

Abstract
Ritonavir diminishes methadone plasma concentrations, attributed to CYP3A, but mechanisms are unknown. We determined short-term (2 day) and steady-state (2 week) ritonavir effects on intestinal/hepatic CYP3A (probed with IV and oral alfentanil, and miosis), P-glycoprotein (fexofenadine) and methadone pharmacokinetics and pharmacodynamics in healthy volunteers. Acute ritonavir increased the AUC0-∞/dose ratio (ritonavir/control) for oral alfentanil 25-fold. Steady-state ritonavir increased the AUC0-∞/dose ratio for intravenous and oral alfentanil 4- and 10-fold, respectively, reduced hepatic extraction (0.26 to 0.07), intestinal extraction (0.51 to zero), and increased bioavailability (37 to 95%). Acute ritonavir inhibits first-pass CYP3A >96%. Chronic ritonavir inhibits hepatic (>70%) and first-pass (>90%) CYP3A. Acute and steady-state ritonavir increased fexofenadine AUC0-∞ 2.8- and 1.4-fold, suggesting P-glycoprotein inhibition. Steady-state ritonavir caused mild apparent induction of P-glycoprotein and hepatic CYP3A, but net inhibition still predominated. Ritonavir inhibits both intestinal and hepatic CYP3A and drug transport. Alfentanil miosis noninvasively determined CYP3A inhibition by ritonavir.
The HIV protease inhibitor ritonavir is the most potent and efficacious clinical inhibitor of CYP3A, and ritonavir is now a standard component of highly active retroviral therapy, included for boosting the systemic exposure of other antiretrovirals, achieved through inhibition of first-pass and hepatic CYP3A activity.1 Ritonavir, both alone and in combination with other antiretrovirals, causes well-documented decreases in both S- and R-methadone (the active enantiomer) plasma concentrations and area under the concentration-time curve, increased methadone clearance, and variably causes methadone withdrawal symptoms.2 Ritonavir and other protease inhibitor effects on methadone disposition and clearance have been attributed to CYP3A induction in some reports.3-7 Nonetheless, the apparent paradox of ritonavir induction of methadone clearance, despite profound inhibition of CYP3A activity, has never been addressed. Methadone is also a substrate for the efflux transporter P-glycoprotein (P-gp) in vitro, and in the intestine and brain of animals in vivo, where it influences methadone absorption, brain access, pharmacodynamics, and analgesic effects.8-11 The role of P-gp in determining human methadone intestinal absorption and bioavailability is unknown. Ritonavir has been reported to inhibit renal tubular and hepatic P-gp in humans in vivo,12-14 and to induce P-gp in a human intestinal cell line, and in rats in vivo,15,16 but the role of P-gp in ritonavir alterations in methadone disposition is unknown. Indeed, mechanism(s) of ritonavir alteration of methadone pharmacokinetics and clinical effects still remain fundamentally unidentified.
The duration of ritonavir administration may affect the degree of CYP3A inhibition. Short-term ritonavir causes inhibition. Longer-term administration has been reported to cause apparent induction, or relative induction compared with acute administration but with net inhibition predominating.17-19 Whether ritonavir interactions occur secondary to modulation of hepatic or intestinal CYP3A is not well understood.20
The purpose of this clinical investigation was to determine: 1) mechanism(s) of acute and steady-state ritonavir alterations in methadone disposition and clinical effect; including the role of CYP3A4/5- and/or P-gp-mediated methadone bioavailability, first-pass metabolism, and systemic clearance; 2) the influence of ritonavir on methadone pharmacodynamics, 3) acute and steady-state ritonavir effects on hepatic CYP3A4/5, first-pass CYP3A4/5, and intestinal P-gp activities using validated in vivo probes; 4) the ability of a noninvasive in vivo CYP3A4/5 probe to detect ritonavir drug interactions and predict methadone disposition.
A comprehensive crossover investigation was conducted in healthy volunteers (Figure 1). An accompanying manuscript describes ritonavir effects on methadone pharmacokinetics and pharmacodynamics.21 This manuscript reports ritonavir effects on CYP3A4/5 and P-gp activities. The P-gp substrate fexofenadine was used as the in vivo P-gp probe.22,23 Hepatic and first-pass CYP3A4/5 activities were evaluated using intravenous and oral alfentanil (ALF) as the in vivo probe.24,25 ALF is metabolized similarly by CYPs 3A4 and 3A5, while CYP3A7 has significantly less activity,26,27 and CYP3A5 polymorphisms have no effect on intravenous or oral ALF clearances,25 hence we consider ALF to be a nonselective CYP3A4/5 (henceforth collectively referred to as CYP3A) probe. Pupil diameter change (miosis) was used as a surrogate for ALF plasma concentrations to noninvasively estimate ALF clearance, and hence CYP3A activity.24,25
Figure 1.
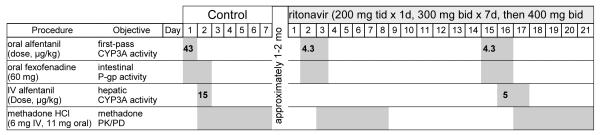
Study protocol for ritonavir-methadone interaction. Shaded boxes show drug administration and/or blood and urine sampling.
Results
The initial protocol design involved fexofenadine, IV ALF, oral ALF and methadone on sequential days, with constant ALF doses (15 μg/kg IV and 43 μg/kg oral) in the control and ritonavir arms. Plasma samples were to be batched and analyzed at the conclusion of the investigation, however pupil diameter results were available in real time. Figure 2 shows IV and oral ALF miosis for the control and ritonavir sessions. Prolonged ALF miosis was apparent during the ritonavir sessions, indicating markedly delayed ALF elimination. Carryover of ALF miosis into subsequent methadone study days confounded the methadone pupil data. Based on this subject’s pupil data, the protocol was redesigned. IV and oral ALF doses for the ritonavir sessions were reduced to 5 and 4.3 μg/kg, which allowed complete ALF elimination and prevented carryover to the next day study session. Coincidentally, simultaneous assessment of first-pass CYP3A and P-gp activity with oral ALF and fexofenadine was validated,28 so the oral ALF and fexofenadine sessions were combined, and scheduled prior to the IV ALF session. The final protocol design is shown in Fig 1. ALF data and methadone pupil data for the first subject were excluded from the final analysis.
