

Abstract
Desferrithiocin (DFT, 1) is a very efficient iron chelator when given orally. However, it is severely nephrotoxic. Structure-activity studies with 1 demonstrated that removal of the aromatic nitrogen to provide desazadesferrithiocin (DADFT, 2) and introduction of either a hydroxyl group or a polyether fragment onto the aromatic ring resulted in orally active iron chelators that were much less toxic than 1. The purpose of the current study was to determine if a comparable reduction in renal toxicity could be achieved by performing the same structural manipulations on 1 itself. Accordingly, three DFT analogues were synthesized. Iron clearing efficiency and ferrokinetics were evaluated in rats and primates; toxicity assessments were carried out in rodents. The resulting DFT ligands demonstrated a reduction in toxicity that was equivalent to that of the DADFT analogues and presented with excellent iron clearing properties.
Introduction
Nearly all life forms require iron as a micronutrient. However, the low solubility of Fe(III) hydroxide (Ksp = 1 × 10−39),1 the predominant form of the metal in the biosphere, required the development of sophisticated iron storage and transport systems in nature. Microorganisms utilize low molecular weight, ferric iron-specific ligands, siderophores;2 eukaryotes tend to employ proteins to transport and store iron.3–5 Humans have evolved a highly efficient iron management system in which we absorb and excrete only about 1 mg of the metal daily; there is no mechanism for the excretion of excess metal.6 Whether derived from transfused red blood cells7–9 or from increased absorption of dietary iron,10,11 without effective treatment, body iron progressively increases with deposition in the liver, heart, pancreas, and elsewhere (iron overload disease).
In patients with iron overload disease, the toxicity derives from iron’s interaction with reactive oxygen species.12–14 For example, in the presence of Fe(II), endogenous H2O2 is reduced to the hydroxyl radical (HO•), a very reactive species, and HO−, the Fenton reaction. The hydroxyl radical reacts very quickly with a variety of cellular constituents and can initiate free radicals and radical-mediated chain processes that damage DNA and membranes as well as produce carcinogens.13,15 The liberated Fe(III) is reduced back to Fe(II) via a variety of biological reductants (e.g., ascorbate, glutathione), a problematic cycle.
Iron-mediated damage can be focal, as in reperfusion damage,16 Parkinson’s,17 Friedreich’s ataxia,18 macular degeneration,19 and hemorrhagic stroke,20 or global, as in transfusional iron overload, e.g., thalassemia.21 sickle cell disease,21, 22 and myelodysplasia,23 with multiple organ involvement. The solution in both scenarios is the same: chelate and promote the excretion of excess unmanaged iron.
Treatment with a chelating agent capable of sequestering iron and permitting its excretion from the body is the only therapeutic approach available.24,25 Some of the iron chelating agents that are now in use or that have been clinically evaluated include desferrioxamine B mesylate (DFOa),26 1,2-dimethyl-3-hydroxy-4-pyridinone (deferiprone, L1),27–30 and 4-[3,5-bis(2-hydroxyphenyl)-1,2,4-triazol-1-yl]benzoic acid (desferasirox, ICL670A).31–34 Each of these ligands presents with shortcomings. DFO must be given subcutaneously (sc) for protracted periods of time, e.g., 12 h a day, five days a week, a serious patient compliance issue.7,35,36 Deferiprone, while orally active, simply does not remove enough iron to maintain patients in a negative iron balance.27–30 Desferasirox did not show noninferiority to DFO and is associated with numerous side effects, including some serious renal toxicity issues.31–34
In addition to the above chelators, two desferrithiocin [(S)-4,5-dihydro-2-(3-hydroxy-2-pyridinyl)-4-methyl-4-thiazolecarboxylic acid (DFT, 1, Chart 1) analogues have made it to clinical trials. As will be described below, although 1 itself is severely nephrotoxic,37 extensive structure activity relationship (SAR) studies,38–40 including simple hydroxylation of the aromatic ring of (S)-4,5-dihydro-2-(2-hydroxyphenyl)-4-methyl-4-thiazolecarboxylic acid (2, Chart 1), to yield (S)-2-(2,4-dihydroxyphenyl)-4,5-dihydro-4-methyl-4-thiazolecarboxylic acid (deferitrin, 3,41 Chart 1), or the introduction of polyether fragments42–43 to 2, e.g., (S)-4,5-dihydro-2-[2-hydroxy-4-(3,6,9-trioxadecyloxy)]-4-methyl-4-thiazolecarboxylic acid (4, Chart 1), led to the discovery of orally active iron chelators that were much less toxic than 1. This begs the question of what the impact of the hydroxylation of DFT itself, or fixing a polyether fragment to DFT, would have on the nephrotoxicity and iron clearance properties of the new analogues.
Chart 1.

Extensive structural alterations of the tridentate chelator (S)-4,5-dihydro-2-(3-hydroxy-2-pyridinyl)-4-methyl-4-thiazolecarboxylic acid (1) were carried out, including simple hydroxylation of the aromatic ring of (S)-4,5-dihydro-2-(2-hydroxyphenyl)-4-methyl-4-thiazolecarboxylic acid (2) to yield (S)-2-(2,4-dihydroxyphenyl)-4,5-dihydro-4-methyl-4-thiazolecarboxylic acid (3), or the introduction of polyether fragments to 2, e.g., (S)-4,5-dihydro-2-[2-hydroxy-4-(3,6,9-trioxadecyloxy)]-4-methyl-4-thiazolecarboxylic acid (4), and (S)-4,5-dihydro-2-[2-hydroxy-3-(3,6,9-trioxadecyloxy)]-4-methyl-4-thiazolecarboxylic acid (5).
