

Synopsis
Progress in our understanding of the molecular biology of neoplasms has been driven by remarkable improvements in molecular biology techniques. This has created a rapidly moving field in which even subspecialists struggle to keep abreast of the current literature. Nowhere is this more clearly demonstrated than in neuro-oncology, wherein molecular diagnostics can now wring more clinically useful information out of very small biopsies than ever before. Herein the biologic and practical aspects of four key molecular biomarkers in gliomas are discussed, including two that have been known for some time (1p/19q codeletion and EGFR amplification) as well as two whose relevance was discovered via advanced whole-genome assays (IDH1/2 mutations and BRAF alterations).
Keywords: Molecular pathology, Glioma, Head and neck cancer, Neuro-oncology, Biomarkers
Histologic review of the most common gliomas
Gliomas are the most common primary brain tumors in both children and adults, though in children the posterior fossa is the most frequent site whereas adult gliomas are much more likely to develop in the supratentorium. The WHO grading scheme ranges from I to IV, with an accompanying increase in severity.1, 2
Pilocytic astrocytomas
Grade I gliomas are usually pilocytic astrocytomas (PAs), which are the most common primary brain tumors in children. About two-thirds are located in the cerebellum but they can occur anywhere in the neuraxis. Key histologic characteristics include biphasic architecture with or without microcysts, bland nuclei, Rosenthal fibres, and eosinophilic granular bodies [virtual slide #1]. Yet the appearance of these tumors can vary greatly, creating diagnostic overlap with other gliomas. In keeping with their WHO grade I designation, most are curable if gross total resection can be achieved.3 However, tumors arising in harder-to-reach areas like the diencephalon (thalamus and hypothalamus) and myelencephalon (brainstem) usually cannot be completely removed and often recur, with worse outcome more likely.
Pilomyxoid astrocytoma
Pilomyxoid astrocytoma (PMA) is an uncommon grade II variant of PA, usually arising in the diencephalon and featuring a looser arrangement of spindly bland tumor cells with a bluish-myxoid background and variable degrees of perivascular orientation [virtual slide #2]. Again, though, there is considerable overlap with PA morphology. In fact, it is possible that many PMAs are simply less “mature” PAs that, because they arise in the biologically sensitive diencephalon, do not have enough time to develop more classic PA histology before being resected.4
Infiltrative glioma
Unlike grade I PAs, the most common grades II—IV gliomas are, by definition, diffusely infiltrative. As a result, surgical debulking can only extend survival. Even radiation and chemotherapy, usually reserved for grades III and IV tumors, are not curative. If a glioma shows signs of being infiltrative (e.g. spreading along neurons and capillaries) yet lacks mitoses, necrosis, and microvascular proliferation, it is WHO grade II. If the tumor nuclei are generally angulated and irregular, the glioma is classified as an astrocytoma, with a median survival of about 5 years.1 Grade II oligodendrogliomas feature round nuclei [virtual slide #3] and have a median survival double that of grade II astrocytomas. All grade II tumors are still considered “low grade” even though they are technically incurable, simply because death is delayed. As a general principle, grade II tumors do not enhance with contrast-inducing agents on radiology. The presence of calcifications should not be used as a discriminator between astrocytomas and oligodendrogliomas; likewise, the absence of “chickenwire” capillaries and “fried egg” artifact do not rule out an oligodendroglioma. The term “oligoastrocytoma” refers to gliomas with mixed features, but while the entity is recognized by the WHO it suffers from poor interobserver consistency.
Mitoses in glioma
The presence of mitoses in a glioma raises the WHO grade to III, though the exact number of mitoses required per microscopic area has never been firmly established. These tumors usually show at least some contrast enhancement on scans. Grade III anaplastic astrocytomas [virtual slide #4] have a median survival of 2 to 3 years, while anaplastic oligodendrogliomas [virtual slide #5] are slightly longer at 3 to 4 years.1 A key diagnostic point is that, while necrosis and/or microvascular proliferation bump an anaplastic astrocytoma up to a grade IV glioblastoma, they do not necessarily increase the WHO grade of an anaplastic oligodendroglioma. There is some controversy about this, though, especially in anaplastic gliomas with necrosis and mixed astrocytic/oligodendroglial morphology.5 Such tumors are sometimes termed “glioblastomas with oligodendroglial component” (GBM-O) or “mixed oligoastrocytoma grade IV” (MOA IV).
Glioblastoma
Glioblastomas (formerly called “glioblastoma multiforme” or “GBM”), are WHO grade IV and have a median survival of 9 to 12 months.1 Unfortunately this is also the most common grade in adults, comprising about 70% of all gliomas. In addition to the aforementioned criteria for grades II and III astrocytomas, GBMs show microvascular proliferation and necrosis, the latter often surrounded by a pseudopalisade of glioma cells [virtual slide #6]. As befits the traditional “multiforme” appellation, though, there is a great deal of microscopic heterogeneity, including gliosarcoma [virtual slide #7], giant cell variant [virtual slide #8], and granular cell variant [virtual slide #9], among others.
Whereas progression to a higher grade is extremely uncommon in PAs, it is the rule in diffusely infiltrative gliomas. Yet most GBMs seem to arise de novo and thus are called “primary” GBMs, in contrast to “secondary” GBMs that developed from known grade II and III astrocytomas.
Glioma molecular biomarkers
The explosion of data in the field of glioma molecular biomarkers has rendered a comprehensive review of all noteworthy markers unwieldy. Instead, discussion is focused on four high-yield genetic biomarkers:
-
Two that have been known to exist for a long time:
1p/19q codeletion
EGFR amplification
-
Two that were discovered only recently as the result of whole genome assays and sophisticated bioinformatics:
IDH1/2 mutation
BRAF fusion/mutation
Careful use of both “old” and “new” biomarkers greatly enhances the practice of surgical neuropathology (Table 1).
Table 1.
Key genetic alterations and their use in glioma diagnostics. 1p/19q codeletion, EGFR amplification, IDH1/2 mutations, and BRAF fusion or V600E mutation are all useful in resolving diagnostic dilemmas and/or refining patient prognosis in gliomas. Each test must be applied judiciously in specific contexts for maximal efficacy, and must always be correlated with light microscopic analysis and radiology. With the exception of BRAF, none of these tests are useful in pre-adolescent patients.
| Genetic alteration | Main gliomas | Diagnostic value | Prognostic value | Predictive value | Most common methods |
|---|---|---|---|---|---|
| 1p/19q codeletion | ~80% of grade II and III oilgodendrogliomas | Differentiates most oligos from astrocytic gliomas and oligo mimickers (e.g neurocytoma, clear cell ependymoma, small cell GBM) | Longer survival | No specific therapy to date, but associated with better response to adjuvant therapy | FISH, PCR-based LOH |
| EGFR amplification | ~40% of GBMs | Often detects scattered tumor cells in undersampled GBMs; differentiates small cell GBM from AO | Controversial | None | FISH |
| IDH1/2 mutation | ~80% of grades II–III astrocytomas and oligodendrogliomas; over 90% of secondary GBMs | Presence strongly suggests an infiltrative glioma; negative in nonneoplastic glioma mimickers and noninfiltrative gliomas (e.g. PA, non-glial tumors) | Longer survival, may even trump WHO grade in some cases | No specific therapy to date, but associated with better response to adjuvant therapy | PCR and sequencing, IHC |
| BRAF fusion or V600E mutation | >75% of PAs (mostly fusions), ~50% PMA, 80% PXA (mostly V600E); 25% GG (mostly V600E) | Fusion suggests a PA or PMA; mutation suggests PXA or GG (though not a perfect discriminator from PAs or diffusely infiltrative gliomas) | May be a favorable marker, but still uncertain | No specific therapy to date; anti-MEK and anti-BRAF V600E clinical trials ongoing | fusions: PCR breakpoint analysis, FISH V600E: PCR and sequencing, IHC |
GBM = glioblastoma; AO = grade III anaplastic oligodendroglioma; PA = pilocytic astrocytoma; PMA = pilomyxoid astrocytoma; PXA = pleomorphic xanthoastrocytoma; GG = ganglioglioma; FISH = fluorescence in situ hybridization; LOH = loss of heterozygosity; IHC = immunohistochemistry.
1p/19q: Some standardization required
Codeletion of the short arm of chromosome 1 and the long arm of chromosome 19 has been known to be a marker of oligodendroglial morphology for nearly 20 years.6, 7 This codeletion is the result of an unbalanced translocation between the two chromosomes with subsequent loss of der(1;19)(p10;q10).8, 9 Despite our karyotype-induced temptation to think of them as separated by vast distances, chromosomes 1 and 19 are actually close together in the nonrandom organization of the nucleus, at least partially explaining why this translocation appears so frequently.
Codeletion and adjuvant therapy
Over the past two decades an abundance of research has shed more light on the clinical and biological implications of 1p/19q codeletion. For example, numerous studies have shown that oligodendrogliomas with 1p/19q codeletion show improved response to adjuvant radiochemotherapy and longer survival, with the survival difference being much stronger in grade III anaplastic oligodendrogliomas than in grade II oligodendrogliomas.10–16 Interestingly, though, the codeletion may impart such a survival difference only if the tumor is treated with adjuvant therapy—i.e. 1p/19q codeletion may not make a difference if the tumor is not challenged with post-surgical radiation and/or chemotherapy.17 An oligodendroglioma that shows textbook-type morphology such as round nuclei, delicate branching vasculature, and microcalcifications is more likely to contain the codeletion, as opposed to gliomas with only a few features suggestive of oligodendroglioma.18, 19 Yet even in gliomas that contain both oligodendroglial and astrocytic features (so-called “oligoastrocytomas”), those with the codeletion tend to behave more like oligodendrogliomas,20–22 whereas strong nuclear p53 staining is more suggestive of astrocytic differentiation.23
Codeletion and reclassification
However, this author’s experience suggests that histologically unequivocal GBMs should not be reclassified based solely on the presence of 1p/19q codeletion, because those tumors will still behave like GBMs and, through extensive genomic instability, produce false-positive results (manuscript under review). Likewise, though isolated loss of 1p has been suggested to be a weakly favorable prognostic factor in GBM,24 this has not proven to be a consistent finding25 (manuscript under review) and is not worth testing for in an otherwise unequivocal GBM.
Codeletion for differentiation among tumors
Testing for 1p/19q codeletion also can help differentiate between oligodendroglioma and its common mimickers, dysembryoplastic neuroepithelial tumor (DNET), clear cell ependymoma, and central neurocytoma, none of which have 1p/19q codeletion.26, 27 However, some case reports have suggested an overlap between oligodendrogliomas and neurocytomas since 1p/19q codeletion is occasionally seen in rare oligo-like tumors that also show unequivocal signs of neurocytic differentiation.28–30
Gliomas with 1p/19q codeletion tend to have a proneural expression profile, MGMT promoter methylation, and IDH1/2 mutations.31–33 In particular, it has been demonstrated that all gliomas with true whole-arm 1p/19q codeletion should also have IDH1/2 mutations.31, 34 Personal experience supports this finding; now this author recommends interrogating IDH1/2 status first, then considering additional 1p/19q testing only if an IDH1/2 mutation is detected. Coexistence of EGFR amplification and 1p/19q codeletion is extremely rare,16, 35, 36 to the point where such cases are probably just false-positive for true whole-arm codeletion. 1p/19q codeletion is about half as frequent in oligodendrogliomas arising in temporal lobe tumors versus those in the frontal, parietal, or occipital lobes, and has been reported to be especially uncommon in tumors from the insula,16, 37, 38 though the latter assertion has been disputed.39 All these observations are applicable only to the adult patient population, since pediatric oligodendrogliomas rarely have 1p/19q codeletion and, even when apparent codeletion is found, it is not clear whether it has any prognostic value in this context.40, 41
Tests for 1p/19q codeletion
There are many ways to test for 1p/19q codeletion in routine clinical samples; two of the most common are fluorescence in situ hybridization (FISH) and polymerase chain reaction (PCR)-based microsatellite loss of heterozygosity (LOH).