Figure 2.

Effect of acute ritonavir on first-pass and hepatic CYP3A activity, assessed with alfentanil as a CYP3A probe. Miosis was used as a surrogate for ALF plasma concentrations. Results are shown as the measured dark-adapted pupil diameter (not baseline subtracted) for the first study subject, who received (A) 15 μg/kg IV ALF and (B) 43 μg/kg oral ALF on sequential days, at the control and ritonavir sessions. Prolonged ALF miosis is apparent during ritonavir treatment, indicating markedly delayed ALF elimination (later verified by plasma analysis at the conclusion of the entire investigation). Carryover of ALF miosis into subsequent methadone study days confounded the methadone pupil data. Based on these pupil data, the protocol was redesigned, with IV and oral ALF dose reduction to 5 and 4.3 μg/kg for ritonavir sessions. This enabled complete ALF elimination and eliminated carryover to the following day’s study session.
Ritonavir caused profound inhibition of hepatic and first-pass CYP3A activity. Initially available data were from ALF miosis, used as a surrogate for ALF plasma concentrations (Fig 3 and Table 1).24,25 Both short-term low-dose, and steady-state higher dose ritonavir prolonged and enhanced miosis. Short-term ritonavir increased maximum dose-adjusted miosis after oral ALF 8-fold. Longer duration ritonavir increased maximum dose-adjusted IV ALF miosis 3-fold, and maximum oral ALF miosis 7-fold. The dose-normalized area under the miosis-time curve (AUEC0-∞/dose) ratio (ritonavir/control) for oral ALF was increased 18-fold by acute ritonavir, and steady-state ritonavir increased the dose-normalized AUEC0-∞ ratio for IV and oral ALF 3- and 7-fold. The effects of ritonavir on ALF plasma concentrations are shown in Fig 4, and pharmacokinetic parameters provided in Table 2. The dose-normalized AUC0-∞ ratio (ritonavir/control) for oral ALF was increased 25-fold by acute ritonavir, and steady-state ritonavir increased the dose-normalized AUC0-∞ ratio for IV and oral ALF 4- and 10-fold, respectively. These results indicate 72% inhibition of hepatic ALF clearance and CYP3A activity by steady-state ritonavir, and 96% and 90% inhibition of first-pass ALF clearance and CYP3A activity by acute low-dose and steady-state higher dose ritonavir, respectively. Steady-state ritonavir reduced mean hepatic ALF extraction from 0.26 to 0.07, intestinal extraction from 0.51 to zero, and increased ALF bioavailability from 37 to 95%.
Figure 3.

Effect of acute and steady-state ritonavir on first-pass and hepatic CYP3A activity, assessed using alfentanil as a CYP3A probe. Pupil diameter change from baseline (miosis) was used as a surrogate for alfentanil plasma concentrations. Shown is dose-adjusted alfentanil miosis after (A) oral and (B) intravenous administration. Subjects received 43 and 4.3 μg/kg oral ALF at the control and ritonavir sessions, respectively, and 15 and 5 μg/kg IV alfentanil at the control and ritonavir sessions, respectively. Each data point is the mean ± SD (n=11).
Table 1.
Ritonavir effect on intravenous and oral alfentanil and methadone pupil effect parameters
| Control | Acute ritonavir | Steady-state Ritonavir |
|
|---|---|---|---|
| IV alfentanil | |||
| Maximum miosis (mm) | 5.4 ± 0.5 | 4.8 ± 0.8 | |
| Maximum miosis/dose (mm/mg) | 5.0 ± 0.7 | 13.8 ± 3.8 a | |
| AUEC0-∞/dose (mm•hr/mg) | 8.4 ± 3.3 | 24.5 ± 17.3 a | |
| AUEC0-∞/dose ratio (ritonavir/control) | 3.1 (1.6, 6.1) | ||
| CLIV, miosis (mg•mm−1•hre−1•kg−1) | 2.3 ± 2.3 | 1.0 ± 0.9 a | |
| Effect t1/2 (hr) | 0.9 ± 0.4 | 1.4 ± 0.8 a | |
| Oral alfentanil | |||
| Maximum miosis (mm) | 3.3 ± 0.7 | 2.7 ± 0.9 | 2.4± 1.0 |
| Maximum miosis/dose (mm/mg) | 1.1 ± 0.2 | 8.9 ± 3.5 a | 8.0 ± 3.7 a |
| AUEC0-∞/dose (mm•hr/mg) | 3.0 ± 1.4 | 62.3 ± 46.1 a | 28.2± 14.6 b |
| AUE0-∞/dose ratio (ritonavir/control) | 18.0 (10.9, 25.4) | 7.3 (4.2, 12.8) | |
| CL/F miosis/F (mg•mm−1•hr−1•kg−1) | 5.8 ± 3.6 | 0.34 ± 0.20 a | 0.65 ± 0.67 a |
| Effect t1/2 (hr) | 1.8 ± 1.0 | 6.5 ± 4.1 a | 2.9 ± 1.4 a |
Subjects received 15 and 5 μg/kg IV ALF at the control and ritonavir sessions, respectively, and 43 and 4.3 μg/kg oral ALF at the control and ritonavir sessions, respectively. Maximum miosis and AUC are shown normalized to dose. All other variables are not dose adjusted. Results (n=11) are the arithmetic mean ± SD, except the AUC0-∞/dose ratio (ritonavir/control), which is the geometric mean (90% CI). b Subjects received 6.0 mg IV and 11.2 mg oral methadone HCl at all sessions. Results are the mean ± SD (n=11) except the AUC0-∞/dose ratio (ritonavir/control), which is the geometric mean (90% CI).
Significantly different from control (p<0.05)
Significantly different from acute ritonavir (p<0.05)
Figure 4.

Effect of acute and steady-state ritonavir on first-pass and hepatic CYP3A activity, assessed using alfentanil as a CYP3A probe. Shown are dose-adjusted alfentanil concentrations after (A) oral and (B) intravenous administration. Subjects received 43 and 4.3 μg/kg oral ALF at the control and ritonavir sessions, respectively, and 15 and 5 μg/kg IV alfentanil at the control and ritonavir sessions, respectively. Each data point is the mean ± SD (n=11).
Table 2.
