1):A1400. [Google Scholar]
- Nikolajczyk BS, Jagannathan-Bogdan M, Denis GV. The outliers become a stampede as immunometabolism reaches a tipping point. Immunol Rev. 2012;249:253–275. doi: 10.1111/j.1600-065X.2012.01142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell J, Lynch L, Hogan A, Cawood TJ, O’Shea D. Preadipocyte factor-1 is associated with metabolic profile in severe obesity. J Clin Endocrinol Metab. 2011;96:E680–E684. doi: 10.1210/jc.2010-2026. [DOI] [PubMed] [Google Scholar]
- Oflaz H, Ozbey N, Mantar F, Genchellac H, Mercanoglu F, Sencer E, Molvalilar S, Orhan Y. Determination of endothelial function and early atherosclerotic changes in healthy obese women. Diabetes Nutr Metab. 2003;16:176–181. [PubMed] [Google Scholar]
- Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, Amstrup Pedersen A, Kalthoff C, Tullin S, Sams A, Summer R, Walsh K. Adiponectin promotes macrophage polarization towards an anti-inflammatory phenotype. J Biol Chem. 2010;285:6153–6160. doi: 10.1074/jbc.M109.088708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki K, Ohnishi Y, Iida A, Sekine A, Yamada R, Tsunoda T, Sato H, Sato H, Hori M, Nakamura Y, Tanaka T. Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat Genet. 2002;32:650–654. doi: 10.1038/ng1047. [DOI] [PubMed] [Google Scholar]
- Ozgen M, Koca SS, Dagli N, Balin M, Ustundag B, Isik A. Serum adiponectin and vaspin levels in rheumatoid arthritis. Arch Med Res. 2010;41:457–463. doi: 10.1016/j.arcmed.2010.08.012. [DOI] [PubMed] [Google Scholar]
- Pasarica M, Gowronska-Kozak B, Burk D, Remedios I, Hymel D, Gimble J, Ravussin E, Bray GA, Smith SR. Adipose tissue collagen VI in obesity. J Clin Endocrinol Metab. 2009;94:5155–5162. doi: 10.1210/jc.2009-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pear R. New York Times. 2006. Jul 24, Obesity surgery often leads to complications, study says. [Google Scholar]
- Perfield JW, III, Lee Y, Shulman GI, Samuel VT, Jurczak MJ, Chang E, Xie C, Tsichlis PN, Obin MS, Greenberg AS. Tumor progression locus 2 (TPL2) regulates obesity-associated inflammation and insulin resistance. Diabetes. 2011;60:1168–1176. doi: 10.2337/db10-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA. 2004;291:1730–1737. doi: 10.1001/jama.291.14.1730. [DOI] [PubMed] [Google Scholar]
- Pouliot MC, Després JP, Nadeau A, Moorjani S, Prud’Homme D, Lupien PJ, Tremblay A, Bouchard C. Visceral obesity in men. Associations with glucose tolerance, plasma insulin and lipoprotein levels. Diabetes. 1992;41:826–834. doi: 10.2337/diab.41.7.826. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell. 1999;4:611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14:1293–1307. [PubMed] [Google Scholar]
- Ruderman NB, Schneider SH, Berchtold P. The “metabolically-obese”, normal-weight individual. Am J Clin Nutr. 1981;34:1617–1621. doi: 10.1093/ajcn/34.8.1617. [DOI] [PubMed] [Google Scholar]
- Ruderman NB, Berchtold P, Schneider SH. Obesity associated disorders in normal weight individuals: some speculations. Int J Obes. 1982;6 (Suppl 1):151–157. [PubMed] [Google Scholar]
- Ruderman N, Chisholm D, Pi-Sunyer X, Schneider S. The metabolically obese, normal-weight individual revisited. Diabetes. 1998;47:699–713. doi: 10.2337/diabetes.47.5.699. [DOI] [PubMed] [Google Scholar]
- Santry HP, Gillen DL, Lauderdale DS. Trends in bariatric surgical procedures. JAMA. 2005;294:1909–1917. doi: 10.1001/jama.294.15.1909. [DOI] [PubMed] [Google Scholar]
- Shaul ME, Bennett G, Strissel KJ, Greenberg AS, Obin MS. Dynamic, M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high fat diet-induced obesity in mice. Diabetes. 2010;59 (5):1171–1181. doi: 10.2337/db09-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:179–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims EAH. Are there persons who are obese, but metabolically healthy? Metabolism. 2001;50:1499–1504. doi: 10.1053/meta.2001.27213. [DOI] [PubMed] [Google Scholar]
- Sinha A, Faller DV, Denis GV. Bromodomain analysis of Brd2-dependent transcriptional activation of cyclin A. Biochem J. 2005;387:257–269. doi: 10.1042/BJ20041793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurk T, Alberti-Huber C, Herder C, Hauner H. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab. 2007;92:1023–1033. doi: 10.1210/jc.2006-1055. [DOI] [PubMed] [Google Scholar]
- Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, Blomqvist L, Hoffstedt J, Näslund E, Britton T, Concha H, Hassan M, Rydén M, Frisén J, Arner P. Dynamics of fat cell turnover in humans. Nature. 2008;453:783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- Spencer M, Yao-Borengasser A, Unal R, Rasouli N, Gurley CM, Zhu B, Peterson CA, Kern PA. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. Am J Physiol Endocrinol Metab. 2010;299:E1016–E1027. doi: 10.1152/ajpendo.00329.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Succurro E, Marini MA, Frontoni S, Hribal ML, Andreozzi F, Lauro R, Perticone F, Sesti G. Insulin secretion in metabolically obese, but normal weight, and in metabolically healthy but obese individuals. Obesity (Silver Spring) 2008;16:1881–1886. doi: 10.1038/oby.2008.308. [DOI] [PubMed] [Google Scholar]