FISH
FISH uses fluorophore-labeled DNA probes to bind targeted regions in tumor nuclei. In the case of 1p/19q, 1p and 19q obviously have to be interrogated, and 1q and 19p are included as internal ploidy controls. The most commonly used probe pairs are commercially available, targeting 1p36/1p25 and 19q13/19p13 [Figure 1]. Because only two unstained slides are needed (one slide per probe pair), and only 60 to 100 tumor nuclei need to be counted to get a reliable score, FISH can be done on very small tissue biopsies—a feature of great importance in neuropathology. Also, a corresponding H&E section can be used to focus analysis on a specific area of interest. Patient-matched nonneoplastic DNA is not needed as an internal control. And FISH is well-suited to detecting polysomy of chromosomes 1 and 19, which has been shown to correlate with shorter progression-free survival in anaplastic oligodendrogliomas that have relative 1p/19q codeletion.42
Figure 1.

Testing for 1p/19q codeletion. (A) Microsatellites spanning 1p and 19q are targeted via PCR-based LOH analysis, whereas the most widely used FISH probes bind to 1p36 and 19q13 (red) and 1q25 and 19p13 (green). (B) Under the microscope, DAPI-counterstained nuclei in a codeleted oligodendroglioma show the same signal pattern for both probe pairs: 2 green signals but only 1 red signal.
There are, however, some drawbacks to FISH in assessing 1p/19q status. Because the goal is to find relative deletions of 1p and 19q, and tissue sections need to be 4–6 microns thick for FISH to work even though nuclei are usually greater than 6 microns in diameter, it follows that partial loss of some tumor nuclei, including their DNA, is inevitable. This issue is addressed by the use of unrelated nonneoplastic brain tissue (e.g. postmortem white matter) as a negative validation control; any case showing fewer 1p and 19q signals than the controls is considered codeleted. Even this approach has problems, though, as there are no universally-adopted cutoff criteria for how many cells have to show codeletion in “true” 1p/19q codeletion.43 For that matter, even the scoring parameters vary between institutions. Some incorporate 1p/1q and 19q/19p signal ratios into the analysis, whereas others base it solely on the percent of tumor cells showing deletion of 1p and 19q signals relative to 1q and 19p signals. Because FISH probes cannot be more than about 1 Mb to hybridize properly, it is impractical to accurately interrogate all of 1p and 19q by FISH. Thus, only small representative areas can be targeted. Prior studies indicated that 1p36 and 19q13 are minimally-deleted regions in diffuse gliomas,44, 45 which is why these loci were initially targeted by commercial developers of FISH probes. However, since it is whole-arm codeletion that has prognostic value, these loci end up being sensitive but not as specific as one would like.46 Even if optimal probes and interpretation criteria were known, 1p/19q FISH would still be unreliable for proving a glioma in the case of a biopsy where only scattered atypical cells are present, as “dilution” with nonneoplastic nuclei would render the data unreliable.
Based on our current outcomes-based work, it appears that if one wishes to adopt signal ratios as the interpretation criteria, 1p36/1q25 and 19q13/19p13 should each be less than 0.75 for optimal prognostic stratification and concordance with PCR-based microsatellite LOH (see below).46 If using percent of tumor nuclei with relative deletion, at least 40% should show relative loss of both 1p36 and 19q13. As mentioned above, IDH1/2 mutation status is a useful prescreening test; IDH1/2 wild-type cases practically never have true whole-arm codeletion. Also, 10q copy number testing can act as a check against 1p/19q FISH, since the presence of 10q deletion should raise suspicion of a false-positive codeletion.46
PCR-based analysis of LOH
The other most commonly used tool for testing 1p/19q status is PCR-based analysis of LOH in DNA repeats known as microsatellites, which are scattered throughout the genome and are used as surrogate markers for nearby regions of interest. Microsatellites vary in the number of repeats they contain, and when both copies of a DNA locus contain the same microsatellite with two different lengths, the PCR products will migrate at different rates. Such a locus is then said to be informative. If a known informative locus shows only one PCR product size, the other is assumed to have been lost. Like FISH, PCR-based LOH can be done on formalin-fixed, paraffin-embedded (FFPE) tissue. Unlike FISH, it is quite feasible to interrogate many regions of DNA at once, allowing for a more complete view of 1p and 19q (Figure 1A). This reduces the risk of scattered interstitial deletions producing false-positive data, a risk especially notable in higher-grade gliomas. Criteria for interpretation of LOH data must be strict, calling codeletion only if all or nearly all of the microsatellites on both 1p and 19q show LOH. While FISH and PCR-based LOH generally show over 90% concordance,45, 47–49 concomitant prospective testing of gliomas with both assays showed PCR-based LOH is better at prognostic stratification in anaplastic oligodendrogliomas.46
Although PCR-based microsatellite LOH analysis is more specific than FISH for true 1p/19q codeletion, it too has its drawbacks. For one thing, it is harder to accurately target subregions on a piece of tissue for analysis, and thus is more susceptible to dilution with nonneoplastic DNA. For optimal results it requires control patient-matched nonneoplastic DNA, usually in the form of white blood cells. Both assays are fairly labor intensive, but PCR-based LOH tends to be more expensive because it needs more sophisticated equipment. Thus, right now FISH is more widely used. In the near future, use of single nucleotide polymorphism (SNP) arrays and array comparative genomic hybridization (aCGH) will probably become more popular since they can, in a single test, scan not just 1p and 19q but also other loci of interest like 7p12 (EGFR), 9p21 (CDKN2A), and 10q.
Targeted therapy for 1p/19q-codeleted glioma
From a clinical perspective there is no targeted therapy for 1p/19q-codeleted gliomas to date—that is, no particular chemotherapy or course of radiation has proven to be specifically more effective in 1p/19q-codeleted gliomas, just that such tumors respond better to any adjuvant therapy compared to their 1p/19q-intact brethren. Thus, the decision as to whether to administer adjuvant therapy, and what kind of therapy to give, is generally predicated more on the WHO grade and location of the tumor, extent of tumor resection, and overall condition of the patient. 1p/19q status is of interest to the clinicians mostly because they want to anticipate how well their patients will respond to treatment.
EGFR: A new twist on an old friend
Amplification of the epidermal growth factor receptor (EGFR) gene on 7p12 has been known to occur in gliomas since the 1980s, preceding even 1p/19q codeletion as a recognized phenomenon in gliomas.50 The transmembrane receptor tyrosine kinase triggers multiple pathways known to promote cell growth, migration, and survival,51–55 and its overexpression broadly correlates with increased WHO grade.
EGFR amplification in diagnosis
From a diagnostic standpoint EGFR amplification occurs in about 40% of all GBMs, specifically primary GBMs but not secondary GBMs.1, 56–58 Amplification is more common in patients 40 years or older, and is only very rarely seen in pediatric high-grade gliomas.59, 60 Nearly half of all GBMs with EGFR amplification (and less than 10% without amplification) contain a truncated variant of the gene, encoding a protein that lacks the extracellular domain and is therefore constitutively active (EGFRvIII).61–63
Because the majority of gliomas with EGFR amplification are histologically GBMs, in cases where the tissue biopsy is not fully representative of the actual tumor, detecting amplification is usually tantamount to a diagnosis of GBM.21, 64 Yet around 15 to 25% of grade III anaplastic astrocytomas also show EGFR amplification,62, 65 and while some studies suggest such tumors will behave more like GBMs,66, 67 others do not.62 Thus, it is not entirely clear whether EGFR amplification in an otherwise grade III glioma always represents an undersampled GBM. The same, however, cannot be said for gliomas that only meet grade II histologic requirements yet have amplification; such cases behave far worse than true grade II gliomas and can be assumed to be undersampled high grade gliomas, especially if the radiologic findings suggest high grade features like contrast enhancement.49
Another feature of EGFR amplification is its tight association with astrocytic gliomas—to wit, tumors that look like oligodendrogliomas but have EGFR amplification lack 1p/19q codeletion and are extremely aggressive, meriting the term “small cell GBM” instead of oligodendroglioma.16, 32, 36, 68, 69 Parenthetically, EGFR amplification and p53 mutation are also nearly mutually exclusive in gliomas,25 although rare cases of dual amplification and mutation have been documented.70
Prognostic significance of EGFR amplification
While testing for EGFR amplification clearly aids the diagnostic side of neuropathology, the prognostic significance of EGFR amplification is ambiguous if one has already reached an unequivocal histologic diagnosis of GBM. Some studies suggest there is no survival difference, while others show worse survival; a few have even suggested an association with longer survival.25, 63, 67, 71–73 Part of the problem may be the common practice of pooling all EGFR-amplified tumors into one large group for analysis, even though the degree of amplification varies widely between tumors. For example, if one is determining EGFR status by FISH (the most common method), the number of EGFR signals and chromosome 7 centromeric enumeration probe (CEP7) signals is expressed as an EGFR:CEP7 ratio. But although most consider a ratio greater than 2 to constitute amplification, the ratio in any given case can easily exceed 20 or more. One might expect a worse prognosis with a higher ratio, but the reality may not be so simple: in a series of 542 GBMs at the University of Pittsburgh, GBMs with EGFR:CEP7 ratios over 20 had longer median survival compared to non-amplified GBMs, which in turn fared better than tumors with EGFR:CEP7 ratios between 2 to 20.74 At first blush such a finding seems counterintuitive, it has been shown that higher ratios of EGFR:CEP7 correlate with more fragments of extrachromosomal DNA in the tumor nuclei (a.k.a. double minutes), whereas lower levels of amplification tend to be interstitial repeats of the gene within an otherwise more stable chromosome.75 Since there is a precedence for “excessive” cancer genomic instability being more prognostically favorable than “moderate” instability,76 perhaps high-amplifiers simply have more fragile genomes and are thus more sensitive to genotoxic therapy. Of note, the size of the 7p12 amplicon can also vary greatly from one GBM to another, sometimes including co-amplified “passenger genes” like LANCL2 and ECOP that themselves could greatly modulate tumor biology.77, 78 However, analysis using The Cancer Genome Atlas Data showed no correlation between amplification of either gene and survival.74
Tests for EGFR amplification
As implied, the most commonly used assay to test for EGFR amplification is FISH, which is particularly advantageous when attempting to spot just a few scattered infiltrating amplified cells in an otherwise under-representative biopsy. Even if there is no reason to suspect EGFR amplification in a particular case, the CEP7 in the EGFR probe cocktail can act as a test for trisomy 7, which helps differentiate between astrocytoma and reactive gliosis.79
Worth noting are the occasional cases of GBM that show only scattered cells with EGFR amplification yet, by both histology and CEP7 hyperploidy, clearly contain many neoplastic cells (Figure 2). Recent work has shown that such cases have heterogeneous amplification of other receptor tyrosine kinases besides EGFR, including MET and PDGFRA.80 Such patterns of amplification could explain why anti-EGFR therapies have been disappointing in EGFR-amplified GBMs.
