

Abstract
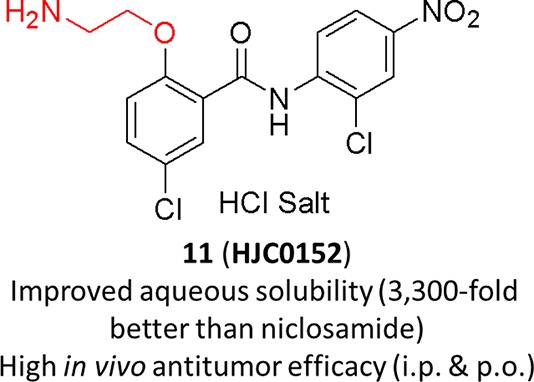
Niclosamide has been identified to potently inhibit the activation, nuclear translocation, and transactivation of STAT3. Nevertheless, the poor aqueous solubility and bioavailability of niclosamide have hindered its further clinical development for cancer therapy. To discover new molecules with enhanced druglike properties, a series of novel O-alkylamino-tethered derivatives of niclosamide have been designed, synthesized, and biologically evaluated. Among them, compound 11 (HJC0152) has been demonstrated to significantly suppress MDA-MB-231 xenograft tumor growth in vivo (ip and po), indicating its great potential as efficacious and orally bioavailable therapeutics for human cancer.
Keywords: niclosamide derivatives, STAT3, water solubility, orally bioavailable
Signal transducers and activators of transcription (STATs) are a family of transcription factors that serve as signaling transmitters for a large number of cytokines and growth factors in the regulation of critical biological processes including cell growth, proliferation, differentiation, and survival.1−6 Accumulating evidence has demonstrated that persistent activation of STAT3 stimulates tumor angiogenesis, promotes tumor immune evasion, and even confers resistance to apoptosis induced by conventional therapies.7−13 Therefore, STAT3 represents an attractive therapeutic target for the treatment of various types of human cancer.14−16
Despite significant advances in recent discovery efforts targeting STAT3, only several STAT3 inhibitors are advanced into early phase clinical trials.16 A poor physicochemical property is one of the most significant obstacles in promoting a potent agent into clinical trials.16 For example, peptide and peptidomimetics inhibitors targeting STAT3 suffer from poor cellular permeability and stability.17−20 Aqueous solubility plays an essential role in drug disposition and is one of the crucial molecular properties for successful drug development. Although an appropriate formulation could be used to enhance solubility and absorption, the stability and manufacturing difficulties should also be taken into consideration. In addition, oral chemotherapy is obviously a preferred administrative route in cancer treatment due to its convenience, patient compliance, and cost effectiveness.21,22 Niclosamide, an FDA-approved anticestodal drug, has recently been identified with significant inhibition of the activation, nuclear translocation, and transactivation of STAT3.19 It inhibits the transcription of STAT3 target genes and induces cell growth inhibition, apoptosis, and cell cycle arrest of cancer cells with constitutively active STAT3. Nevertheless, niclosamide does not have an ideal pharmacokinetic profile due to its poor water solubility and low oral bioavailability.23,24 It was of our interest to utilize rational chemical approaches to generate and identify novel derivatives of niclosamide with improved aqueous solubility and bioavailability as potential clinical candidates for cancer therapy. Herein, we report our chemical structural modifications of niclosamide to discover novel molecules aiming at enhanced efficacy and improved druglike properties.
We first directed our optimization effort involving modification of the hydroxyl group on the phenol ring of niclosamide by introduction of an O-alkylamino side chain. The amino group-containing scaffolds are important motifs of structural tuning with the capability to form hydrogen bonding like the phenol group for selective protein binding and druglikeness enhancement.25 These new O-alkylamino tethered analogues were expected to provide efficient target binding for a better potency, and as free bases, these molecules could also form salts for the final target compounds to have a better aqueous solubility. As shown in Scheme 1, analogues 2–6 were conveniently prepared by Mitsunobu reaction. Alkylation of the bromide intermediate 2 with a variety of amines including heterocyclic moieties introduced basic functionalities into the molecules, providing niclosamide derivatives 7 and 8. Mitsunobu coupling of niclosamide with N-Boc-protected amino alcohols followed by the Boc deprotection afforded analogues 9–15 with diversified O-alkyamino side chains. Both linear alkyl amines and heterocyclic alkyl amines have been explored for the purpose of comparison.
