

Abstract
ASA404, a flavonoid tumor-vascular disrupting agent (Tumor-VDA), is in clinical trial for the treatment of non-small cell lung cancer. Its action differs from both that of the tubulin binding class of Tumor-VDAs and antiangiogenic agents. In mice, ASA404 induces a sequence of changes in tumor tissue, starting within one hour with increased vascular permeability, increased endothelial apoptosis and decreased blood flow. Later effects include the release of serotonin, induction of tumor necrosis factor and other cytokines and chemokines, as well as induction of nitric oxide. This cascade of events induces sustained effects on blood flow, tumor hypoxia, vascular failure, inflammatory responses and, in some tumors, complete regression. One feature of the action of ASA404 against murine tumors is its ability to potentiate the effects of radiation and a variety of chemotherapeutic agents. The flavonoid class appears to be unique because of its dual action on vascular endothelial function and innate immunity. The translation of preclinical to clinical results demands an understanding of both the mechanisms underlying the dual effects and the species differences in ASA404 activity. Clinical trials indicate that the future of ASA404 as an effective agent relies on a deep appreciation of its cellular action.
Keywords: DMXAA, ASA404, vadimezan, antivascular, tumor necrosis factor, toll-like receptors, serotonin, ceramide
1. Introduction
Tumor vasculature, like that of normal tissues, is essential for the supply of oxygen, nutrients and signaling molecules. However, tumor vasculature differs from normal vasculature in number of ways including the presence of disorganized architecture, increased vascular permeability, inefficient and sometimes intermittent blood flow, and areas of angiogenesis. Many of the features of tumor vasculature are similar to those seen during inflammation associated with the process of wound healing, giving rise to the concept of cancer as “a wound that does not heal” (1). Consideration of the properties of tumor vasculature has given rise to two main rationales for selective anticancer therapy. The first, developed by Folkman and others, is based on the concept that tumor growth requires the initiation and concomitant growth and remodeling of an accompanying vascular network, and that inhibition of vasculogenesis and angiogenesis will therefore impede tumor growth (2). The second, proposed by Denekamp and others, is that tumor vasculature, because of its distinctive properties, can be targeted for selective disruption (3). It is this second rationale that is relevant to this review on the action of the tumor vascular disrupting agent (Tumor-VDA) ASA404, whose action is shown diagrammatically in Figure 1. Changes in the shape and connectivity of tumor vascular endothelial cells, as well as loss of these cells by apoptosis, lead not only to a reduction in tumor blood flow but also to disruption of the vessel itself, allowing extravasation of blood cells into the surrounding tissue and irreversible arrest of blood flow. Tumor cells become hypoxic and survive in the absence of oxygen for at least 2 hours, but continued inhibition of blood flow leads after approximately 5 hours to loss of tumor cell viability (4) and at later times to the progressive development of tumor hemorrhagic necrosis.
Figure 1.

Conceptual illustration of temporal changes in tumor vasculature following administration of a Tumor-VDA.
Antiangiogenic agents and Tumor-VDAs are often considered together and it is important to differentiate their actions. Blood flow within a tumor blood vessel is a function of the pressure difference along the vessel, vessel diameter and blood viscosity (5) and increased tumor vascular permeability leads to leakage of plasma, increased blood viscosity, reduction in the diameters of individual blood vessels and consequently reduced blood flow. As shown in Figure 2, tumor vasculature is partially compromised because of the presence of vascular endothelial growth factor (VEGF) and other inflammatory molecules. Antiangiogenic agents are thought to normalize the vasculature, for instance by reducing VEGF concentrations, decreasing permeability and increasing blood flow (6). In contrast, Tumor-VDAs further increase vascular permeability and decrease blood flow, sometimes inducing loss of vascular endothelial cells by apoptosis. Such processes can lead to catastrophic vascular failure and tumor necrosis.
Figure 2.

Theoretical relationship between vascular permeability and blood flow. Tumor vasculature is already partially compromised because of increased vascular permeability and angiogenesis. An anti-angiogenic drug acts to decrease angiogenesis and normalize vascular permeability, while a Tumor-VDA acts by further compromising permeability, leading to further reduction of blood flow and possible catastrophic failure.
Antiangiogenic agents have an established role in clinical cancer; two examples are bevacizumab, a humanized monoclonal antibody to VEGF (7) and everolimus, an inhibitor of the mammalian target of rapamycin (mTOR) pathway (8). Tumor-VDAs are at a slightly earlier stage of clinical development but several classes are known (9). Among small molecule Tumor-VDAs, two classes have been developed to clinical trial, each of which has a distinctive action. The tubulin-binding class, typified by the drug combretastatin-A-4 phosphate (10, 11) inhibits the polymerization of tubulin, altering the cytoskeleton of vascular endothelial cells and increasing vascular permeability. The flavonoid class, named after flavone acetic acid as will be described later in the review, appears to act through targeted induction of an inflammatory response within the tumor microenvironment, inducing parallel changes in vascular endothelium and cells of the innate immune system. This review commences with a brief description of the development of ASA404, the most active of the flavonoid class found to date, and follows with a more detailed description of the time-course of the effect of ASA404 on tumor tissue and discussion of possible mechanisms of its action.
2. Development of ASA404
The discovery at the US National Cancer Institute that the drug flavone acetic acid (FAA; structure in Figure 3) had unexpectedly high experimental activity against murine colon tumors (12) led to the anticipation of a new type of cytotoxic drug. However, negative results in Phase I clinical trials in a large number of cancer patients (13) combined later with a reported lack of activity in a rat tumor model (14) raised the question of whether FAA was inactive because of a species difference. On the other hand, studies in the Auckland Cancer Society Research Centre, while confirming the activity of FAA against murine tumors, showed that FAA had an unexpected and novel antitumor mechanism, inducing widespread hemorrhagic necrosis of murine colon and lung tumors within 24 hours of administration of a single dose (15, 16). A search for other drugs inducing similar tumor hemorrhagic necrosis responses in this tumor model led to the identification of a diverse series of compounds including the antibiotic fostriecin, the plant product homoharringtonine, and the plant-derived mitotic poisons colchicine, podophyllotoxin, vinblastine and vincristine (17, 18). A parallel search for active drugs with structures related to that of FAA led to the identification of the tricyclic analogue xanthenone-4-acetic acid (XAA; Figure 3) (19). Good synthetic routes were available to prepare ring-substituted derivatives of XAA and, although most of these were inactive, derivatives substituted at the 3-, 5-, and 6-positions (Figure 3) were active. Disubstitution with methyl groups led to 5,6-dimethyllxanthenone-4-acetic acid (DMXAA), now known as ASA404, which displayed both increased dose potency and activity as compared to FAA and XAA, and was also active against a rat mammary carcinoma (20).
Figure 3.
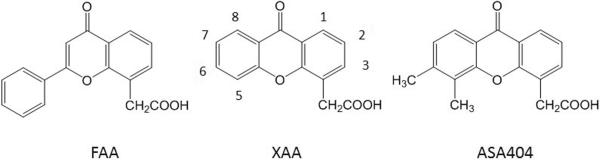
Chemical structures of FAA, XAA and ASA404.
An important feature of ASA404 was its ability to combine productively with other agents including radiation, hyperthermia, photodynamic therapy and a range of chemotherapeutic drugs (21–23). A striking example of this combination was provided by the drug paclitaxel, which by itself induced a tumor growth delay of 0.3 days; co-administration of ASA404 extended the median tumor growth delay to 80 days with three of seven animals cured (24).