Results and Discussion
Design Concept
DFT, 1, Table 1, is a natural product iron chelator isolated from Streptomyces antibioticus.44 It forms a 2:1 complex with Fe(III) with a cumulative formation constant of 4 × 1029 M−1.45,46 Although the compound was shown to be an excellent deferration agent when administered orally (po) to rats47 and primates,48,49 it caused severe nephrotoxicity in rats.37 However, the compound’s oral activity spurred SAR studies focused on the DFT platform aimed at identifying an orally active and safe DFT analogue.38–40
Table 1.
Iron-Clearing Efficiency of Desferrithiocin Analogues Administered to Rodents and Primates with the Respective Log Papp values.
| Chelator | Comp. No. |
Rodent Iron-Clearing Efficiencya (%) |
Primate Iron-Clearing Efficiencyc (%) |
Log Papp | PRd |
|---|---|---|---|---|---|
 |
1 | 5.5 ± 3.2 [93/7] |
16.1 ± 8.5 [78/22] |
−1.77 | 2.9 |
 |
2 | 2.7 ± 0.5 [100/0] |
21.5 ± 12.0 [76/24] |
−0.34 | 8.0 |
 |
3 | 1.1 ± 0.8 [100/0] |
16.8 ± 7.2 [88/12] |
−1.05 | 15.3 |
 |
6 | 9.0 ± 3.8 [97/3] |
10.0 ± 2.9 [58/42] |
−1.68 | 1.1 |
 |
7 | 26.7 ± 4.7b [97/3] |
26.3 ± 9.9 [93/7] (capsule) 28.7 ± 12.4 [83/17] (sodium salt) |
−0.89 | 1.0 1.1 |
 |
8 | 11.7 ± 1.2 [97/3] |
18.0 ± 5.2 [63/37] |
−1.59 | 1.5 |
 |
9 | 15.1 ± 2.0b [99/1] |
22.5 ± 6.4 [86/14] |
−0.96 | 1.5 |
 |
10 | 14.2 ± 2.4 [98/2] |
6.1 ± 1.8 (po) [40/60] 16.9 ± 7.3 (sc) [64/36] |
−1.38 | 0.4 1.2 |
In the rodents [n = 3 (7), 4 (2, 10), 5 (1, 6, 8, 9), or 8 (3)], the drugs were given po at a dose of 150 µmol/kg (1–2) or 300 µmol/kg (3, 6–10). The drugs were administered in capsules (7), solubilized in 40% Cremophor RH-40/water (1, 2), or were given as their monosodium salts, prepared by the addition of 1 equiv of NaOH to a suspension of the free acid in distilled water (3, 6, 8–10). The efficiency of each compound was calculated by subtracting the 24 or 48-h iron excretion of control animals from the iron excretion of the treated animals. The number was then divided by the theoretical output; the result is expressed as a percent. The relative percentages of the iron excreted in the bile and urine are in brackets. The ICE data for: 1 is from ref 47; 2 is from ref 38; 3 is from ref 51; 7 is from ref 54; 9 is from ref 55.
ICE is based on a 48 h sample collection period.
In the primates [n = 4 (1, 2, 6, 7 in capsules, 8–10), or 6 (3), or 7 (7 as the monosodium salt)], the chelators were given po at a dose of 75 µmol/kg (2, 6–10) or 150 µmol/kg (1, 3). Ligand 10 was also given to the primates sc at a dose of 75 µmol/kg. The drugs were administered in capsules (7), solubilized in 40% Cremophor RH-40/water (1, 2), or were given as their monosodium salts, prepared by the addition of 1 equiv of NaOH to a suspension of the free acid in distilled water (1, 2, 3, 6–10). The efficiency was calculated by averaging the iron output for 4 days before the drug, subtracting these numbers from the 2-day iron clearance after the administration of the drug, and then dividing by the theoretical output; the result is expressed as a percent. The ICE data for: 1–3 are from ref 38; 7 is from ref 54; 9 is from ref 55. The relative percentages of the iron excreted in the feces and urine are in brackets.