Figure 2.

Unusual EGFR amplification pattern in a GBM. By FISH, EGFR amplification in tumor nuclei is readily appreciated as an excess of red EGFR probe signals relative to green centromeric enumeration probe 7 signals. However, in this case the surrounding nuclei are also neoplastic, as evidenced by trisomy 7 (arrowheads).
Before doing EGFR FISH an effective initial screen is EGFR immunohistochemistry; gliomas that are negative-to-weak do not need FISH since amplification in such cases is extremely rare.81 This same immunostain is also quite useful in oligodendrogliomas, with moderate-to-strong expression being an unfavorable prognostic marker in grade II oligodendrogliomas and, paradoxically, a favorable marker in grade III anaplastic oligodendrogliomas even after adjusting for 1p/19q status.81–83
Targeted therapy for glioblastoma with EGFR amplification
As mentioned, anti-EGFR drugs have been tried in GBMs.84–86 Results have been mixed thus far, not only because of the heterogeneous nature of receptor tyrosine kinase amplification, but also in part because alterations in other downstream proteins can thwart EGFR inhibition (e.g. loss of PTEN and/or high pAkt).85, 87 As of now there is no data to suggest that GBMs with or without EGFR amplification should be treated differently. For the time being, then, EGFR testing is solely for diagnostic (and perhaps prognostic) purposes.
IDH1/2: Metabolism’s never been hotter
One of the most recent and exciting events in neuro-oncology has been the discovery of mutations in isocitrate dehydrogenase types 1 and 2 (IDH1/2) in gliomas, found via whole-genome sequencing in GBMs.88 Both IDH1 and IDH2 convert isocitrate to alpha-ketoglutarate, in the process reducing nicotinamide adenine dinucleotide phosphate to NADPH. Mutations in these enzymes are clinically remarkable for many reasons.
First, IDH1 or IDH2 mutations are present in about 80% of astrocytomas and oligodendrogliomas, and in 15% of GBMs (practically all of which are secondary GBMs).88–90
Second, they are always point mutations, restricted to the arginine codons encoding isocitrate binding sites for both enzymes (R132 in IDH1 and R172 in IDH2, with very rare R100, R49, and G97 mutations in IDH1).91, 92
Third, only one of the two alleles is mutated in a tumor, with the other allele remaining wild-type.
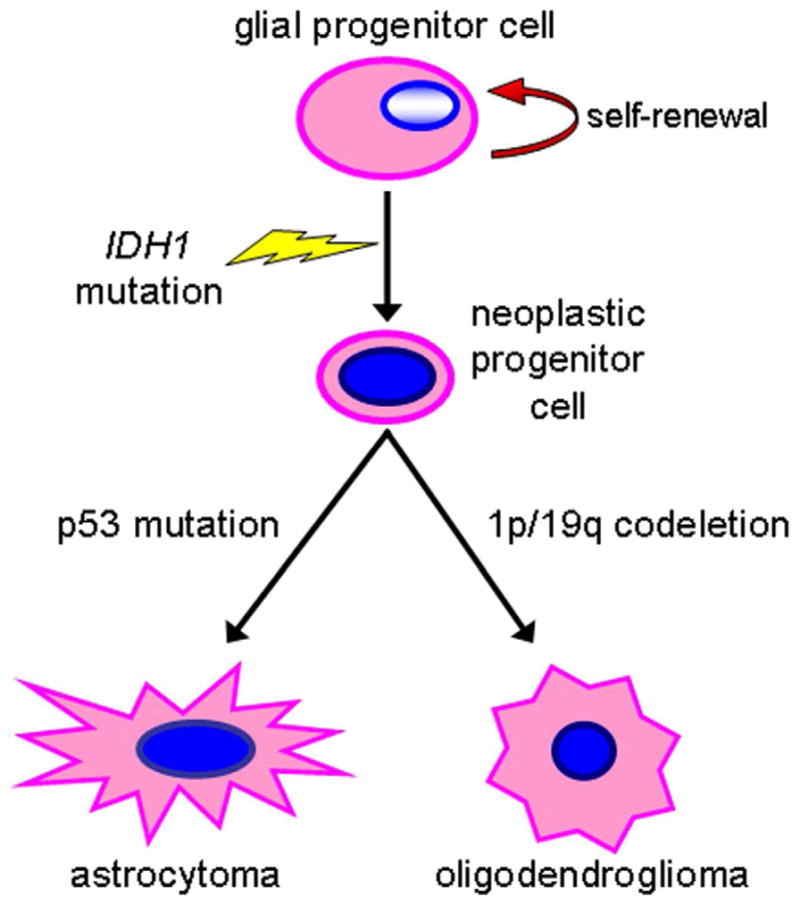
Fourth, the similarly high rate of IDH1/2 mutation in both astrocytic and oligodendroglial tumors, and its tight association with TP53 mutations in the former and 1p/19q codeletion in the latter, suggests that it is an early step in gliomagenesis, perhaps in a glial cell precursor (Figure 3).31, 88, 93–96
Fifth, these mutations appear to be relatively specific to gliomas versus most other solid neoplasms,89, 97, 98 though about half of all cartilaginous tumors, some intrahepatic cholangiocarcinomas, a minority of melanomas and thyroid carcinomas, and very rare prostate and colon cancers also contain IDH1/2 mutations.99–105
Finally, high-grade gliomas that harbor IDH1/2 mutations are less aggressive than their grade-matched wild-type counterparts.88, 89, 95, 106–109 In fact, IDH1/2 mutation status may be the most powerful independent molecular prognostic marker identified to date in high-grade gliomas,22, 110 though whether it is the most powerful marker has been disputed.96, 111 One study even showed that grade IV GBMs with IDH1/2 mutations fared better than grade III anaplastic astrocytomas without the mutation, and that this mutation, preferentially occurring in younger adults, accounts for the well-known correlation of younger patient age with longer survival.112 In contrast, 15 to 30% of acute myeloid leukemias have IDH1/2 mutations, with either no effect or an adverse effect on prognosis.101, 113–118
Figure 3.
IDH1/2 mutations are early events in gliomas. The presence of IDH1 (or IDH2) mutations in most WHO grade II and III astrocytomas and oligodendrogliomas suggests that they arise in glial progenitors, with p53 mutation or 1p/19q codeletion occurring at a later step.
IDH1/2 mutations are practically always heterozygous, meaning that there is always one wild-type allele in the tumor. Likewise, mutation of both IDH1 and IDH2 in the same tumor is exceedingly rare.119 These findings indicate a dominant gain-of-function, which was proven when studies showed that mutant IDH1 enzyme no longer performs its regular function, but instead converts alpha-ketoglutarate to D-2-hydroxyglutarate (D-2-HG), which is normally present in only trace amounts but is greatly increased in mutant gliomas.120–122 While the full consequences of D-2-HG are still being characterized (and beyond the scope of this discussion), one noteworthy effect appears to be promotion of a hypermethylator profile by competitively inhibiting alpha-ketoglutarate-dependent demethylases.123–128
Interestingly, there exist very rare organic acidurias that usually contain defects in D- or L-2-HG dehydrogenase, with consequent elevation of D-2-HG or L-2-HG, respectively. Both types of acidurias include seizures and severe retardation in their phenotypes, but only L-2-HG aciduria patients have developed brain tumors, and not all are gliomas.129 Even more provocative was the finding that D-2-HG aciduria is sometimes caused by a germline IDH2 mutation at R140, with the same biochemical result as the R172 mutation, yet to date no D-2-HG aciduria patients have been diagnosed with any kind of cancer130 other than chondromatosis.131 Along those lines, it is remarkable that somatic mosaic mutations in IDH1/2 have recently been shown to be present in Ollier disease and Mafucci syndrome.132
IDH1/2 mutations for differentiation among tumors
The role of IDH1/2 mutations and D-2-HG in gliomas will obviously be the subject of intense investigation for quite some time, but testing for the mutation has immediate practical relevance in surgical neuropathology. Because of its relative specificity in gliomas, it can be used to differentiate between reactive gliosis and an undersampled diffusely infiltrative glioma,79, 133–135 as well as to distinguish an incurable diffusely infiltrative glioma from a potentially curable mimicker like ganglioglioma or dysembryoplastic neuroepithelial tumor.136, 137 It has been proposed to help distinguish between a diffusely infiltrative glioma and a PA, the latter being negative for IDH1/2 mutations but usually positive for BRAF mutations or rearrangements (see below).138 However, since IDH1/2 mutations are rare in diffuse gliomas from preadolescent patients,139 and the rate of BRAF fusions in PAs to declines with patient age,140 the utility of dual IDH/BRAF testing to differentiate the two is unclear. Another topic of ambiguity is the prognostic power of IDH1/2 mutations in grade II gliomas; some have shown the mutations to be favorable markers,141 but others have not.96, 142–144 Considering that true grade I gliomas do not have IDH1/2 mutations, one wonders whether some gliomas that appear to be grade II histologically but are IDH1/2 wild-type might actually represent “overgraded” grade I gliomas. If so, inclusion of such cases in survival analyses of wild-type grade II gliomas would mask the prognostic effects of IDH1/2 mutations in true diffusely infiltrative grade II gliomas.145 At this point, it remains a matter of speculation.
Tests for IDH1/2 mutation
The gold standard for IDH1/2 mutation testing is obviously PCR amplification and sequencing of the tumor, which is quite feasible in routine FFPE tissues.134 But while PCR and conventional Sanger sequencing will detect as little as 20% mutant alleles in a specimen, real-time PCR with fluorescence melting curve analysis (RT-PCR/FMCA) is faster and twice as sensitive.135 The latter technique is predicated on the fact that even just one base pair mismatch between the fluorescent probe and the target sequence will lower the PCR product melting temperature, which can be detected by a change in fluorescence. In the case of IDH-mutant gliomas, two melting peaks will be produced, a higher peak for the wild-type allele and a lower peak for the mutant allele.
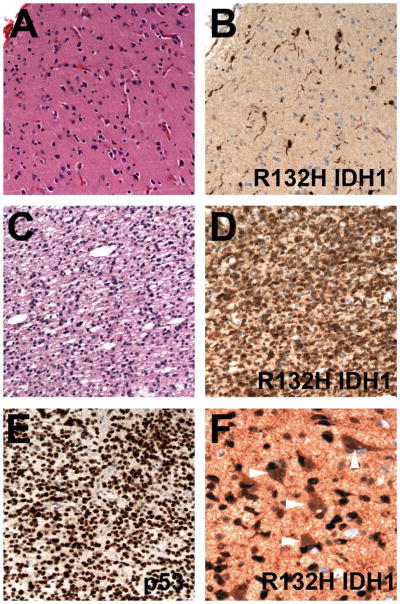
However, the development of an R132H-specific IDH1 mutant antibody has already rendered upfront PCR/sequencing unnecessary, to be used only if the immunostain is negative or equivocal. This is because about 95% of all IDH1/2 mutations are in IDH1, and among those over 90% are arginine-to-histidine R132H, making an R132H-specific antibody an excellent screening test.137, 146–148 So far a couple of monoclonal antibodies exist, with comparable efficacy.149 One that is commercially available is very specific and robust in FFPE tissues, although at times the background staining can be rather high, including occasionally prominent edge artifact. Also, some cells can show spurious reactivity, most notably neurons and red blood cells (Figure 4); such artifacts are generally readily discerned from true positivity. In the author’s experience, a true positive reaction in an immunoperoxidase system should be solid brown staining, including clear visualization of tumor cell processes. (In fact, this author never appreciated just how long glioma cell processes could grow in vivo before looking at R132H antibody results.) Sometimes staining heterogeneity is seen, where some areas are nicely positive while other areas of obvious tumor are negative; such cases are still always positive for the R132H IDH1 mutation on sequencing.149
Figure 4.