Intravenous and oral alfentanil pharmacokinetic parameters
| Control | Acme ritonavir | Sieauy-siaie Ritonavir |
|
|---|---|---|---|
| IV alfentanil | |||
| Cmax (ng/ml) | 115 ± 32 | 28 ± 5a | |
| Cmax/dose (ng/ml/mg) | 109 ± 40 | 78 ± 17 a | |
| AUC0-∞ (ng •hr •ml−1) | 73 ± 25 | 86 ± 26 | |
| AUC0-∞/dose (ng•hr•ml−1/mg) | 67 ± 23 | 237±62a | |
| AUC0-∞/dose ratio (ritonavir/control) |
3.6 (2.8, 4.7) | ||
| CLIV (ml•kg−1•min−1) | 3.91 ± 1.59 | 1.05 ± 0.28 a | |
| Elimination t1/2 (hr) | 1.1 ± 0.2 | 4.9 ± 1.2 a | |
| EH | 0.26 ± 0.10 | 0.07 ± 0.02 a | |
| Oral alfentanil | |||
| Cmax (ng/ml) | 43 ± 17 | 15 ± 5 a | 12 ± 3 a |
| Cmax/dose (ng/ml/mg) | 14 ± 5 | 46 ± 14 a | 38 ± 9 a b |
| AUC0-∞ (ng •hr •ml−1) | 78 ± 36 | 182±48a | 74 ± 21 |
| AUC0-∞/dose (ng•hr•ml−1/mg) | 25 ± 9 | 573 ±107a | 236±62ab |
| AUC0-∞/dose ratio (ritonavir/control) |
24.8 (15.2, 40.4) | 10.0 (6.7, 15.1) | |
| CL/F (ml•kg−1•min−1) | 11.5 ± 6.3 | 0.4 ± 0.1 a | 1.1 ± 0.3 a b |
| Elimination t1/2 (hr) | 1.1 ± 0.3 | 12.1 ± 1.7 a | 6.1 ± 2.9 a b |
| Foral | 0.37 ± 0.08 | 0.95 ± 0.08 a | |
| EG | 0.51 ± 0.12 | 0.00 ± 0.08 a |
Subjects received 15 and 5 μg/kg IV ALF at the control and ritonavir sessions, respectively, and 43 and 4.3 μg/kg oral ALF at the control and ritonavir sessions, respectively. One subject received 15 μg/kg IV ALF and 43 μg/kg oral ALF at control and ritonavir sessions, and was omitted from the analysis of ALF pharmacokinetics. Cmax and AUC are shown normalized to dose. All other variables are not dose adjusted. Results are the arithmetic mean ± SD (n=11), except the AUC0-∞/dose ratio (ritonavir/control), which is the geometric mean (90% CI).
Significantly different from control (p<0.05)
Significantly different from acute ritonavir (p<0.05)
Ritonavir had time-and dose-dependent effects on plasma fexofenadine concentrations (Fig 5). Short-term low dose ritonavir increased plasma concentrations, Cmax (by 60%), and the AUC0-∞ ratio (ritonavir/control) nearly 3-fold (Table 3). Steady-state higher dose ritonavir had a diminished but still significant effect, increasing the AUC0-∞ ratio 1.4-fold. Ritonavir had no effect on fexofenadine elimination half-life.
Figure 5.

Effect of acute and steady-state ritonavir on intestinal transporter activity, assessed using fexofenadine as a transporter probe. Shown are plasma fexofenadine concentrations after 3d and 2.5 wk ritonavir. Each subject received 60 mg oral fexofenadine on all occasions. Each data point is the mean ± SD (n=12).
Table 3.
Fexofenadine pharmacokinetic parameters
| Control | Acute ritonavir | Steady-state Ritonavir |
|
|---|---|---|---|
| Cmax (ng/ml) | 194 ± 116 | 311±102a | 191±95 b |
| AUC0-∞ (ng •hr •ml−1) | 1010±460 | 2780±920a | 1400 ± 700 b |
| AUC0-∞ ratio (ritonavir/control) |
2.8 (2.2,3.6) | 1.4 (1.1,1.7) | |
| CL/F (ml•kg−1•mm−1) | 15.5 ± 5.6 | 5.4 ± 1.9 a | 10.7 ± 6.5 a b |
| Elimination t1/2 (hr) | 10.6 ± 1.9 | 11.3 ± 2.5 | 10.4 ± 3.9 |
Results are the arithmetic mean ± SD (n=12), except the AUC0-∞ ratio (ritonavir/control), which is the geometric mean (90% CI)
Significantly different from control (p<0.05)
Significantly different from acute ritonavir (p<0.05)
Discussion
One purpose of this investigation was to quantify the effects of short-term and steady-state ritonavir on hepatic and first-pass CYP3A activity. The major finding, as expected, was that ritonavir profoundly inhibited CYP3A. These results are consistent with known effects of ritonavir on the pharmacokinetics of CYP3A substrates. Short-term ritonavir inhibited 96% of first-pass CYP3A, and steady-state ritonavir inhibited 72% and 90% of hepatic and first-pass activities, respectively, as assessed using ALF clearance is an in vivo probe. Steady-state ritonavir reduced ALF hepatic extraction from 0.26 to 0.07, intestinal extraction from 0.51 to zero, and increased bioavailability from 37 to 95%. This is the first investigation to evaluate ritonavir effects on both hepatic and first-pass CYP3A, and to show that ritonavir inhibits both intestinal and hepatic CYP3A activity.
CYP3A inhibition by steady-state higher-dose ritonavir was moderately diminished compared with short-term low-dose ritonavir. The dose-normalized ALF AUC0-∞ ratio (ritonavir/control) was 25 and 10 after short-term and steady-state ritonavir, and mean ALF CL/F was 0.4 and 1.1 ml•kg−1•min−1 (vs 12 in controls). Similar long- vs short-term ritonavir effects on the oral clearance of other CYP3A substrates have been reported previously, and attributed to longer-term CYP3A induction, although the site of induction (intestine vs liver) was not identified, and the potential contribution of intestinal transporters could not be excluded.17-19,29 In the present investigation, since ALF intestinal extraction was zero after steady-state ritonavir, this cannot represent the site of effective CYP3A upregulation compared with short-term ritonavir. Thus it is more likely that the liver was the site of the mild functional upregulation of CYP3A. Direct comparison of short-term and steady-state ritonavir effects on hepatic and intestinal extraction coefficients was not possible, since this investigation did not evaluate acute ritonavir effects on IV ALF clearance. Overall, while mild CYP3A induction by steady-state ritonavir was apparent, this was clinically inconsequential, since profound ritonavir inhibition of CYP3A activity was still dominant.