Scheme 1.

To examine whether the substitution of moiety groups affected biological activities of newly synthesized analogues and explore the structure–activity relationship (SAR), we first evaluated the in vitro anticancer effects of the compounds 3–15 on the proliferation of breast cancer cell lines MCF-7 (ER-positive) and MDA-MB-231 (ER-negative and triple-negative), as well as pancreatic cancer cell lines AsPC1 and Panc-1 using MTS assays. The ability of these new analogues to inhibit the growth of cancer cells is summarized in Table 1. The results revealed that analogues 10–15 bearing the terminal amino group-containing side chain at the phenol moiety showed promising antiproliferative activities with low micromolar to nanomolar IC50 values, while the alkyl-substituted derivatives 5–9 at the terminal amino group on the same position displayed moderate to low activities. For example, new analogues 10 and 11 exhibited a similar or significantly higher potency than niclosamide. However, compound 4 with a methylated terminal amino group in 4-O-piperidinyl moiety displayed 10-fold loss of antiproliferative activity in comparison with analogue 10. The same trend of SAR was also found for compounds 5–8, in which the dialkylsubstitution at the terminal amino group resulted in significant loss of activities, while O-ethylpiperazinyl derivative 9 in contrast with 8 regained antiproliferative activity with IC50 values of 12.4 and 8.7 μM against two breast cancer cell lines MCF-7 and MDA-MB-231, respectively. The fluorinated compound 3 that was initially designed for potential PET imaging studies displayed no significant effects against the tested cancer cells.
Table 1. Effects of New Niclosamide Analogues on Proliferation of Human Breast and Pancreatic Cancer Cell Lines.
| IC50 (μM) |
|||||
|---|---|---|---|---|---|
| breast cancer |
pancreatic
cancer |
||||
| compd | cLogPa | MCF-7 | MDA-MB-231 | AsPC1 | Panc-1 |
| 1 | 4.05 | 1.06 | 0.79 | 1.47 | 1.73 |
| 3 | 4.26 | >10 | >10 | NDb | ND |
| 4 | 4.22 | 4.11 | 2.69 | 3.51 | >10 |
| 5 | 3.77 | >10 | >10 | ND | ND |
| 6 | 3.47 | 9.87 | 18.0 | 72.2 | >10 |
| 7 | 4.73 | >10 | >10 | ND | ND |
| 8 | 3.56 | >10 | >10 | ND | ND |
| 9 | 3.20 | 12.4 | 8.7 | ND | ND |
| 10 | 3.91 | 0.25 | 0.29 | 2.76 | 0.54 |
| 11 | 3.01 | 0.91 | 1.64 | 1.9 | 1.08 |
| 12 | 3.33 | 3.11 | 2.61 | 4.2 | 3.11 |
| 13 | 3.76 | 3.7 | 2.37 | 7.4 | >10 |
| 14 | 4.15 | 3.49 | 3.1 | 2.08 | 4.67 |
| 15 | 2.87 | 9.37 | 5.3 | >10 | >10 |
| 19 | 2.95 | >10 | >10 | ND | ND |
| 20 | 3.13 | >10 | 5.87 | ND | ND |
ND, not determined.
Further modifications of nitro group in compound 10, one of the most potent analogues identified from our first SAR exploration, were also investigated. As outlined in Scheme 2, reduction of the nitro group of the intermediate 17 with zinc dust provided the corresponding amine 18. Further treatment of 18 with methanesulfonyl chloride or acetyl chloride followed by removal of the Boc group afforded the desired products 19 and 20, respectively. As shown in Table 1, replacement of the nitro group of compound 10 with methanesulfonamide (19) or acetamide (20) resulted in a significant loss of the antiproliferative activity, indicating that the nitro moiety is fairly important for potency.