R132H IDH1 immunohistochemistry. (A) In a 30 year-old male with a right frontal brain lesion, superficial cortical tissue is nondiagnostic for infiltrating glioma. (B) R132H IDH1 immunostain, however, highlights scattered tumor cells with long processes. (C) Deeper areas of the same tissue showed unequivocal anaplastic astrocytoma, which was also positive for R132H IDH1 (D) as well as having strong nuclear p53 immunostaining (E). In a different case, neurons show a fair amount of nonspecific staining (arrowheads) but are still readily distinguished from infiltrating glioma cells that are strongly positive for R132H IDH1.
The practical aspects of screening all gliomas and suspected gliomas via R132H IDH1 antibodies are obvious:
Faster turnaround time
Lower cost
Ability to detect just a few scattered positive cells that even the most sensitive PCR tests would miss
Thus, upfront R132H IDH1 immunostaining is now done routinely on all our gliomas, including suboptimal biopsies suspected of harboring glioma cells. Any case that is immunonegative or equivocal is reflex-tested for less common IDH1 and IDH2 mutations by RT-PCR/FMCA. And although IDH1/2-mutations are often thought to be restricted to adult patients, as mentioned, they can be seen in some gliomas from pediatric patients.139 Testing for IDH1/2 mutations is therefore advisable on all infiltrative gliomas or suspected gliomas, especially in patients approximately age 14 years and up.
BRAF rearrangement and V600E in pilocytic astrocytomas: Six of one, half dozen of the other?
Just like the discovery of IDH1/2 mutations, cutting-edge molecular technologies have shed additional light on PA biology. Conventional comparative genomic hybridization showed no large cytogenetic abnormalities in most of these tumors,150 but array comparative genomic hybridization identified a biologically active, low-level copy number gain of the BRAF gene on 7q34 in a significant proportion of PAs.151, 152 BRAF is part of the raf/mil family of serine/threonine protein kinases. It is a proto-oncogene that participates in the MAPK/ERK signaling pathway, which regulates cellular differentiation, proliferation, and migration, among other things.153 In PAs the BRAF copy gain is due to a tandem duplication producing a KIAA1549:BRAF fusion protein with loss of the Ras-binding domain on BRAF,154, 155 though a minority of cases can involve RAF1 fusion with SRGAP or FAM131B.154, 156, 157 Either way the Ras-binding domain in BRAF is lost, resulting in constitutive BRAF activity.
While constitutive BRAF activity is sufficient to induce PA-like tumors in xenografted mice, it eventually leads to tumor senescence.158–161 In contrast, WHO grades II-IV gliomas also have some form of MAPK activation, but in those tumors it is accompanied by impairment of p53/Rb cell cycle pathways, which might explain why those higher-grade tumors undergo progression rather than senescence. In fact, loss of p16 in PAs may correlate with higher risk of aggressive behavior.158, 162
BRAF fusions are present in nearly 70% of all PAs, compared to about 15% of all other low-grade gliomas in the differential (e.g. diffuse grade II astrocytoma, ganglioglioma, etc.).151, 152, 160, 163–167 They are hardly ever seen in high-grade pediatric gliomas.165, 167 There is a difference by location, as BRAF fusions are seen in approximately 75 to 80% of cerebellar PAs and 55% of noncerebellar (mostly supratentorial) PAs.82, 151, 152, 157, 160, 163–165, 167 The frequency of BRAF fusions appears to decline with patient age, from about 80% in the first decade to 50% in the second decade, with less than 10% of PAs in patients over 40 having the fusion.140 About half of all pilomyxoid astrocytomas contain BRAF fusions,160, 163 underscoring the link between the two tumors but making a distinction based on BRAF fusion impossible.
Another BRAF alteration is the constitutively active V600E point mutation, previously known to be important in melanomas, colonic adenocarcinomas, and papillary thyroid carcinomas. It is present in roughly 15% of grade II–IV diffusely infiltrative pediatric astrocytomas168–170 but less than 2% of comparable adult gliomas.169, 171–173 In both children and adults about 10% of PAs have the mutation (but only 2% of cerebellar tumors).152, 157, 160, 163, 165, 167, 169 Sometimes the mutation can occur in conjunction with a BRAF fusion in the same tumor.160, 162 Although present in tumors with PA-like morphology, V600E is more common in other tumors in the PA differential, including 25% of pediatric and adult gangliogliomas and 80% of pleomorphic xanthoastrocytomas.160, 167, 169, 173, 174
Thus, over 75% of sporadic PAs have some sort of BRAF alteration, compared to about 40 to 50% of all other pooled tumors in the PA differential. Less than 10% of NF1-associated PAs contain a BRAF fusion or mutation, but such cases do occur.157, 160, 162, 175–177 From a diagnostics standpoint the presence of a BRAF fusion is therefore most suggestive of a low-grade glioma, especially a PA, but whether it can be used to definitively prove PA is still unclear. In contrast, detecting a V600E mutation is more associated with other tumors in the PA differential, but again cannot be used to definitively prove one type of tumor versus another.
From an outcome-based perspective, recent work showed that supratentorial low-grade pediatric gliomas with BRAF fusion usually behave as typical grade I PAs regardless of what they look like under the microscope or whether they are completely resected, whereas PAs without BRAF fusion are more likely to behave like grade II diffuse astrocytomas.160 A similar trend was seen in PAs in the cerebellum.164 In another study, 6 of 118 WHO grade II diffuse astrocytomas and oligodendrogliomas in children had a BRAF fusion; all 6 tumors were in the posterior fossa of pediatric patients and were extremely indolent despite their grade II microscopic appearance.165 However, two studies that included pediatric low-grade gliomas from both the supratentorium and posterior fossa did not show BRAF fusion to be an independent prognostic variable on multivariate analysis.162, 178 Nevertheless, while there is not yet a consensus on the prognostic impact of BRAF fusion in gliomas, as of now it appears to be at least a neutral biomarker, and perhaps even a favorable marker in certain contexts. In contrast, data on BRAF V600E mutations and outcome are very sparse. Our recent work comparing both types of BRAF alterations in the same cohort of pediatric low-grade gliomas suggested that there is a trend toward divergence in prognosis—i.e. BRAF fusions tend to be associated with longer PFS, while BRAF V600E mutations suggest shorter PFS. However, both markers pale in comparison to tumor location, and by extension the degree of tumor resection, as independent prognostic variables.162
Regarding BRAF-specific treatments, clinical trials aimed at blocking the downstream effects of BRAF fusion via MEK inhibition are still in accrual as of the time of this writing. Likewise, comparable anti V600E clinical trials in low grade pediatric gliomas are still in progress, but if animal model data and experience with melanomas is any indication, such therapies are worth pursuing.170, 179
BRAF fusion and mutation detection
Detection of BRAF fusion is feasible via several strategies, including RT-PCR breakpoint analysis, fusion transcript RT-PCR assay, and a three-probe FISH cocktail that encompasses the entire BRAF gene (Figure 5A).180, 181 Using this cocktail, a normal cell will show two signals while a cell with BRAF rearrangement will also show a third (often smaller) signal near one of the other signals, corresponding to the additional activation segment and kinase domain 164(Figure 5B). Such a pattern should be clearly seen in at least 10% of tumor cells138, 164 and will detect both common and uncommon rearrangements.182 A two-color probe cocktail containing a green probe (KIAA-1549) and red probe (BRAF RP4-726N20) has also been used, with a positive signal being yellow and indicating fusion of red and green signals.138, 160 Although there is good correlation between BRAF FISH, RT-PCR breakpoint analysis, and array CGH,160 from personal experience the detection of BRAF fusion via FISH is visually challenging, especially if the tumor has high overall ploidy. At this time the optimal method is probably RT-PCR aimed at detecting breakpoints or fusion transcript.165 FISH could still be used if there is not enough tissue for PCR analysis.
Figure 5.
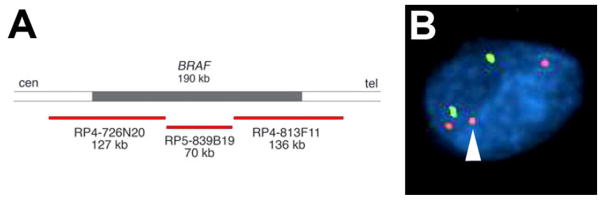
BRAF rearrangement can be detected by FISH. (A) Schematic of a three-probe cocktail spanning the entire BRAF gene [adapted with permission from Ciampi et al,, J Clin Invest. Jan 2005;115(1):94–101]. (B) Abnormal BRAF is detected by the presence of an excess red signal relative to centromeric enumeration probe 7 (green).
Naturally the standard way to detect BRAF V600E point mutation is via PCR and sequencing, but recently the same group that developed the aforementioned R132H IDH1-specific antibody has produced a V600E BRAF-specific antibody.183 While the antibody will not work in tissues subjected to surgical coagulation or pathologic freezing, and staining intensity varies greatly between mutated cases, it does appear promising as a first-line screening test.
When to test for BRAF
In summary, testing for BRAF fusion and mutation is recommended in cases where the differential is between a PA and one of its mimickers, especially if there is concern that the tumor could be a grade II–IV diffusely infiltrative glioma. The presence of the fusion or point mutation, however, might not unequivocally diagnose one type of tumor versus another, and is less useful in non-pediatric patients. Similarly, the absence of a BRAF abnormality does not rule out a grade I glioma. Reflex testing of all gliomas for BRAF alterations is therefore not yet recommended, though this could quickly change depending on additional data, especially clinical trials.
Final thoughts on molecular pathology for gliomas
Strictly speaking there is no predictive molecular biomarker in gliomas because, unlike Herceptin and HER2 in breast cancer, no therapy has yet been developed that is only effective against tumors with a particular marker. But in certain circumstances, testing for 1p/19q codeletion, EGFR amplification, IDH1/2 mutations, and/or BRAF alterations can help refine the patient’s diagnosis and prognosis (Table 1, Figure 6). Wise and cost-effective use of these markers is still predicated on histologic evaluation of the tissue, as is WHO grading. Yet based on the rates of EGFR amplification and IDH1/2 mutations in the adult population, as well as the frequencies of grades II–IV gliomas, tumor cells can now be detected in about 60% of suboptimally-biopsied diffusely infiltrative gliomas, even in cases where the diagnosis of glioma cannot be made based on light microscopy alone.49, 79, 133, 134
Figure 6.

Example of a possible diagnostic algorithm incorporating molecular biomarkers in the workup of gliomas. Judicious use of 1p/19q testing, EGFR FISH, IDH1/2 mutation screening, and BRAF analysis requires a logical series of decisions based on clinical and histologic factors. In most circumstances the best use of molecular tests are to support a favored diagnosis or help the neuropathologist get “off the fence” in difficult cases. The tests can also refine prognosis or detect scattered glioma cells in undersampled tumors. Of note, patient age plays a large role in determining which tests are likely to help (see text for details). See Table 1 legend for abbreviations.