A second major purpose of this investigation was to assess the ability of the noninvasive in vivo probe ALF miosis to detect alterations in CYP3A activity and drug interactions. Results show that ALF miosis was an excellent predictor of ritonavir effects on both hepatic and first-pass CYP3A activity. From the immediately available miosis data, long before that of plasma ALF concentrations and formal clearance determinations, it was clear that ritonavir profoundly increased ALF AUEC0-∞ and prolonged elimination of ALF effects. Changes in miosis were representative of alterations in CYP3A activity, with increases in AUEC0-∞ ratios reflecting those in ALF plasma AUC. Moreover, the early availability of miosis data enabled a change in the protocol, to prevent ALF carryover during ritonavir inhibition. Had a different CYP3A probe been used, with conventional batch plasma analysis after conclusion of the entire investigation, such carryover would not have been known, and the results of the entire investigation would have been invalid. No other CYP3A probe offers such real-time assessment of CYP3A activity.
A third major purpose of this investigation was to determine the effect of ritonavir on gastrointestinal P-gp activity. The major finding was that ritonavir significantly inhibited P-gp activity. Short-term and steady-state ritonavir significantly increased apparent fexofenadine bioavailability, without altering its elimination. Fexofenadine is a well-characterized substrate for human P-gp (ABCB1).22,30 The well-known P-gp inhibitors ketoconazole,31 verapamil,23,32,33 itraconazole,34,35 and erythromycin,36 increased plasma fexofenadine concentrations, and the P-gp inducers rifampin and St. John’s wort decreased plasma fexofenadine,37,38 effects largely although not exclusively attributed to inhibition and upregulation of intestinal and/or hepatic P-gp. Hence fexofenadine has been used frequently as an in vivo probe to assess P-gp pharmacogenetics and drug interactions.23,31-33,35,37-40 In the present investigation, short-term and steady-state ritonavir caused 2.8- and 1.4-fold increases in fexofenadine AUC. The extent of ritonavir alterations of fexofenadine AUC and oral clearance is similar to that caused by other P-gp inhibitors, such as itraconazole and verapamil. It is well-established that CYP3A-mediated metabolism accounts for less than 1% of fexofenadine elimination, renal excretion accounts for less than 5-10%, and changes in renal fexofenadine excretion do not affect plasma concentrations because it is a minor route of elimination.23,33,37 Hence it is unlikely that the observed effects of ritonavir on fexofenadine are attributable to changes in CYP3A or renal P-gp activity. Rather, they suggest ritonavir inhibition of intestinal and/or hepatic P-gp. P-gp is expressed on the luminal aspect of enterocytes and the canalicular aspect of hepatocytes. The site of action (intestinal vs hepatic) of other P-gp inhibitors (verapamil, erythromycin, ketoconazole) on first-pass fexofenadine extraction remains controversial,23,31,32,36 hence the present results do not discriminate the site of P-gp inhibition by ritonavir. Previous studies have also shown an effect of ritonavir on drug transport. Ritonavir (300 mg twice daily for 3d) doubled the plasma AUC of intravenous digoxin, also a P-gp substrate, and decreased both nonrenal and renal clearances by 40-50%, which was ascribed to inhibition of renal tubular P-gp.12 In contrast, 2 weeks of lower-dose (200 mg twice daily) ritonavir caused a 22% increase in oral digoxin AUC and 30% reduction in oral clearance, without altering digoxin renal clearance, effects which were attributed to inhibition of hepatic P-gp and reduced biliary fexofenadine excretion.13 Single-dose ritonavir (100 mg) or lopinavir/ritonavir (400/100 mg) increased the fexofenadine plasma AUC 2- and 4-fold, without affecting fexofenadine elimination half-life, which was also attributed to hepatic P-gp inhibition.14 Ritonavir has been shown to induce P-gp protein expression and transport activity in a human intestinal cell line, and in rats in vivo.15,16
Fexofenadine is not exclusively transported by P-gp, and absorption has been suggested to involve active intestinal uptake transport, as well as passive diffusion and active efflux.31,32,36 Fexofenadine is a substrate for the uptake transporters OATP1A2,22 OATP2B1,41 and the liver-specific OATP1B1 and OATP1B3.42,43 Decreased fexofenadine bioavailability by citrus and apple juices was attributed to inhibition of intestinal OATP1A2-mediated uptake.39 Increased fexofenadine bioavailability by verapamil and ketoconazole, which did not increase apparent intestinal permeability and absorption,31,32 were attributed instead to inhibition of hepatic first-pass extraction, specifically decreased OATP-mediated fexofenadine uptake across the sinusoidal membrane and/or inhibition of P-gp-mediated canalicular secretion.31,32 Thus a potential role for both intestinal P-gp efflux and OATP-mediated absorption in fexofenadine bioavailability has been proposed,31,32 and some may consider fexofenadine an in vivo probe for OATP as well as P-gp. Ritonavir has been shown to be an inhibitor of OATP-mediated fexofenadine transport, however such effects were small (13%).22 Ritonavir inhibition of OATP in vivo would not explain the observed increase in fexofenadine bioavailability, hence P-gp inhibition remains the more likely explanation.
Ritonavir effects on fexofenadine AUC were biphasic. The 2-fold increase in AUC by short-term ritonavir (600 mg/d) was partially attenuated to a 1.4-fold increase after 2 weeks of ritonavir (800 mg/d). The phenomenon of short-term inhibition of fexofenadine oral clearance, which abates over time despite continued inhibitor administration, has been previously described. Single dose verapamil or St John’s wort caused an approximately 20-40% decrease in fexofenadine oral clearance, which slowly abated over the course of 2 (St John’s wort) or 5 (verapamil) weeks.23,38 Long-term effects were attributed to P-gp induction,38 supported by reported St. John’s wort induction of immunodetectable duodenal P-gp expression.44 Similar effects of short- (1 wk) and longer-term (6 wk) verapamil on the clearance of the P-gp substrate digoxin, were also reported.45 Single-dose lopinavir/ritonavir increased the fexofenadine AUC 4-fold, while steady-state lopinavir/ritonavir effects were attenuated to a non-significant 3-fold increase, attributed to mild steady-state P-gp induction (although net P-gp inhibition prevailed).14 Ritonavir effects in the present investigation could be explained by acute P-gp inhibition, partially counteracted by long-term P-gp induction, albeit with net strong inhibition prevailing. Alternatively, these results could be explained by P-gp inhibition followed by concomitant OATP induction. Nevertheless, although ritonavir moderately induced P-gp mRNA expression, it had little or no effect on OATP1B1 or OATP1B3.46 Ritonavir effects on human intestinal and hepatic P-gp and OATP expression in vivo have not been reported.
