

Abstract
Nanogold and undecagold are covalently linked gold cluster labels which enable the identification and localization of biological components with molecular precision and resolution. They can be prepared with different reactivities, which means they can be conjugated to a wide variety of molecules, including nucleic acids, at specific, unique sites. The location of these sites can be synthetically programmed in order to preserve the binding affinity of the conjugate and impart novel characteristics and useful functionality. Methods for the conjugation of undecagold and Nanogold to DNA and RNA are discussed, and applications of labeled conjugates to the high-resolution microscopic identification of binding sites and characterization of biological macromolecular assemblies are described. In addition to providing insights into their molecular structure and function, high-resolution microscopic methods also show how Nanogold and undecagold conjugates can be synthetically assembled, or self-assemble, into supramolecular materials to which the gold cluster labels impart useful functionality.
Keywords: Gold label, Nucleic acid, Undecagold, Electron microscopy, Nanogold, Nanostructured materials
1. Introduction
Gold nanoparticle-conjugated oligonucleotides are of particular interest as microscopic probes for localizing nucleic acid targets, and for studying the structure of and function of biological systems at macromolecular or molecular resolution. They are also of importance for the synthesis of nanostructured materials to which the gold nanoparticles impart useful functionality. Gold nanoparticles possess a number of size-dependent physical and chemical properties. When they are incorporated into biomolecular structures at specific sites, these properties can enable the labeled molecules to recognize, detect or even intervene in a biochemical process. This provides novel approaches to the diagnosis and even therapy of disease.
In this review, we will discuss the site-specific labeling of oligonucleotides with the 1.4 nm Nanogold and 0.8 nm undecagold nanoparticles. These are both cluster compounds of gold stabilized by well-defined coordination shells of organic ligands that control the chemical properties of the nanoparticles. The use of combinations of ligands with different chemical modifications affords control over the surface character of the particles, and enables the incorporation of well-defined chemical reactivity for selective conjugation to biomolecules. We will review methods for the conjugation of Nanogold and undecagold to nucleic acids, techniques for examination of the resulting species by high-resolution electron microscopy, and the applications of labeled nucleic acids to localize targets with molecular precision. Particular emphasis is given to methods for elucidating how Nanogold and undecagold-labeled nucleic acids can undergo supramolecular organization to give new hybrid nanostructures, and identifying potentially useful properties of these materials.
2. The development and advantages of gold cluster labeling
Colloidal gold has been the electron microscopic label of choice, because it is electron dense and may be prepared in a monodisperse form in a choice of sizes from 1 to 30 nm or larger that are readily visualized by electron microscopy (Handley, 1989a). Because it is punctate, it may be used to identify and localize discrete sites. Gold particles conjugated to targeting biomolecules can provide sufficient resolution to identify and localize targets with macromolecular precision (Handley, 1989b). Colloidal gold is typically conjugated through electrostatic (Weiser, 1933) or hydrophobic interactions, and this has limited the range of conjugates to antibodies, certain other proteins and lectins (Bendayan and Garzon, 1988; Simmons and Albrecht, 1989; Roth et al., 1996; Bendayan, 2000), and a few peptides (Mentlein et al., 1990; Krisch et al., 1993): neither is applicable to conjugation to nucleic acids.
Thiols (sulfhydryls) have a unique affinity for gold, and this affords a mechanism for the conjugation of thiol-functionalized molecules to gold particles with greater specificity (Mrksich and Whitesides, 1996; Ackerson et al., 2006; Ackerson et al., 2005). The direct coordination of thiolated oligonucleotides to gold nanoparticles has acquired wide utility for the preparation of conjugates using thiolated oligonucleotides (Giljohann et al., 2010). Hybridization of such oligonucleotide–gold conjugates may be used to prepare supramolecular assemblies of gold nanoparticles (Mirkin et al., 1996) in which the gold nanoparticle spacing and interaction may be controlled. Gold nanoparticles linked to thiolated oligonucleotides have been used in a variety of methods for the detection and assay of biological targets (Thaxton et al., 2006), as well as the preparation of self-assembled materials (Claridge et al., 2008), and multiplexed detection through ‘biobarcoding’ (Stoeva et al., 2006). This chemistry is reviewed elsewhere (Giljohann et al., 2010; Rosi and Mirkin, 2005). The present discussion will focus on the applications of gold cluster complexes and nanoparticles which can be site-specifically attached to oligonucleotides through the formation of an identifiable, covalent cross-link to a specific chemical group. In this discussion, a gold cluster complex will refer to a gold particle with a discrete molecular structure in which a gold core is coordinated by a layer of small organic molecules.
Although chemically specific, direct coordination of thiolated nucleic acids to gold does not afford easy control over conjugate stoichiometry. A large number of thiolated molecules are required to effectively coat and passivate the gold; because the resulting conjugate is multifunctional, control of the stoichiometry of target binding can be problematic. If targets are mobile or closely spaced, they may be cross-linked by multifunctional gold probes, and the preparation of individual labeled duplexes for assembly into more complex structures may be impossible. For these applications, discrete molecular labeling requires the preparation of gold labeled conjugates with a 1:1 ratio of gold label: conjugate biomolecule.
This was first realized with the advent of undecagold (Lipka et al., 1983; Reardon and Frey, 1984; Hainfeld, 1987), and more particularly Nanogold (Hainfeld and Furuya, 1992; Hainfeld and Powell, 1997). This is similar to undecagold, but contains a larger gold core 1.4 nm in diameter, thought to contain between 55 and 75 gold atoms. Compared with undecagold, Nanogold can be more easily visualized directly in the transmission electron microscope. Other covalently linked metal cluster labels have also been described, including heteropolytungstates (Hainfeld et al., 1990), tetratiridium (Weinstein et al., 1989; Furuya et al., 1988; Cheng et al., 1999), and larger platinum clusters (Powell et al., 1999). These are all cluster compounds containing discrete numbers of heavy metal atoms, which are functionalized using synthetic organic ligands. Ligands with different terminal functionalities are used to impart solubility and biocompatibility to the cluster, or to introduce a functional group for selective cross-linking. Synthetic tuning of ligand properties has resulted in the almost complete elimination of non-specific protein and biomolecule interactions: thus, conjugation is controlled entirely by the specific reactivity.
For most current biomolecule labeling applications, undecagold and Nanogold are prepared in a form containing close to one primary aliphatic amine. This is then activated for selective reaction using a cross-linking reagent, which introduces either a maleimide group for selective reaction with thiols, or a sulfo-N-hydroxyxuccinimide (sulfo-NHS) group for labeling amines. In a typical preparation, a mixture of solubilizing and functionalizing ligands is used in which the proportion of the ligand bearing the cross-linkable functionality is calculated to introduce approximately one amine per cluster. The resulting gold cluster complex is then purified by ion exchange chromatography, which separates species bearing different numbers of amines (Hainfeld, 1989): the first major species to elute after non-binding fractions have cleared typically reacts in a 1:1 manner with a wide variety of biomolecules, and is assumed to have one or close to one reactive group.
The use of covalent cross-linking has expanded the applications of gold clusters far beyond those of colloidal gold, making possible a variety of labeling strategies similar to those enabled in past decades by reactive fluorescent probes (Hainfeld and Powell, 1997, 2000; Hainfeld et al., 2004). Conjugates prepared using Nanogold include antibody IgG molecules, Fab′ and Fv fragments (Hainfeld and Furuya, 1992; Ribrioux et al., 1996), proteins (Yonekura et al., 2006; Rappas et al., 2005) and peptides (Gregori et al., 1997; Segond von Banchet et al., 1999), as well as lipids, which can be used to prepare gold-decorated liposomes or “metallosomes” (Hainfeld et al., 1999a). Both Undecagold (Kessler et al., 1994) and Nanogold (Jones et al., 2004) have been used to label toxins in order to study protein binding. Metal cluster labels can also be targeted to genetically encoded tags. A 1.8 nm Nanogold cluster label, functionalized with a nickel (II) chelate derivative of nitrilotriacetic acid (NTA), binds very strongly to polyhistidine (His) tags (Hainfeld et al., 1999b), and has been extensively used to study the localization and orientation of His-tagged subunits in protein complexes (Buchel et al., 2001; Bumba et al., 2005; Adami et al., 2007). The wider range of biomolecule labeling applications of gold clusters has been reviewed previously (Hainfeld and Powell, 1997, 2000; Hainfeld et al., 2004; Ackerson et al., in press), and will not be further discussed here. Gold cluster labeling is also a route to the preparation of nanostructured materials and devices, including “nanowires” or one-dimensional electrical conductors with widths on the nanometer scale (Scheibel et al., 2003), and “biocatalytic inks” in which enzymes, conjugated with Nanogold and developed with mixtures of enzyme substrate with tetrachloroauric acid, have been used to make nanowires by dip-pen nanolithography (Basnar et al., 2006).
In addition, reactive, undecagold and Nanogold also enable the site-specific labeling of nucleic acids in a controlled manner. Labeling may be directed to any position in an oligonucleotide, and stoichiometry is readily controlled to yield conjugates containing one gold label per conjugate nucleic acid. When combined with the opportunities for programmed supramolecular assembly presented by oligonucleotide hybridization, this enables the preparation of complex, nanostructured materials, in which gold clusters are incorporated at specific sites where they can potentially impart useful functionality: for example, they can act as electron relays to increase the reaction rate of an enzyme (Xiao et al., 2003).