2.1 Phase I clinical trials of ASA404
Among drug molecules found to induce tumor hemorrhagic necrosis in murine tumors, FAA, XAA, ASA404 and certain other XAA derivatives were distinguished from the tubulin-binding class of Tumor-VDAs (10, 11) by their ability to induce the cytokine TNF in mice (25). However, ASA404 was essentially a more dose-potent analogue of FAA, which had previously been shown to be inactive in Phase I trials, and a new argument was required for its advancement to a clinical trial. This was provided by the observation that while both ASA404 and FAA induced cultured murine monocytes to synthesize TNF, only ASA404 induced cultured human monocytes to synthesize TNF (26). The novelty of this action, combined with interest at that time in targeting tumor vasculature, led the Cancer Research Campaign (now Cancer Research UK) to support Phase I clinical trials of ASA404 in New Zealand and the United Kingdom. Two different single-agent administration schedules were used and the maximum tolerated dose was considerably higher than in mice, although slightly lower than that in rats. Two unconfirmed responses were observed, one in cervical cancer (27) and one in melanoma (28). However, the trial allowed the use of a new technique, dynamic contrast-enhanced magnetic resonance imaging using a gadolinium-labeled probe, which provided evidence for a reduction of tumor blood flow at a dose comparable to that used in preclinical studies (29). These two trials did not provide an unequivocal value for a recommended dose for combination Phase II trials and a third Phase I trial was instituted in 15 patients with refractory tumors who were allocated randomly to receive six sequential doses of DMXAA (300, 600, 1,200, 1,800, 2,400, and 3,000 mg/m2), each given weekly as a 20-minute intravenous infusion. This established an optimal initial Phase II dose of 1200 mg/m2 (calculated as the sodium salt) (30).
2.2 Phase II clinical trials of ASA404
The preclinical demonstration of the activity of combinations of ASA404 with taxanes and platinum-based anticancer drugs (24) provided a rationale for combination clinical trials. Three Phase II trials were designed for patients with non-small cell lung cancer (NSCLC), ovarian cancer and prostate cancer. Each had two arms, the first employing the recommended best cytotoxic therapy and the second with additional ASA404 administration. In the NSCLC trial, standard therapy comprised carboplatin (AUC; area under the concentration-time curve = 6) and paclitaxel (175 mg/m2) administered every three weeks for up to six cycles, and resulted in a response rate of 29% and median survival of 8.8 months. When ASA404 (1200 mg/m2) was added to the combination of cytotoxic drugs, the response rate was 34.4% and the median survival increased to 14 months (HR (95% CI) = 0.73 (0.39, 1.38)) (31). In a single-arm extension to this trial, a higher ASA404 dose (1800 mg/m2) was administered; the response rate was 37.9% and median survival increased to 14.9 months (32). However, these increases did not reach statistical significance. The ovarian cancer trial employed the same drug administration schedule as the NSCLC trial. The response rates were 48.6% for standard therapy and 63.9% for the ASA404 (1200 mg/m2) arm, and the times to tumor progression were 8.6 and 9.0 months, respectively (33). In the prostate cancer trial the standard therapy was docetaxel (75 mg/m2), supplemented in the second arm with ASA404 (1200 mg/m2). The prostate specific antigen response rates were 36.8% for standard therapy and 59.4% for the ASA404 arm; the times to tumor progression were 8.7 and 8.4 months, respectively (34). Thus, all three trials showed increased response rates when ASA404 was included in the treatment but did not prolong survival in ovarian and prostate cancer (35).
2.3 Phase III clinical trials of ASA404
The promising Phase II results with ASA404 led to its advancement to two randomized, double blind, placebo-controlled Phase III trials in advanced NSCLC, the first (ATTRACT-1) as first-line therapy and the second (ATTRACT-2) as second-line therapy (35). Both combinations employed ASA404 at a dose of 1800 mg/m2 (calculated as the free base) for the second treatment arm and were administered every three weeks for up to six cycles. The ATTRACT-1 study utilized paclitaxel (200 mg/m2) and carboplatin (AUC 6 mg/ml.min) as standard therapy. There were no safety concerns and no unexpected adverse effects when compared to the Phase II data, but the trial was halted when interim data analysis failed to show a survival advantage (http://www.antisoma.com). The ATTRACT-2 trial, which utilizes docetaxel (75 mg/m2) as standard therapy in patients with Stage IIIb/IV disease with no restrictions on histology, remains ongoing.
3. Early events in the action of ASA404
The action of ASA404 is very different from that of other agents and its biochemical target is currently not known. However, it is useful to discuss our current knowledge of ASA404 in terms of the time course of its action, which is illustrated conceptually in Figure 4. It is clear from what has been discussed above that for tumor vascular disruption to be successful, it must be maintained for at least 6 hours to allow the transition from hypoxia induction to the onset of hemorrhagic necrosis. Experimental data obtained in mice show that three interrelated events are detected within one hour of drug administration - increased tumor vascular permeability (36, 37), increased tumor endothelial cell apoptosis (38, 39) and decreased tumor blood flow (40, 41). A key feature of this action is its relationship to the expression of tumor necrosis factor (TNF). Increased tumor endothelial apoptosis, increased vascular permeability and decreased tumor blood flow are all greatly attenuated in mice lacking expression either of TNF itself (42) or of the TNFR1 receptor (43). Interferon-β may also play a role since the action of ASA404 is attenuated in mice lacking expression of this cytokine (44). ASA404 also results in an increase in VEGF production (45) and since the potential of VEGF to increase endothelial cell permeability is increased by TNF (46), the action of ASA404 in tumor tissue may involve the intersection of the action of these key cytokines.
Figure 4.

Scheme showing the approximate time course of ASA404 action in mice; the vertical axis indicates the approximate time scale and the shaded boxes represent the effects that contribute to the cascade of vascular disruption. ASA404 facilitates changes in both tumor endothelium and innate immune cells such as macrophages. Its direct effect is limited by a relatively short plasma half-life but leads in vascular endothelial cells to decreased blood flow, increased tumor hypoxia and vascular injury. Early effects on innate immune cells appear to involve increased ceramide production (BC Baguley, unpublished) and later effects involved the production of inflammatory cytokines and nitric oxide. The induction of hypoxia may potentiate responses of innate immune cells and can also induce the cytokine VEGF, which increases vascular permeability. Serotonin produced by platelets in response to vascular injury can potentiate inflammatory cytokine production, as can the appearance of necrotic tumor cells. Co-administration of cytotoxic drugs can also induce tumor cells to induce HMGB1 and other mediators that further stimulate host cytokine responses.
It is important to note that ASA404 induces endothelial cell apoptosis in addition to increasing vascular permeability (38). This action contrasts with those of physiological regulators of vascular permeability which do not induce apoptosis (47), but is similar to that induced by pathological conditions such as gram-negative bacterial infection and hemorrhagic shock. Bacterial lipopolysaccharide, acting through the toll-like receptor TLR4, induces endothelial apoptosis with early onset and with maximal effect by 6 hours (48). The induction of hemorrhagic shock, for instance following rapid blood loss, also involves activation of toll-like receptors such as TLR4 (49). These observations suggest that toll-like receptors on vascular endothelial cells could be involved in the response to ASA404.
One of the earliest biochemical changes in tumor tissue following administration of ASA404 is an increase in tumor concentrations of ceramides and a consequent rise in plasma concentrations of the ceramide metabolite sphingosine (50, 51). Ceramide plays an important role in the induction of apoptosis by many different agents (52) and appears to mediate the induction of endothelial cell apoptosis in response to bacterial lipopolysaccharide (53). Ceramide not only has multiple intracellular targets, including protein phosphatase 2A and protein kinase Cζ, which contribute to the induction of apoptosis, but is also active in the conversion of lipid rafts on the plasma membrane to active signaling platforms (54). Thus, ASA404-induced increases in tumor tissue ceramide concentrations could play a role in the induction of endothelial apoptosis.