Array-based technologies that screen for copy number aberrations across the entire genome are likely to gain in popularity as their cost goes down and their ability to interpret FFPE tissues goes up.184 FFPE-based platforms for screening mutations in multiple genes is also promising.185 Implementation of both copy number and mutation platforms may someday obviate the need for parsimonious, sequential targeting of specific biomarkers. But for the foreseeable future, and especially when tissue is inadequate for arrays, microscopic analysis of the venerable H&E-stained slide will be essential for selective testing of specific molecular biomarkers, both old standbys like 1p/19q and EGFR as well as new ones like IDH1/2 and BRAF.
Acknowledgments
The author wishes to thank Clayton Wiley, M.D., Ph.D., for access to the virtual slides, and Marina Nikiforova, M.D., for her assistance with Figure 1A.
Footnotes
Disclosures/Conflicts of Interest: The author has nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 2005 Jun;64(6):479–489. doi: 10.1093/jnen/64.6.479. [DOI] [PubMed] [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. In: WHO Classification of Tumors of the Central Nervous System. 4. Ohgaki H, editor. Lyon: IARC; 2007. World Health Organization Classification of Tumors. [Google Scholar]
- 3.Campbell JW, Pollack IF. Cerebellar astrocytomas in children. J Neurooncol. 1996 May-Jun;28(2–3):223–231. doi: 10.1007/BF00250201. [DOI] [PubMed] [Google Scholar]
- 4.Johnson MW, Eberhart CG, Perry A, et al. Spectrum of pilomyxoid astrocytomas: intermediate pilomyxoid tumors. Am J Surg Pathol. 2011 Dec;34(12):1783–1791. doi: 10.1097/PAS.0b013e3181fd66c3. [DOI] [PubMed] [Google Scholar]
- 5.Miller CR, Dunham CP, Scheithauer BW, Perry A. Significance of necrosis in grading of oligodendroglial neoplasms: a clinicopathologic and genetic study of newly diagnosed high-grade gliomas. J Clin Oncol. 2006 Dec 1;24(34):5419–5426. doi: 10.1200/JCO.2006.08.1497. [DOI] [PubMed] [Google Scholar]
- 6.Reifenberger J, Reifenberger G, Liu L, James CD, Wechsler W, Collins VP. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol. 1994 Nov;145(5):1175–1190. [PMC free article] [PubMed] [Google Scholar]
- 7.Kraus JA, Koopmann J, Kaskel P, et al. Shared allelic losses on chromosomes 1p and 19q suggest a common origin of oligodendroglioma and oligoastrocytoma. J Neuropathol Exp Neurol. 1995 Jan;54(1):91–95. doi: 10.1097/00005072-199501000-00011. [DOI] [PubMed] [Google Scholar]
- 8.Jenkins RB. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006;66:9852–9861. doi: 10.1158/0008-5472.CAN-06-1796. [DOI] [PubMed] [Google Scholar]
- 9.Griffin CA. Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol. 2006;65:988–994. doi: 10.1097/01.jnen.0000235122.98052.8f. [DOI] [PubMed] [Google Scholar]
- 10.Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998 Oct 7;90(19):1473–1479. doi: 10.1093/jnci/90.19.1473. [DOI] [PubMed] [Google Scholar]
- 11.Smith JS, Perry A, Borell TJ, et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol. 2000 Feb;18(3):636–645. doi: 10.1200/JCO.2000.18.3.636. [DOI] [PubMed] [Google Scholar]
- 12.Perry A, Fuller CE, Banerjee R, Brat DJ, Scheithauer BW. Ancillary FISH analysis for 1p and 19q status: preliminary observations in 287 gliomas and oligodendroglioma mimics. Front Biosci. 2003 Jan 1;8(8):a1–9. doi: 10.2741/896. [DOI] [PubMed] [Google Scholar]
- 13.van den Bent MJ, Looijenga LH, Langenberg K, et al. Chromosomal anomalies in oligodendroglial tumors are correlated with clinical features. Cancer. 2003 Mar 1;97(5):1276–1284. doi: 10.1002/cncr.11187. [DOI] [PubMed] [Google Scholar]
- 14.Fallon KB. Prognostic value of 1p, 19q, 9p, 10q, and EGFR-FISH analyses in recurrent oligodendrogliomas. J Neuropathol Exp Neurol. 2004;63:314–322. doi: 10.1093/jnen/63.4.314. [DOI] [PubMed] [Google Scholar]
- 15.Felsberg J, Erkwoh A, Sabel MC, et al. Oligodendroglial tumors: refinement of candidate regions on chromosome arm 1p and correlation of 1p/19q status with survival. Brain Pathol. 2004 Apr;14(2):121–130. doi: 10.1111/j.1750-3639.2004.tb00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kouwenhoven MC, Gorlia T, Kros JM, et al. Molecular analysis of anaplastic oligodendroglial tumors in a prospective randomized study: A report from EORTC study 26951. Neuro Oncol. 2009 Dec;11(6):737–746. doi: 10.1215/15228517-2009-011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weller M, Berger H, Hartmann C, et al. Combined 1p/19q loss in oligodendroglial tumors: predictive or prognostic biomarker? Clin Cancer Res. 2007 Dec 1;13(23):6933–6937. doi: 10.1158/1078-0432.CCR-07-0573. [DOI] [PubMed] [Google Scholar]
- 18.Giannini C, Burger PC, Berkey BA, et al. Anaplastic oligodendroglial tumors: refining the correlation among histopathology, 1p 19q deletion and clinical outcome in Intergroup Radiation Therapy Oncology Group Trial 9402. Brain Pathol. 2008 Jul;18(3):360–369. doi: 10.1111/j.1750-3639.2008.00129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDonald JM, See SJ, Tremont IW, et al. The prognostic impact of histology and 1p/19q status in anaplastic oligodendroglial tumors. Cancer. 2005 Oct 1;104(7):1468–1477. doi: 10.1002/cncr.21338. [DOI] [PubMed] [Google Scholar]
- 20.Eoli M, Bissola L, Bruzzone MG, et al. Reclassification of oligoastrocytomas by loss of heterozygosity studies. Int J Cancer. 2006 Jul 1;119(1):84–90. doi: 10.1002/ijc.21759. [DOI] [PubMed] [Google Scholar]
- 21.Fuller CE, Schmidt RE, Roth KA, et al. Clinical utility of fluorescence in situ hybridization (FISH) in morphologically ambiguous gliomas with hybrid oligodendroglial/astrocytic features. J Neuropathol Exp Neurol. 2003 Nov;62(11):1118–1128. doi: 10.1093/jnen/62.11.1118. [DOI] [PubMed] [Google Scholar]
- 22.Wick W, Hartmann C, Engel C, et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol. 2009 Dec 10;27(35):5874–5880. doi: 10.1200/JCO.2009.23.6497. [DOI] [PubMed] [Google Scholar]
- 23.Gupta M, Djalilvand A, Brat DJ. Clarifying the diffuse gliomas: an update on the morphologic features and markers that discriminate oligodendroglioma from astrocytoma. Am J Clin Pathol. 2005 Nov;124(5):755–768. doi: 10.1309/6JNX-4PA6-0TQ5-U5VG. [DOI] [PubMed] [Google Scholar]
- 24.Homma T, Fukushima T, Vaccarella S, et al. Correlation among pathology, genotype, and patient outcomes in glioblastoma. J Neuropathol Exp Neurol. 2006 Sep;65(9):846–854. doi: 10.1097/01.jnen.0000235118.75182.94. [DOI] [PubMed] [Google Scholar]
- 25.Houillier C, Lejeune J, Benouaich-Amiel A, et al. Prognostic impact of molecular markers in a series of 220 primary glioblastomas. Cancer. 2006 May 15;106(10):2218–2223. doi: 10.1002/cncr.21819. [DOI] [PubMed] [Google Scholar]
- 26.Fouladi M, Helton K, Dalton J, et al. Clear cell ependymoma: a clinicopathologic and radiographic analysis of 10 patients. Cancer. 2003 Nov 15;98(10):2232–2244. doi: 10.1002/cncr.11783. [DOI] [PubMed] [Google Scholar]
- 27.Tong CY, Ng HK, Pang JC, Hu J, Hui AB, Poon WS. Central neurocytomas are genetically distinct from oligodendrogliomas and neuroblastomas. Histopathology. 2000 Aug;37(2):160–165. doi: 10.1046/j.1365-2559.2000.00977.x. [DOI] [PubMed] [Google Scholar]
- 28.Makuria AT, Henderson FC, Rushing EJ, Hartmann DP, Azumi N, Ozdemirli M. Oligodendroglioma with neurocytic differentiation versus atypical extraventricular neurocytoma: a case report of unusual pathologic findings of a spinal cord tumor. J Neurooncol. 2007 Apr;82(2):199–205. doi: 10.1007/s11060-006-9268-0. [DOI] [PubMed] [Google Scholar]
- 29.Mrak RE, Yasargil MG, Mohapatra G, Earel J, Jr, Louis DN. Atypical extraventricular neurocytoma with oligodendroglioma-like spread and an unusual pattern of chromosome 1p and 19q loss. Hum Pathol. 2004 Sep;35(9):1156–1159. doi: 10.1016/j.humpath.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 30.Perry A, Scheithauer BW, Macaulay RJ, Raffel C, Roth KA, Kros JM. Oligodendrogliomas with neurocytic differentiation. A report of 4 cases with diagnostic and histogenetic implications. J Neuropathol Exp Neurol. 2002 Nov;61(11):947–955. doi: 10.1093/jnen/61.11.947. [DOI] [PubMed] [Google Scholar]
- 31.Labussiere M, Idbaih A, Wang XW, et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology. 2010 Jun 8;74(23):1886–1890. doi: 10.1212/WNL.0b013e3181e1cf3a. [DOI] [PubMed] [Google Scholar]
- 32.van den Bent MJ, Dubbink HJ, Marie Y, et al. IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res. 2010 Mar 1;16(5):1597–1604. doi: 10.1158/1078-0432.CCR-09-2902. [DOI] [PubMed] [Google Scholar]
- 33.Ducray F, Idbaih A, de Reynies A, et al. Anaplastic oligodendrogliomas with 1p19q codeletion have a proneural gene expression profile. Mol Cancer. 2008;7(41):41. doi: 10.1186/1476-4598-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yip S, Butterfield YS, Morozova O, et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol. 2012 Jan;226(1):7–16. doi: 10.1002/path.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Idbaih A, Marie Y, Lucchesi C, et al. BAC array CGH distinguishes mutually exclusive alterations that define clinicogenetic subtypes of gliomas. Int J Cancer. 2008 Apr 15;122(8):1778–1786. doi: 10.1002/ijc.23270. [DOI] [PubMed] [Google Scholar]
- 36.Idbaih A, Dalmasso C, Kouwenhoven M, et al. Genomic aberrations associated with outcome in anaplastic oligodendroglial tumors treated within the EORTC phase III trial 26951. J Neurooncol. 2011 Jun;103(2):221–230. doi: 10.1007/s11060-010-0380-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zlatescu MC, TehraniYazdi A, Sasaki H, et al. Tumor location and growth pattern correlate with genetic signature in oligodendroglial neoplasms. Cancer Res. 2001 Sep 15;61(18):6713–6715. [PubMed] [Google Scholar]
- 38.Goze C, Rigau V, Gibert L, Maudelonde T, Duffau H. Lack of complete 1p19q deletion in a consecutive series of 12 WHO grade II gliomas involving the insula: a marker of worse prognosis? J Neurooncol. 2009 Jan;91(1):1–5. doi: 10.1007/s11060-008-9680-8. [DOI] [PubMed] [Google Scholar]
- 39.Wu A, Aldape K, Lang FF. High rate of deletion of chromosomes 1p and 19q in insular oligodendroglial tumors. J Neurooncol. 2009 Aug;99(1):57–64. doi: 10.1007/s11060-009-0100-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raghavan R, Balani J, Perry A, et al. Pediatric oligodendrogliomas: a study of molecular alterations on 1p and 19q using fluorescence in situ hybridization. J Neuropathol Exp Neurol. 2003 May;62(5):530–537. doi: 10.1093/jnen/62.5.530. [DOI] [PubMed] [Google Scholar]