3. Preparation and microscopic applications of gold cluster labeled nucleic acids
Both because of their small size and because of their site-selective conjugation, the initial application of gold labeled nucleic acids was for the localization of specific significant binding sites or subunits within large biomolecules or macromolecular complexes using high-resolution electron microscopic methods, such as scanning transmission electron microscopy (STEM). However, nucleotides require modification in order to be labeled. Undecagold and Nanogold are mostly used in the form of maleimido- or sulfo-NHS- derivatives, which selectively react with thiols and aliphatic amines respectively. Naturally occurring nucleic acids do not contain thiols or aliphatic amines that will react directly. Therefore these must be chemically or biochemically introduced.
Site-specific methods for labeling nucleic acids with gold clusters were first developed to take advantage of the very high resolution afforded by the scanning transmission electron microscope (STEM) to localize specific sites within biological macromolecules. STEM allows the resolution of macromolecules as small as 25 kDa, and its high efficiency in the detection of elastically scattered electrons enables visualization of unstained, freeze-dried specimens at low radiation doses. The first such application to be described in detail was the enzymatic incorporation of modified nucleosides into tRNA, with undecagold labeling for STEM localization of a specific nucleotide. Hainfeld et al. (1991) selectively labeled the 3’ end of yeast tRNA(Phe) with undecagold at a sulfhydryl group on 2-thiocytidine, enzymatically inserted using t-nucleotidyl transferase at position 75 (Sprinzl et al., 1973). This was labeled using either Monomaleimido undecagold, or an iodoacetate derivative prepared by the reaction of monoamino undecagold with iodoacetic acid N-hydroxysuccinimido ester. Labeled tRNA was separated using gel filtration. 5 μL of labeled tRNA, at a concentration of 10 μg/mL in 0.1 M ammonium acetate at pH 6.8, was injected into a 5 μL droplet of 0.1 M sodium phosphate buffer at pH 7.5 and deposited onto a thin carbon-coated support grid. This washed several times with 20 mM ammonium acetate containing 2 mM magnesium acetate, partially blotted, rapidly frozen in liquid nitrogen slush, then lyophilized overnight. The volatile ammonium acetate is removed during this process, leaving minimal residue to obscure the individual labeled tRA molecules. Specimens were observed unstained on a high-vacuum STEM stage at −160 °C at a direct magnification of 250,000 or 500,000 and an accelerating voltage of 40 kV.
An undesirable consequence of labeling at the 75 position was loss of reactivity towards enzymatic acylation by tRNAPhe synthetase. The labeled tRNA(Phe) no longer bound to the elongation factor (EF) Tu.GTP complex of Thermus thermophilus, which prevented is use as a probe to localize its binding sites. However, Blechschmidt et al. (1993, 1994) reported an alternative approach: they used Phe-tRNA(Phe) containing 3-(3-amino-3-carboxypropyl) uridine at position 47, converted to a thiol derivative using 2-iminothiolane (Traut’s reagent). This was labeled with maleimido undecagold. Enzymatically aminoacylated Phe-tRNA(Phe) was then mixed with EF-Tu.GTP and ternary complex formation was observed by STEM; specimens were prepared as described above, and observed at −150 °C at a direct magnification of 500,000 and an accelerating voltage of 40 kV. The formation of ternary complexes with 1:1 EF-Tu.GTP: Phe-tRNA(Phe) was demonstrated by the results. STEM images of both forms of undecagold-labeled Phe-RNA(Phe) and the ternary EF-Tu.GTP:Phe-tRNA(Phe) complex, together with a projection of the molecular structure of Phe-tRNA(Phe) for comparison, are shown in Fig. 1. Undecagold-labeled Phe-tRNA(Phe) could also be used as a heavy atom derivatization reagent for crystallographic studies of the 70S ribosome at different stages of elongation.
Fig. 1.

Scanning transmission electron microscopy (STEM) with undecagold-labeled Phe-tRNA(Phe). (A) STEM micrograph of freeze-dried unstained molecules of yeast Phe-tRNA(Phe) derivatized at position 75 using maleimido undecagold; particles with the undecagold label are denoted by arrows (scale bar = 50 nm). (B) Structure of Phe-tRNA(Phe) from X-ray crystallography data (Sussman et al., 1978), with undecagold cluster schematically inserted at position 75. Inset: StEM image of a single yeast Phe-tRNA(Phe) molecule (framed in (A)) smoothed by low pass filtering and displayed in simulated colors: the bright area represents the undecagold cluster. (C) STEM micrograph of freeze-dried unstained molecules of Phe-tRNA(Phe) labeled with an undecagold cluster on acp3U47, denoted by arrowheads (scale bar = 50 nm). (D) Freeze-dried unstained particles of the ternary complex of acp3U47 undecagold-labeled Phe-tRNA(Phe) with T. thermophilus EF Tu.GTP (scale bar = 50 nm). (A and B) From Hainfeld et al., 1991; (C and D) from Blechschmidt et al. (1993) (used with permission).
In a similar reaction, Medalia et al. (1999) used ribonucleoside triphosphate analogs with a terminal thiol group attached to the heterocyclic ring for in vitro transcription of RNAs carrying free thiol groups; these were then labeled with maleimido Nanogold. The larger size of the Nanogold cluster enabled direct visualization in the transmission electron microscope (TEM). RNA was dissolved in water and deposited on ultrathin carbon films on holey carbon-coated copper grids; excess solution was blotted and the grids were imaged in a Philips CM-12 TEM operating at 100 kV.
Another technique which has proven useful for the characterization of Nanogold labeled macromolecules is atomic force microscopy (AFM), which combines very high lateral spatial sensitivity with both very high-resolution height measurements, and the capability to differentiate sites with differing degrees of rigidity or compressibility, where the more rigid sites correspond to gold cluster cores. Direct evidence for the attachment of gold clusters at random locations on the RNA was achieved by simultaneous visualization of RNA and Nanogold by AFM imaging. Diluted RNA samples were deposited on freshly cleaved mica chips, dried in air, and imaged in air. Single-stranded gold-tagged b-globin RNA molecules yielded images in which knob-like structures are clearly seen along the RNA chain. The height of the brightest knobs ranged between 1.7 and 2.5 nm, which is in agreement with the height observed for free Nanogold (2.0–2.6 nm). A comparison of the images obtained by TEM and AFM is shown in Fig. 2.
Fig. 2.
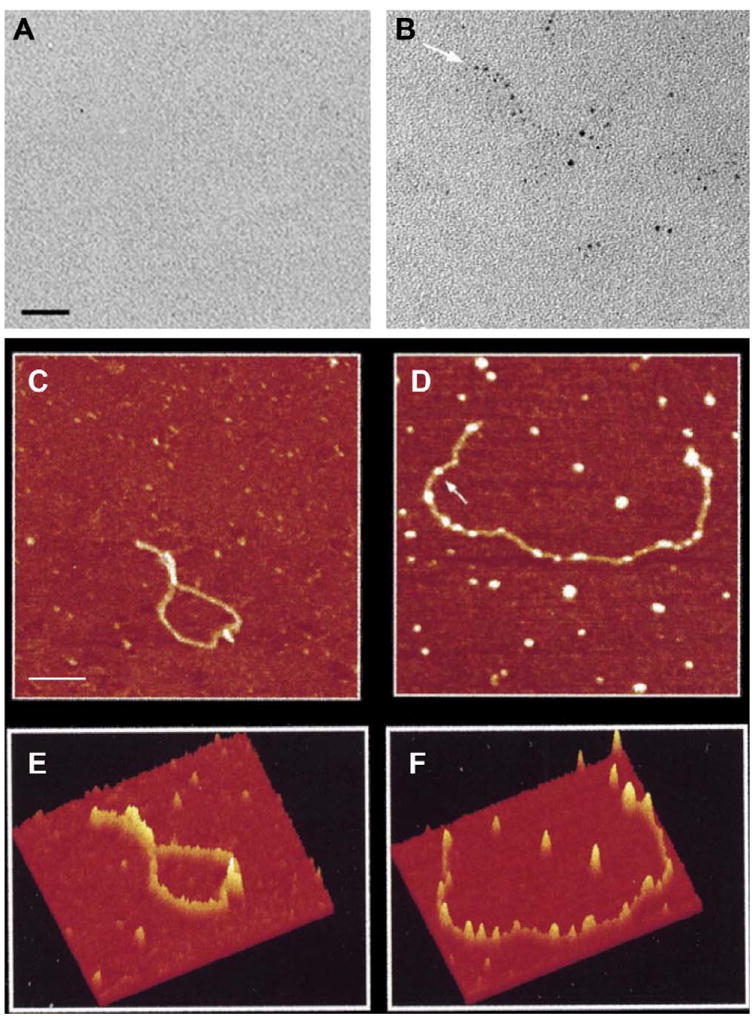
TEM and AFM of β-globin pre-mRNA transcripts. RNA was transcribed in vitro, treated with monomaleimido Nanogold. (A) Bright-field TEM of unmodified RNA; (B) RNAtranscribed in the presence of UTP-SH (50% of total input UTP). Arrows indicate the RNA termini (Scale.bar, 15 nm). (C) – (F) Atomic force microscopy image of β-globin pre-mRNA. (C) Unmodified single-stranded RNA; (D) gold-labeled single-stranded RNA transcribed in the presence of ATP-SH (5% of total input ATP); black to white spans 6 nm (scale bar, 100 nm). (E) Surface plot of AFM image drawn from unlabeled RNA (C), and (F) surface plot of AFM image drawn from Nanogold-labeled RNA (D) using the “surface plot” function of the NIH Image package (from Medalia et al., 1999; used with permission).