There are recognized potential limitations with this investigation. First, the ritonavir dose differs from that used in typical contemporary “boosted” antiretroviral regimens (100-200 mg).1. The protocol was designed, however, as a mechanistic investigation, specifically to provide insights into the ritonavir reduction of methadone plasma concentrations first reported when higher ritonavir doses were used clinically. Nonetheless, CYP induction did occur after only 3 days of twice daily 200-300 mg ritonavir, as has been observed after longer periods at lower (100 mg twice daily) doses.47 Thus the present investigation is of both mechanistic and clinical relevance. Second, ritonavir effects were evaluated in healthy volunteers, rather than HIV-infected patients. This was deliberate, because standard antiretroviral therapy involves several drugs, thereby precluding a careful mechanistic evaluation and attribution of results to any one specific drug.
In summary, acute and steady-state ritonavir inhibited >90% of first-pass CYP3A4/5 activity, and steady-state ritonavir inhibited >70% of hepatic CYP3A4/5 activity. Acute ritonavir inhibited apparent gastrointestinal P-gp activity, and steady-state ritonavir caused mild apparent induction of P-gp relative to acute ritonavir, but net inhibition still predominated. Ritonavir inhibits both intestinal and hepatic CYP3A and drug transport. Alfentanil miosis was an excellent noninvasive probe for determining CYP3A4/5 inhibition by ritonavir.
Methods
Clinical Protocol
The investigation was approved by the University of Washington Institutional Review Board and each subject provided written informed consent. The study population was normal healthy volunteers. Eligibility criteria included 1) age 18-40 yr, 2) within 25% of ideal body weight (body mass index <30). Exclusion criteria were 1) major medical problems, 2) history of hepatic or renal disease, 3) family history of type 2 diabetes, 4) use of medications or nonprescription preparations known to alter CYP3A activity, 5) a known history of addiction to drugs or alcohol, or 6) access to and routine handling of addicting drugs in the regular course of employment. Females taking hormonal contraceptives were excluded from enrollment. Both smokers and nonsmokers were enrolled. Subjects underwent a screening visit during which blood samples for fasting glucose concentration and HIV serologic status were obtained. Subjects were excluded if their glucose exceeded 110 mg/dl (because ritonavir can cause glucose intolerance) or they were HIV seropositive (since monotherapy can cause HIV resistance). The final study population was twelve healthy subjects (six men, six women; 25 ± 5 yr, range 19-34; 74 ± 13 kg, range 55-99). Subjects were CYP3A5 genotyped at the conclusion of the study. Since, however, since this does not affect alfentanil clearance,25 enrollment was not affected by CYP3A5 genotype and it was not considered in the data analysis.
The protocol used a 3-session sequential crossover design (control session first, due to logistical considerations). Each subject served as their own control. The study protocol is shown in Figure 1. Subjects were instructed to consume no food or beverages that contain grapefruit, apples or oranges for 7d before any study day, no alcohol or caffeine for 1d before each study session and on the study day, and no food or water after midnight before each study session. For each session, a catheter was placed in an arm vein for blood sampling and (if needed) a second catheter was placed for drug administration. Subjects (supine) were monitored with a pulse oximeter and automated blood pressure cuff, and received supplemental oxygen for saturations less than 94%.
Hepatic CYP3A activity was evaluated using intravenous ALF as an in vivo probe, as described previously.24,25,28,48 Subjects received ondansetron (4 mg IV) for antinausea prophylaxis, followed 30 min later by ALF bolus (15 and 5 μg/kg at control and ritonavir sessions, respectively). Subjects received a standard breakfast 4 hr after ALF, and free access to food and water thereafter. Venous blood was sampled for 24 hr after ALF dosing, and plasma stored at −20°C for later analysis. Coincident with blood sampling, dark-adapted pupil diameter was measured using a Pupilscan Model 12A infrared pupillometer with 0.1 mm resolution (Keeler USA) as described previously.24,25 Each recorded value was the mean of triplicate measurements, which typically agreed to within 0.1-0.3 mm. First-pass CYP3A activity and intestinal P-gp (and other transporters) activity were evaluated using oral ALF and fexofenadine as in vivo probes, as described previously.24,25,28 Subjects received ondansetron (4 mg IV) for antinausea prophylaxis, followed 30 min later by oral ALF (43 and 4.3 μg/kg at control and ritonavir sessions, respectively) with 100 cc water. Fexofenadine (60 mg) was administered with 100 cc water 1 hr after ALF. Subjects received a standard breakfast and lunch 3 and 6 hr, respectively, after ALF. Venous blood was sampled for 48 hr after ALF dosing, and dark-adapted pupil diameter was measured coincident with blood sampling. The protocol for methadone administration is described in an accompanying manuscript.21
Hepatic and first-pass CYP3A, intestinal transporters activity and methadone disposition were assessed at baseline. Subjects then began taking ritonavir, 200 mg three times daily for 1 day, 300 mg twice daily for 6 days, then 400 mg twice daily for 2 weeks.17,18,49 Initial reports of reduced methadone plasma concentrations and withdrawal occurred with ritonavir 400 mg twice daily,3 hence this was the final target dose chosen for this investigation. A ritonavir dose-escalation lead-in is clinically standard, and 1-2 days of a lead-in dose were shown to inhibit CYP3A activity.18 First-pass CYP3A and transporter activities and methadone disposition were determined on the second and third day of low-dose ritonavir, when CYP3A inhibition was expected. Hepatic and first-pass CYP3A, intestinal transporters activity, and methadone disposition were again assessed at steady-state ritonavir. Previous reports suggested a potential for CYP3A induction during steady-state ritonavir.18,19 Since the time course of potential CYP3A induction by ritonavir was unknown, and time to evaluate hepatic CYP3A, first-pass CYP3A, and P-gp activities potentially not sufficient before commencement of induction, only first-pass CYP3A and P-gp activities were evaluated in the acute ritonavir phase. First-pass is a greater determinant than hepatic metabolism of oral drug disposition, and hence the more important parameter to evaluate.