4. Effects on innate immunity
One of the most intriguing properties of the flavonoid Tumor-VDAs is their ability to stimulate innate immune cells to induce a range of inflammatory cytokines that are indicative of a targeted pro-inflammatory response within tumor tissue. The lack of TNF induction by tubulin-binding Tumor-VDAs indicates that cytokine induction is not simply a consequence of tumor blood flow reduction or of hypoxia induction. Within 4 hours of administration of ASA404 to mice, detectable increases are observed in tumor concentrations of TNF, interleukin-6 (IL-6), granulocyte colony stimulating factor (GCSF) and several chemokines, including KC (CXCL1), IP-10 (CXCL10), MCP-1 (CCL2), MIP-1α (CCL3) and RANTES (CCL5) (43, 55, 56). These cytokines appears to originate mainly from CD11b+ macrophages and CD45R+ B-lymphocytes. Chemokines function as chemo-attractants and may explain the increase in tumor-associated neutrophils in mice during the first 24 hours after treatment (56). ASA404-induced increases in cytokine concentrations are much higher in tumor tissue than in plasma, consistent with intra-tumor synthesis. Rats treated with ASA404 also produced elevated tumor tissue concentrations of cytokines and chemokines, including TNF, VEGF, IL-6 and IL-1α, MIP-1α (CCL3), KC (CXCL1) and IP-10 (CXCL10) (20). However, chemokine concentrations in rat tumor tissue were typically 100-fold lower than those of mouse tumor tissue, and plasma TNF was not detected in rat tumors.
Nitric oxide induction, as indicated by plasma nitrite/nitrate concentrations increases from 4 hours to a maximum 12 hours after treatment and is larger in mice with tumors than those without (57). The results are consistent with a rise in macrophage inducible nitric oxide synthase (iNOS) activity to a peak 6 hours after ASA404 administration (58). TNF and nitric oxide have vascular disruptive properties in their own right; consistent with the hypothesis that ASA404 induces a cascade of tumor vascular disrupting effects involving direct effects, serotonin induction, TNF induction and nitric oxide induction. This occurs over a time scale of several hours, leading to long-term arrest of blood flow (Figure 4).
Administration of ASA404 to mice at a potential toxic dose leads to a decrease in haematocrit and reduction of both skin and body temperature (37). These responses appear to be mediated by TNF production because the maximum tolerated dose (MTD) of ASA404 in mice lacking expression of either TNF or its TNFR1 receptor (>100 mg/kg) was higher than that in wild-type mice (27.5 mg/kg; 82 mg/m2) (42, 43). The MTD of ASA404 in rats (350 mg/kg; 1,800 mg/m2) is much higher than that in mice, consistent with the hypothesis that induced TNF is not the dose limiting toxicity in this species.
Bacterial toxins induce cytokines and chemokines through binding to toll-like receptors, and ASA404 and LPS induce cross-resistance to each other (59), raising the question of whether ASA404 acts by directly stimulating toll-like receptors such as TLR2 and TLR4. However, many observations argue against this hypothesis. The time course of TNF induction by LPS is much earlier than that of ASA404 with an early rise (within 1 hour) to maximal activity. The distribution of TNF production is also different with ASA404, which induces much higher levels of tumor-associated TNF (60). LPS also has minimal antitumor effects on the Colon 38 tumor and does not potentiate the activity of ASA404 (61).
5. Tumor responses to vascular disruption
The disruption of tumor blood flow by ASA404 induces a variety of effects, including responses to both vascular injury and the reduced supply of oxygen, between 1 and 4 hours after drug administration. Vascular injury, possibly from loss of apoptotic endothelial cells from the vascular sleeve and consequent activation of platelets by exposed collagen, leads to the activation of platelets within 3 hours with consequent release of serotonin and von Willebrand factor (62, 63). Serotonin is a Tumor-VDA in its own right, reducing blood flow and inducing hemorrhagic necrosis (64). Co-administration of serotonin with ASA404 potentiates its antitumor activity (41, 64) while co-administration of a serotonin receptor-2 antagonist delays the induction of TNF by 1 hour (65). Moreover, serotonin stimulates vascular endothelial cells to secrete the cytokine HMGB1 (66), which may also contribute to the overall tumor response through induction of cytokines. Free serotonin in plasma is converted by the liver to 5-hydroxyindole-3-acetic acid (5-HIAA), which rises between one and 4 hours after administration and is a useful marker for the action of ASA404 (67). ASA404 induces a dose-dependent increase of 5-HIAA in mice (68) which correlates with both increased tumor vascular permeability and decreased tumor blood flow (36).
The induction of hypoxia is detectable in mice within 3 hours of ASA404 treatment, as measured with a technetium-labeled marker (69). Hypoxia leads to the induction of the HIF-1α transcription factor, changes in glucose metabolism and the induction of VEGF (70). Experiments in mice have shown increased mRNA for VEGF-A in tumor tissue (45) and experiments in rats have shown increased tumor-associated VEGF (20). Although the induction of hypoxia in itself does not result in cytokine synthesis, it can potentiate the ability of LPS to induce inflammatory cytokine production (71). TNF production in tumor tissue in mice occurs at a later time (3–4 hours) after ASA404 administration than that (1 hour) after LPS administration (72) and one possible reason for this delay is that it is consequent to the development of hypoxia.
Feedback on endothelial function is provided by the synthesis of cytokines such as TNF by innate immune cells, as well as the induction of inducible nitric oxide synthase and its product nitric oxide. The result is an increase in tumor vascular permeability and inhibition of blood flow. The production of VEGF (45), as well as increasing vascular permeability, could potentially stimulate angiogenesis and decrease the overall antitumor effect. However, induced angiogenesis might be prevented by the induction in innate immune cells of chemokines such as IP-10 (73).
6. Interaction with cytotoxic therapy
The ability of ASA404 to induce vascular disruption by multiple pathways suggests that it should induce tumor regression as a single agent, but while some murine tumors (such as Colon 38) are highly responsive to ASA404, others show only small responses. On the other hand, numerous studies have demonstrated potentiation of the antitumor activity of cytotoxic drugs and radiotherapy by ASA404 (21–24). At least two mechanisms can be hypothesized to account for this synergy and both may contribute to the efficacy of combination therapy.
The first mechanism, related to tumor architecture, is based on the principle that cytotoxic therapy is more effective against tumor cells with good vascular access, while ASA404 is effective on tumor areas with compromised vascular function. ASA404 has minimal effects on normal vasculature and only at toxic doses does it lead to coldness of skin and increased blood haematocrit, indicative of increased normal vascular permeability (37). Blood vessels at the tumor periphery may have a more normal phenotype and therefore reduced response to ASA404, and as a consequence tumor cells associated with the tumor periphery survive ASA404 treatment, giving rise to a phenomenon reported for all Tumor-VDAs and known as the “viable ring” effect (9–11).
A second potential mechanism for interaction between ASA404 and cytotoxic therapy is that it combines with cytotoxic therapy to induce immunogenic cell death. Injury of tumor cells by some cancer chemotherapeutic agents leads to the release of cytokines such as HMGB1 and interleukin-1α, with resultant activation of corresponding receptors to activate innate immune cells (74, 75). Cytotoxic drugs such as doxorubicin and oxaliplatin (76) can also induce the translocation, by a caspase-8 mediated mechanism, of calreticulin from the endoplasmic reticulum to the cell surface, where it can cooperate with cytokines such as HMGB1 in to induce tumor cell death (77).
Administration of ASA404 in combination with cytotoxic drugs sets up two sequences of signals, the first affecting the tumor vasculature function and the second affecting innate immune cells, and optimization of both the intensity and the duration of these signals is important for effective tumor cell killing. Tumor vascular disruption must be maintained for at least 6 hours and since the plasma half-life of ASA404 in mice at the maximum tolerated dose (MTD) is less than 1 hour (78), one approach to optimize drug administration is to design schedules that to maintain a relatively constant plasma concentration of ASA404 over a number of hours. This has been demonstrated to be effective in a tumor xenograft model (79). In addition, interactions with cytotoxic therapy may be strongly dependent on the schedule of administration. In rodent (KHT sarcoma) and human (SKBR3 breast and OW-1 ovarian) tumor models, ASA404 enhanced the activity of cisplatin but was most effective when administered 1–3 hours after chemotherapy (22). Murine C3H mammary carcinomas and KHT sarcomas were found to be treated effectively with radiotherapy and ASA404 when both were administered together, or when ASA404 was administered 1–3 hours after irradiation (80). In another study, combination of radiotherapy with ASA404 was found to be most effective against RIF-1 and MDAH-MCa-4 tumors when the two were administered together, or when radiation was administered 12–48 hours after ASA404 (81).