Naturally occurring oligonucleotide structures may be labeled via the enzymatic incorporation of a modified nucleotide bearing a reactive functionality during plasmid preparation. A variety of modified bases are available that can be incorporated into plasmids or other nucleic acid structures by polymerases or other enzymes. Weizmann et al. (2004) used a 10:1 mixture of unmodified dUTP with an amino-modified form of dUTP, 5-[3-Aminoallyl]-2′-deoxyuridine 5′-triphosphate (amino-dUTP), to prepare amino-modified DNA in cancer cells. This introduces a primary aliphatic amine, which was labeled using sulfo-NHS-Nanogold. The labeled DNA was deposited from solution onto freshly cleaved mica surfaces and observed using AFM; the gold particles were then catalytically enlarged as an approach to making gold nanowires. AFM and high-resolution TEM (HRTEM) observation revealed nanowires up to 1–3 μm long with heights of about 10 nm, composed of individual gold crystallites which did not always contact each other. Nick translation with a biotin-conjugated dUTP has been used to incorporate biotin; the biotinylated DNA was then incubated with Nanogold-streptavidin as part of the transfection complex (Malecki, 1996). Incorporation of a limited amount of the modified nucleotide into a plasmid preparation mixture results in the introduction of a limited number of reactive sites for labeling; this facilitates lower-density labeling which has less likelihood of affecting transfection efficiency.
Oligonucleotides may be modified with photoreactive cross-linking reagents which introduce a label or reactive group for labeling, or a hapten such as biotin, which is detectable using gold-labeled streptavidin. In one study, psoralen was conjugated to Nanogold and used as a photoreactive labeling reagent. Excess amino psoralen was reacted with sulfo-NHS-Nanogold in HEPES buffer solution at pH 8.0 for 2 h at room temperature, then 12 h at 4 °C; the conjugate was purified using a microspin column (spin column 30, Sigma) and dialyzed against 10 mM HEPES buffer solution. Psoralen intercalates into DNA, then upon 360 nm ultraviolet light irradiation, undergoes a photoinduced 2π+ 2π cycloaddition. The psoralen-Nanogold was intercalated with a double-stranded polyA/polyT duplex approximately 900 nm in length, and the resulting assembly irradiated to give a covalent DNA complex. AFM observation on a freshly cleaved mica surface showed the formation of a gold-labeled DNA ‘nanowire’ more than 600 nm in length with a width of approximately 3.5–8 nm (Patolsky et al., 2002). Although this approach does not require chemical modification during nucleotide synthesis, the convenience comes at the expense of control over labeling location.
Synthetic oligonucleotides afford the most control over label attachment, because reactive groups can be introduced at any position during synthesis. Amines or thiol may be incorporated using modified phosphoramidites (available from a number of sources, including Glen Research, Sterling, VA). Synthetic approaches to modification and Nanogold attachment are shown in Fig. 3. Thiols may be introduced at the 3’position using a modified CPG (Controlled Pore Glass) support linked through a cleavable disulfide, or at the 5’ position with a cleavable disulfide linked via a C6 or C12 alkyl chain. Once synthesis is complete, the oligonucleotide is cleaved and deprotected. Thiol-modified oligonucleotides may require reduction to activate the thiol group; the reducing agent (dithiothreitol, DTT, or mercaptoethylamine hydrochloride, MEA) must be completely separated from the reduced oligonucleotide prior to labeling, otherwise it will react with the maleimido Nanogold and prevent labeling. The oligonucleotide is then mixed with reconstituted maleimido Nanogold at a pH between 6.0 and 7.0, incubated overnight at 4 °C, then separated next day chromatographically (gel filtration, reverse-phase or hydrophobic interaction chromatography), or by gel electrophoresis.
Fig. 3.
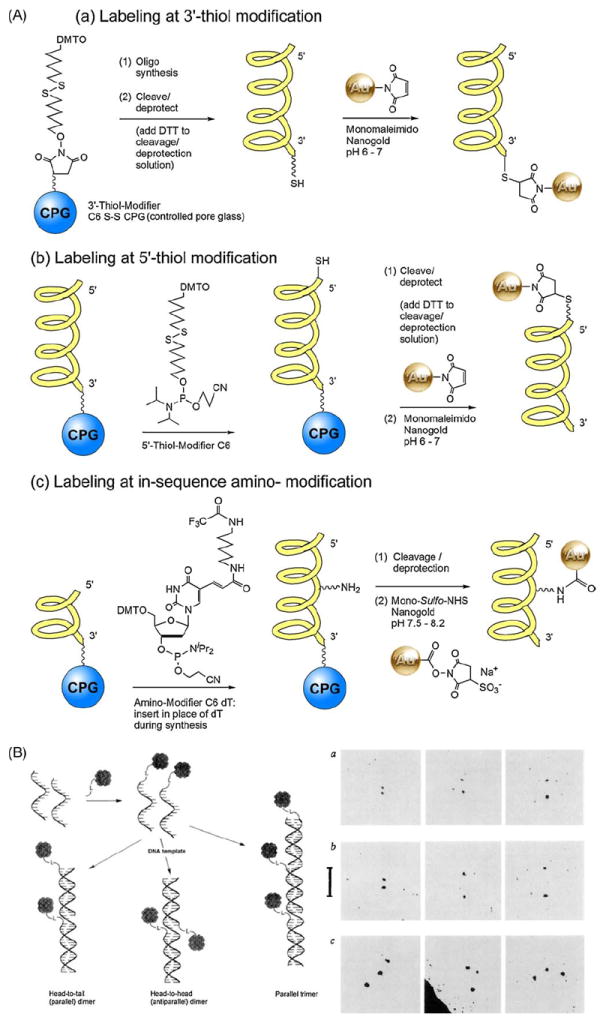
(A) Reaction schemes for the introduction of thiols and amines at specific sites using modified phosphoramidites during the synthesis of oligonucleotides, and labeling of modified oligonucleotides with Nanogold reagents. (B) Left: Assembly of 3′ and 5′-Nanogold labeled oligonucleotides based on Watson-crick base pairing. Right: TEM images of Nanogold-labeled oligonucleotides corresponding to schematic. From left to right are representative images of the nearest, average and furthest separations of the Nanogold particles: (a) Head-to-head dimer: gold separation 2.0 ± 0.6–6.3 ± 0.6 nm. (b) Head-to-tail dimer: gold separation 2.9 ± 0.6–10.2 ± 0.6 nm. (c) Linear equidistant trimer ((B) from Alivisatos et al., 1996; used with permission).
Alivisatos first used this approach to label 17-mer oligonucleotides containing 3′- or 5′-thiol modifications with maleimido Nanogold (Alivisatos et al., 1996). This labeling procedure was adopted by several other groups in subsequent studies. Thiol-modified oligonucleotides were reacted with a 10-fold excess of maleimido Nanogold in 20 mM sodium phosphate buffer with 150 mM NaCl and 1 mM ethylenediamine tetraethyl acetate (EDTA) at pH 6.5, containing 10% isopropanol, at 4 °C for 24 h. The labeled oligonucleotides were then hybridized with unlabeled 35-mer or 51-mer complementary strands, and the doubly or triply labeled duplexes separated by gel filtration over Superose 12 10/30 media (GE Healthcare) eluted with 5 mM NaH2PO4, 150 mM NaCl buffer with 1 mM ethylenediamine tetraethyl acetate (EDTA), pH 6.5. An excess of Nanogold was used for labeling because excess, unbound Nanogold is considerably smaller than the labeled duplexes and is easily separated by gel filtration, whereas unlabeled duplexes would be much closer in size to the labeled ones and less easily separated. Gel electrophoresis on 10% crosslinked polyacrylamide gels in 1× TBE (Tris/Borate/EDTA) buffer at 80 mV resulted in effective separation of several different species: an unlabeled 35-nt (nucleotide) template hybridized with two unlabeled 18-nt oligomers; a 35-nt template hybridized with a labeled 17-nt oligomer and an unlabeled 18-nt oligomer (singly labeled construct); and a 35-mer template hybridized with two labeled 17-nt oligomers (doubly labeled construct). The unlabeled construct was visualized with ethidium bromide staining and the labeled constructs with silver enhancement. Specific labeling was confirmed by transmission electron microscopy. Samples were deposited into 400 mesh copper grids with ultrathin (2–3 nm) holey carbon film that had been exposed briefly to air plasma then treated with polylysine at pH 7.5 and observed at an accelerating voltage 80 kV. Separation of the gold particles for doubly and triply labeled constructs in which the gold particles were attached in different positions agreed with that predicted from structural models. Assembly of labeled nucleotides and TEM images are shown in Fig. 3B. In another study, 3′-fluorescently labeled 25-mer molecular beacons bearing a 5′-thiol modification were reacted with between 16- and 160-fold excess of maleimido Nanogold under identical buffer and temperature conditions (Dubertret et al., 2001).
Protected aliphatic amines may be introduced at the 3′ and 5′ terminals using equivalent amino-modifiers, but may also be introduced at any position using an amino-C6 modifier (available for all four bases). Amino-modified oligonucleotides are mixed with reconstituted sulfo-NHS-Nanogold at a pH between 7.5 and 8.2, incubated overnight at 4 °C, then separated next day by gel filtration, an alternative chromatographic method, or by gel electrophoresis. Hamad-Schifferli et al. (2002) successfully labeled two amino-modified oligonucleotides using sulfo-NHS-Nanogold: a 12-nt oligomer bearing an amine at the 3′ end, and a 38-nt molecular beacon containing a modified thymine bearing a C6 primary amine at the 5 position of the base, located in the loop region. Both were incubated with a 10-fold or greater excess of sulfo-NHS-Nanogold in the reconstituted buffer from which the reagent had been lyophilized (0.02 M HEPES, pH 7.5, at room temperature for 1 h). Excess Nanogold was removed by 70% ethanol precipitation on ice and repeated washes. Nanogold is soluble in ethanol, but the DNA or species attached to it precipitates (Hamad-Schifferli et al., 2002).