Scheme 2.

From the in vitro screening of all tested compounds, analogues 10 (HJC0125) and 11 (HJC0152) stood out with an attractive anticancer profile and were therefore subjected to further biological evaluation. To study the effects of these two compounds on cell growth, cellular morphological changes were examined in MDA-MB-231 breast cancer cells treated with compounds 10, 11, or niclosamide for 48 h, under light microscopy. Like niclosamide, both compounds 10 and 11 significantly inhibited cell proliferation and induced apoptosis accompanying cellular morphological changes at concentrations of 1, 5, and 10 μM, respectively (Supporting Information).
Because one of major goals for our discovery effort was to identify compounds with improved water solubility and oral bioavailability, aqueous solubility of the new analogues 10 and 11 was determined by an HPLC method.26 As expected, introduction of the O-alkylamino-tethered moiety not only enhanced anticancer activity but also significantly increased the aqueous solubility. Both compounds 10 and 11 (in the form of HCl salt) demonstrated excellent water solubility, with a saturated concentration of 248 and 762 μg/mL, respectively (Figure 1). The O-ethylamino derivative 11 showed the most favorable solubility, which is about 3300-fold improvement in comparison with that of niclosamide (0.23 μg/mL).27 The significant improvement of the aqueous solubility is expected to facilitate these compounds to be more orally bioavailable and efficacious in vivo than niclosamide, which is substantially water insoluble.
Figure 1.
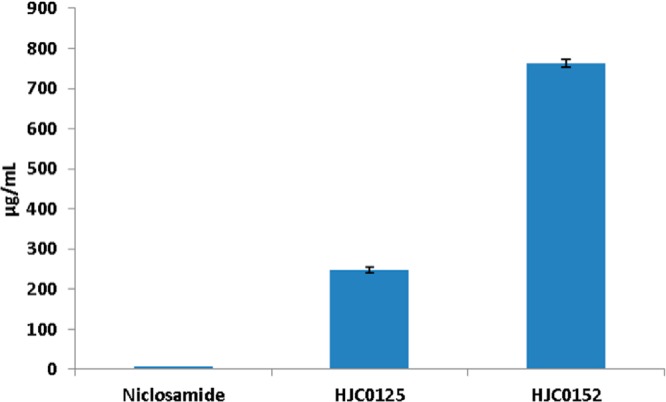
Aqueous solubility of novel niclosamide analogues. Compounds 10 (HJC0125) and 11 (HJC0152) showed significantly improved solubility as compared with niclosamide.
Previous studies have demonstrated that niclosamide is a potential inhibitor of STAT3.19 To determine whether the new derivatives act as potent small-molecule inhibitors of STAT3 activation, we measured the effect of niclosamide and compound 11 on promoter activity using the cell-based transient transfection and dual luciferase reporter assays. MDA-MB-231 cells were pretreated with niclosamide or 11 at different concentrations for 24 h. The STAT3 promoter activity in MDA-MB-231 cells was determined after transient transfecting with pSTAT3-Luc vector. As shown in Figure 2A, treatment with 10 μM compound 11 decreased the STAT3 promoter activity in MDA-MB-231 cells by approximately 32%, and increasing the dose of compound 11 to 20 μM further decreased STAT3 promoter activity by 62% as compared with control. Similarly, STAT3 promoter activity in MDA-MB-231 cells was suppressed after treatment with 20 μM niclosamide, while proliferation of MDA-MB-231 cells (MTT assay)19 showed comparable to slightly more potent reduction (Figure 2B). These results demonstrate that compound 11 inhibits STAT3 promoter activity in MDA-MB-231 cells in a dose-dependent manner and has a similar effect as niclosamide.
Figure 2.

(A) HJC0152 inhibited the STAT3-mediated luciferase reporter activity in MDA-MB-231 cells. (B) Proliferation of MDA-MB-231 cells treated with HJC0152 and niclosamide for 24 h.