Performance ratio (PR) is defined as the mean ICEprimates/ICErodents
Our approach first entailed simplifying the platform. Removal of the pyridine nitrogen of 1 provided 2 (Chart 1, Table 1), the parent ligand of the desazadesferrithiocin (DADFT) series.40 Interestingly, although 2 was not overtly nephrotoxic, it elicited serious gastrointestinal (GI) problems.37,38,40 In spite of its GI toxicity, the ligand’s excellent ICE and the absence of nephrotoxicity prompted further SAR studies predicated on this pharmacophore. This led to the discovery that the lipophilicity (partition between octanol and water, expressed as the log of the fraction in the octanol layer, log Papp)50 of the DADFT analogues could have a profound effect on the ligand's iron-clearing efficiency (ICE), organ distribution, and toxicity profile.38,51,52
Ultimately, it was determined that hydroxylation of DADFT and a number of different analogues in the 3′-, 4′-, or 5′- position allowed for ligands that were very efficient, orally active iron chelators with profoundly less toxicity than 1 or 2.38,51 Clearly, hydroxylation had a significant effect on toxicity reduction. One of these ligands, 3, was taken into human clinical trials by Genzyme and was moving forward until the once daily dosing was changed to twice daily.41 Patients presented with increases in blood urea nitrogen (BUN) and serum creatinine (SCr) levels and the trial was terminated.41
The molecule (3) was reengineered, introducing a 3,6,9-trioxadecyloxy group in the 4′- position of 2 (4, Chart 1).42 This provided a remarkably efficient orally active iron chelator which, given to rats po once or twice daily, was virtually nephrotoxicity-free.42 This turned out to also be true when a variety of polyether backbones were fixed at the 3′-, 4′-, or 5′- position of the DADFT pharmacophore.43,53–55 In fact, (S)-4,5-dihydro-2-[2-hydroxy-3-(3,6,9-trioxadecyloxy)]-4-methyl-4-thiazolecarboxylic acid (5, Chart 1) has now been moved forward to clinical trials. Thus, it appeared as though fixing a polyether fragment to the DADFT framework was also a uniformly effective tool in further reducing 3-induced nephrotoxicity. These observations drive the current study.
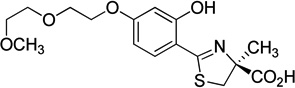
A series of questions based on the results of the SAR studies of DADFT analogues are addressed: 1) How does hydroxylation of 1 itself affect its iron clearing properties? 2) How does hydroxylation of 1 affect its renal toxicity? 3) What is the effect of fixing polyether fragments to 1 on its iron clearance? 4) What is the effect of fixing polyether fragments to 1 on its renal toxicity? In order to answer these questions, three ligands were assembled (Table 1), (S)-4,5-dihydro-2-(3,5-dihydroxy-2-pyridinyl)-4-methyl-4-thiazolecarboxylic acid, (6), (S)-4,5-dihydro-2-[3-hydroxy-5-(3,6-dioxaheptyloxy)-2-pyridinyl]-4-methyl-4-thiazolecarboxylic acid, (8), and (S)-4,5-dihydro-2-[3-hydroxy-4-(3,6-dioxaheptyloxy)-2-pyridinyl]-4-methyl-4-thiazolecarboxylic acid, (10).
Synthesis
The preparation of 5′-hydroxydesferrithiocin (6) and its 5′-nor polyether (8) (Table 1) began with 2-cyano-3,5-difluoropyridine (11), which was converted to 2-cyano-3,5-dihydroxypyridine (13) in two steps (Scheme 1). Heating 11 with 4-methoxybenzyl alcohol in the presence of NaH (2.5 equivalents each) in DMF56,57 at 95 °C for 18 h gave protected diol 12 in 73% yield. Removal of the 4-methoxybenzyl groups of 12 using excess trifluoroacetic acid (TFA)58 and pentamethylbenzene59 at room temperature for 22 h provided nitrile 13 in quantitative yield. Cyclocondensation of 13 with (S)-2-methyl cysteine (14) in aqueous CH3OH buffered at pH 6 at 75 °C for 45 h followed by esterification of crude acid 6 with iodoethane and N,N-diisopropylethylamine (DIEA) (1.5 equivalents each) in DMF produced ethyl (S)-4,5-dihydro-2-(3,5-dihydroxy-2-pyridinyl)-4-methyl-4-thiazolecarboxylate (15) in 70% yield. Hydrolysis of 15 with aqueous NaOH in CH3OH at room temperature generated (S)-4,5-dihydro-2-(3,5-dihydroxy-2-pyridinyl)-4-methyl-4-thiazolecarboxylic acid (6) as a solid in 96% yield. Also, ester 15 was alkylated at the less hindered phenol54 in the presence of the pyridine nitrogen with tosylate 16 and K2CO3 in refluxing acetone, affording ligand precursor 17 in 65% yield. The carboxylate was unmasked under alkaline conditions to give 8 in 97% yield as an oil.
Scheme 1.

Synthesis of 6 and 8a
aReagents and conditions: (a) 4-methoxybenzyl alcohol, 60% NaH (2.5 equiv each), DMF, 95–100 °C, 18 h, 73%; (b) TFA, pentamethylbenzene, 22 h, quantitative; (c) CH3OH, 0.1 M pH 6 buffer, NaHCO3, 73–76 °C, 45 h; (d) EtI, DIEA (1.5 equiv each), DMF, 47 h, 70%; (e) K2CO3 (1.6 equiv), acetone, reflux, 1 d, 65%; (f) 50% NaOH (aq), CH3OH, then HCl, 96% (6), 97% (8).