Gold cluster labels may be conjugated to nucleic acids through other modifications. RNA differs from DNA in that it contains a cis-1,2-dihydroxy moiety. This may be readily oxidized using periodate to give a dialdehyde, which then reacts directly with monoamino Nanogold; this has been used to label RNA (Gazitt, Y: private communication). This can be used to direct labeling to the 3′ end of the molecule, and two mRNAs have been labeled with monoamino undecagold in this manner (Skripkin et al., 1993). mRNA (5 pM) in 10 mM sodium acetate buffer (pH 4.5) containing 10 mM sodium periodate (NaIO4) was incubated for 1 h at 20 °C in the dark, then precipitated with ethanol and treated with ethylene glycol to remove the periodate. The oxidized mRNA was dissolved in 50 mM sodium borate buffer (pH 9.0) to a final concentration of 25 pM, and monoamino undecagold added to a concentration of 1 mM. The reaction mixture was allowed to stand in the dark for 12–14 h at 4 °C. This reaction produces a Schiff base linkage: this was reduced to the amine using sodium borohydride (NaBH4) in 50 mM sodium borate buffer (pH 9.0). Labeled mRNA was precipitated with ethanol and purified by electrophoresis in 10% polyacrylamide gel (19/1 bis) 50 mM Tris-borate buffer (pH 8.3) without urea and EDTA.
Undecagold-ATP was prepared using 1,1′-carbonyldiimidazole (CDI)-mediated esterification of per-carboxylated undecagold, followed by reaction with ATP, thus attaching the undecagold to the ribose. In an alternative approach, an undecagold cluster containing a small number of carboxyl functionalities was converted to an activated ester form and reacted with a modified ATP containing an amine, linked to the N6 of the adenine base through a 10-atom linker arm through amide bond formation (Hainfeld et al., 1999c). In addition, decoration of double stranded DNA (from T7 bacteriophage) using polyamino (‘Positively Charged’) Nanogold results in the formation of densely labeled strands, visualized by STEM (Hainfeld et al., 2001): this indicates labeling by charge-based interaction of the positively ionizing amines with the negatively charged nucleic acids, and combined with autometallography (Powell and Hainfeld, 2002), offers another route to the fabrication of nanowires.
A recent report describes the preparation of gold-labeled DNA nanowires using ‘click chemistry,’ or the reaction between azide-functionalized gold nanoparticles and alkyne-modified DNA duplex using the copper (I)-catalyzed Huisgen cycloaddition (Fischler et al., 2008). Azide-functionalized gold nanoparticles were prepared by the reduction of the gold-precursor, tetrachloroauric acid, by sodium naphthalenide, followed by the stabilization of the gold particles by the addition of azide-modified glutathione. Alkyne-modified DNA was synthesized using an alkyne-modified triphoshate; this was then reacted with the azide-terminated nanoparticles via a ‘click’ reaction using copper (I) complexes of the ligand TBTA (tris((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)amine) as the catalyst. TEM examination of the resulting one dimensional nanoparticle arrangement revealed a nearly equidistant interparticle spacing of approximately 2.8 ± 0.5 nm, consistent with twice the calculated ligand shell thickness of approximately 1.4 nm. The particles also exhibited a high degree of size monodispersity, being close to 1.6 nm in diameter.
4. Supramolecular assemblies of gold-labeled nucleic acids: nanomaterials and nanodevices
4.1. Preparation and properties of gold-labeled oligonucleotides
The principle first set forth by Alivisatos, of programmed assembly of gold nanoparticles through oligonucleotide hybridization, has since been applied to the preparation of a range of nanostructured materials. Nanogold-labeled 2-D arrays have been constructed by tiling together rigid DNA motifs composed of double-crossover (DX) molecules containing DNA hairpins, to form programmable molecular scaffolding (Xiao et al., 2002). The use of 2D DNA crystals offers potentially significant advantages over other self-assembly approaches for the precision, rigidity, and programmability of the assembled nanostructures. DNA-Nanogold conjugates were prepared from trityl-protected 5′-thiol-modified C6 oligonucleotides, which were deprotected and reacted with Monomaleimido Nanogold: 6 nmol of deprotected oligonucleotide was combined with 6–12 nmol of Monomaleimido Nanogold in 200 μmol of reaction buffer (20 mM NaH2PO4, 150 mM NaCl, 1 mM disodium ethylenediaminetetraacetic acid (EDTA), pH 6.8): after incubation at room temperature for 1 h, unreacted Nanogold was separated from the unreacted DNA and DNA–gold conjugate by addition of 3 M sodium acetate (20 μL) and ice cold absolute ethanol (500 μL). After incubation at −20 °C for 1.2 h, the mixture was centrifuged at 4 °C in a microcentrifuge (13,000 rpm) for 15 min. The Nanogold conjugate was then isolated by C4 reversed-phase HPLC with an acetonitrile gradient in 0.1 M aqueous triethylammonium acetate at pH 6.5. DNA:Nanogold labeling stoichiometry of the purified conjugate was estimated spectroscopically to be very close to the desired 1:1 product. The Nanogold-labeled oligonucleotides were incorporated into a mixture of four different rigid tile motifs with complementary edges, which were then crystallized at elevated temperature, resulting in regular arrays in which the tiles were assembled in repeating patterns. TEM images were obtained using a JEOL 1210 instrument operated at 120 kV; samples were prepared on 400 mesh copper grids with an ultra thin carbon film. Dried carbon grids were floated on a 2 μL drop of the DNA crystal solution, then stained by sequentially floating the grid on four 50 μL drops of 0.1% uranyl acetate for 30 s each, and then wicking off the solution. Stained bands were observed, with their separation in agreement with the 64 nm value predicted from the tile design.
Results were confirmed by AFM: samples were prepared by spotting 3–5 μL of solution on freshly cleaved mica, waiting 2 min for adsorption to the surface, then removing buffer salts with 5–10 drops of distilled water which were removed by compressed air blowing. The sample was imaged in contact mode in an isopropanol fluid cell at a scanning frequency of 6.1 Hz using a Digital Instruments NanoScope III with a JV-4042 scanning head with commercial Model NPS (Digital Instruments) Si3N4 cantilevers. Higher resolution STEM images were acquired of the assembled material on thin 0.2 nm carbon film over a holey substrate on an EM grid. Samples were rinsed several times with 20 mM ammonium acetate, then rapidly frozen in liquid nitrogen slush and freeze dried overnight. Grids were viewed in dark field with the high-performance field-emission Brookhaven STEM operating at 40 keV. The results showed that the gold particles formed 2-D arrays with interparticle spacings of 4 and 64 nm, as predicted from the design. The system is shown schematically, with TEM, AFM and STEM micrographs of the assembled structure showing the Nanogold spacings, in Fig. 4.
Fig. 4.

Upper: Sequences of the four DX molecules (A–D) and the tiling pattern used to assemble 2D DNA crystals. The sequences of the 22 DNA strands including the DNA–gold conjugate (35-mer present in tile B, showing covalently attached Nanogold) are shown along with symbolic representations to the left. The 3′ end of each strand is indicated by an arrowhead. Molecules B and D each contain two protruding structures that extend out of the crystal plane, one above and one below the plane. The open and filled circles in the figure represent the protruding structures on one side (e.g. above) the plane. Upper right: Final DNA assembly showing that the spacing between these features is 32 nm in the horizontal direction and 4 nm in the vertical direction. Lower: (A) TEM image and (B) AFM contact-mode image of 2D DNA crystals without gold nanoparticles (NP). (C) Dark field unstained STEM image of a DNA crystal incorporating the DNA–Nanogold conjugate, showing 64 and 4 nm interparticle spacings (from Xiao et al., 2002; used with permission).
Templating of gold nanoparticles into periodic arrays, with a repeat distance of 38 nm, has also been achieved using 2-dimensional DNA nanogrids (Park et al., 2005; Zhang et al., 2006). The DNA nanogrid structure was generated by utilizing a family of DNA tiles that resemble a cross structure, each composed of four-arm DNA branch junctions, designed with appropriate complementary sequences at the end of each arm. Hybridization resulted in the assembly of the DNA into a two-dimensional array. The sequences for the two DNA tiles were modified from that used for similar tiles constructed previously, by incorporating a 15-base polyadenine tag (A15) into the A tile as a hybridization site for the gold particle; the gold nanoparticles were then imaged using AFM in a similar manner to the RNA–gold complexes described above.