To further investigate the inhibitory activity of compound 11 against the STAT3 pathway, we examined STAT3 phosphorylation and expression of the known STAT3 target genes in MDA-MB-231 cell line. The cells were treated with different doses of compound 11 for 24 h, and levels of total STAT3 and phosphorylated STAT3 at Tyr-705 were then examined by Western blot. As shown in Figure 3, total STAT3 was reduced after treatment with 11 or niclosamide, suggesting that these compounds may alter transcription and/or translation of STAT3. Similarly, phosphorylated STAT3 (p-STAT3) at Tyr-705 is suppressed by 11 or niclosamide, suggesting that compound 11 has a comparable potency in downregulating STAT3 protein production and phosphorylation at the Tyr-705 site. Blocking STAT3 signaling in many different tumor cells leads to induce growth arrest and apoptosis.28−30 We observed that compound 11 induced cleaved caspase-3 and downregulated cyclin D1 in MDA-MB-231 cells. These results suggest that compound 11 inhibits cell cycle progression and promotes apoptosis. We further confirmed these results using annexin V-based measurement using flow cytometry. As depicted in Figure S2 in the Supporting Information, compound 11 and niclosamide activated apoptosis in MDA-MB-231 breast cancer cells in a dose-dependent manner. Other than targeting STAT3, niclosamide has also been found to inhibit NF-kB and Wnt/β-catenin signaling.19,31,32 Currently, we are investigating the mechanisms of how compound 11 regulates STAT3 upstream targets and related signaling pathways and transcriptional/translational regulation.
Figure 3.

(A) Western blot analysis of biochemical markers for apoptosis induction and inhibition of STAT3 activity by HJC0152 in the MDA-MB-231 cell line. (B) Densitometric analysis of three independent experiments for the expression level of total STAT3, phospho-STAT3, cyclin D1, and cleaved caspase 3. * represents p < 0.05, and ** represents p < 0.01.
Furthermore, compound 11 was further evaluated for its antitumor activity in inhibition of tumor growth in the MDA-MB-231 xenograft model. As expected, mice treated with 7.5 mg/kg of compound 11 via ip showed a better effect in inhibiting tumor growth as compared to the mice treated with 12.5 mg/kg niclosamide (Figure 4A). Similarly, we treated the MDA-MB-231 xenograft mice with oral administration of compound 11 and found that the growth of xenograft tumors in mice was significantly reduced by compound 11 at a dose of 25 mg/kg and even more efficacious than niclosamide at 75 mg/kg (Figure 4B). This observation might be attributed to the superior solubility of compound 11, which might have resulted in an improved oral bioavailability33−37 and, consequently, an enhanced suppression of tumor growth in mice. It is also noteworthy that compound 11 did not show significant signs of toxicity at a dose of 75 mg/kg. These results have demonstrated that compound 11 (HJC0152) is a promising anticancer drug candidate with excellent aqueous solubility and have significant potential to be developed as an orally bioavailable anticancer agent.
Figure 4.

In vivo efficacy of compound 11 (HJC0152) in inhibiting growth of xenograft tumors (breast cancer MDA-MB-231) in mice. (A) ip and (B) po.
In summary, a series of novel O-alkylamino tethered derivatives of niclosamide have been designed, synthesized, and biologically evaluated in this work. New analogues 10 and 11 were identified to exhibit a similar or significantly higher potency than niclosamide against human breast and pancreatic cancer cells. Both compounds 10 and 11 have demonstrated a superior aqueous solubility, especially the compound 11, which has about 3300-fold improvement in water solubility in comparison with niclosamide. In MDA-MB-231 cells, compound 11 inhibited STAT3 promoter activity, increased the expression of active caspase-3, inhibited cell cycle progression, and promoted apoptosis. In nude mice bearing breast tumor xenografts, compound 11 significantly suppressed MDA-MB-231 xenograft tumor growth in vivo (both ip and po). These results suggest that compound 11 (HJC0152) with remarkably improved aqueous solubility may have great potential to be developed as an orally bioavailable agent for cancer therapy.