Ritonavir was administered at 7 am, 3 pm, and 10 pm on the first day, and at 7am and 7 pm thereafter. Dosing was adjusted on study days to preclude an acute inhibitor effect from the morning dose. Subjects received their morning dose at lunch, and took their evening dose at 11 pm.
Sample size was determined using a simplified analysis (paired t-test) for comparing the primary outcome variable, methadone systemic clearance. A previous study found 22 and 33% interday/intrasubject variability in IV and oral methadone clearances, respectively.50 To detect a 30% change in clearance, using a paired t-test, with 33% variability, ß=0.8, α=0.05, would require 12 subjects.
Analytical Methods
Plasma ALF and fexofenadine concentrations were simultaneously quantified using solid-phase extraction and electrospray liquid chromatography-mass spectrometry as described previously.28
Data Analysis
ALF plasma data were analyzed by noncompartmental methods as described previously.24,25 Systemic clearance of intravenous ALF (CLIV)=doseIV/AUCIV, apparent oral clearance of ALF was (CL/F)=doseoral/AUCoral, bioavailability was (Foral)= (AUCoral/doseoral) x (doseIV/AUCIV), volume of distribution based on the terminal phase was (Vz)=Dose/(AUC x λ) where λ is the terminal elimination rate constant, and steady-state volume of distribution was (Vss)=CL x mean residence time. ALF effect (miosis) vs time curves were treated analogously to conventional plasma concentration curves, with similar noncompartmental analysis, to yield effect parameters similar to conventional pharmacokinetic parameters.24,25 Hence, the area under the effect curve (AUEC) was obtained. ALF miosis was treated similarly to plasma concentration to obtain an effect clearance (CLmiosis, dose/AUEC), analogous to plasma clearance (dose/AUC). Effect clearance and dose-adjusted AUEC were used when subjects received different ALF doses.24,25
Statistical Analysis
One-way repeated measures analysis of variance followed by the Student-Newman-Keuls test for multiple comparisons, or paired t-tests, were used as appropriate to assess the significance of differences between groups for pharmacokinetic and effect parameters (SigmaStat 3.5, Systat Corp). Non-normal data were log transformed for analysis, but reported as the non-transformed results. Statistical significance was assigned at p< 0.05. Results are reported as the arithmetic mean ± standard deviation (SD). Plasma AUC and urine data were also assessed as ratios (ritonavir/control) and the geometric mean and 90% confidence interval of the geometric mean. Confidence intervals excluding 1.0 were considered statistically significant.
ACKNOWLEDGEMENTS
Supported by NIH grants R01-GM63674, R01-DA14211 and K24-DA00417 (to EDK), and M01-RR00037 to the University of Washington General Clinical Research Center. Ritonavir was the generous gift of Abbott Laboratories, Abbott Park, IL
Footnotes
No author has any conflict of interest.
REFERENCES
- 1.Piacenti FJ. An update and review of antiretroviral therapy. Pharmacotherapy. 2006;26:1111–33. doi: 10.1592/phco.26.8.1111. [DOI] [PubMed] [Google Scholar]
- 2.Bruce RD, Altice FL, Gourevitch MN, Friedland GH. Pharmacokinetic drug interactions between opioid agonist therapy and antiretroviral medications: implications and management for clinical practice. J Acquir Immune Defic Syndr. 2006;41:563–72. doi: 10.1097/01.qai.0000219769.89679.ec. [DOI] [PubMed] [Google Scholar]
- 3.Geletko SM, Erickson AD. Decreased methadone effect after ritonavir initiation. Pharmacotherapy. 2000;20:93–4. doi: 10.1592/phco.20.1.93.34654. [DOI] [PubMed] [Google Scholar]
- 4.Bart PA, et al. Methadone blood concentrations are decreased by the administration of abacavir plus amprenavir. Ther Drug Monit. 2001;23:553–5. doi: 10.1097/00007691-200110000-00010. [DOI] [PubMed] [Google Scholar]
- 5.McCance-Katz EF, Rainey PM, Friedland G, Jatlow P. The protease inhibitor lopinavir-ritonavir may produce opiate withdrawal in methadone-maintained patients. Clin Infect Dis. 2003;37:476–82. doi: 10.1086/376907. [DOI] [PubMed] [Google Scholar]
- 6.Hendrix CW, et al. Pharmacokinetics and pharmacodynamics of methadone enantiomers after coadministration with amprenavir in opioid-dependent subjects. Pharmacotherapy. 2004;24:1110–21. doi: 10.1592/phco.24.13.1110.38091. [DOI] [PubMed] [Google Scholar]
- 7.McCance-Katz EF. Treatment of opioid dependence and coinfection with HIV and hepatitis C virus in opioid-dependent patients: the importance of drug interactions between opioids and antiretroviral agents. Clin Infect Dis. 2005;41(Suppl 1):S89–95. doi: 10.1086/429503. [DOI] [PubMed] [Google Scholar]
- 8.Thompson SJ, Koszdin KK, Bernards CM. Opiate-induced analgesia as increased and prolonged in mice lacking P-glycoprotein. Anesthesiology. 2000;92:1392–9. doi: 10.1097/00000542-200005000-00030. [DOI] [PubMed] [Google Scholar]
- 9.Wang JS, Ruan Y, Taylor RM, Donovan JL, Markowitz JS, DeVane CL. Brain penetration of methadone (R)- and (S)-enantiomers is greatly increased by P-glycoprotein deficiency in the blood-brain barrier of Abcb1a gene knockout mice. Psychopharmacology (Berl) 2004;173:132–8. doi: 10.1007/s00213-003-1718-1. [DOI] [PubMed] [Google Scholar]