This approach has been extended to three dimensions by the use of triple-crossover DNA tiles, modified with thiols, to form self-assembled DNA nanotubes; these were then metallated to prepare conductive nanowires (Yan et al., 2003). The structural motifs were constructed using oligonucleotides about 50 bases in length, modified with thiols in such a way that the thiol-bearing portion projected out of the plane of the tile. These were reduced with DTT before use and exchanged into annealing buffer using gel permeation over a NAP5 column. Oligonucleotides were annealed to form tiles and lattices in stoichiometric mixtures by heating to 95 °C for 5 min, then slowly cooling to room temperature to produce a nanotube lattice. This was metallated using a two-step procedure. First, the TX nanotube lattice was seeded with silver using the glutaraldehyde method: annealed DNA was incubated with 0.2% glutaraldehyde in 1× TAE/Mg2+ buffer (40 mM Tris acetate, pH 7.6/2 mM EDTA/10 mM MgCl2) on ice for 20 min, then room temperature for 20 min, then dialyzed overnight at 4 °C in 1× TAE/Mg2+ buffer. The resulting aldehyde-derivatized DNA was incubated in the dark with an equal volume of 0.1 M silver nitrate in 25% ammonia buffer (pH 10.5) at room temperature, then automet-allographically enhanced (HQ Silver, Nanoprobes) for 5 min to give metallic nanowires. Thiol-specific labeling with maleimido Nanogold was attempted, in order to confirm the locations of thiols: however, this did not result in labeling. When the thiol modification was replaced with an amine configured to project in the opposite direction, the amines were successfully labeled with sulfo-NHS-Nanogold. This confirmed the location of the thiols on the inner tube surface. AFM images of specimens on freshly cleaved mica, and SEM (taken at 10–15 kV accelerating voltage, with chamber pressure was between 1.0 × 10−7 and 1.0 × 10−6 mbar; samples were prepared on doped n-type silicon substrate with resistivity = 2.0 × 10−5 Ωm and 500 nm thermal oxide layer prepared using the RCA cleaning process without oxide) indicated that metallation produced nanowires about 40 nm in diameter, 35 nm in height, and up to 5 μm in length.
Nanowires in which Nanogold forms the basic conductive unit were prepared using telomerase from HeLa cancer cells to generate repeated DNA sequences, which were labeled using two different approaches: (a) sulfo-NHS-Nanogold labeling of a 12-nt oligomer complementary to the telomer, which was then hybridized with the telomerase-produced DNA strand spacing the Nanogold particles at short intervals along the chain; and (b) enzymatic telomer generation using a 1:10 mixture of unmodified dUTP and the amino-modified form, 5-[3-Aminoallyl]-2′-deoxyuridine 5′-triphosphate (amino-dUTP), followed by treatment of the resulting amino-functionalized oligonucleotide with excess sulfo-NHS-Nanogold. The metal nanoparticles were then enlarged using a catalytic gold-based autometallography procedure until AFM and TEM measurements indicated that they touched (Weizmann et al., 2004). Using this method, nanowires up to 3.5 μm in length could be prepared. A parallel approach using a genetically engineered form of the nucleoprotein RecA with a surface-accessible cysteine, labeled using maleimido Nanogold, then gold or silver enhanced for different times, was used to generate protein-based nanowires which were observed using similar methods (Nishinaka et al., 2005).
4.2. Functional nanodevices
The most exciting area of this field is the use of gold nanoparticles to impart functionality and construct working nanodevices. Perhaps the simplest are molecular beacons: these are hairpin loops of DNA whose ends are complementary, enclosing a loop which is complementary to a target sequence (Tyagi and Kramer, 1996). One end is labeled with a fluorophore, the other with a quencher. In the absence of target, the ends are hybridized together, bringing the fluorophore and quencher into proximity and quenching fluorescence; when the loop binds the target, the ends are forced apart and fluorescence appears. Nanogold has been found to be a highly effective quencher, yielding signal-to-noise ratios (the ratio of fluorescence intensity when the beacon is open to that when it is closed) of up to 2940, whereas the conventional organic quencher, 4-(dimethylaminoazo) benzene-4-carboxylic acid (DABCYL), produces 100 at most (Dubertret et al., 2001). Further reports indicate that in addition to conventional Förster energy transfer, additional short-range interactions increase quenching when the fluorophore and gold nanoparticles are close together (Dulkeith et al., 2002, 2005).
Subsequently, an RNA beacon has been described, using Nanogold as the quencher, in which the fluorescence is activated by cleavage rather than unfolding (Jennings et al., 2006). Nanometal surface energy transfer, which describes an energy transfer event from optically excited organic fluorophores to small metal nanoparticles, was used to measure Mg2+-induced conformational changes for a hammerhead ribozyme, and these measurements confirmed using fluorescence resonance energy transfer (FRET). To prepare the hammerhead complex, purified RNA strands were deprotected and desalted, and 50 mM tris (carboxyethyl) phosphine was added to regenerate free thiols from any disulfide bonds formed by thiol oxidation. Maleimido Nanogold was reconstituted in RNase-free ultrapure water, mixed with the RNA, vortexed, and placed in the refrigerator at 4 °C for at least 24 h to allow coupling. The Nanogold-labeled hammerhead ribosome was analyzed by polyacrylamide gel electrophoresis (PAGE) to confirm labeling and separate the labeled complex. Equimolar amounts of Nanogold-ribozyme (NG-Rib) and FAM-bound substrate strand were combined in a microcentrifuge tube and heated to 95 °C for 2 min, allowed to cool to room temperature for ten minutes, then cooled with ice for an additional 10 min to allow formation of the Nanogold-hammerhead complex (NG-HHComp). Gel electrophoresis on 15% polyacrylamide gel at 4 °C was used to separate the conjugate. PAGE and optical analyses of Nanogold-labeled ribosome were correlated to derive values for the rate constants for the reaction and its rate determining step, the cleavage reaction. Photoluminescence data showed that upon rapid mixing of Mg2+ with the Nanogold-ribosome complex, the intensity of the FAM decreases immediately as the complex folds to bring the FAM into closer proximity of the NG. After 1–2 min, the intensity of the FAM photoluminescence begins to increase as the cleavage reaction progresses, approaching a maximum value near t = 250 min. After the initial incubation (mixing) period, an increase in the fluorescence of FAM is observed both in the PA gel and in the emission intensity plot, which correlates directly with the kinetics of the cleavage reaction (k2) and can be fit to a first-order reaction for substrate at the limit of low ribozyme concentration. Nanogold also produced quenching equal to DABCYL when used to label quantum dot (semiconductor nanoparticle) molecular beacons (Cady et al., 2007).
Hamad-Schifferli et al. (2002) used Nanogold-labeled DNA beacons to demonstrate that the highly localized temperature rise, produced by a conjugated Nanogold particle inductively coupled to a pulsed radio frequency magnetic field may be used for the remote control of DNA hybridization. Sulfo-NHS Nanogold was used to label molecular beacons with primary amino-group synthetically inserted into the loop; fluorescence was used to determine the extent of the local temperature rise: a surface-bound Nanogold-oligonucleotide antenna was hybridized to a fluorescently labeled complementary strand. The effect was highly localized: neighboring molecules lacking the Nanogold antenna were unaffected. This is shown schematically in Fig. 5.
Fig. 5.
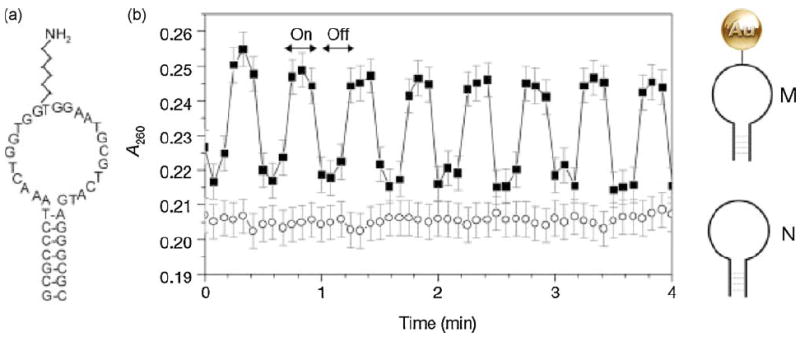
Inductive coupling to Nanogold linked to DNA and evidence of dehybridization. (a) Sequence of the hairpin molecular beacon, M. The molecule is self-complementary at the ends for 7 bases, with a primary amine in the loop section, to which the 1.4-nm Nanogold is covalently linked by reaction with sulfo-NHS-Nanogold. (b) Absorbance at 260 nm (A260) of a solution of M in a radio-frequency magnetic field (RFMF) (squares). Arrows indicate when the RFMF is on/off. Circles show response of N (unlabeled beacon: no Nanogold) in RFMF. Nanogold also shows an absorbance increase under RFMF, qualitatively attributed to the change in the optical absorption due to eddy currents. However, the absorbance increases nearly uniformly over 200 ± 300 nm with RFMF and is small relative to the oligonucleotide change in absorbance. To obtain the data shown here, the nanocrystal contribution was subtracted (see Hamad-Schifferli et al., 2002 for details). M with a fluorophore/quencher pair on the ends shows fluorescence increasing reversibly with RFMF (not shown), confirming dehybridization (from Hamad-Schifferli et al., 2002; used with permission).
The ability of programmed DNA assembly to organize multiple biochemical or chemical events has been illustrated by Wilner et al. (2009). It had been previously demonstrated that reconstitution of apo-glucose oxidase with Nanogold-labeled cofactor, flavin adenine dinucleotide, ‘nanowires’ the enzyme to give a seven-fold increase in reaction rate (Xiao et al., 2003). Subsequently, a DNA template prepared by rolling circle amplification was used to organize a ‘cascade’ of enzymatic reactions, and as a scaffold for bio-catalytic nanowire formation through the action of glucose oxidase activated by Nanogold. A continuous oligonucleotide containing two alternating discrete binding sequences and an intervening spacer unit was synthesized by rolling circle amplification with a circular primer. Glucose oxidase (GOx) and horseradish peroxidase (HRP) were derivatized with oligonucleotides complementary to the two binding sequences, then hybridized to the template. This assembly then conducted two sequential reactions: The GOx-catalyzed oxidation of glucose yielded gluconic acid and H2O2, and the resulting H2O2 oxidized 2,2′-azino-bis[3-ethylbenzthiazoline-6-sulfonic-acid] (ABTS2-) in the presence of HRP. This cascade was not activated in the absence of the organizing DNA template or in the presence of non-specific DNA. Biocatalytic formation of metallic nanowires was conducted using GOx conjugated with sulfo-NHS-Nanogold, separated from excess Nanogold using a 50,000 MW cut-off centrifuge filter, then functionalized with oligonucleotides complementary to the two binding sequences on the template, as shown in Fig. 6.