Synthesis of the 4′-nor polyether desferrithiocin analogue 10, an isomer of iron chelator 8, (Table 1) started with 2-methyl-3-(benzyloxy)-4-pyridone (18), available in two steps from maltol60 (Scheme 2). O-Alkylation of 18 with tosylate 16 and K2CO3 in refluxing acetonitrile61 afforded 2-methyl-3-(benzyloxy)-4-(3,6-dioxaheptyloxy)pyridine (19) in 68% yield. The methyl group of 19 was oxidized by known methodology,60 providing aldehyde 21. Specifically, 19 was treated with 3-chloroperbenzoic acid (m-CPBA) in CH2Cl2, and the resulting N-oxide was heated at reflux in acetic anhydride. Cleavage of the acetate ester with base gave the 2-pyridinemethanol 20 in 87% overall yield. Primary alcohol 20 was further oxidized to aldehyde 21 in 83% yield with sulfur trioxide-pyridine complex and NEt3 in DMSO and CHCl3. The oxime 22, generated in 90% yield under standard conditions,42 was heated at reflux with acetic anhydride, furnishing the corresponding nitrile 23 in 94% yield. Removal of the benzyl-protecting group from 23 by hydrogenolysis (1 atm, 10% Pd-C, CH3OH) in the presence of the cyano group and pyridine ring produced 4-(3,6-dioxaheptyloxy)-3-hydroxy-2-pyridinecarbonitrile (24) in 85% yield. Heating 24 with amino acid 14 in aqueous CH3OH buffered at pH 6 generated (S)-4,5-dihydro-2-[3-hydroxy-4-(3,6-dioxaheptyloxy)-2-pyridinyl]-4-methyl-4-thiazolecarboxylic acid (10) in 95% yield.
Scheme 2.

Synthesis of 10a
aReagents and conditions: (a) K2CO3 (2 equiv), CH3CN, 68%; (b) m-CPBA, CH2Cl2; (c) Ac2O, reflux; (d) NaOH (aq), EtOH, reflux, 4h, 87%; (e) SO3˙pyridine, NEt3, DMSO, CHCl3, 16 h, 83%; (f) H2NOH˙HCl, NaOAc, CH3OH, reflux, 2 h, 90%; (g) Ac2O, reflux, 94%; (h) H2, 10% Pd-C, CH3OH, 85%; (i) CH3OH, 0.1 M pH 6 buffer, NaHCO3, 75 °C, 48 h, 95%.
Stoichiometry of the Ligand-Fe(III) Complexes
Earlier studies with 1 by Anderegg and Räber showed the chelator to form a 2:1 complex with Fe(III).46 The cumulative formation constant for this complex was determined to be 4 × 1029 M−1. Hahn et al. were ultimately able to isolate both the Δ and λ 1-Cr(III) complexes, with chromium serving as a surrogate for Fe(III).45 As expected, the crystal structures of the complexes unequivocally demonstrated a 2:1 ligand to metal ratio. In later studies in our laboratories, Job’s plots with 338 (Table 1) and the corresponding desmethyl analogue,40 also showed that these ligands formed 2:1 complexes with Fe(III). This is of course in keeping with the fact that the donor groups of the chelators, the aromatic hydroxyl, the thiazoline nitrogen, and the carboxylate are the same as in 1 itself. Furthermore, the cumulative distance (9.77 Å) between the donor groups in our x-ray crystal structure of free ligand 362 is identical to the corresponding value in the Cr(III) complex of 1.45 In the current study, Job’s plots were run on 6, 8, and 10 (Figure 1). In each instance, the ligands formed 2:1 complexes with Fe(III).
Figure 1.

Job’s plots of the Fe(III) complex of ligand 6, 8, and 10. Solutions containing different ligand/Fe(III) ratios were prepared such that [ligand] + [Fe(III)] = 1.0 mM in Tris-HCl buffer at pH 7.4. The theoretical mole fraction maximum for a 2:1 ligand:Fe complex is 0.667 (arrow). The observed maxima for 6, 8, and 10 are 0.669, 0.676, and 0.677, respectively. Optical density (y-axis) was determined at 498, 484, and 485 nm for 6, 8, and 10, respectively.
Partition Properties
The partition values between octanol and water (at pH 7.4, Tris buffer) were determined using a “shake flask” direct method of measuring log Papp values.50 The fraction of drug in the octanol is then expressed as log Papp. While the values vary widely (Table 1), one observation stands out: DFT and its analogues are always more hydrophilic than their DADFT counterparts, i.e., 1 vs 2, 6 vs 3, 8 vs 7, 10 vs 9. This, of course, is likely due to the presence of the aromatic nitrogen, a moderately good hydrogen bond acceptor, on the DFT analogues. Relative to the differences in lipophilicity between DFT and DADFT, fixing a polyether backbone to either the DFT or the DADFT pharmacophore had a much more moderate effect (Table 1).
Chelator-Induced Iron Clearance in Non-Iron Overloaded Bile Duct-Cannulated Rodents
A measure of the amount of iron excretion induced by a chelator is best described by its iron clearing efficiency (ICE). The ICE, expressed as a percent, is calculated as (ligand-induced iron excretion/theoretical iron excretion) × 100. To illustrate, the theoretical iron excretion after administration of one millimole of DFO, a hexadentate chelator that forms a 1:1 complex with Fe(III), is one milli-g-atom of iron. Two millimoles of desferrithiocin (DFT, 1, Table 1), a tridentate chelator that forms a 2:1 complex with Fe(III), are required for the theoretical expression of one milli-g-atom of iron.
The ICE values for compounds 1–3, 7 and 9 (Table 1) are historical and included for comparative purposes.38,42,47,54,55 The biliary ferrokinetics profiles of ligands 3 and 6–10 are presented in Figure 2. Each of the rats in these studies was given a single po dose of the chelator at 300 µmol/kg. Note that the biliary ferrokinetics data for compounds 1 and 2 are not included, simply because these animals were dosed at 150 µmol/kg and the curves are not strictly comparable with the 300 µmol/kg data. Although these results are published elsewhere,38,47 we will comment on this briefly.
Figure 2.