7. In vitro studies
Research with cultured cells has provided further insights into the action of ASA404 and the relationship between effects on murine and human host cells. Early studies established that ASA404 (200 μg/ml) induced mRNA for TNF in murine splenocytes, the murine J774 macrophage cell line and the human HL-60 myelomonocytic leukemia cell line (26). ASA404 (82) and related compounds (83) also induced murine peritoneal macrophages to kill target tumor cells through a mechanism involving cell-cell contact. Later studies showed that ASA404 (10–100 μg/ml) stimulated production of TNF mRNA in murine macrophages but that its action could be distinguished from that of LPS because of a different spectrum of induced cytokines (84). Cellular signaling was also found to be different to that induced by LPS, with minimal dependence on the NF-κB transcription factor (59). However, ASA404 (10–100 μg/ml) potentiated TNF production in response to stimulation of TLR4 receptors of cultured murine splenocytes by LPS (85).
Studies using cultured human peripheral blood monocytes showed that ASA404 did not in itself induce TNF. However, as shown in Figure 5, ASA404 at a high concentration (800 μg/ml) potentiated TNF production in response to a range of agents including LPS, an antibody to CD-14 receptor, interleukin-1 and phorbol myristate acetate (86). The above agents, although diverse in structure and in target signaling pathways, share the property of inducing the enzyme acid sphingomyelinase (54, 87). ASA404 also had effects on cultured human endothelial cells, inducing apoptosis in a mechanism that was independent of NF-κB activation (88). A further study showed that endothelial cells underwent apoptosis in a dose-dependent fashion following exposure for 24 hours to ASA404 concentrations between 25 and 200 μM (89). Subsequent studies showed that in addition to inducing apoptosis, ASA404 induced alterations in the rate of endothelial cell network formation, changes in the cytoskeleton and increases in ceramide (45, 51).
Figure 5.

A. A model indicating the possible involvement of the ceramide family of lipid signaling molecules in the action of ASA404. A diverse range of agents is known to simulate the ability of acid sphingomyelinase to synthesize ceramides from sphingomyelin. ASA404 is known to potentiate the ability of several of these agents to stimulate TNF production by cultured human peripheral blood leucocytes (HPBL) (86); two illustrative graphs redrawn from this publication are shown in B and C. Increased concentrations of cellular ceramides facilitate the activation of signaling platforms that can lead to physiological changes in both the tumor vascular endothelium and cells of the innate immune system (see text). B. TNF concentrations of supernatants of HPBL cultures incubated for 8 hours with the indicated concentrations of interleukin-1 alone (∘) or in combination with ASA404 (■). C. TNF concentrations of supernatants of HPBL cultures incubated for 8 hours with the indicated concentrations of phorbol myristate acetate (PMA) alone (∘) or in combination with ASA404 (■). Vertical lines represent the ranges of duplicate cultures.
Taken together with in vivo studies, these observations suggest that ASA404 has the capacity to potentiate the capacity of diverse agents to modify both endothelial and macrophage function. A possible mechanism, as shown in Figure 5, is that ASA404 potentiates the action of agents that induce acid sphingomyelinase. It is known that ionizing radiation, cisplatin and doxorubicin induce acid sphingomyelinase (54, 90) and that the resulting increases in ceramide concentrations promote the conversion of lipid rafts to signaling platforms (54, 91). Potentiation of such signaling effects by ASA404 could mediate its effects on both endothelial cells and cells of the innate immune system. In the case of innate immune cells, which release TNF in response to ASA404, signaling is likely to involve stress-induced protein kinase pathways (92) rather than the pathway involving the NF-κB transcription factor previously proposed (93).
8. Expert opinion
We stand at a cross-road in making decisions on the further development of ASA404 and other members of the flavonoid class of Tumor-VDAs. In NSCLC, one Phase II combination trial showed extreme promise, one Phase III combination trial in first-line therapy showed no advantage, and one Phase III trial as second-line therapy is still in progress. The clinical results do not parallel the dramatic results obtained in preclinical studies, and the first question that needs to be addressed is whether the action of ASA404 in humans is similar to that in mice. Several early events observed in mice, including inhibition of tumor blood flow, increased tumor vascular permeability, induction of tumor endothelial cell apoptosis and increases in the plasma concentrations of the serotonin metabolite 5-HIAA, have been reported in cancer patients within 24 hours of drug administration (39, 94, 95). On the other hand, while large increases in tumor concentrations of the cytokine TNF were observed in mice in response to ASA404, the limited data available for cancer patients indicate only a small increase in tumor tissue TNF (96). Nevertheless, despite leading to only small increases in tumor tissue TNF in rats, ASA404 treatment resulted in significant antitumor effects, albeit at higher doses than those used in human studies (20). Finally, ASA404 stimulates cultured human peripheral blood leucocytes to produce TNF in vitro (26, 86). Taken together, these observations suggest that ASA404 should be active in humans as it is in rats and mice.
The second question is whether the conditions of drug administration used in clinical trials are similar to those employed in mice. Carboplatin, paclitaxel and docetaxel were chosen for use in combination with ASA404 in clinical studies and there is good supporting evidence for the choice of these chemotherapeutic agents from results observed in mice (24). However, one potentially critical difference in the treatment protocol is that corticosteroids such as dexamethasone have been utilized to counteract side-effects (emesis and hypersensitivity reactions) of the cytotoxic agents in clinical but not in preclinical investigations. Co-administration of cortisone is known to inhibit the antitumor activity of the Tumor-VDA FAA in a number of murine tumors (97) and both cortisone and dexamethasone inhibit the macrophage tumoricidal activity of ASA404 in vitro (82). Further clinical studies to test the hypothesis that the use of corticosteroids in clinical trials may compromise the antitumor action of ASA404 appear to be warranted.
The third question is whether the temporal aspects of action of ASA404 are adequately reflected in clinical trial design. Preclinical studies have emphasized the importance of administering cytotoxic drugs prior to or simultaneously with ASA404 to prevent vascular disruption from inhibiting delivery of cytotoxic drugs; in fact vascular disruption might have a role in preventing clearance of these drugs from tumors (98). However, over a longer time scale, vascular disruption will lead to hypoxia and subsequent stimulation of angiogenesis, including the induction of VEGF (45). This raises the question as to whether Tumor-VDA and angiogenic treatment should be given in tandem.
The fourth question is whether the choice of tumor for the clinical trials is optimal for the application of a Tumor-VDA. As shown in Fig. 2, the vascular stability of tumor tissue varies according to the microenvironment and particularly as a function of production of vasoactive mediators such as VEGF, or of other properties that cause a tumor to have a high degree of hypoxia. For example, renal cell carcinomas that produce large amounts of VEGF as a consequence of genetic or epigenetic changes in the expression of the von Hippel-Landau gene (VHL), are highly vascularized and could be more susceptible to the induction of vascular failure; they might constitute an interesting target for the application of ASA404-based therapy (99).
In conclusion, the studies described in this review indicate that the mechanism of action of ASA404 is quite unlike that of other classes of anticancer drugs and further work is required to delineate its underlying molecular mechanisms. In mice, ASA404 orchestrates a temporal series of events involving both endothelial cells and immune cells in the tumor microenvironment, leading to sustained vascular disruption and tumor regression; the coordinated vascular and innate immune responses are reminiscent of those occurring in response to infection. The optimization of the ability of ASA404 to induce such a targeted pro-inflammatory response within tumor tissue may therefore be an important aim of combination anticancer therapy. It is worth noting that many elements of current clinical cancer therapy, particularly in taxane-based chemotherapy protocols, involve suppression of inflammation, and this should be carefully considered when applying flavonoid Tumor-VDAs in a clinical setting.