Fig. 6.
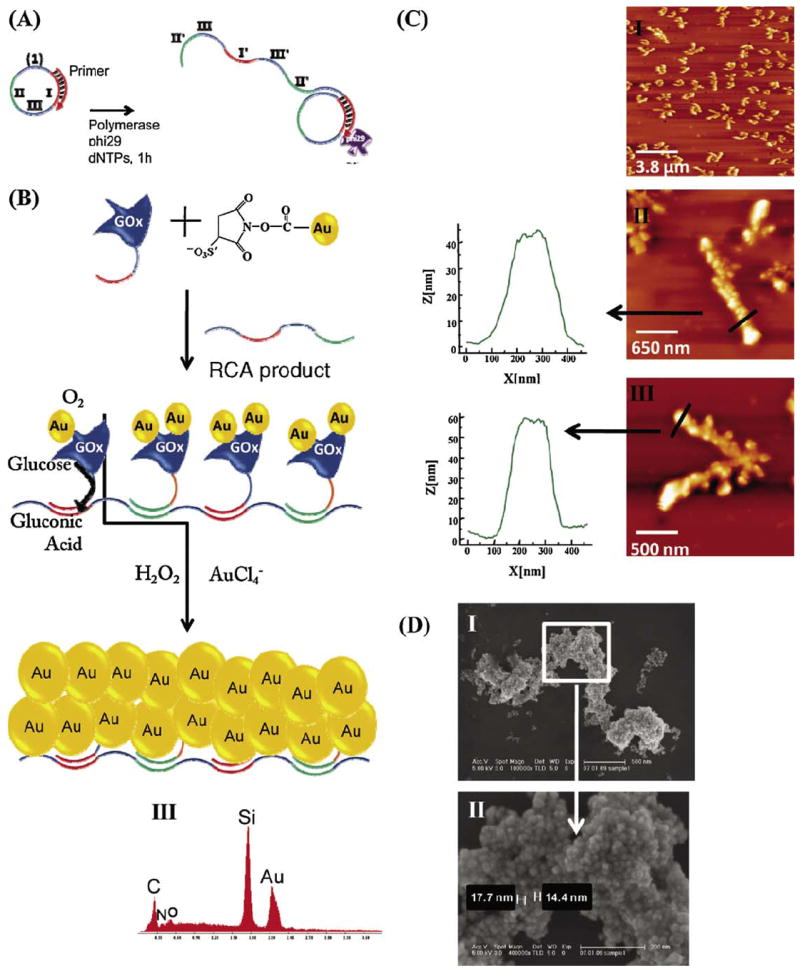
(A) Sequence-specific programmed DNA strands using rolling circle amplification (RCA) to generate a template for enzyme attachment. (B) Assembly of Au-NP-functionalized glucose oxidase (GOx) on the RCA-synthesized template, and the biocatalytic enlargement of Nanogold to gold nanowire structures. (C) AFM images of the resulting gold nanostructures: (I) a large 12 μm2 image; (II and III) images of different single wires and the respective cross-section analyses. (D) (I) Scanning electron microscopy image of the biocatalytically generated gold nanowires and (II) the enlargement of one domain of the wire. (III) Energy-dispersive spectrometry analysis of the resulting gold nanowire (from Wilner et al., 2009; used with permission).
Gold nanowire formation was accomplished by incubation with glucose followed by a mixture of 8 × 10−2 M glucose and 0.3 × 10−3 M HAuCl4, resulting in the formation of gold nanowires 1–5 μm in length, and ca. 150 nm in width. Nanowire formation was visualized by AFM and SEM. For AFM, specimens were diluted 50-fold in deionized water, then applied to freshly cleaved mica surfaces activated with MgCl2, rinsed and dried under a gentle argon flow before observation in tapping mode using NSC 15 AFM tips (Mikromasch, Germany) using the tapping mode at their resonant frequency. The height of the Nanogold-GOx-functionalized DNA scaffold prior to biocatalytic growth of the gold nanowires was found to correspond to ca. 6.5 nm; after biocatalytic growth, heights were ca. 50 nm. SEM imaging was carried out on silicon slides, washed with distilled water and ethanol, and then UV/ozone cleaned using a HR-SEM Sirion (FEI) microscope operated at 5 kV accelerating voltage. Images were taken on silicon slides (Virginia semiconductor, Inc.): slides were first washed with distilled water and ethanol, and then UV/ozone cleaned before application of specimens.
5. Conclusions
Electron microscopy provides high inherent resolution, and gold nanoparticles, because they are point labels, provide the highest resolution of any electron microscopic technique, combined with the ability to directly marked and quantitate target sites. The gold cluster labels undecagold and Nanogold, which are conjugated through specific, bond-forming reactions, have two important advantages over conventional colloidal gold labels: they allow control over the stoichiometry of labeling, and they may be conjugated via different specific chemical reactions to different sites thus providing a high level of labeling resolution, and selective labeling at a site that usually does not perturb binding or allow multiple cross-links which can lead to aggregation. Undecagold and Nanogold labels confer their greatest advantages when used in unstained, molecular specimens with high-resolution electron microscopic methods such as STEM, TEM, and cryoEM where they provide unsurpassed resolution for the localization not just of macromolecules, but of unique sites within molecules. In addition, atomic force microscopy (AFM) has demonstrated great utility for mapping gold cluster labeled macromolecules: It can detect and differentiate an unlabeled macromolecule, such as DNA, from gold cluster labels both linked to the macromolecule and unbound. Using undecagold and Nanogold, reactive sites have been localized within macromolecules as small as the 25 kDa Phe-tRNA(Phe); the labeled RNA was successfully used to probe the localization of binding sites in a ternary complex with T. thermophilus EF-Tu.GTP.
The sequence-specific hybridization of oligonucleotides means that an almost unlimited number of unique recognition or attachment sites can be incorporated into sets of components, which can then be assembled through a series of unique molecular recognition events. This provides an inherent programmability, and combinations of such interactions have even been proposed as the basis for a method of organic computing (Fu, 2007). Covalent gold nanoparticle conjugation complements this programmability. Gold nanoparticles may be attached at unique sites where their chemical or physical properties impart useful functionality, and because gold cluster labels can be prepared with different functionalities, it may be possible to attach different particles to different sites within a supramolecular construct in order to construct novel nano-materials or perform more complex tasks. This approach holds the promise of great advances in research tools, medicine, and technology. Methods for the synthesis and characterization of components from which such hybrid materials may be constructed are therefore of fundamental importance to this field. High-resolution microscopy is of particular importance because it can help bridge the gap between the molecular and the macroscale structures of these materials, and help elucidate how their structures and properties are related.
The tools available for preparing such constructs will be further expanded in the future through the introduction of larger gold nanoparticle labels with the same covalent reactivity (Gutierrez et al., 1999) but different properties, such as surface plasmon interactions and capacitance. These novel, useful properties may enable the preparation of improved probes and materials with nanoscale functionality.