Biliary ferrokinetics of DFT analogues (6, 8, 10) and DADFT analogues (3, 7, 9) in bile duct-cannulated rats. The compounds were given po at 300 µmol/kg. The iron excretion (y-axis) is reported as µg of iron per kg body weight.
DFT (1) given to the rats po at a dose of 150 µmol/kg had an ICE of 5.5 ± 3.2%.47 Maximum iron clearance (MIC) occurred at 3 h, but deferration had returned to baseline levels by 12 h. The desaza analogue of DFT, 2, at 150 µmol/kg had an ICE of 2.7 ± 0.5%.38 The ligand reached MIC at 6 h and had returned to baseline iron excretion by 12 h post-drug. Compound 3 was the least effective ligand, with an ICE of 1.1 ± 0.8%.42 It presented with an MIC at 3 h; deferration was virtually over at 9 h (Figure 2). The DFT analogue of 3, ligand 6, had an ICE that was significantly better than 3, 9.0 ± 3.8% (p < 0.005). MIC occurred at 6 h and its iron decorporation slowly dropped to near baseline levels by 24 h. The most efficient chelator, 7, had an ICE of 26.7 ± 4.7%.54 The ligand also had a very protracted iron clearance; even though its MIC occurred at around 12 h, it was still active at 48 h. Note that although the biliary ferrokinetics curve of 7 may appear to be biphasic (Figure 2), the reason for this unusual line shape is that several animals had temporarily obstructed bile flow. While the concentration of iron in the bile remained the same, the bile volume, and thus overall iron excretion, decreased. Once the obstruction was resolved, bile volume and overall iron excretion normalized.
The DFT analogue 8 had an ICE that was significantly less than that of 7 (11.7 ± 1.2% vs 26.7 ± 4.7% for 8 and 7, respectively, p < 0.02). Ligand 8 also achieved MIC earlier than 7, 6 h vs 12 h (Figure 2) and the iron clearance induced by 8 was basically over by 21 h. In addition, DADFT analogue 955 and its corresponding DFT analogue 10 presented with similar ICEs (~ 15%), but with very different biliary ferrokinetics (Figure 2). Both ligands achieved MIC at 6 h. However, while ligand 9-induced iron clearance had returned to baseline by 24 h, 10 was still quite active.
Finally, there is an excellent correlation between ICE and log Papp in rodents amongst the DFT analogues 6, 8, and 10 (Figure 3). The most lipophilic ligands are also the most active. We have observed this ICE vs log Papp pattern time and again.38,51,52
Figure 3.

Iron-clearing efficiency, expressed as a percentage (y-axis), versus log Papp for DFT analogues 6, 8, and 10 in bile duct-cannulated rats given po at a dose of 300 µmol/kg.
Chelator-Induced Iron Clearance in Iron Overloaded Primates
The primate iron clearance data are provided in Table 1. The ICE values for compounds 1, 2, 3, 7, and 9 are again historical and included for comparative purposes.38,54,55 The chelators were given to the primates po at a dose of 75 µmol/kg (2, 6–10) or 150 µmol/kg (1, 3); 10 was also given to the primates sc at a dose of 75 µmol/kg. Ligand 1 was found to have an ICE of 16.1 ± 8.5%.38 Removal of the pyridine nitrogen to yield 2 increased the ICE to 21.5 ± 12%.38 However, the increase in ICE was not significant (p > 0.05). The introduction of a hydroxyl group at the 4′-position of 2 to provide analogue 3 resulted in a chelator with an ICE of 16.8 ± 7.2%,38 which is within error of the ICE found for 1 and 2 (p > 0.05). The reintroduction of the pyridine nitrogen into DADFT ligand 3 to provide DFT analogue 6 (Table 1) decreased the ICE to 10.0 ± 2.9%, significantly less than its DADFT counterpart, 3, (p < 0.05). When a polyether fragment was attached to the 5′-position of 6 to yield 8, the ICE increased to 18.0 ± 5.2%, again, less than that achieved by the corresponding DADFT ligand 7. Likewise, the ICE of 10 given po was also less than that of DADFT analogue 9 (Table 1). In fact, the DADFT analogues were consistently better deferrating agents in the primates than the corresponding DFT ligands (Table 1).
Several generalizations can be derived from Table 1. The performance ratios, PR values, ICEprimate/ICErodent (Table 1), show that the ligands are either as effective or better at iron clearance in primates than in rodents. The exception to this is ligand 10. The ICE of this drug given po to the primates is 6.1 ± 1.8%, while in the rats is 14.2 ± 2.4%. Its PR ratio was 0.4, showing it to be far less efficient in primates than rodents. The poor iron clearance in primates relative to rodents was surprising. Two scenarios were evaluated in search of an explanation: ligand-plasma binding, and a potential GI absorption problem.
A ligand-plasma binding experiment was performed in which rodent and primate plasma were incubated separately with chelator 10 at 37 °C for 4 h. Each sample was then passed through a Millipore Amicon Ultra regenerated cellulose filter (3,000 MWCO). The filtrate was assayed for 10. The results indicated there was little, if any, binding of the ligand to either the rodent or the primate plasma. This suggests that ligand-plasma binding does not explain the difference in the rodent vs primate ICE values. However, when primates were given ligand 10 sc, the ICE rose to 16.9 ± 7.3%, which is similar to what was seen in rodents given 10 orally (Table 1). This observation is consistent with the idea that the primates do not absorb 10 well when the drug is administered orally.