- 10.Dagenais C, Graff CL, Pollack GM. Variable modulation of opioid brain uptake by P-glycoprotein in mice. Biochem Pharmacol. 2004;67:269–76. doi: 10.1016/j.bcp.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 11.Bauer B, et al. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up-regulation. Mol Pharmacol. 2006;70:1212–9. doi: 10.1124/mol.106.023796. [DOI] [PubMed] [Google Scholar]
- 12.Ding R, et al. Substantial pharmacokinetic interaction between digoxin and ritonavir in healthy volunteers. Clin. Pharmacol. Ther. 2004;76:73–84. doi: 10.1016/j.clpt.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 13.Penzak SR, Shen JM, Alfaro RM, Remaley AT, Natarajan V, Falloon J. Ritonavir decreases the nonrenal clearance of digoxin in healthy volunteers with known MDR1 genotypes. Ther Drug Monit. 2004;26:322–30. doi: 10.1097/00007691-200406000-00018. [DOI] [PubMed] [Google Scholar]
- 14.van Heeswijk RP, et al. Time-dependent interaction between lopinavir/ritonavir and fexofenadine. J Clin Pharmacol. 2006;46:758–67. doi: 10.1177/0091270006288733. [DOI] [PubMed] [Google Scholar]
- 15.Perloff MD, Von Moltke LL, Marchand JE, Greenblatt DJ. Ritonavir induces P-glycoprotein expression, multidrug resistance-associated protein (MRP1) expression, and drug transporter-mediated activity in a human intestinal cell line. J Pharm Sci. 2001;90:1829–37. doi: 10.1002/jps.1133. [DOI] [PubMed] [Google Scholar]
- 16.Perloff MD, von Moltke LL, Greenblatt DJ. Ritonavir and dexamethasone induce expression of CYP3A and P-glycoprotein in rats. Xenobiotica. 2004;34:133–50. doi: 10.1080/00498250310001630215. [DOI] [PubMed] [Google Scholar]
- 17.Hsu A, Granneman GR, Bertz RJ. Ritonavir. Clinical pharmacokinetics and interactions with other anti-HIV agents. Clin. Pharmacokinet. 1998;35:275–91. doi: 10.2165/00003088-199835040-00002. [DOI] [PubMed] [Google Scholar]
- 18.Greenblatt DJ, von Moltke LL, Daily JP, Harmatz JS, Shader RI. Extensive impairment of triazolam and alprazolam clearance by short-term low-dose ritonavir: the clinical dilemma of concurrent inhibition and induction. J. Clin. Psychopharmacol. 1999;19:293–6. doi: 10.1097/00004714-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Greenblatt DJ, et al. Alprazolam-ritonavir interaction: implications for product labeling. Clin. Pharmacol. Ther. 2000;67:335–41. doi: 10.1067/mcp.2000.105757. [DOI] [PubMed] [Google Scholar]
- 20.Lu J-F, Blaschke TF, Flexner C, Rosenkranz SL, Sheiner LB. Model-based analysis of the pharmacokinetic interactions between ritonavir, nelfinavir, and saquinavir after simultaneous and staggered oral administration. Drug. Metab. Dispos. 2002;30:1455–61. doi: 10.1124/dmd.30.12.1455. [DOI] [PubMed] [Google Scholar]
- 21.Kharasch ED, Bedynek PS, Park S, Whittington D, Walker A, Hoffer C. Mechanism of ritonavir changes in methadone pharmacokinetics and pharmacodynamics. I. Evidence against CYP3A mediation of methadone clearance. Clin Pharmacol Ther. 2008 doi: 10.1038/clpt.2008.104. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cvetkovic M, Leake B, Fromm MF, Wilkinson GR, Kim RB. OATP and P-glycoprotein transporters mediate the cellular uptake and excretion of fexofenadine. Drug Metab. Dispos. 1999;27:866–71. [PubMed] [Google Scholar]
- 23.Lemma GL, Wang Z, Hamman MA, Zaheer NA, Gorski JC, Hall SD. The effect of short- and long-term administration of verapamil on the disposition of cytochrome P450 3A and P-glycoprotein substrates. Clin Pharmacol Ther. 2006;79:218–30. doi: 10.1016/j.clpt.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Kharasch ED, Walker A, Hoffer C, Sheffels P. Intravenous and oral alfentanil as in vivo probes for hepatic and first-pass CYP3A activity. Noninvasive assessment using pupillary miosis. Clin Pharmacol Ther. 2004;76:452–66. doi: 10.1016/j.clpt.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 25.Kharasch ED, et al. Influence of CYP3A5 genotype on the pharmacokinetics and pharmacodynamics of the cytochrome P4503A probes alfentanil and midazolam. Clin Pharmacol Ther. 2007;82:410–26. doi: 10.1038/sj.clpt.6100237. [DOI] [PubMed] [Google Scholar]
- 26.Klees TM, Sheffels P, Dale O, Kharasch ED. Metabolism of alfentanil by cytochrome P4503A (CYP3A) enzymes. Drug Metab Dispos. 2005;33:303–11. doi: 10.1124/dmd.104.002709. [DOI] [PubMed] [Google Scholar]
- 27.Klees TM, Sheffels P, Thummel KE, Kharasch ED. Pharmacogenetic determinants of human liver microsomal alfentanil metabolism and the role of cytochrome P450 3A5. Anesthesiology. 2005;102:550–6. doi: 10.1097/00000542-200503000-00012. [DOI] [PubMed] [Google Scholar]
- 28.Kharasch ED, Walker A, Hoffer C, Sheffels P. Evaluation of first-pass cytochrome P4503A (CYP3A) and P-glycoprotein activities using alfentanil and fexofenadine in combination. J Clin Pharmacol. 2005;45:79–88. doi: 10.1177/0091270004269705. [DOI] [PubMed] [Google Scholar]
- 29.Culm-Merdek KE, et al. Effect of extended exposure to grapefruit juice on cytochrome P450 3A activity in humans: comparison with ritonavir. Clin Pharmacol Ther. 2006;79:243–54. doi: 10.1016/j.clpt.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 30.Perloff MD, von Moltke LL, Greenblatt DJ. Fexofenadine transport in Caco-2 cells: inhibition with verapamil and ritonavir. J Clin Pharmacol. 2002;42:1269–74. doi: 10.1177/009127002762491370. [DOI] [PubMed] [Google Scholar]