References
- Ackerson CJ, Powell RD, Hainfeld JF. Site-specific biomolecule labeling with gold clusters. Meth Enzymol. doi: 10.1016/S0076-6879(10)81009-2. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerson CJ, Jadzinsky PD, Jensen GJ, Kornberg RD. Rigid, specific, and discrete gold nanoparticle/antibody conjugates. J Am Chem Soc. 2006;128:2635–2640. doi: 10.1021/ja0555668. [DOI] [PubMed] [Google Scholar]
- Ackerson CJ, Sykes MT, Kornberg RD. Defined DNA/nanoparticle conjugates. Proc Natl Acad Sci U S A. 2005;102:13383–13385. doi: 10.1073/pnas.0506290102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adami A, Garcia-Alvarez B, Arias-Palomo E, Barford D, Llorca O. Structure of TOR and its complex with KOG1. Mol Cell. 2007;27:509–516. doi: 10.1016/j.molcel.2007.05.040. [DOI] [PubMed] [Google Scholar]
- Alivisatos AP, Johnsson KP, Peng X, Wilson TE, Loweth CJ, Bruchez MP, Jr, Schultz PG. Organization of ‘Nanocrystal Molecules’ using DNA. Nature. 1996;382:609–611. doi: 10.1038/382609a0. [DOI] [PubMed] [Google Scholar]
- Basnar B, Weizmann Y, Cheglakov Z, Willner I. Synthesis of nanowires using dip-pen nanolithography and biocatalytic inks. Adv Mater. 2006;18:713–718. [Google Scholar]
- Bendayan M. A review of the potential and versatility of colloidal gold cytochemical labeling for molecular morphology. Biotechnol Histochem. 2000;75:203–242. [PubMed] [Google Scholar]
- Bendayan M, Garzon S. Protein G-gold complex: comparative evaluation with protein A-gold for high-resolution immunocytochemistry. J Histochem Cytochem. 1988;36:597–607. doi: 10.1177/36.6.2452843. [DOI] [PubMed] [Google Scholar]
- Blechschmidt B, Jahn W, Hainfeld JF, Sprinzl M, Boublik M. Visualization of a ternary complex of the Escherichia coli Phe-tRNAPhe and TU, GTP from Thermus thermophilus by scanning electron microscopy. J Struct Biol. 1993;110:84–89. doi: 10.1006/jsbi.1993.1007. [DOI] [PubMed] [Google Scholar]
- Blechschmidt B, Shirokov V, Sprinzl M. Undecagold cluster modified tRNA (Phe) from Escherichia coli and its activity in the protein elongation cycle. Eur J Biochem. 1994;219:65–71. doi: 10.1111/j.1432-1033.1994.tb19915.x. [DOI] [PubMed] [Google Scholar]
- Buchel C, Morris E, Orlova E, Barber J. Localisation of the PsbH subunit in photosystem II: a new approach using labelling of His-tags with a Ni(2+)-NTA gold cluster and single particle analysis. J Mol Biol. 2001;312:371–379. doi: 10.1006/jmbi.2001.4951. [DOI] [PubMed] [Google Scholar]
- Bumba L, Tichy M, Dobakova M, Komenda J, Vacha F. Localization of the PsbH subunit in photosystem II from the Synechocystis 6803 using the His-tagged Ni–NTA Nanogold labeling. J Struct Biol. 2005;152:28–35. doi: 10.1016/j.jsb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Cady NC, Strickland AD, Batt CA. Optimized linkage and quenching strategies for quantum dot molecular beacons. Mol Cell Probes. 2007;21:116–124. doi: 10.1016/j.mcp.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Cheng N, Conway JF, Watts NR, Hainfeld JF, Joshi V, Powell RD, Stahl SJ, Wingfield PE, Steven AC. Tetrairidium, a 4-atom cluster, is readily visible as a density label in 3D Cryo-EM maps of proteins at 10–25 Å resolution. J Struct Biol. 1999;127:169–176. doi: 10.1006/jsbi.1999.4120. [DOI] [PubMed] [Google Scholar]
- Claridge SA, Mastroianni AJ, Au YB, Liang HW, Micheel CM, Fréchet JM, Alivisatos AP. Enzymatic ligation creates discrete multinanoparticle building blocks for self-assembly. J Am Chem Soc. 2008;130:9598–9605. doi: 10.1021/ja8026746. [DOI] [PubMed] [Google Scholar]
- Dubertret B, Calame M, Libchaber A. Single-mismatch detection using gold-quenched fluorescent oligonucleotides. Nat Biotechnol. 2001;19:365–370. doi: 10.1038/86762. [DOI] [PubMed] [Google Scholar]
- Dulkeith E, Morteani AC, Niedereichholz T, Klar TA, Feldmann J, Levi SA, van Veggel FCJM, Reinhoudt DN, Enschede AE, Möller M, Gittins DI. Fluorescence quenching of dye molecules near gold nanoparticles: radiative and nonradiative effects. Phys Rev Lett. 2002;89:203002. doi: 10.1103/PhysRevLett.89.203002. [DOI] [PubMed] [Google Scholar]
- Dulkeith E, Ringler M, Klar TA, Feldmann J, Munoz Javier A, Parak WJ. Gold nanoparticles quench fluorescence by phase induced radiative rate suppression. Nano Lett. 2005;5:585–589. doi: 10.1021/nl0480969. [DOI] [PubMed] [Google Scholar]
- Fischler M, Sologubenko A, Mayer J, Clever G, Burley G, Gierlich J, Carell T, Simon U. Chain-like assembly of gold nanoparticles on artificial DNA templates via ‘click chemistry’. JCS Chem Commun (Camb) 2008:169–171. doi: 10.1039/b715602b. [DOI] [PubMed] [Google Scholar]
- Fu P. Biomolecular computing: is it ready to take off? Biotechnol J. 2007;2:91–101. doi: 10.1002/biot.200600134. [DOI] [PubMed] [Google Scholar]
- Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC, Mirkin CA. Gold nanoparticles for biology and medicine. Angew Chem Int Ed Eng. 2010;49:3280–3294. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregori L, Hainfeld JF, Simon MN, Goldgaber D. Binding of amyloid beta protein to the 20 S proteasome. J Biol Chem. 1997;272:58–62. doi: 10.1074/jbc.272.1.58. [DOI] [PubMed] [Google Scholar]
- Gutierrez E, Powell RD, Hainfeld JF, Takvorian PM. A covalently linked 10 nm gold immunoprobe. In: Bailey GW, Jerome WG, McKernan S, Mans-field JF, Price RL, editors. Microsc Microanal. Suppl 2. Vol. 5. Springer–Verlag; New York, NY: 1999. pp. 1324–1325. Proceedings. [Google Scholar]
- Hainfeld JF. A small gold-conjugated antibody label: improved resolution for electron microscopy. Science. 1987;236:450–453. doi: 10.1126/science.3563522. [DOI] [PubMed] [Google Scholar]
- Furuya FR, Miller LL, Hainfeld JF, Christopfel WC, Kenny PW. Use of Ir4 (CO)11 to measure the lengths of organic molecules with a scanning transmission electron microscope. J Am Chem Soc. 1988;110:641–643. [Google Scholar]
- Hainfeld JF. Undecagold-antibody method. In: Hayat MA, editor. Colloidal Gold: Principles, Methods, and Applications. Vol. 2. Academic Press; San Diego: 1989. pp. 413–429. [Google Scholar]
- Hainfeld JF, Foley CJ, Maelia LE, Lipka JJ. Eleven tungsten atom cluster labels: high-resolution, site-specific probes for electron microscopy. J Histochem Cytochem. 1990;38:1787–1793. doi: 10.1177/38.12.1701458. [DOI] [PubMed] [Google Scholar]
- Hainfeld JF, Sprinzl M, Mandiyan V, Tumminia SJ, Boublik M. Localization of a specific nucleotide in yeast tRNA by scanning transmission electron microscopy using an undecagold cluster. J Struct Biol. 1991;107:1–5. doi: 10.1016/1047-8477(91)90024-q. [DOI] [PubMed] [Google Scholar]
- Hainfeld JF, Furuya FR. A 1.4 nm gold cluster covalently attached to antibodies improves immunolabeling. J Histochem Cytochem. 1992;40:177–184. doi: 10.1177/40.2.1552162. [DOI] [PubMed] [Google Scholar]
- Hainfeld JF, Powell RD. Nanogold technology: new frontiers in gold labeling. Appl Immunohistochem Mol Morphol (Cell Vision) 1997;4:408–432. [Google Scholar]
- Hainfeld JF, Furuya FR, Powell RD. Metallosomes. J Struct Biol. 1999a;127:152–160. doi: 10.1006/jsbi.1999.4145. [DOI] [PubMed] [Google Scholar]
- Hainfeld JF, Liu W, Halsey CMR, Freimuth P, Powell RD. Ni-NTA-gold clusters target his-tagged proteins. J Struct Biol. 1999b;127:185–198. doi: 10.1006/jsbi.1999.4149. [DOI] [PubMed] [Google Scholar]
- Hainfeld JF, Liu W, Barcena M. Gold-ATP. J Struct Biol. 1999c;127:120–134. doi: 10.1006/jsbi.1999.4146. [DOI] [PubMed] [Google Scholar]
- Hainfeld JF, Powell RD. New frontiers in gold labeling. J Histochem Cytochem. 2000;48:471–480. doi: 10.1177/002215540004800404. [DOI] [PubMed] [Google Scholar]
- Hainfeld JF, Furuya FR, Powell RD, Liu W. DNA nanowires. In: Bailey GW, Price RL, Voelkl E, Musselman IH, editors. Microsc Microanal. Suppl 2. Vol. 7. Springer–Verlag; New York, NY: 2001. pp. 1034–1035. Proceedings. [Google Scholar]
- Hainfeld JF, Powell RD, Hacker GW. Nanoparticle molecular labels. In: Mirkin CA, Niemeyer CM, editors. Nanobiotechnology. Chapter 23 Wiley-VCH; Weinheim, Germany: 2004. pp. 353–386. [Google Scholar]
- Hamad-Schifferli K, Schwartz JJ, Santos AT, Zhang S, Jacobson JM. Remote electronic control of DNA hybridization through inductive coupling to an attached metal nanocrystal antenna. Nature. 2002;415:152–155. doi: 10.1038/415152a. [DOI] [PubMed] [Google Scholar]
- Handley DA. The development and application of colloidal gold as a microscopic probe. In: Hayat MA, editor. Colloidal Gold: Principles, Methods and Applications. Chapter 1 Academic Press; San Diego, CA: 1989a. pp. 1–11. [Google Scholar]
- Handley DA. Methods for synthesis of colloidal gold. In: Hayat MA, editor. Colloidal Gold: Principles, Methods and Applications. Chapter 2 Academic Press; San Diego, CA: 1989b. pp. 12–22. [Google Scholar]