The Effect of Hydroxylation or Introduction of a Polyether Fragment on DFT Toxicity
In a previous study, 1 was given to rats with normal iron stores po once daily at a dose of 384 µmol/kg/d (100 mg/kg/d). All of the rats were dead by day 5 of a planned 10-d experiment. The chelator was found to be severely nephrotoxic.37 The pathologist noted vacuolar changes of the proximal tubules that were diffuse and severe, with multifocal vaculolar degeneration and necrosis. Nevertheless, the drug’s remarkable oral activity initiated a series of SAR studies aimed at the development of orally active, nontoxic DFT analogues. This led to the development of 3, Table 1, which made it to clinical trials.41 This chelator, when given once daily, cleared iron from the patients and was proceeding forward. Unfortunately, it was discovered that the drug induced proximal tubule nephrotoxicity when it was administered twice daily, and the trial was halted.41 The chelator was reengineered, and it was determined that, by fixing polyether fragments to the 3′- or 4′- position of 2, e.g., 5 and 4 (Chart 1), 7 (Table 1), the renal toxicity virtually disappeared.42,53–55 This outcome suggested that the introduction of either a hydroxyl group or a polyether fragment directly into DFT itself might reduce the drug’s nephrotoxicity. Accordingly, ligands 6, 8 and 10 were synthesized and evaluated for their toxicity in rodents relative to 1.
Assessment of chelator-induced impaired renal function has traditionally relied on the detection of a rise in blood urea nitrogen (BUN) and/or serum creatinine (SCr). However, because of the functional reserve of the kidney, these parameters are often unreliable indicators of acute kidney injury; the ultimate answer requires histopathology. The Critical Path Institute’s Preventive Safety Testing Consortium (PSTC) has identified kidney injury molecule-1 (Kim-1, rat) or (KIM-1, human) as an early diagnostic biomarker for monitoring acute kidney tubular toxicity.63 Kim-1 is a type 1 transmembrane protein located in the epithelial cells of proximal tubules.64,65 After injury, e.g., exposure to a nephrotoxic agent or ischemia, the ectodomain of Kim-1 is shed from the proximal tubular kidney epithelial cells into the urine.66–68 BioAssay Works has recently developed RenaStick, a direct lateral flow immunochromato-graphic assay, which allows for the rapid detection (less than 30 minutes) and quantitation of urinary Kim-1 (rat) or KIM-1 (human) excretion.69 In the current study, rats were treated with 1, 6, 8, and 10 given po twice daily at a dose of 237 µmol/kg/dose (474 µmol/kg/d) for up to 7 d. Urinary Kim-1 levels were assessed at 24-h intervals (Figure 4). The studies were performed on rats with normal iron stores; each animal served as its own control. The data for ligands 3 and 7 are historical and are included for comparative purposes.55
Figure 4.

Urinary Kim-1 excretion (y-axis) is expressed as Kim-1 (ng/kg/24 h) of rats treated with DFT (1), DFT analogues 6, 8, and 10 or DADFT analogues 3 and 7. The rodents were given the drugs po twice daily (b.i.d.) at a dose of 237 µmol/kg/dose (474 µmol/kg/d) for up to 7 d. Note that none of the rats survived the planned 7-d exposure to 1. N = 5 for 1, 6–8, and 10; N = 3 for ligand 3.
None of the 1-treated rats (n = 5) survived the planned 7-d exposure to the drug. Two rats became moribund and were sacrificed after being given the drug for four days. The three remaining animals were found dead the morning of day 6; they had received the chelator for 5 days. None of the rodents produced any urine on day 5. The 1-treated rats’ baseline (day 0) urinary Kim-1 value was < 20 ng/kg/24 h (Figure 4). After one day of 1, the Kim-1 had increased nearly 10-fold, to 192 ± 316 ng/kg/24 h. After three days of 1, the Kim-1 had further increased to 1528 ± 539 ng/kg/24h. Blood was taken from the two moribund animals immediately prior to sacrifice; the serum was assessed for its BUN and SCr content. The rats’ BUN was 139 ± 8 mg/dl (normal 9–30 mg/dl),70 while their SCr was 5.1 ± 0.3 mg/dl (normal 0.4–1 mg/dl).70 In addition, as no blood was obtained from the three animals that were found dead, these values likely underestimate the actual impact of 1 on these parameters.
In contrast, in a previous study assessing the impact of 3 po on urinary Kim-1 excretion,55 all of the treated rats (n = 3) survived the 237 µmol/kg twice daily (474 µmol/kg/d) × 7 d dosing period. The rats’ baseline (day 0) urinary Kim-1 content was < 20 ng/kg/24 h (Figure 4). After three days of exposure to 3, the urinary Kim-1 had increased to 69 ± 47 ng/kg/24 h. At the end of the 7 d dosing period, the urinary Kim-1 had further increased to 189 ± 187 ng/kg/24 h (Figure 4). The rats were euthanized on day 8; their BUN at that time was 32 ± 13 mg/dl, while their SCr was 1.3 ± 1.0 mg/dl.