Supporting Information Available
Supplemental data, spectroscopic data with methods of synthesis for all compounds, and methods of biological evaluation. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ These authors contribute equally to this work.
This work was supported by Grants P50 CA097007, P30DA028821, and R21MH093844 (J.Z.) from the National Institute of Health, R. A. Welch Foundation Chemistry and Biology Collaborative Grant from Gulf Coast Consortia (GCC), John Sealy Memorial Endowment Fund (J.Z.), the Duncan Family Institute (DFI) Seed Funding Program, and startup funds from MD Anderson Cancer Center (Q.S.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Darnell J. E. Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [DOI] [PubMed] [Google Scholar]
- Bowman T.; Garcia R.; Turkson J.; Jove R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [DOI] [PubMed] [Google Scholar]
- Bromberg J.; Darnell J. E. Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–24673. [DOI] [PubMed] [Google Scholar]
- Darnell J. E. Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [DOI] [PubMed] [Google Scholar]
- Buettner R.; Mora L. B.; Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 2002, 8, 945–954. [PubMed] [Google Scholar]
- Yu H.; Jove R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [DOI] [PubMed] [Google Scholar]
- Becker S.; Groner B.; Müller C. W. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature 1998, 394, 145–151. [DOI] [PubMed] [Google Scholar]
- Bromberg J. F.; Wrzeszczynska M. H.; Devgan G.; Zhao Y.; Pestell R. G.; Albanese C.; Darnell J. E. Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [DOI] [PubMed] [Google Scholar]
- Siddiquee K.; Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008, 18, 254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell J. E. Jr. Validating Stat3 in cancer therapy. Nat. Med. 2005, 11, 595–596. [DOI] [PubMed] [Google Scholar]
- Yue P.; Turkson J. Targeting STAT3 in cancer: how successful are we?. Expert Opin. Invest. Drugs 2009, 18, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haftchenary S.; Avadisian M.; Gunning P. T. Inhibiting aberrant Stat3 function with molecular therapeutics: A progress report. Anticancer Drugs 2011, 22, 115–127. [DOI] [PubMed] [Google Scholar]
- Yu H.; Pardoll D.; Jove R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J.; Grande F.; Neamati N. Small molecule inhibitors of Stat3 signaling pathway. Curr. Cancer Drug Targets 2007, 7, 91–107. [DOI] [PubMed] [Google Scholar]
- Page B. D.; Ball D. P.; Gunning P. T. Signal transducer and activator of transcription 3 inhibitors: A patent review. Expert Opin. Ther. Pat. 2011, 21, 65–83. [DOI] [PubMed] [Google Scholar]
- Debnath B.; Xu S.; Neamati N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J. Med. Chem. 2012, 55, 6645–6668. [DOI] [PubMed] [Google Scholar]
- Mandal P. K.; Ren Z.; Chen X.; Xiong C.; McMurray J. S. Structure–affinity relationships of glutamine mimics incorporated into phosphopeptides targeted to the SH2 domain of signal transducer and activator of transcription 3. J. Med. Chem. 2009, 52, 6126–6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Bai L.; Bernard D.; Nikolovska-Coleska Z.; Gomez C.; Zhang J.; Yi H.; Wang S. Structure-based design of conformationally constrained, cell-permeable STAT3 inhibitors. ACS Med. Chem. Lett. 2010, 1, 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X.; Duan L.; He Q.; Zhang Z.; Zhou Y.; Wu D.; Pan J.; Pei D.; Ding K. Identification of niclosamide as a new small-molecule inhibitor of the STAT3 signaling pathway. ACS Med. Chem. Lett. 2010, 1, 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schust J.; Sperl B.; Hollis A.; Mayer T. U.; Berg T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [DOI] [PubMed] [Google Scholar]