- Tan CY, Vidal-Puig A. Adipose tissue expandability: the metabolic problems of obesity may arise from the inability to become more obese. Biochem Soc Trans. 2008;36:935–940. doi: 10.1042/BST0360935. [DOI] [PubMed] [Google Scholar]
- Tarkun I, Arslan BC, Cantürk Z, Türemen E, Sahin T, Duman C. Endothelial dysfunction in young women with polycystic ovary syndrome: relationship with insulin resistance and low-grade chronic inflammation. J Clin Endocrinol Metab. 2004;89:5592–5596. doi: 10.1210/jc.2004-0751. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Ann Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- Toulis KA, Goulis DG, Farmakiotis D, Georgopoulos NA, Katsikis I, Tarlatzis BC, Papadimas I, Panidis D. Adiponectin levels in women with polycystic ovary syndrome: a systematic review and a meta-analysis. Hum Reprod Update. 2009;15:297–307. doi: 10.1093/humupd/dmp006. [DOI] [PubMed] [Google Scholar]
- Trayhurn P, Wang B, Wood IS. Hypoxia in adipose tissue: a basis for the dysregulation of tissue function in obesity? Br J Nutr. 2008;100:227–235. doi: 10.1017/S0007114508971282. [DOI] [PubMed] [Google Scholar]
- Tsukamoto O, Fujita M, Kato M, Yamazaki S, Asano Y, Ogai A, Okazaki H, Asai M, Nagamachi Y, Maeda N, Shintani Y, Minamino T, Asakura M, Kishimoto I, Funahashi T, Tomoike H, Kitakaze M. Natriuretic peptides enhance the production of adiponectin in human adipocytes and in patients with chronic heart failure. J Am Coll Cardiol. 2009;53:2070–2077. doi: 10.1016/j.jacc.2009.02.038. [DOI] [PubMed] [Google Scholar]
- Vague J. La différentiation sexuelle, facteur déterminant des formes de l’obésité. Presse Méd. 1947;55:339–340. [PubMed] [Google Scholar]
- Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- Vougioukalaki M, Kanellis DC, Gkouskou K, Eliopoulos AG. Tpl2 kinase signal transduction in inflammation and cancer. Cancer Lett. 2011;304:80–89. doi: 10.1016/j.canlet.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Vrbíková J, Cibula D, Dvoráková K, Stanická S, Sindelka G, Hill M, Fanta M, Vondra K, Skrha J. Insulin sensitivity in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2004;89:2942–2945. doi: 10.1210/jc.2003-031378. [DOI] [PubMed] [Google Scholar]
- Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev. 2000;21:697–738. doi: 10.1210/edrv.21.6.0415. [DOI] [PubMed] [Google Scholar]
- Wang F, Liu H, Blanton WP, Belkina A, LeBrasseur NK, Denis GV. Brd2 disruption in mice causes severe obesity without Type 2 diabetes. Biochem J. 2009;425:71–83. doi: 10.1042/BJ20090928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Deeney JT, Denis GV. Brd2 gene disruption causes ‘metabolically healthy’ obesity: epigenetic and chromatin-based mechanisms that uncouple obesity from Type 2 diabetes. In: Litwack G, editor. Vit Horm. Vol. 91. Elsevier Academic Press; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasko MC, Kay J, Hsia EC, Rahman MU. Diabetes mellitus and insulin resistance in patients with rheumatoid arthritis: risk reduction in a chronic inflammatory disease. Arthritis Care Res (Hoboken) 2011;63:512–521. doi: 10.1002/acr.20414. [DOI] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westlake SL, Colebatch AN, Baird J, Curzen N, Kiely P, Quinn M, Choy E, Ostor AJ, Edwards CJ. Tumour necrosis factor antagonists and the risk of cardiovascular disease in patients with rheumatoid arthritis: a systematic literature review. Rheumatology (Oxford) 2011;50:518–531. doi: 10.1093/rheumatology/keq316. [DOI] [PubMed] [Google Scholar]
- Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- Wildman RP, Muntner P, Reynolds K, McGinn AP, Rajpathak S, Wylie-Rosett J, Sowers MR. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004) Arch Intern Med. 2008;168:1617–1624. doi: 10.1001/archinte.168.15.1617. [DOI] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Sukumaran S, Nie J, Jusko WJ, DuBois DC, Almon RR. Adipose tissue deficiency and chronic inflammation in diabetic Goto-Kakizaki rats. PLoS ONE. 2011;6:e17386. doi: 10.1371/journal.pone.0017386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara E, Fujimoto S, Inagaki N, Okawa K, Masaki S, Yodoi J, Masutani H. Disruption of TBP-2 ameliorates insulin sensitivity and secretion without affecting obesity. Nat Commun. 2010;1:127. doi: 10.1038/ncomms1127. [DOI] [PMC free article] [PubMed] [Google Scholar]