In the current study, all of the animals treated po with the corresponding hydroxylated DFT analogue, 6, at 237 µmol/kg twice daily (474 µmol/kg/day) also survived the full 7 d of treatment. The rats’ baseline (day 0) urinary Kim-1 content was < 20 ng/kg/24 h and remained < 50 ng/kg/24h until day 5 (Figure 4). On day 6, the Kim-1 increased to 125 ± 48 ng/kg/24 h, and further increased to 435 ± 269 on day 7 (Figure 4). Although the increase in Kim-1 is greater with the 6-treated rats than with the 3-treated animals, the increase is not statistically significant (p > 0.05). The animals were euthanized on day 8; their BUN at that time was 13 ± 2 mg/dl, while their SCr was 0.5 ± 0.1 mg/dl. Thus, simple hydroxylation of the aromatic ring of both DADFT and DFT, e.g., 3 and 6, respectively, resulted in ligands that were much less toxic than DFT itself.
We previously demonstrated that introducing a polyether fragment in the 3′-, or 4′-position of the DADFT pharmacophore provided remarkably efficient orally active iron chelators that were much less toxic than 3.42,43,53–55 For example, the impact of ligand 7 on urinary Kim-1 excretion was determined when the drug was given po: 1) once daily during the course of 28 d toxicity trials; 2) once daily at a dose of 384 µmol/kg/d × 10 d, and 3) twice daily at a dose of 237 µmol/kg/dose (474 µmol/kg/d) × 7 d.55 All of the rats survived the dosing period. The rats’ baseline (day 0) urinary Kim-1 content was < 20 ng/kg/24 h and stayed within error of this value for the duration of the drug exposure. The data from the 237 µmol/kg twice daily (474 µmol/kg/d) × 7 d regimen55 are depicted in Figure 4. In addition, the BUN and SCr of all of the 7-treated rats were well within the normal range.
In the current study, we evaluated the impact that affixing a polyether fragment to DFT itself would have on nephrotoxicity. Accordingly, groups of rats (n = 5) were given 8 or 10 po twice daily at 237 µmol/kg/dose (474 µmol/kg/d) × 7 d. All of the animals survived the drug dosing regimen. The animals’ urinary Kim-1 excretion remained within error of that of the baseline (day 0) levels (Figure 4). In addition, the BUN and SCr of all of the 8- or 10-treated rats were well within the normal range. Thus, as with the DADFT pharmacophore, fixing a polyether fragment to the DFT framework was an effective tool in further reducing nephrotoxicity.
Conclusion
We previously demonstrated that the severe nephrotoxicity associated with 1 could be ameliorated by the removal of the pyridine nitrogen of 1 to provide 2, and simple hydroxylation of the aromatic ring of 2 to yield 3 (Chart 1).38,51 Further reduction in 3-induced nephrotoxicity, observed when the chelator was given po at 237 µmol/kg twice daily, was accomplished by the addition of polyether fragments, e.g., 4, 5, and 7 (Chart 1, Table 1).42,53–55 The purpose of the current study was to determine how these same structural modifications to DFT itself would impact the new ligands’ ICE and nephrotoxicity. Accordingly, three DFT analogues, 6, 8, and 10, were synthesized and assessed for their lipophilicity, ICE properties in rats and primates, and for their toxicity in rats.
DFT (1, Table 1) and its analogues were all significantly more water-soluble (lower log Papp) than the corresponding DADFT analogues, e.g., 1 vs 2, 6 vs 3, 8 vs 7, and 10 vs 9. There was an excellent correlation between ICE and log Papp in rodents amongst the DFT analogues 6, 8, and 10 (Figure 3), with the more lipophilic ligands having a greater ICE. This trend is in keeping with previous observations that more lipophilic ligands have better ICE properties. The biliary ferrokinetics of the DFT and DADFT ligands in the bile duct-cannulated rats (Figure 2) have similar temporal properties, except for ligand 7. This drug has the highest ICE and a very protracted iron clearance time.
In the primates, the DADFT analogues were consistently better deferration agents than the corresponding DFT ligands (Table 1). The most unusual finding was with ligand 10, a DFT analogue with a 4′-(3,6-dioxaheptyloxy) ether functionality fixed to the 4′-position of 1. When the drug was given po to the primates, its ICE was only 6.1 ± 1.8% vs 14.2 ± 2.4% in the rats, and a PR value of 0.4 (Table 1). However, when the monkeys were given the chelator sc, its ICE increased to 16.9 ± 7.3%, with a PR value now at 1.2. This is consistent with the idea that ligand 10 simply was not absorbed well orally in primates.
The effects of structural modification of DFT on its renal toxicity were assessed in rats using a urinary Kim-1 (kidney injury molecule) assay,69 as well as monitoring BUN and SCr. The most notable finding was that fixing a hydroxyl group or a polyether fragment to the DFT aromatic ring resulted in a nearly identical reduction in renal toxicity as seen after the same modification to DADFT (Figure 4). Although some nephrotoxicity was noted with both hydroxylated DADFT and DFT analogues, 3 and 6, respectively, the introduction of polyether groups into either pharmacophore resulted in ligands with little to no impact on renal function, e.g., 7, 8, and 10 (Figure 4).
In summary, rather simple manipulation of the DFT aromatic ring, e.g., hydroxylation, or the introduction of a polyether functionality, can have a marked effect on the ligand’s ICE and renal toxicity (Table 1, Figure 4). Although the resulting DFT chelators were generally as effective in the rodents as their DADFT counterparts, they were less active in the primates. However, the tissue distribution of 6, 8, and 10 in rodents remains to be elucidated. Higher levels of these analogues in the critical target organs, i.e., the liver, heart, and pancreas, could easily compensate for their somewhat lower ICE values. Nevertheless, at least one of the DFT polyethers (8) was sufficiently effective at iron clearance in rodents (ICE 11.7 ± 1.2%) and primates (ICE 18.0 ± 5.2%) and had an acceptable toxicity profile to merit further studies.