- Jennings TL, Schlatterer JC, Singh MP, Greenbaum NL, Strouse GF. NSET molecular beacon analysis of hammerhead RNA substrate binding and catalysis. Nano Lett. 2006;6:1318–1324. doi: 10.1021/nl052458a. [DOI] [PubMed] [Google Scholar]
- Jones IW, Barik J, O’ Neill MJ, Wonnacott S. Alpha bungarotoxin-1.4 nm gold: a novel conjugate for visualising the precise subcellular distribution of alpha 7* nicotinic acetylcholine receptors. J Neurosci Methods. 2004;134:65–74. doi: 10.1016/j.jneumeth.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Kessler P, Kotzyba-Hilbert F, Leonetti M, Bouet F, Ringler P, Brisson A, Mendez A, Goeldner MP, Hirth C. Synthesis of an acetylcoline receptor-specific toxin derivative regioselectively labeled with an undecagold cluster. Bioconjug Chem. 1994;5:199–204. doi: 10.1021/bc00027a004. [DOI] [PubMed] [Google Scholar]
- Krisch B, Buchholz C, Mentlein R, Turzynski A. Visualization of neuropeptide-binding sites on individual telencephalic neurons of the rat. Cell Tissue Res. 1993;272:523–531. doi: 10.1007/BF00318559. [DOI] [PubMed] [Google Scholar]
- Lipka JJ, Hainfeld JF, Wall JS. Undecagold labeling of a glycoprotein: STEM visualization of an undecagoldphosphine cluster labeling the carbohydrate sites of human haptoglobin-hemoglobin complex. J Ultrastruct Res. 1983;84:120–129. doi: 10.1016/s0022-5320(83)90123-5. [DOI] [PubMed] [Google Scholar]
- Malecki M. Preparation of plasmid DNA in transfection complexes for fluorescence and spectroscopic imaging. In: Malecki M, Roomans GM, editors. Scanning Microsc Suppl; Proc. 14th Pfefferkorn Conf.; Chicago, IL: Scanning Microscopy International; 1996. pp. 1–16. [PubMed] [Google Scholar]
- Medalia O, Heim M, Guckenberger R, Sperling R, Sperling J. Gold-tagged RNA—a probe for macromolecular assemblies. J Struct Biol. 1999;127:113–119. doi: 10.1006/jsbi.1999.4134. [DOI] [PubMed] [Google Scholar]
- Mentlein R, Buchholz C, Krisch B. Binding and internalization of gold-conjugated somatostatin and growth hormone-releasing hormone in cultured rat somatotropes. Cell Tissue Res. 1990;262:431–442. doi: 10.1007/BF00239451. [DOI] [PubMed] [Google Scholar]
- Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature. 1996;382:607–609. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- Mrksich M, Whitesides GM. Using self-assembled monolayers to understand the interactions of man-made surfaces with proteins and cells. Annu Rev Biophys Biomol Struct. 1996;25:55–78. doi: 10.1146/annurev.bb.25.060196.000415. [DOI] [PubMed] [Google Scholar]
- Nishinaka T, Takano A, Doi Y, Hashimoto M, Nakamura A, Matsushita Y, Kumaki J, Yashima E. Conductive metal nanowires templated by the nucleoprotein filaments. Complex of DNA and RecA protein. J Am Chem Soc. 2005;127:8120–8125. doi: 10.1021/ja050487h. [DOI] [PubMed] [Google Scholar]
- Patolsky F, Weizmann Y, Lioubashevski O, Willner I. Au-nanoparticle nanowires based on DNA and polylysine templates. Angew Chem Int Ed Eng. 2002;41:2323–2327. doi: 10.1002/1521-3773(20020703)41:13<2323::AID-ANIE2323>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Park SH, Yin P, Liu Y, Reif JH, LaBean TH, Yan H. Programmable DNA self-assemblies for nanoscale organization of ligands and proteins. Nano Lett. 2005;5:729–733. doi: 10.1021/nl050175c. [DOI] [PubMed] [Google Scholar]
- Powell RD, Halsey CMR, Liu W, Joshi VN, Hainfeld JF. Giant platinum clusters: 2 nm covalent metal cluster labels. J Struct Biol. 1999;127:177–184. doi: 10.1006/jsbi.1999.4153. [DOI] [PubMed] [Google Scholar]
- Powell RD, Hainfeld JF. Silver- and gold-based autometallography of Nanogold. In: Hacker GW, Gu J, editors. Gold and Silver Staining: Techniques in Molecular Morphology. Chapter 3 CRC Press; Boca Raton, FL: 2002. pp. 29–46. [Google Scholar]
- Rappas M, Schumacher J, Beuron F, Niwa H, Bordes P, Wigneshweraraj S, Keetch CA, Robinson CV, Buck M, Zhang X. Structural insights into the activity of enhancer-binding proteins. Science. 2005;307:1972–1975. doi: 10.1126/science.1105932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JE, Frey PA. Synthesis of undecagold cluster molecules as biochemical labeling reagents. 1 Monoacyl and mono[N-(succinimidooxy)succinyl] undecagold clusters. Biochemistry. 1984;23:3849–3856. doi: 10.1021/bi00312a009. [DOI] [PubMed] [Google Scholar]
- Ribrioux S, Kleymann G, Haase W, Heitmann K, Ostermeier C, Michel H. Use of Nanogold- and fluorescent-labeled antibody Fv fragments in immunocytochemistry. J Histochem Cytochem. 1996;44:207–213. doi: 10.1177/44.3.8648079. [DOI] [PubMed] [Google Scholar]
- Rosi NL, Mirkin CA. Nanostructures in biodiagnostics. Chem Rev. 2005;105:1547–1562. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]
- Roth J, Zuber C, Komminoth P, Sata T, Li WP, Heitz PU. Applications of immunogold and lectin-gold labeling in tumor research and diagnosis. Histochem Cell Biol. 1996;106:131–148. doi: 10.1007/BF02473207. [DOI] [PubMed] [Google Scholar]
- Scheibel T, Parthasarathy R, Sawicki G, Lin XM, Jaeger H, Lindquist SL. Conducting nanowires built by controlled self-assembly of amyloid fibers and selective metal deposition. Proc Natl Acad Sci U S A. 2003;100:4527–4532. doi: 10.1073/pnas.0431081100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segond von Banchet G, Schindler M, Hervieu GJ, Beckmann B, Emson PC, Heppelmann B. Distribution of somatostain receptor subtypes in rat lumbar spinal cord examined with gold-labelled somatostatin and anti-receptor antibodies. Brain Res. 1999;816:254–257. doi: 10.1016/s0006-8993(98)01226-8. [DOI] [PubMed] [Google Scholar]
- Simmons SR, Albrecht RM. Probe size and bound label conformation in colloidal gold-ligand labels and gold-immunolabels. Scanning Microsc. 1989;3(Suppl):27–33. [PubMed] [Google Scholar]
- Skripkin E, Yusupova G, Yusupov M, Kessler P, Ehresmann C, Ehresmann B. Synthesis and ribosome binding properties of model mRNAs modified with undecagold cluster. Bioconjug Chem. 1993;4:549–553. doi: 10.1021/bc00024a019. [DOI] [PubMed] [Google Scholar]
- Sprinzl M, Scheit K-H, Cramer F. Preparation in vitro of a 2-thiocytidine-containing yeast tRNA Phe-A 73-C 74-S 2C 75-A 76 and its interaction with p-hydroxymercuribenzoate. Eur J Biochem. 1973;34:306–310. doi: 10.1111/j.1432-1033.1973.tb02759.x. [DOI] [PubMed] [Google Scholar]
- Stoeva SI, Lee JS, Thaxton CS, Mirkin CA. Multiplexed DNA detection with biobarcoded nanoparticle probes. Angew Chem Int Ed Engl. 2006;45:3303–3306. doi: 10.1002/anie.200600124. [DOI] [PubMed] [Google Scholar]
- Sussman JL, Holbrook SR, Warrant RW, Chruch G, Kim SH. Crystal structure of yeast phenylalanine transfer RNA. I. Crystallographic refinement. J Mol Biol. 1978;123:607–630. doi: 10.1016/0022-2836(78)90209-7. [DOI] [PubMed] [Google Scholar]
- Thaxton CS, Georganopoulou DG, Mirkin CA. Gold nanoparticle probes for the detection of nucleic acid targets. Clin Chim Acta. 2006;363:120–126. doi: 10.1016/j.cccn.2005.05.042. [DOI] [PubMed] [Google Scholar]
- Tyagi S, Kramer FR. Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- Weinstein S, Jahn W, Hansen H, Wittmann HG, Yonath A. Novel procedures for derivatization of ribosomes for crystallographic studies. J Biol Chem. 1989;264:19138–19142. [PubMed] [Google Scholar]
- Weiser HB. Inorganic Colloid Chemistry. Vol. 1. Wiley; New York, NY: 1933. pp. 21–57. [Google Scholar]
- Weizmann Y, Patolsky F, Popov I, Willner I. Telomerase-generated templates for the growing of metal nanowires. Nano Lett. 2004;4:787–792. [Google Scholar]
- Wilner OI, Shimron S, Weizmann Y, Wang ZG, Willner I. Self-assembly of enzymes on DNA scaffolds: en route to biocatalytic cascades and the synthesis of metallic nanowires. Nano Lett. 2009;9:2040–2043. doi: 10.1021/nl900302z. [DOI] [PubMed] [Google Scholar]
- Xiao S, Liu F, Rosen AE, Hainfeld JF, Seeman NC, Musier-Forsyth K, Kiehl RA. Selfassembly of metallic nanoparticle arrays by DNA scaffolding. J Nanoparticle Res. 2002;4:313–317. [Google Scholar]
- Xiao Y, Patolsky F, Katz E, Hainfeld JF, Willner I. Plugging into enzymes: nanowiring of redox enzymes by a gold nanoparticle. Science. 2003;299:1877–1881. doi: 10.1126/science.1080664. [DOI] [PubMed] [Google Scholar]
- Yan H, Park SH, Finkelstein G, Reif JH, LaBean TH. DNA-templated self-assembly of protein arrays and highly conductive nanowires. Science. 2003;301:1882–1884. doi: 10.1126/science.1089389. [DOI] [PubMed] [Google Scholar]
- Yonekura K, Yakushi T, Atsumi T, Maki-Yonekura S, Homma M, Namba K. Electron cryomicroscopic visualization of PomA/B stator units of the sodium-driven flagellar motor in liposomes. J Mol Biol. 2006;357:73–81. doi: 10.1016/j.jmb.2005.12.041. [DOI] [PubMed] [Google Scholar]
- Zhang J, Liu Y, Ke Y, Yan H. Periodic square-like gold nanoparticle arrays templated by self-assembled 2D DNA nanogrids on a surface. Nano Lett. 2006;6:248–251. doi: 10.1021/nl052210l. [DOI] [PubMed] [Google Scholar]