Article Highlights
Tumor vasculature is different to normal vasculature and can be targeted with Tumor-VDAs.
ASA404 is the most active of the flavonoid class of Tumor-VDAs, demonstrating a survival advantage in Phase II NSCLC studies, and is currently in clinical trial for second-line treatment of the disease.
Early effects of ASA404 include increased tumor endothelial apoptosis, increased vascular permeability and decreased tumor blood flow, and these effects may intersect with the actions of the cytokines TNF and VEGF.
Flavonoid Tumor-VDAs stimulate innate immune cells to induce a range of inflammatory cytokines within a few hours of administration.
ASA404-induced tumor vascular disruption leads to serotonin release, hemorrhagic necrosis and induction of hypoxia within 3 hours.
ASA404 potentiates the antitumor activity of both cytotoxic drugs and radiotherapy, by targeting areas with compromised vascular function that are resistant to these modalities.
The effects of ASA404 on both endothelial and innate immune cells, involves indirect TNF production, possibly linked to induction of ceramides.
The series of events involving endothelial and immune cells that leads to vascular disruption and tumor regression, involves coordinated vascular and innate immune responses – this targeted pro-inflammatory response in combination anticancer therapy should be balanced with any concomitant suppression of inflammation.
Acknowledgement
Dr Baguley is supported by the Auckland Division of the Cancer Society of New Zealand. Dr Siemann is supported in part by a grant from the National Cancer Institute (Public Health Service Grant CA084408); the funding sponsor had no involvement in the preparation of this manuscript. The authors would like to thank Articulate Science, London, UK (funded by Novartis Pharmaceuticals Corporation) for writing assistance.
Footnotes
Declaration of Interest Dr BC Baguley has received funding for research on ASA404 from Novartis.
References
- 1.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–9. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 3.Denekamp J. Vascular endothelium as the vulnerable element in tumours. Acta Radiol Oncol. 1984;23:217–25. doi: 10.3109/02841868409136015. [DOI] [PubMed] [Google Scholar]; *Pioneering work on selective targeting of tumor vasculature
- 4.Zwi LJ, Baguley BC, Gavin JB, Wilson WR. Blood flow failure as a major determinant in the antitumor action of flavone acetic acid. J Natl Cancer Inst. 1989;81:1005–13. doi: 10.1093/jnci/81.13.1005. [DOI] [PubMed] [Google Scholar]
- 5.Milosevic MF, Fyles AW, Hill RP. The relationship between elevated interstitial fluid pressure and blood flow in tumors: a bioengineering analysis. Int J Radiat Oncol Biol Phys. 1999;43:1111–23. doi: 10.1016/s0360-3016(98)00512-4. [DOI] [PubMed] [Google Scholar]
- 6.Dickson PV, Hamner JB, Sims TL, et al. Bevacizumab-induced transient remodeling of the vasculature in neuroblastoma xenografts results in improved delivery and efficacy of systemically administered chemotherapy. Clin Cancer Res. 2007;13:3–3950. doi: 10.1158/1078-0432.CCR-07-0278. [DOI] [PubMed] [Google Scholar]
- 7.Mukherji SK. Bevacizumab (Avastin) Amer J Neuroradiol. 2010;31:235–6. doi: 10.3174/ajnr.A1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulders P. Vascular endothelial growth factor and mTOR pathways in renal cell carcinoma: differences and synergies of two targeted mechanisms. BJU Int. 2009;104:1585–9. doi: 10.1111/j.1464-410X.2009.08987.x. [DOI] [PubMed] [Google Scholar]
- 9.Siemann DW, Horsman MR. Vascular targeted therapies in oncology. Cell Tissue Res. 2009;335:241–8. doi: 10.1007/s00441-008-0646-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Comprehensive review on targeting tumor blood vessels
- 10.Siemann DW, Chaplin DJ, Horsman MR. Vascular-targeting therapies for treatment of malignant disease. Cancer. 2004;100:2491–9. doi: 10.1002/cncr.20299. [DOI] [PubMed] [Google Scholar]
- 11.Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer. 2005;5:423–35. doi: 10.1038/nrc1628. [DOI] [PubMed] [Google Scholar]; *Comprehensive review on targeting tumor blood vessels
- 12.Plowman J, Narayanan VL, Dykes D, et al. Flavone acetic acid: a novel agent with preclinical antitumor activity against colon adenocarcinoma 38 in mice. Cancer Treat Rep. 1986;70:631–5. [PubMed] [Google Scholar]
- 13.Kerr DJ, Kaye SB, Cassidy J, et al. Phase I and pharmacokinetic study of flavone acetic acid. Cancer Res. 1987;47:6776–81. [PubMed] [Google Scholar]
- 14.Kal HB, de Graaff E, Van Berkel AH, Goedoen HH. Responses of experimental rat tumours and a mouse colon tumour to flavone acetic acid. In Vivo. 1992;6:73–5. [PubMed] [Google Scholar]
- 15.Smith GP, Calveley SB, Smith MJ, Baguley BC. Flavone acetic acid (NSC 347512) induces haemorrhagic necrosis of mouse colon 26 and 38 tumours. Eur J Cancer Clin Oncol. 1987;23:1209–11. doi: 10.1016/0277-5379(87)90157-x. [DOI] [PubMed] [Google Scholar]; *First report suggesting that flavone acetic acid selectively disrupted tumor vasculature
- 16.Finlay GJ, Smith GP, Fray LM, Baguley BC. Effect of flavone acetic acid on Lewis lung carcinoma: evidence for an indirect effect. J Natl Cancer Inst. 1988;80:2410245. doi: 10.1093/jnci/80.4.241. [DOI] [PubMed] [Google Scholar]
- 17.Baguley BC, Calveley SB, Crowe KK, et al. Comparison of the effects of flavone acetic acid, fostriecin, homoharringtonine and tumour necrosis factor alpha on colon 38 tumours in mice. Eur J Cancer Clin Oncol. 1989;25:263–9. doi: 10.1016/0277-5379(89)90018-7. [DOI] [PubMed] [Google Scholar]
- 18.Baguley BC, Holdaway KM, Thomsen LL, Zhuang L, Zwi LJ. Inhibition of growth of colon 38 adenocarcinoma by vinblastine and colchicine: evidence for a vascular mechanism. Eur J Cancer. 1991;27:482–7. doi: 10.1016/0277-5379(91)90391-p. [DOI] [PubMed] [Google Scholar]
- 19.Rewcastle GW, Atwell GJ, Baguley BC, Calveley SB, Denny WA. Potential antitumor agents. 58. Synthesis and structure-activity relationships of substituted xanthenone-4-acetic acids active against the colon 38 tumor in vivo. J Med Chem. 1989;32:793–9. doi: 10.1021/jm00124a012. [DOI] [PubMed] [Google Scholar]; *Discovery of DMXAA and its anticancer potential
- 20.Liu J, Ching LM, Goldthorpe N, et al. Antitumour action of 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in rats bearing chemically-induced primary mammary tumours. Cancer Chemother Pharmacol. 2007;59:661–9. doi: 10.1007/s00280-006-0321-7. [DOI] [PubMed] [Google Scholar]; *First report of the activity of ASA404 against rat tumors
- 21.Baguley BC, Wilson WR. Potential of DMXAA combination therapy for solid tumors. Expert Rev Anticancer Ther. 2002;2:593–603. doi: 10.1586/14737140.2.5.593. [DOI] [PubMed] [Google Scholar]; *Review of the action of DMXAA in combination therapy