- 41.Kreiger PA, Okada Y, Simon S, Rorke LB, Louis DN, Golden JA. Losses of chromosomes 1p and 19q are rare in pediatric oligodendrogliomas. Acta Neuropathol. 2005 Apr;109(4):387–392. doi: 10.1007/s00401-004-0976-2. [DOI] [PubMed] [Google Scholar]
- 42.Snuderl M. Polysomy for chromosomes 1 and 19 predicts earlier recurrence in anaplastic oligodendrogliomas with concurrent 1p/19q loss. Clin Cancer Res. 2009;15:6430–6437. doi: 10.1158/1078-0432.CCR-09-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woehrer A, Sander P, Haberler C, et al. FISH-based detection of 1p 19q codeletion in oligodendroglial tumors: procedures and protocols for neuropathological practice - a publication under the auspices of the Research Committee of the European Confederation of Neuropathological Societies. Clin Neuropathol. 2011 Mar;30(2):47–55. doi: 10.5414/npp30047. [DOI] [PubMed] [Google Scholar]
- 44.Barbashina V, Salazar P, Holland EC, Rosenblum MK, Ladanyi M. Allelic losses at 1p36 and 19q13 in gliomas: correlation with histologic classification, definition of a 150-kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene. Clin Cancer Res. 2005;11:1119–1128. [PubMed] [Google Scholar]
- 45.Smith JS, Alderete B, Minn Y, et al. Localization of common deletion regions on 1p and 19q in human gliomas and their association with histological subtype. Oncogene. 1999 Jul 15;18(28):4144–4152. doi: 10.1038/sj.onc.1202759. [DOI] [PubMed] [Google Scholar]
- 46.Horbinski C, Nikiforova MN, Hobbs J, et al. The importance of 10q status in an outcomes-based comparison between 1p/19q fluorescence in situ hybridization and polymerase chain reaction-based microsatellite loss of heterozygosity analysis of oligodendrogliomas. J Neuropathol Exp Neurol. 2012 Jan;71(1):73–82. doi: 10.1097/NEN.0b013e318240fa65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jha P, Sarkar C, Pathak P, et al. Detection of allelic status of 1p and 19q by microsatellite-based PCR versus FISH: limitations and advantages in application to patient management. Diagn Mol Pathol. 2011 Mar;20(1):40–47. doi: 10.1097/PDM.0b013e3181e961e9. [DOI] [PubMed] [Google Scholar]
- 48.Broholm H, Born PW, Guterbaum D, Dyrbye H, Laursen H. Detecting chromosomal alterations at 1p and 19q by FISH and DNA fragment analysis--a comparative study in human gliomas. Clin Neuropathol. 2008 Nov-Dec;27(6):378–387. doi: 10.5414/npp27378. [DOI] [PubMed] [Google Scholar]
- 49.Horbinski C. Practical molecular diagnostics in neuropathology: making a tough job a little easier. Semin Diagn Pathol. 2010 May;27(2):105–113. doi: 10.1053/j.semdp.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 50.Wong AJ, Bigner SH, Bigner DD, Kinzler KW, Hamilton SR, Vogelstein B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci U S A. 1987 Oct;84(19):6899–6903. doi: 10.1073/pnas.84.19.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nagane M, Coufal F, Lin H, Bogler O, Cavenee WK, Huang HJ. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res. 1996 Nov 1;56(21):5079–5086. [PubMed] [Google Scholar]
- 52.Chakravarti A, Chakladar A, Delaney MA, Latham DE, Loeffler JS. The epidermal growth factor receptor pathway mediates resistance to sequential administration of radiation and chemotherapy in primary human glioblastoma cells in a RAS-dependent manner. Cancer Res. 2002 Aug 1;62(15):4307–4315. [PubMed] [Google Scholar]
- 53.Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 2010 Sep;12(9):675–684. doi: 10.1593/neo.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li L, Dutra A, Pak E, et al. EGFRvIII expression and PTEN loss synergistically induce chromosomal instability and glial tumors. Neuro Oncol. 2009 Feb;11(1):9–21. doi: 10.1215/15228517-2008-081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mazzoleni S, Politi LS, Pala M, et al. Epidermal growth factor receptor expression identifies functionally and molecularly distinct tumor-initiating cells in human glioblastoma multiforme and is required for gliomagenesis. Cancer Res. 2010 Oct 1;70(19):7500–7513. doi: 10.1158/0008-5472.CAN-10-2353. [DOI] [PubMed] [Google Scholar]
- 56.Biernat W, Tohma Y, Yonekawa Y, Kleihues P, Ohgaki H. Alterations of cell cycle regulatory genes in primary (de novo) and secondary glioblastomas. Acta Neuropathol. 1997 Oct;94(4):303–309. doi: 10.1007/s004010050711. [DOI] [PubMed] [Google Scholar]
- 57.Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004 Oct 1;64(19):6892–6899. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 58.Ohgaki H, Kleihues P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009 Aug 6; doi: 10.1111/j.1349-7006.2009.01308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007 May;170(5):1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pollack IF, Hamilton RL, James CD, et al. Rarity of PTEN deletions and EGFR amplification in malignant gliomas of childhood: results from the Children’s Cancer Group 945 cohort. J Neurosurg. 2006 Nov;105(5 Suppl):418–424. doi: 10.3171/ped.2006.105.5.418. [DOI] [PubMed] [Google Scholar]
- 61.Aldape KD, Ballman K, Furth A, et al. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J Neuropathol Exp Neurol. 2004 Jul;63(7):700–707. doi: 10.1093/jnen/63.7.700. [DOI] [PubMed] [Google Scholar]
- 62.Smith JS, Tachibana I, Passe SM, et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst. 2001 Aug 15;93(16):1246–1256. doi: 10.1093/jnci/93.16.1246. [DOI] [PubMed] [Google Scholar]
- 63.Shinojima N, Tada K, Shiraishi S, et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003 Oct 15;63(20):6962–6970. [PubMed] [Google Scholar]
- 64.Fuller CE, Perry A. Fluorescence in situ hybridization (FISH) in diagnostic and investigative neuropathology. Brain Pathol. 2002;12:67–86. doi: 10.1111/j.1750-3639.2002.tb00424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bigner SH, Burger PC, Wong AJ, et al. Gene amplification in malignant human gliomas: clinical and histopathologic aspects. J Neuropathol Exp Neurol. 1988 May;47(3):191–205. doi: 10.1097/00005072-198805000-00001. [DOI] [PubMed] [Google Scholar]
- 66.Dehais C, Laigle-Donadey F, Marie Y, et al. Prognostic stratification of patients with anaplastic gliomas according to genetic profile. Cancer. 2006 Oct 15;107(8):1891–1897. doi: 10.1002/cncr.22211. [DOI] [PubMed] [Google Scholar]
- 67.Liu L, Backlund LM, Nilsson BR, et al. Clinical significance of EGFR amplification and the aberrant EGFRvIII transcript in conventionally treated astrocytic gliomas. J Mol Med. 2005 Nov;83(11):917–926. doi: 10.1007/s00109-005-0700-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perry A, Aldape KD, George DH, Burger PC. Small cell astrocytoma: an aggressive variant that is clinicopathologically and genetically distinct from anaplastic oligodendroglioma. Cancer. 2004 Nov 15;101(10):2318–2326. doi: 10.1002/cncr.20625. [DOI] [PubMed] [Google Scholar]
- 69.Idbaih A, Criniere E, Marie Y, et al. Gene amplification is a poor prognostic factor in anaplastic oligodendrogliomas. Neuro Oncol. 2008 Aug;10(4):540–547. doi: 10.1215/15228517-2008-022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gil-Benso R, Lopez-Gines C, Benito R, et al. Concurrent EGFR amplification and TP-53 mutation in glioblastomas. Clin Neuropathol. 2007 Sep-Oct;26(5):224–231. doi: 10.5414/npp26224. [DOI] [PubMed] [Google Scholar]
- 71.Benito R, Gil-Benso R, Quilis V, et al. Primary glioblastomas with and without EGFR amplification: relationship to genetic alterations and clinicopathological features. Neuropathology. 2009 Aug;30(4):392–400. doi: 10.1111/j.1440-1789.2009.01081.x. [DOI] [PubMed] [Google Scholar]
- 72.Diedrich U, Lucius J, Baron E, Behnke J, Pabst B, Zoll B. Distribution of epidermal growth factor receptor gene amplification in brain tumours and correlation to prognosis. J Neurol. 1995 Oct;242(10):683–688. doi: 10.1007/BF00866920. [DOI] [PubMed] [Google Scholar]
- 73.Korshunov A, Sycheva R, Golanov A. The prognostic relevance of molecular alterations in glioblastomas for patients age < 50 years. Cancer. 2005 Aug 15;104(4):825–832. doi: 10.1002/cncr.21221. [DOI] [PubMed] [Google Scholar]
- 74.Hobbs J, Nikiforova MN, Fardo DW, et al. Paradoxical Relationship Between the Degree of EGFR Amplification and Outcome in Glioblastomas. Am J Surg Pathol. 2012 Mar 31; doi: 10.1097/PAS.0b013e3182518e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lopez-Gines C, Gil-Benso R, Ferrer-Luna R, et al. New pattern of EGFR amplification in glioblastoma and the relationship of gene copy number with gene expression profile. Mod Pathol. 2010 Jun;23(6):856–865. doi: 10.1038/modpathol.2010.62. [DOI] [PubMed] [Google Scholar]
- 76.Birkbak NJ, Eklund AC, Li Q, et al. Paradoxical Relationship between Chromosomal Instability and Survival Outcome in Cancer. Cancer Res. 2011 May 15;71(10):3447–3452. doi: 10.1158/0008-5472.CAN-10-3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eley GD, Reiter JL, Pandita A, et al. A chromosomal region 7p11.2 transcript map: its development and application to the study of EGFR amplicons in glioblastoma. Neuro Oncol. 2002 Apr;4(2):86–94. doi: 10.1093/neuonc/4.2.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Park S, James CD. ECop (EGFR-coamplified and overexpressed protein), a novel protein, regulates NF-kappaB transcriptional activity and associated apoptotic response in an IkappaBalpha-dependent manner. Oncogene. 2005 Apr 7;24(15):2495–2502. doi: 10.1038/sj.onc.1208496. [DOI] [PubMed] [Google Scholar]
- 79.Camelo-Piragua S, Jansen M, Ganguly A, et al. A sensitive and specific diagnostic panel to distinguish diffuse astrocytoma from astrocytosis: chromosome 7 gain with mutant isocitrate dehydrogenase 1 and p53. J Neuropathol Exp Neurol. 2011 Feb;70(2):110–115. doi: 10.1097/NEN.0b013e31820565f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Snuderl M, Fazlollahi L, Le LP, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011 Dec 13;20(6):810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 81.Horbinski C, Hobbs J, Cieply K, Dacic S, Hamilton RL. EGFR Expression Stratifies Oligodendroglioma Behavior. Am J Pathol. 2011:11. doi: 10.1016/j.ajpath.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang H, Okamoto Y, Yokoo H, et al. Gene expression profiling and subgroup identification of oligodendrogliomas. Oncogene. 2004 Aug 5;23(35):6012–6022. doi: 10.1038/sj.onc.1207781. [DOI] [PubMed] [Google Scholar]