- 31.Tannergren C, Knutson T, Knutson L, Lennernas H. The effect of ketoconazole on the in vivo intestinal permeability of fexofenadine using a regional perfusion technique. Br J Clin Pharmacol. 2003;55:182–90. doi: 10.1046/j.1365-2125.2003.01722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tannergren C, Petri N, Knutson L, Hedeland M, Bondesson U, Lennernas H. Multiple transport mechanisms involved in the intestinal absorption and first-pass extraction of fexofenadine. Clin. Pharmacol. Ther. 2003;74:423–36. doi: 10.1016/S0009-9236(03)00238-8. [DOI] [PubMed] [Google Scholar]
- 33.Yasui-Furukori N, Uno T, Sugawara K, Tateishi T. Different effects of three transporting inhibitors, verapamil, cimetidine, and probenecid, on fexofenadine pharmacokinetics. Clin Pharmacol Ther. 2005;77:17–23. doi: 10.1016/j.clpt.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 34.Shon JH, et al. Effect of itraconazole on the pharmacokinetics and pharmacodynamics of fexofenadine in relation to the MDR1 genetic polymorphism. Clin Pharmacol Ther. 2005;78:191–201. doi: 10.1016/j.clpt.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 35.Shimizu M, Uno T, Sugawara K, Tateishi T. Effects of itraconazole and diltiazem on the pharmacokinetics of fexofenadine, a substrate of P-glycoprotein. Br J Clin Pharmacol. 2006;61:538–44. doi: 10.1111/j.1365-2125.2006.02613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petri N, Borga O, Nyberg L, Hedeland M, Bondesson U, Lennernas H. Effect of erythromycin on the absorption of fexofenadine in the jejunum, ileum and colon determined using local intubation in healthy volunteers. Int J Clin Pharmacol Ther. 2006;44:71–9. doi: 10.5414/cpp44071. [DOI] [PubMed] [Google Scholar]
- 37.Hamman MA, Bruce MA, Haehner-Daniels BD, Hall SD. The effect of rifampin administration on the disposition of fexofenadine. Clin. Pharmacol. Ther. 2001;69:114–21. doi: 10.1067/mcp.2001.113697. [DOI] [PubMed] [Google Scholar]
- 38.Wang Z, Hamman MA, Huang SM, Lesko LJ, Hall SD. Effect of St John’s wort on the pharmacokinetics of fexofenadine. Clin. Pharmacol. Ther. 2002;71:414–20. doi: 10.1067/mcp.2002.124080. [DOI] [PubMed] [Google Scholar]
- 39.Dresser GK, et al. Fruit juices inhibit organic anion transporting polypeptide-mediated drug uptake to decrease the oral availability of fexofenadine. Clin. Pharmacol. Ther. 2002;71:11–20. doi: 10.1067/mcp.2002.121152. [DOI] [PubMed] [Google Scholar]
- 40.Dresser GK, Kim RB, Bailey DG. Effect of grapefruit juice volume on the reduction of fexofenadine bioavailability: possible role of organic anion transporting polypeptides. Clin Pharmacol Ther. 2005;77:170–7. doi: 10.1016/j.clpt.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 41.Nozawa T, Imai K, Nezu J, Tsuji A, Tamai I. Functional characterization of pH-sensitive organic anion transporting polypeptide OATP-B in human. J Pharmacol Exp Ther. 2004;308:438–45. doi: 10.1124/jpet.103.060194. [DOI] [PubMed] [Google Scholar]
- 42.Niemi M, Kivisto KT, Hofmann U, Schwab M, Eichelbaum M, Fromm MF. Fexofenadine pharmacokinetics are associated with a polymorphism of the SLCO1B1 gene (encoding OATP1B1) Br J Clin Pharmacol. 2005;59:602–4. doi: 10.1111/j.1365-2125.2005.02354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimizu M, et al. Contribution of OATP (organic anion-transporting polypeptide) family transporters to the hepatic uptake of fexofenadine in humans. Drug Metab Dispos. 2005;33:1477–81. doi: 10.1124/dmd.105.004622. [DOI] [PubMed] [Google Scholar]
- 44.Dürr D, et al. St John’s Wort induces intestinal P-glycoprotein/MDR1 and intestinal and hepatic CYP3A4. Clin. Pharmacol. Ther. 2000;68:598–604. doi: 10.1067/mcp.2000.112240. [DOI] [PubMed] [Google Scholar]
- 45.Pedersen KE, Dorph-Pedersen A, Hvidt S, Klitgaard NA, Pedersen KK. The long-term effect of verapamil on plasma digoxin concentration and renal digoxin clearance in healthy subjects. Eur J Clin Pharmacol. 1982;22:123–7. doi: 10.1007/BF00542456. [DOI] [PubMed] [Google Scholar]
- 46.Dixit V, Hariparsad N, Li F, Desai P, Thummel KE, Unadkat JD. Cytochrome P450 enzymes and transporters induced by anti-human immunodeficiency virus protease inhibitors in human hepatocytes: implications for predicting clinical drug interactions. Drug Metab Dispos. 2007;35:1853–9. doi: 10.1124/dmd.107.016089. [DOI] [PubMed] [Google Scholar]
- 47.Hogeland GW, Swindells S, McNabb JC, Kashuba AD, Yee GC, Lindley CM. Lopinavir/ritonavir reduces bupropion plasma concentrations in healthy subjects. Clin Pharmacol Ther. 2007;81:69–75. doi: 10.1038/sj.clpt.6100027. [DOI] [PubMed] [Google Scholar]
- 48.Kharasch ED, Thummel KE, Watkins PB. CYP3A probes can quantitatively predict the in vivo kinetics of other CYP3A substrates and can accurately assess CYP3A induction and inhibition. Mol Interv. 2005;5:151–3. doi: 10.1124/mi.5.3.3. [DOI] [PubMed] [Google Scholar]
- 49.Olkkola KT, Palkama VJ, Neuvonen PJ. Ritonavir’s role in reducing fentanyl clearance and prolonging its half-life. Anesthesiology. 1999;91:681–5. doi: 10.1097/00000542-199909000-00020. [DOI] [PubMed] [Google Scholar]
- 50.Kharasch ED, Hoffer C, Whittington D, Sheffels P. Role of hepatic and intestinal cytochrome CYP3A and CYP2B6 in the metabolism, disposition and miotic effects of methadone. Clin Pharmacol Ther. 2004;76:250–69. doi: 10.1016/j.clpt.2004.05.003. [DOI] [PubMed] [Google Scholar]