- 22.Siemann DW, Mercer E, Lepler S, Rojiani AM. Vascular targeting agents enhance chemotherapeutic agent activities in solid tumor therapy. Int J Cancer. 2002;99:1–6. doi: 10.1002/ijc.10316. [DOI] [PubMed] [Google Scholar]
- 23.Siemann DW, Horsman MR. Enhancement of radiation therapy by vascular targeting agents. Curr Opin Investig Drugs. 2002;3:1660–5. [PubMed] [Google Scholar]
- 24.Siim BG, Lee AE, Shalal-Zwain S, et al. Marked potentiation of the antitumour activity of chemotherapeutic drugs by the antivascular agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA) Cancer Chemother Pharmacol. 2003;51:43–52. doi: 10.1007/s00280-002-0529-0. [DOI] [PubMed] [Google Scholar]; **First preclinical evidence of synergy of ASA404 with taxanes
- 25.Mace KF, Hornung RL, Wiltrout RH, Young HA. Correlation between in vivo induction of cytokine gene expression by flavone acetic acid and strict dose dependency and therapeutic efficacy against murine renal cancer. Cancer Res. 1990;50:1742–7. [PubMed] [Google Scholar]
- 26.Ching LM, Joseph WR, Crosier KE, Baguley BC. Induction of tumor necrosis factor-alpha messenger RNA in human and murine cells by the flavone acetic acid analogue 5,6-dimethylxanthenone- 4-acetic acid (NSC 640488) Cancer Res. 1994;54:870–2. [PubMed] [Google Scholar]; *First report of induction of tumor necrosis factor-alpha by DMXAA
- 27.Jameson MB, Thompson PI, Baguley BC, et al. Clinical aspects of a phase I trial of 5,6-dimethylxanthenone-4-acetic acid (DMXAA), a novel antivascular agent. Br J Cancer. 2003;88:1844–50. doi: 10.1038/sj.bjc.6600992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rustin GJ, Bradley C, Galbraith S, et al. 5,6-dimethylxanthenone-4-acetic acid (DMXAA), a novel antivascular agent: phase I clinical and pharmacokinetic study. Br J Cancer. 2003;88:1160–7. doi: 10.1038/sj.bjc.6600885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rustin G, Galbraith S, Taylor N, et al. Impact on tumour perfusion measured by dynamic magnetic resonance imaging (MRI) in the phase I trial of 5,6-dimethylxanthenone-4-acetic acid (DMXAA) Ann Oncol. 1998;9(Suppl 2):126. [Google Scholar]; *First report of effect if ASA404 on tumor blood flow in clinical studies
- 30.McKeage MJ, Fong P, Jeffery M, et al. 5,6-Dimethylxanthenone-4-acetic acid in the treatment of refractory tumors: a phase I safety study of a vascular disrupting agent. Clin Cancer Res. 2006;12:1776–84. doi: 10.1158/1078-0432.CCR-05-1939. [DOI] [PubMed] [Google Scholar]
- 31.McKeage MJ, von Pawel J, Reck M, et al. Randomised phase II study of ASA404 combined with carboplatin and paclitaxel in previously untreated advanced non-small cell lung cancer. Br J Cancer. 2008;99:2006–12. doi: 10.1038/sj.bjc.6604808. [DOI] [PMC free article] [PubMed] [Google Scholar]; **Phase II trial report on ASA404 1200 mg/m2 in combination with carboplatin and paclitaxel
- 32.McKeage MJ, Reck M, Jameson MB, et al. Phase II study of ASA404 (vadimezan, 5,6-dimethylxanthenone-4-acetic acid/DMXAA) 1800mg/m2 combined with carboplatin and paclitaxel in previously untreated advanced non-small cell lung cancer. Lung Cancer. 2009;65:192–7. doi: 10.1016/j.lungcan.2009.03.027. [DOI] [PubMed] [Google Scholar]
- 33.Gabra H, Jameson MB. Update of phase II study of DMXAA (AS1404) combined with carboplatin and paclitaxel in recurrent ovarian cancer. EJC Supplements. 2007;5:319. [Google Scholar]
- 34.Pili R, Rosenthal MA, Mainwaring PN, et al. Phase II study on the addition of ASA404 (vadimezan; 5,6-dimethylxanthenone-4-acetic acid) to docetaxel in CRMPC. Clin Cancer Res. 2010;16:2906–14. doi: 10.1158/1078-0432.CCR-09-3026. [DOI] [PubMed] [Google Scholar]
- 35.McKeage MJ, Baguley BC. Disrupting established tumor blood vessels: an emerging therapeutic strategy for cancer. Cancer. 2010;116:1859–71. doi: 10.1002/cncr.24975. [DOI] [PubMed] [Google Scholar]
- 36.Zhao L, Ching LM, Kestell P, Kelland LR, Baguley BC. Mechanisms of tumor vascular shut-down induced by 5,6-dimethylxanthenone-4-acetic acid (DMXAA); increased tumor vascular permeability. Int J Cancer. 2005;116:322–6. doi: 10.1002/ijc.21005. [DOI] [PubMed] [Google Scholar]
- 37.Chung F, Liu J, Ching LM, Baguley BC. Consequences of increased vascular permeability induced by treatment of mice with 5,6-dimethylxanthenone-4-acetic acid (DMXAA) and thalidomide. Cancer Chemother Pharmacol. 2008;61:497–502. doi: 10.1007/s00280-007-0495-7. [DOI] [PubMed] [Google Scholar]
- 38.Ching LM, Zwain S, Baguley BC. Relationship between tumour endothelial cell apoptosis and tumour blood flow shutdown following treatment with the antivascular agent DMXAA in mice. Br J Cancer. 2004;90:906–10. doi: 10.1038/sj.bjc.6601606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ching LM, Cao Z, Kieda C, et al. Induction of endothelial cell apoptosis by the antivascular agent 5,6-dimethylxanthenone-4-acetic acid. Br J Cancer. 2002;86:1937–42. doi: 10.1038/sj.bjc.6600368. [DOI] [PMC free article] [PubMed] [Google Scholar]; *First report of induction of endothelial cell apoptosis by DMXAA
- 40.Zwi LJ, Baguley BC, Gavin JB, Wilson WR. Correlation between immune and vascular activities of xanthenone acetic acid antitumor agents. Oncol Res. 1994;6:79–85. [PubMed] [Google Scholar]
- 41.Lash CJ, Li AE, Rutland M, et al. Enhancement of the anti-tumour effects of the antivascular agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA) by combination with 5- hydroxytryptamine and bioreductive drugs. Br J Cancer. 1998;78:439–45. doi: 10.1038/bjc.1998.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ching LM, Goldsmith D, Joseph WR, et al. Induction of intratumoral tumor necrosis factor (TNF) synthesis and hemorrhagic necrosis by 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in TNF knockout mice. Cancer Res. 1999;59:3304–7. [PubMed] [Google Scholar]; *First genetic evidence for the importance of TNF in the action of ASA404
- 43.Zhao L, Ching LM, Kestell P, Baguley BC. The antitumour activity of 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in TNF receptor-1 knockout mice. Br J Cancer. 2002;87:465–70. doi: 10.1038/sj.bjc.6600479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roberts ZJ, Ching LM, Vogel SN. IFN-beta-dependent inhibition of tumor growth by the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA) J Interferon Cytokine Res. 2008;28:133–9. doi: 10.1089/jir.2007.0992. [DOI] [PubMed] [Google Scholar]
- 45.Baguley BC, Zhan X, Finlay GJ, et al. The antitumor action of ASA404 (vadimezan; DMXAA); potential involvement of vascular endothelial growth factor (VEGF) Proc AACR. 2010;101:#1660. [Google Scholar]
- 46.Clauss M, Sunderkotter C, Sveinbjornsson B, et al. A permissive role for tumor necrosis factor in vascular endothelial growth factor-induced vascular permeability. Blood. 2001;97:1321–9. doi: 10.1182/blood.v97.5.1321. [DOI] [PubMed] [Google Scholar]