- 83.Zhou Y-H, Hess KR, Raj VR, et al. Establishment of Prognostic Models for Astrocytic and Oligodendroglial Brain Tumors with Standardized Quantification of Marker Gene Expression and Clinical Variables. Biomarker Insights. 2010;5:153–168. doi: 10.4137/BMI.S6167. 2420-BMI-Establishment-of-Prognostic-Models-for-Astrocytic-and-Oligodendroglial.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brandes AA, Franceschi E, Tosoni A, Hegi ME, Stupp R. Epidermal growth factor receptor inhibitors in neuro-oncology: hopes and disappointments. Clin Cancer Res. 2008 Feb 15;14(4):957–960. doi: 10.1158/1078-0432.CCR-07-1810. [DOI] [PubMed] [Google Scholar]
- 85.Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005 Nov 10;353(19):2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 86.Prados MD, Chang SM, Butowski N, et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J Clin Oncol. 2009 Feb 1;27(4):579–584. doi: 10.1200/JCO.2008.18.9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Haas-Kogan DA, Prados MD, Tihan T, et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005 Jun 15;97(12):880–887. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- 88.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008 Sep 26;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009 Feb 19;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008 Dec;116(6):597–602. doi: 10.1007/s00401-008-0455-2. [DOI] [PubMed] [Google Scholar]
- 91.Pusch S, Sahm F, Meyer J, Mittelbronn M, Hartmann C, von Deimling A. Glioma IDH1 mutation patterns off the beaten track. Neuropathol Appl Neurobiol. 2011 Jun;37(4):428–430. doi: 10.1111/j.1365-2990.2010.01127.x. [DOI] [PubMed] [Google Scholar]
- 92.Ward PS, Cross JR, Lu C, et al. Identification of additional IDH mutations associated with oncometabolite R(−)-2-hydroxyglutarate production. Oncogene. 2011 Sep 26;(26):416. doi: 10.1038/onc.2011.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ichimura K, Pearson DM, Kocialkowski S, et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 2009 Aug;11(4):341–347. doi: 10.1215/15228517-2009-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009 Apr;174(4):1149–1153. doi: 10.2353/ajpath.2009.080958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weller M, Felsberg J, Hartmann C, et al. Molecular Predictors of Progression-Free and Overall Survival in Patients With Newly Diagnosed Glioblastoma: A Prospective Translational Study of the German Glioma Network. J Clin Oncol. 2009 Oct 5; doi: 10.1200/JCO.2009.23.0805. [DOI] [PubMed] [Google Scholar]
- 96.Kim YH, Nobusawa S, Mittelbronn M, et al. Molecular classification of low-grade diffuse gliomas. Am J Pathol. 2010 Dec;177(6):2708–2714. doi: 10.2353/ajpath.2010.100680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bleeker FE, Lamba S, Leenstra S, et al. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat. 2009 Jan;30(1):7–11. doi: 10.1002/humu.20937. [DOI] [PubMed] [Google Scholar]
- 98.Park SW, Chung NG, Han JY, et al. Absence of IDH2 codon 172 mutation in common human cancers. Int J Cancer. 2009 Nov 15;125(10):2485–2486. doi: 10.1002/ijc.24647. [DOI] [PubMed] [Google Scholar]
- 99.Gaal J, Burnichon N, Korpershoek E, et al. Isocitrate Dehydrogenase Mutations Are Rare in Pheochromocytomas and Paragangliomas. Clin Endocrinol Metab. 2009 Nov 13; doi: 10.1210/jc.2009-2170. [DOI] [PubMed] [Google Scholar]
- 100.Kang MR, Kim MS, Oh JE, et al. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer. 2009 Jul 15;125(2):353–355. doi: 10.1002/ijc.24379. [DOI] [PubMed] [Google Scholar]
- 101.Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009 Sep 10;361(11):1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lopez GY, Reitman ZJ, Solomon D, et al. IDH1 mutation identified in human melanoma. Biochem Biophys Res Commun. 2010 Jul 2;:2. doi: 10.1016/j.bbrc.2010.06.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shibata T, Kokubu A, Miyamoto M, Sasajima Y, Yamazaki N. Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am J Pathol. 2011 Mar;178(3):1395–1402. doi: 10.1016/j.ajpath.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Amary MF, Bacsi K, Maggiani F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011 Apr 7;(7):2913. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 105.Borger DR, Tanabe KK, Fan KC, et al. Frequent Mutation of Isocitrate Dehydrogenase (IDH)1 and IDH2 in Cholangiocarcinoma Identified Through Broad-Based Tumor Genotyping. Oncologist. 2011 Dec 16;:16. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dubbink HJ, Taal W, van Marion R, et al. IDH1 mutations in low-grade astrocytomas predict survival but not response to temozolomide. Neurology. 2009 Nov 24;73(21):1792–1795. doi: 10.1212/WNL.0b013e3181c34ace. [DOI] [PubMed] [Google Scholar]
- 107.Wick W, Hartmann C, Engel C, et al. NOA-04 Randomized Phase III Trial of Sequential Radiochemotherapy of Anaplastic Glioma With Procarbazine, Lomustine, and Vincristine or Temozolomide. J Clin Oncol. 2009 Nov 9; doi: 10.1200/JCO.2009.23.6497. [DOI] [PubMed] [Google Scholar]
- 108.Sanson M, Marie Y, Paris S, et al. Isocitrate Dehydrogenase 1 Codon 132 Mutation Is an Important Prognostic Biomarker in Gliomas. J Clin Oncol. 2009 Jul 27; doi: 10.1200/JCO.2009.21.9832. [DOI] [PubMed] [Google Scholar]
- 109.Nobusawa S, Watanabe T, Kleihues P, Ohgaki H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res. 2009 Oct 1;15(19):6002–6007. doi: 10.1158/1078-0432.CCR-09-0715. [DOI] [PubMed] [Google Scholar]
- 110.Sanson M, Marie Y, Paris S, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009 Sep 1;27(25):4150–4154. doi: 10.1200/JCO.2009.21.9832. [DOI] [PubMed] [Google Scholar]
- 111.Ichimura K. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 2009;11:341–347. doi: 10.1215/15228517-2009-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hartmann C, Hentschel B, Wick W, et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol. 2010 Nov 19;:19. doi: 10.1007/s00401-010-0781-z. [DOI] [PubMed] [Google Scholar]
- 113.Abbas S, Lugthart S, Kavelaars FG, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010 Sep 23;116(12):2122–2126. doi: 10.1182/blood-2009-11-250878. [DOI] [PubMed] [Google Scholar]
- 114.Chou WC, Hou HA, Chen CY, et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood. 2010 Apr 8;115(14):2749–2754. doi: 10.1182/blood-2009-11-253070. [DOI] [PubMed] [Google Scholar]
- 115.Green CL, Evans CM, Hills RK, Burnett AK, Linch DC, Gale RE. The prognostic significance of IDH1 mutations in younger adult patients with acute myeloid leukemia is dependent on FLT3/ITD status. Blood. 2010 Jul 22;2010:22. doi: 10.1182/blood-2010-02-270926. [DOI] [PubMed] [Google Scholar]
- 116.Thol F, Damm F, Wagner K, et al. Prognostic impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood. 2010 Jul 29;116(4):614–616. doi: 10.1182/blood-2010-03-272146. [DOI] [PubMed] [Google Scholar]
- 117.Boissel N, Nibourel O, Renneville A, et al. Prognostic Impact of Isocitrate Dehydrogenase Enzyme Isoforms 1 and 2 Mutations in Acute Myeloid Leukemia: A Study by the Acute Leukemia French Association Group. J Clin Oncol. 2010 Jul 12;:12. doi: 10.1200/JCO.2010.28.2285. [DOI] [PubMed] [Google Scholar]
- 118.Marcucci G, Maharry K, Wu YZ, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010 May 10;28(14):2348–2355. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hartmann C, Meyer J, Balss J, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009 Jun 25; doi: 10.1007/s00401-009-0561-9. [DOI] [PubMed] [Google Scholar]
- 120.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009 Nov 22; doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gross S, Cairns RA, Minden MD, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010 Feb 15;207(2):339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ward PS, Patel J, Wise DR, et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting alpha-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell. 2010 Feb 17;:17. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Toedt G, Barbus S, Wolter M, et al. Molecular signatures classify astrocytic gliomas by IDH1 mutation status. Int J Cancer. 2010 May 12;:12. doi: 10.1002/ijc.25448. [DOI] [PubMed] [Google Scholar]
- 124.Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010 May 18;17(5):510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Xu W, Yang H, Liu Y, et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of alpha-Ketoglutarate-Dependent Dioxygenases. Cancer Cell. 2010 Jan 18;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chowdhury R, Yeoh KK, Tian YM, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011 Apr 1;:1. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell. 2010 Dec 2;:2. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Christensen BC, Smith AA, Zheng S, et al. DNA Methylation, Isocitrate Dehydrogenase Mutation, and Survival in Glioma. J Natl Cancer Inst. 2011 Dec 16;:16. doi: 10.1093/jnci/djq497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Aghili M, Zahedi F, Rafiee E. Hydroxyglutaric aciduria and malignant brain tumor: a case report and literature review. J Neurooncol. 2009;91:233–236. doi: 10.1007/s11060-008-9706-2. [DOI] [PubMed] [Google Scholar]
- 130.Kranendijk M, Struys EA, van Schaftingen E, et al. IDH2 Mutations in Patients with D-2-Hydroxyglutaric Aciduria. Science. 2010 Sep 16;:16. doi: 10.1126/science.1192632. [DOI] [PubMed] [Google Scholar]
- 131.Vissers LE, Fano V, Martinelli D, et al. Whole-exome sequencing detects somatic mutations of IDH1 in metaphyseal chondromatosis with D-2-hydroxyglutaric aciduria (MC-HGA) Am J Med Genet A. 2011 Nov;155A(11):2609–2616. doi: 10.1002/ajmg.a.34325. [DOI] [PubMed] [Google Scholar]
- 132.Amary MF, Damato S, Halai D, et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet. 2011 Nov 6;(6):994. doi: 10.1038/ng.994. [DOI] [PubMed] [Google Scholar]
- 133.Camelo-Piragua S, Jansen M, Ganguly A, Kim JC, Louis DN, Nutt CL. Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol. 2010 Apr;119(4):509–511. doi: 10.1007/s00401-009-0632-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN. Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol. 2009;68:1319–1325. doi: 10.1097/NEN.0b013e3181c391be. [DOI] [PubMed] [Google Scholar]
- 135.Horbinski C, Kelly L, Nikiforov YE, Durso MB, Nikiforova MN. Detection of IDH1 and IDH2 mutations by fluorescence melting curve analysis as a diagnostic tool for brain biopsies. J Mol Diagn. 2010 Jul;12(4):487–492. doi: 10.2353/jmoldx.2010.090228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Horbinski C, Kofler J, Yeaney G, et al. Isocitrate Dehydrogenase 1 Analysis Differentiates Gangliogliomas from Infiltrative Gliomas. Brain Pathol. 2011 Feb 14;2011(14):1750–3639. doi: 10.1111/j.1750-3639.2011.00480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Capper D, Reuss D, Schittenhelm J, et al. Mutation-specific IDH1 antibody differentiates oligodendrogliomas and oligoastrocytomas from other brain tumors with oligodendroglioma-like morphology. Acta Neuropathol. 2011 Feb;121(2):241–252. doi: 10.1007/s00401-010-0770-2. [DOI] [PubMed] [Google Scholar]