- O’Neill V. J.; Twelves C. J. Oral cancer treatment: Developments in chemotherapy and beyond. Br. J. Cancer 2002, 87, 933–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott S.; Schneeweiss A.; Reinhardt J.; Bruckner T.; Domschke C.; Sohn C.; Eichbaum M. H. Acceptance of oral chemotherapy in breast cancer patients—A survey study. BMC Cancer 2011, 11, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkihel L.; Loiseau P. M.; Bourass J.; Gayral P.; Letourneux Y. Synthesis and orally macrofilaricidal evaluation of niclosamide lymphotropic prodrugs. Arzneimittelforschung 1994, 44, 1259–1264. [PubMed] [Google Scholar]
- Navab M.; Ruchala P.; Waring A. J.; Lehrer R. I.; Hama S.; Hough G.; Palgunachari M. N.; Anantharamaiah G. M.; Fogelman A. M. A novel method for oral delivery of apolipoprotein mimetic peptides synthesized from all L-amino acids. J. Lipid Res. 2009, 50, 1538–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y.; Teegarden B. R.; Choi J. S.; Strah-Pleynet S.; Decaire M.; Jayakumar H.; Dosa P. I.; Casper M. D.; Pham L.; Feichtinger K.; Ullman B.; Adams J.; Yuskin D.; Frazer J.; Morgan M.; Sadeque A.; Chen W.; Webb R. R.; Connolly D. T.; Semple G.; Al-Shamma H. Discovery and structure-activity relationship of 3-methoxy-N-(3-(1-methyl-1H-pyrazol-5-yl)-4-(2-morpholinoethoxy)phenyl)benzamide (APD791): a highly selective 5-hydroxytryptamine2A receptor inverse agonist for treatment of arterial thrombosis. J. Med. Chem. 2010, 53, 4412–4421. [DOI] [PubMed] [Google Scholar]
- Vogel G. H.Determination of solubility by hyphenated HPLC methods. Drug Discovery and Evaluation: Safety and Pharmacokinetics Assay; Springer: New York, 2006; pp 400–402. [Google Scholar]
- The Merck Index, 13th ed.; Merck: Rahway, NY, 2001. [Google Scholar]
- Turkson J.; Jove R. STAT proteins: Novel molecular targets for cancer drug discovery. Oncogene 2000, 19, 6613–6626. [DOI] [PubMed] [Google Scholar]
- Bromberg J. Stat proteins and oncogenesis. J. Clin. Invest. 2002, 109, 1139–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Yue P.; Page B. D.; Li T.; Zhao W.; Namanja A. T.; Paladino D.; Zhao J.; Chen Y.; Gunning P. T.; Turkson J. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 9623–9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y.; Lu Z.; Ding K.; Li J.; Du X.; Chen C.; Sun X.; Wu Y.; Zhou J.; Pan J. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: Inactivation of the NF-kB pathway and generation of reactive oxygen species. Cancer Res. 2010, 70, 2516–2527. [DOI] [PubMed] [Google Scholar]
- Osada T.; Chen M.; Yang X. Y.; Spasojevic I.; Vandeusen J. B.; Hsu D.; Clary B. M.; Clay T. M.; Chen W.; Morse M. A.; Lyerly H. K. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011, 71, 4172–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [DOI] [PubMed] [Google Scholar]
- Leach A. G.; Jones H. D.; Cosgrove D. A.; Kenny P. W.; Ruston L.; MacFaul P.; Wood J. M.; Colclough N.; Law B. Matched molecular pairs as a guide in the optimization of pharmaceutical properties; a study of aqueous solubility, plasma protein binding and oral exposure. J. Med. Chem. 2006, 49, 6672–6682. [DOI] [PubMed] [Google Scholar]
- Bergström C. A.; Wassvik C. M.; Johansson K.; Hubatsch I. Poorly soluble marketed drugs display solvation limited solubility. J. Med. Chem. 2007, 50, 5858–5862. [DOI] [PubMed] [Google Scholar]
- Ishikawa M.; Hashimoto Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 2011, 54, 1539–1554. [DOI] [PubMed] [Google Scholar]
- Hann M. M.; Keserü G. M. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discovery 2012, 11, 355–365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.