- 47.Komarova YA, Mehta D, Malik AB. Dual regulation of endothelial junctional permeability. Sci STKE. 2007;2007:re8. doi: 10.1126/stke.4122007re8. [DOI] [PubMed] [Google Scholar]
- 48.Bannerman DD, Sathyamoorthy M, Goldblum SE. Bacterial lipopolysaccharide disrupts endothelial monolayer integrity and survival signaling events through caspase cleavage of adherens junction proteins. J Biol Chem. 1998;273:35371–80. doi: 10.1074/jbc.273.52.35371. [DOI] [PubMed] [Google Scholar]
- 49.Benhamou Y, Favre J, Musette P, et al. Toll-like receptors 4 contribute to endothelial injury and inflammation in hemorrhagic shock in mice. Crit Care Med. 2009;37:1724–8. doi: 10.1097/CCM.0b013e31819da805. [DOI] [PubMed] [Google Scholar]
- 50.Baguley BC, Ding Q, Kestell P, Alix S. Potential importance of the ceramide pathway in the action of the tumour vascular disrupting agent ASA404 (DMXAA, 5,6-dimethylxanthenone-4-acetic acid) Eur J Cancer. 2008;6:32. [Google Scholar]; *Recent evidence for the importance of the ceramide pathway in the action of ASA404
- 51.Huang A, Chen Y, Li X, et al. Molecular mechanistic study of ASA404 (vadimezan)-induced endothelial cell death. Proc AACR. 2010;101:#4443. [Google Scholar]
- 52.Andrieu-Abadie N, Levade T. Sphingomyelin hydrolysis during apoptosis. Biochim Biophys Acta. 2002;1585:126–34. doi: 10.1016/s1388-1981(02)00332-3. [DOI] [PubMed] [Google Scholar]
- 53.Haimovitz-Friedman A, Cordon-Cardo C, Bayoumy S, et al. Lipopolysaccharide induces disseminated endothelial apoptosis requiring ceramide generation. J Exp Med. 1997;186:1831–41. doi: 10.1084/jem.186.11.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bollinger CR, Teichgraber V, Gulbins E. Ceramide-enriched membrane domains. Biochim Biophys Acta. 2005;1746:284–94. doi: 10.1016/j.bbamcr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 55.Philpott M, Baguley BC, Ching LM. Induction of tumour necrosis factor-alpha by single and repeated doses of the antitumour agent 5,6-dimethylxanthenone-4-acetic acid. Cancer Chemother Pharmacol. 1995;36:143–8. doi: 10.1007/BF00689199. [DOI] [PubMed] [Google Scholar]
- 56.Wang LC, Thomsen L, Sutherland R, et al. Neutrophil influx and chemokine production during the early phases of the antitumor response to the vascular disrupting agent DMXAA (ASA404) Neoplasia. 2009;11:793–803. doi: 10.1593/neo.09506. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Recent preclinical data on ASA404 mechanism of action
- 57.Thomsen LL, Ching LM, Zhuang L, Gavin JB, Baguley BC. Tumor-dependent increased plasma nitrate concentrations as an indication of the antitumor effect of flavone-8-acetic acid and analogues in mice. Cancer Res. 1991;51:77–81. [PubMed] [Google Scholar]
- 58.Veszelovsky E, Holford NH, Thomsen LL, Knowles RG, Baguley BC. Plasma nitrate clearance in mice: modeling of the systemic production of nitrate following the induction of nitric oxide synthesis. Cancer Chemother Pharmacol. 1995;36:155–9. doi: 10.1007/BF00689201. [DOI] [PubMed] [Google Scholar]
- 59.Roberts ZJ, Goutagny N, Perera PY, et al. The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J Exp Med. 2007;204:1559–69. doi: 10.1084/jem.20061845. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Recent preclinical mechanistic study
- 60.Joseph WR, Cao Z, Mountjoy KG, et al. Stimulation of tumors to synthesize tumor necrosis factor-alpha in situ using 5,6-dimethylxanthenone-4-acetic acid: a novel approach to cancer therapy. Cancer Res. 1999;59:633–8. [PubMed] [Google Scholar]; *First report of in situ TNF mRNA synthesis by both tumor and host cells
- 61.Ching LM, Joseph WR, Zhuang L, Baguley BC. Interaction between endotoxin and the antitumour agent 5,6- dimethylxanthenone-4-acetic acid in the induction of tumour necrosis factor and haemorrhagic necrosis of colon 38 tumours. Cancer Chemother Pharmacol. 1994;35:153–60. doi: 10.1007/BF00686639. [DOI] [PubMed] [Google Scholar]
- 62.Baguley BC, Zhuang L, Kestell P. Increased plasma serotonin following treatment with flavone-8-acetic acid, 5,6-dimethylxanthenone-4-acetic acid, vinblastine, and colchicine: relation to vascular effects. Oncol Res. 1997;9:55–60. [PubMed] [Google Scholar]; *First report of serotonin and 5-hydroxyindoleacetic acid as a potential biomarker
- 63.Siim BG, Baguley BC. Flavones and xanthenones as vascular disrupting agents. In: Sieman DW, editor. Vascular-targeted Therapies in Oncology. John Wiley & Sons Ltd; London: 2006. pp. 159–77. [Google Scholar]
- 64.Baguley BC, Cole G, Thomsen LL, Li Z. Serotonin involvement in the antitumour and host effects of flavone-8- acetic acid and 5,6-dimethylxanthenone-4-acetic acid. Cancer Chemother Pharmacol. 1993;33:77–81. doi: 10.1007/BF00686027. [DOI] [PubMed] [Google Scholar]
- 65.Zhao L, Kestell P, Philpott M, et al. Effects of the serotonin receptor antagonist cyproheptadine on the activity and pharmacokinetics of 5,6-dimethylxanthenone-4-acetic acid (DMXAA) Cancer Chemother Pharmacol. 2001;47:491–7. doi: 10.1007/s002800000267. [DOI] [PubMed] [Google Scholar]
- 66.Kawahara K, Hashiguchi T, Kikuchi K, et al. Induction of high mobility group box 1 release from serotonin-stimulated human umbilical vein endothelial cells. Int J Mol Med. 2008;22:639–44. [PubMed] [Google Scholar]
- 67.Kestell P, Zhao L, Jameson MB, et al. Measurement of plasma 5-hydroxyindoleacetic acid as a possible clinical surrogate marker for the action of antivascular agents. Clin Chim Acta. 2001;314:159–66. doi: 10.1016/s0009-8981(01)00692-1. [DOI] [PubMed] [Google Scholar]; *First demonstration of 5-hydroxyindoleacetic acid as a potential clinical biomarker
- 68.Zhao L, Kestell P, Ching LM, Baguley BC. Oral activity and pharmacokinetics of 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in mice. Cancer Chemother Pharmacol. 2002;49:20–6. doi: 10.1007/s00280-001-0377-3. [DOI] [PubMed] [Google Scholar]
- 69.Siim BG, Laux WT, Rutland MD, Palmer BN, Wilson WR. Scintigraphic imaging of the hypoxia marker (99m)technetium-labeled 2,2'-(1,4-diaminobutane)bis(2-methyl-3-butanone) dioxime (99mTc-labeled HL-91; prognox): noninvasive detection of tumor response to the antivascular agent 5,6-dimethylxanthenone-4-acetic acid. Cancer Res. 2000;60:4582–8. [PubMed] [Google Scholar]
- 70.Raghunand N, Gatenby RA, Gillies RJ. Microenvironmental and cellular consequences of altered blood flow in tumours. Br J Radiol. 2003;76(Spec No 1):S11–S22. doi: 10.1259/bjr/12913493. [DOI] [PubMed] [Google Scholar]
- 71.Meng X, Ao L, Shames BD, Harken AH. Inhibition of cyclic-3',5'-nucleotide phosphodiesterase abrogates the synergism of hypoxia with lipopolysaccharide in the induction of macrophage TNF-alpha production. J Surg Res. 2001;101:210–5. doi: 10.1006/jsre.2001.6290. [DOI] [PubMed] [Google Scholar]