- 138.Korshunov A, Meyer J, Capper D, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009 Sep;118(3):401–405. doi: 10.1007/s00401-009-0550-z. [DOI] [PubMed] [Google Scholar]
- 139.Pollack IF, Hamilton RL, Sobol RW, et al. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children’s Oncology Group. Childs Nerv Syst. 2010 Aug 20;:20. doi: 10.1007/s00381-010-1264-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hasselblatt M, Riesmeier B, Lechtape B, et al. BRAF-KIAA1549 fusion transcripts are less frequent in pilocytic astrocytomas diagnosed in adults. Neuropathol Appl Neurobiol. 2011 Jun 23;(23):1365–2990. doi: 10.1111/j.1365-2990.2011.01193.x. [DOI] [PubMed] [Google Scholar]
- 141.Metellus P, Coulibaly B, Colin C, et al. Absence of IDH mutation identifies a novel radiologic and molecular subtype of WHO grade II gliomas with dismal prognosis. Acta Neuropathol. 2010 Dec;120(6):719–729. doi: 10.1007/s00401-010-0777-8. [DOI] [PubMed] [Google Scholar]
- 142.Goze C, Bezzina C, Goze E, et al. 1P19Q loss but not IDH1 mutations influences WHO grade II gliomas spontaneous growth. J Neurooncol. 2012 May;108(1):69–75. doi: 10.1007/s11060-012-0831-6. [DOI] [PubMed] [Google Scholar]
- 143.Juratli TA, Kirsch M, Robel K, et al. IDH mutations as an early and consistent marker in low-grade astrocytomas WHO grade II and their consecutive secondary high-grade gliomas. J Neurooncol. 2012 Mar 13;:13. doi: 10.1007/s11060-012-0844-1. [DOI] [PubMed] [Google Scholar]
- 144.Takano S, Kato Y, Yamamoto T, et al. Immunohistochemical detection of IDH1 mutation, p53, and internexin as prognostic factors of glial tumors. J Neurooncol. 2012 Mar 7;:7. doi: 10.1007/s11060-012-0837-0. [DOI] [PubMed] [Google Scholar]
- 145.Ahmadi R, Stockhammer F, Becker N, et al. No prognostic value of IDH1 mutations in a series of 100 WHO grade II astrocytomas. J Neurooncol. 2012 Apr 19; doi: 10.1007/s11060-012-0863-y. [DOI] [PubMed] [Google Scholar]
- 146.Capper D, Weissert S, Balss J, et al. Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol. 2009 Jan;20(1):245–254. doi: 10.1111/j.1750-3639.2009.00352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Capper D, Zentgraf H, Balss J, Hartmann C, von Deimling A. Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol. 2009;118:599–601. doi: 10.1007/s00401-009-0595-z. [DOI] [PubMed] [Google Scholar]
- 148.Kato Y, Jin G, Kuan CT, McLendon RE, Yan H, Bigner DD. A monoclonal antibody IMab-1 specifically recognizes IDH1(R132H), the most common glioma-derived mutation. Biochem Biophys Res Commun. 2009 Oct 7; doi: 10.1016/j.bbrc.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Preusser M, Wohrer A, Stary S, Hoftberger R, Streubel B, Hainfellner JA. Value and Limitations of Immunohistochemistry and Gene Sequencing for Detection of the IDH1-R132H Mutation in Diffuse Glioma Biopsy Specimens. J Neuropathol Exp Neurol. 2011 Jul 12; doi: 10.1097/NEN.0b013e31822713f0. [DOI] [PubMed] [Google Scholar]
- 150.Sanoudou D, Tingby O, Ferguson-Smith MA, Collins VP, Coleman N. Analysis of pilocytic astrocytoma by comparative genomic hybridization. Brit J Cancer. 2000 Mar;82(6):1218–1222. doi: 10.1054/bjoc.1999.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol. 2008;67:878–887. doi: 10.1097/NEN.0b013e3181845622. [DOI] [PubMed] [Google Scholar]
- 152.Pfister S. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008;118:1739–1749. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007 May 14;26(22):3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 154.Forshew T, Tatevossian RG, Lawson AR, et al. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol. 2009 Mar 20; doi: 10.1002/path.2558. [DOI] [PubMed] [Google Scholar]
- 155.Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008 Nov 1;68(21):8673–8677. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 2009 Apr 13; doi: 10.1038/onc.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cin H, Meyer C, Herr R, et al. Oncogenic FAM131B-BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol. 2011 Jun;121(6):763–774. doi: 10.1007/s00401-011-0817-z. [DOI] [PubMed] [Google Scholar]
- 158.Raabe EH, Lim KS, Kim JM, et al. BRAF Activation Induces Transformation and Then Senescence in Human Neural Stem Cells: A Pilocytic Astrocytoma Model. Clin Cancer Res. 2011 Jun 1;17(11):3590–3599. doi: 10.1158/1078-0432.CCR-10-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jacob K, Quang-Khuong DA, Jones DT, et al. Genetic Aberrations Leading to MAPK Pathway Activation Mediate Oncogene-Induced Senescence in Sporadic Pilocytic Astrocytomas. Clin Cancer Res. 2011 Jul 12;:12. doi: 10.1158/1078-0432.CCR-11-0127. [DOI] [PubMed] [Google Scholar]
- 160.Hawkins C, Walker E, Mohamed N, et al. BRAF-KIAA1549 Fusion Predicts Better Clinical Outcome in Pediatric Low-Grade Astrocytoma. Clin Cancer Res. 2011 Jul 5;:5. doi: 10.1158/1078-0432.CCR-11-0034. [DOI] [PubMed] [Google Scholar]
- 161.Gronych J, Korshunov A, Bageritz J, et al. An activated mutant BRAF kinase domain is sufficient to induce pilocytic astrocytoma in mice. J Clin Invest. 2011 Mar 14;(14) doi: 10.1172/JCI44656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Horbinski C, Nikiforova MN, Hagenkord JM, Hamilton RL, Pollack IF. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro Oncol. 2012 Apr 5;:5. doi: 10.1093/neuonc/nos077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Forshew T, Tatevossian RG, Lawson AR, et al. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol. 2009 Jun;218(2):172–181. doi: 10.1002/path.2558. [DOI] [PubMed] [Google Scholar]
- 164.Horbinski C, Hamilton RL, Nikiforov Y, Pollack IF. Association of molecular alterations, including BRAF, with biology and outcome in pilocytic astrocytomas. Acta Neuropathol. 2010 May;119(5):641–649. doi: 10.1007/s00401-009-0634-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jones DT. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–8677. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lawson AR, Tatevossian RG, Phipps KP, et al. RAF gene fusions are specific to pilocytic astrocytoma in a broad paediatric brain tumour cohort. Acta Neuropathol. 2010 Aug;120(2):271–273. doi: 10.1007/s00401-010-0693-y. [DOI] [PubMed] [Google Scholar]
- 167.Sievert AJ, Jackson EM, Gai X, et al. Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain Pathol. 2009 Jul;19(3):449–458. doi: 10.1111/j.1750-3639.2008.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Schiffman JD, Hodgson JG, VandenBerg SR, et al. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res. 2010 Jan 15;70(2):512–519. doi: 10.1158/0008-5472.CAN-09-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Schindler G, Capper D, Meyer J, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011 Mar;121(3):397–405. doi: 10.1007/s00401-011-0802-6. [DOI] [PubMed] [Google Scholar]
- 170.Nicolaides TP, Li H, Solomon DA, et al. Targeted therapy for BRAFV600E malignant astrocytoma. Clin Cancer Res. 2011 Dec 15;17(24):7595–7604. doi: 10.1158/1078-0432.CCR-11-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Basto D, Trovisco V, Lopes JM, et al. Mutation analysis of B-RAF gene in human gliomas. Acta Neuropathol. 2005 Feb;109(2):207–210. doi: 10.1007/s00401-004-0936-x. [DOI] [PubMed] [Google Scholar]
- 172.Knobbe CB, Reifenberger J, Reifenberger G. Mutation analysis of the Ras pathway genes NRAS, HRAS, KRAS and BRAF in glioblastomas. Acta Neuropathol. 2004 Dec;108(6):467–470. doi: 10.1007/s00401-004-0929-9. [DOI] [PubMed] [Google Scholar]
- 173.Dias-Santagata D, Lam Q, Vernovsky K, et al. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One. 2011;6(3):17948. doi: 10.1371/journal.pone.0017948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Dougherty MJ, Santi M, Brose MS, et al. Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro Oncol. 2010 Feb 14;:14. doi: 10.1093/neuonc/noq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Yu J, Deshmukh H, Gutmann RJ, et al. Alterations of BRAF and HIPK2 loci predominate in sporadic pilocytic astrocytoma. Neurology. 2009 Nov 10;73(19):1526–1531. doi: 10.1212/WNL.0b013e3181c0664a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Eisenhardt AE, Olbrich H, Roring M, et al. Functional characterization of a BRAF insertion mutant associated with pilocytic astrocytoma. Int J Cancer. 2010 Dec 28;:28. doi: 10.1002/ijc.25893. [DOI] [PubMed] [Google Scholar]
- 177.Jentoft M, Giannini C, Cen L, et al. Phenotypic variations in NF1-associated low grade astrocytomas: possible role for increased mTOR activation in a subset. Int J Clin Exp Pathol. 2010;4(1):43–57. [PMC free article] [PubMed] [Google Scholar]
- 178.Tihan T, Ersen A, Qaddoumi I, et al. Pathologic Characteristics of Pediatric Intracranial Pilocytic Astrocytomas and Their Impact on Outcome in 3 Countries: A Multi-institutional Study. Am J Surg Pathol. 2011 Oct 10;:10. doi: 10.1097/PAS.0b013e3182329480. [DOI] [PubMed] [Google Scholar]
- 179.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011 Jun 30;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ciampi R, Knauf JA, Kerler R, et al. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J Clin Invest. 2005 Jan;115(1):94–101. doi: 10.1172/JCI23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tian Y, Rich BE, Vena N, et al. Detection of KIAA1549-BRAF fusion transcripts in formalin-fixed paraffin-embedded pediatric low-grade gliomas. J Mol Diagn. 2011 Nov;13(6):669–677. doi: 10.1016/j.jmoldx.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 2009 May 21;28(20):2119–2123. doi: 10.1038/onc.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Capper D, Preusser M, Habel A, et al. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol. 2011 Jul;122(1):11–19. doi: 10.1007/s00401-011-0841-z. [DOI] [PubMed] [Google Scholar]
- 184.Mohapatra G, Engler DA, Starbuck KD, et al. Genome-wide comparison of paired fresh frozen and formalin-fixed paraffin-embedded gliomas by custom BAC and oligonucleotide array comparative genomic hybridization: facilitating analysis of archival gliomas. Acta Neuropathol. 2011 Apr;121(4):529–543. doi: 10.1007/s00401-010-0773-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Dias-Santagata D, Akhavanfard S, David SS, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010 May;2(5):146–158. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]