- 72.Cao Z, Joseph WR, Browne WL, et al. Thalidomide increases both intra-tumoural tumour necrosis factor-alpha production and anti-tumour activity in response to 5,6-dimethylxanthenone-4-acetic acid. Br J Cancer. 1999;80:716–23. doi: 10.1038/sj.bjc.6690415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cao Z, Baguley BC, Ching LM. Interferon-inducible protein 10 induction and inhibition of angiogenesis in vivo by the antitumor agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA) Cancer Res. 2001;61:1517–21. [PubMed] [Google Scholar]
- 74.Apetoh L, Tesniere A, Ghiringhelli F, Kroemer G, Zitvogel L. Molecular interactions between dying tumor cells and the innate immune system determine the efficacy of conventional anticancer therapies. Cancer Res. 2008;68:4026–30. doi: 10.1158/0008-5472.CAN-08-0427. [DOI] [PubMed] [Google Scholar]
- 75.Cohen I, Rider P, Carmi Y, et al. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci U S A. 2010;107:2574–9. doi: 10.1073/pnas.0915018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Apetoh L, Mignot G, Panaretakis T, Kroemer G, Zitvogel L. Immunogenicity of anthracyclines: moving towards more personalized medicine. Trends Mol Med. 2008;14:141–51. doi: 10.1016/j.molmed.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 77.Tesniere A, Schlemmer F, Boige V, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–91. doi: 10.1038/onc.2009.356. [DOI] [PubMed] [Google Scholar]
- 78.McKeage MJ, Kestell P, Denny WA, Baguley BC. Plasma pharmacokinetics of the antitumour agents 5,6-dimethylxanthenone-4-acetic acid, xanthenone-4-acetic acid and flavone-8-acetic acid in mice. Cancer Chemother Pharmacol. 1991;28:409–13. doi: 10.1007/BF00685815. [DOI] [PubMed] [Google Scholar]
- 79.Zhao L, Ching LM, Kestell P, Baguley BC. Improvement of the antitumor activity of intraperitoneally and orally administered 5,6-dimethylxanthenone-4-acetic acid by optimal scheduling. Clin Cancer Res. 2003;9:6545–50. [PubMed] [Google Scholar]
- 80.Murata R, Siemann DW, Overgaard J, Horsman MR. Improved tumor response by combining radiation and the vascular-damaging drug 5,6-dimethylxanthenone-4-acetic acid. Radiat Res. 2001;156:503–9. doi: 10.1667/0033-7587(2001)156[0503:itrbcr]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 81.Wilson WR, Li AE, Cowan DS, Siim BG. Enhancement of tumor radiation response by the antivascular agent 5,6- dimethylxanthenone-4-acetic acid. Int J Radiat Oncol Biol Phys. 1998;42:905–8. doi: 10.1016/s0360-3016(98)00358-7. [DOI] [PubMed] [Google Scholar]; *Definitive study on the enhancement of radiation response by DMXAA
- 82.Ching LM, Joseph WR, Baguley BC. Stimulation of macrophage tumouricidal activity by 5,6-dimethyl- xanthenone-4-acetic acid, a potent analogue of the antitumour agent flavone-8-acetic acid. Biochem Pharmacol. 1992;44:192–5. doi: 10.1016/0006-2952(92)90058-q. [DOI] [PubMed] [Google Scholar]
- 83.Ching LM, Finlay GJ, Joseph WR, Baguley BC. In vitro methods for screening agents with an indirect mechanism of antitumour activity: xanthenone analogues of flavone acetic acid. Eur J Cancer. 1991;27:1684–9. doi: 10.1016/0277-5379(91)90446-k. [DOI] [PubMed] [Google Scholar]
- 84.Perera PY, Barber SA, Ching LM, Vogel SN. Activation of LPS-inducible genes by the antitumor agent 5,6-dimethylxanthenone-4-acetic acid in primary murine macrophages. Dissection of signaling pathways leading to gene induction and tyrosine phosphorylation. J Immunol. 1994;153:4684–93. [PubMed] [Google Scholar]
- 85.Wang LC, Reddy CB, Baguley BC, et al. Induction of tumour necrosis factor and interferon-gamma in cultured murine splenocytes by the antivascular agent DMXAA and its metabolites. Biochem Pharmacol. 2004;67:937–45. doi: 10.1016/j.bcp.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 86.Philpott M, Ching L, Baguley BC. The antitumour agent 5,6-dimethylxanthenone-4-acetic acid acts in vitro on human mononuclear cells as a co-stimulator with other inducers of tumour necrosis factor. Eur J Cancer. 2001;37:1930–7. doi: 10.1016/s0959-8049(01)00210-6. [DOI] [PubMed] [Google Scholar]
- 87.Zeidan YH, Hannun YA. The acid sphingomyelinase/ceramide pathway: biomedical significance and mechanisms of regulation. Curr Mol Med. 2009 doi: 10.2174/156652410791608225. in press. [DOI] [PubMed] [Google Scholar]
- 88.Woon ST, Hung SS, Wu DC, et al. NF-kappaB-independent induction of endothelial cell apoptosis by the vascular disrupting agent DMXAA. Anticancer Res. 2007;27:327–34. [PubMed] [Google Scholar]
- 89.Barbera M, Kettunen MI, Caputo A, et al. Immune-modulating and anti-vascular activities of two xanthenone acetic acid analogues: A comparative study to DMXAA. Int J Oncol. 2009;34:273–9. [PubMed] [Google Scholar]
- 90.Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155–9. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- 91.Cuschieri J, Bulger E, Billgrin J, Garcia I, Maier RV. Acid sphingomyelinase is required for lipid Raft TLR4 complex formation. Surg Infect (Larchmt) 2007;8:91–106. doi: 10.1089/sur.2006.050. [DOI] [PubMed] [Google Scholar]
- 92.Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–46. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 93.Baguley BC. Antivascular therapy of cancer: DMXAA. Lancet Oncol. 2003;4:141–8. doi: 10.1016/s1470-2045(03)01018-0. [DOI] [PubMed] [Google Scholar]; *Comprehensive early review on the action of ASA404
- 94.Galbraith SM, Rustin GJ, Lodge MA, et al. Effects of 5,6-dimethylxanthenone-4-acetic acid on human tumor microcirculation assessed by dynamic contrast-enhanced magnetic resonance imaging. J Clin Oncol. 2002;20:3826–40. doi: 10.1200/JCO.2002.09.144. [DOI] [PubMed] [Google Scholar]
- 95.Li J, Jameson MB, Baguley BC, Pili R, Baker SD. Population pharmacokineticpharmacodynamic model of the vascular-disrupting agent 5,6-dimethylxanthenone-4-acetic acid in cancer patients. Clin Cancer Res. 2008;14:2102–10. doi: 10.1158/1078-0432.CCR-07-1475. [DOI] [PubMed] [Google Scholar]
- 96.Jameson MB. A Phase I trial of DMXAA. The University of Auckland; 2005. pp. 1–386. [Google Scholar]
- 97.Ching LM, Joseph WR, Baguley BC. Inhibition of antitumor effects of flavone acetic acid by cortisone. Anticancer Res. 1993;13:1139–41. [PubMed] [Google Scholar]
- 98.Pruijn FB, van Daalen M, Holford NHG, Wilson WR. Mechanisms of enhancement of the antitumour activity of melphalan by the tumour blood flow inhibitor 5,6-dimethylxanthenone-4-acetic acid. Cancer Chemother Pharmacol. 1997;39:541–6. doi: 10.1007/s002800050611. [DOI] [PubMed] [Google Scholar]
- 99.Flaherty KT, Puzanov I. Building on a foundation of VEGF and mTOR targeted agents in renal cell carcinoma. Biochem Pharmacol. 2010;80:638–46. doi: 10.1016/j.bcp.2010.04.005. [DOI] [PubMed] [Google Scholar]