Experimental Section
Materials
Reagents were purchased from Aldrich Chemical Co. (Milwaukee, WI). Compound 11 was obtained from Matrix Scientific (Columbia, SC). Fisher Optima grade solvents were routinely used. Reactions were run under a nitrogen atmosphere, and organic extracts were dried with sodium sulfate. Silica gel 40–63 from SiliCycle, Inc. (Quebec City, Quebec, Canada) was used for column chromatography. Melting points are uncorrected. Glassware that was presoaked in 3 N HCl for 15 min, washed with distilled water and distilled EtOH, and oven-dried was used during the isolation of 6, 8 and 10. Optical rotations were run at 589 nm (sodium D line) and 20 °C on a Perkin-Elmer 341 polarimeter, with c being concentration in grams of compound per 100 mL of solvent (CHCl3, not indicated). 1H NMR spectra were run in CDCl3 (not indicated) at 400 MHz, and chemical shifts (δ) are given in parts per million downfield from tetramethylsilane. 13C NMR spectra were measured at 100 MHz, and chemical shifts (δ) are referenced to the residual solvent resonance of δ 77.16 for CDCl3 (not indicated) or δ 39.52 for DMSO-d6. Coupling constants (J) are in hertz. ESI-FTICR mass spectra are reported. The iron content of the Fe(III)-NTA solution was verified using a Perkin Elmer 5100 PC Atomic Absorption Spectrophotometer (AAS). Data for the Job’s plots were recorded on a UV-2550 UV-VIS spectrophotometer. Elemental analyses were performed by Atlantic Microlabs (Norcross, GA) and were within ± 0.4% of the calculated values. Purity of the compounds is supported by high pressure liquid chromatography (HPLC) (≥ 95% for 6, 8, and 10) and by elemental analyses.
Male Sprague-Dawley rats were procured from Harlan Sprague-Dawley (Indianapolis, IN). Male Cebus apella monkeys (3.5–4 kg) were obtained from World Wide Primates (Miami, FL). Ultrapure salts were obtained from Johnson Matthey Electronics (Royston, UK). All hematological and biochemical studies were performed by Antech Diagnostics (Tampa, FL). Atomic absorption (AA) measurements were made on a Perkin-Elmer model 5100 PC (Norwalk, CT). A R-Rena-strip Lateral-flow Kit for the detection of Kim-1 in rat urine was obtained from BioAssay Works (Ijamsville, MD). A Chromatoreader ReaScan (Otsuka Electronics Co., Japan) was utilized to read the test strips and to allow for the quantitation of Kim-1 in rat urine.
Synthetic Methods. (S)-4,5-Dihydro-2-(3,5-dihydroxy-2-pyridinyl)-4-methyl-4-thiazolecarboxylic Acid (6)
A solution of 50% (w/w) NaOH (13.7 g, 0.171 mol) in CH3OH (135 mL) was added to 15 (4.85 g, 17.2 mmol) in CH3OH (125 mL) over 13 min at 0 °C. The reaction mixture was warmed to rt over 19 h, and the bulk of the solvent was removed by rotary evaporation. The concentrate was treated with 3 M aqueous NaCl (150 mL) and was extracted with Et2O (2 × 100 mL). The aqueous layer was cooled in ice, acidified with cold 6 N HCl (30 mL), and extracted with EtOAc (250 mL, 2 × 100 mL). The EtOAc extracts were washed with saturated NaCl (80 mL). Solvent was removed in vacuo, providing 4.18 g of 6 (96%) as an off white solid, mp 226–227 °C (dec): [α] +46.0° (c 0.82, DMF). 1H NMR (DMSO-d6) δ 1.58 (s, 3 H), 3.27 (d, 1 H, J = 11.3), 3.69 (d, 1 H, J = 11.7), 6.72 (d, 1 H, J = 2.4), 7.80 (d, 1 H, J = 2.0), 10.82 (s, 1 H), 12.32 (s, 1 H), 13.20 (s, 1 H). 13C NMR (DMSO-d6) δ 24.32, 38.48, 82.98, 108.59, 125.88, 131.13, 156.68, 157.58, 173.30, 173.74. HRMS m/z calcd for C10H11N2O4S, 255.0434 (M + H), 277.0253 (M + Na), 299.0073 (M – H + 2Na), 320.9892 (M – 2H + 3Na); found, 255.0439, 277.0255, 299.0077, 320.9899. Anal. (C10H10N2O4S) C, H, N.
(S)-4,5-Dihydro-2-[3-hydroxy-5-(3,6-dioxaheptyloxy)-2-pyridinyl]-4-methyl-4-thiazolecarboxylic Acid (8)
A solution of 50% (w/w) NaOH (1.46 mL, 47.0 mmol) in CH3OH (40 mL) was added dropwise to a solution of 17 (1.66 g, 4.32 mmol) in CH3OH (20 mL) at 0 °C. The reaction mixture was stirred at rt for 6 h, and the bulk of the solvent was removed under reduced pressure. The residue was treated with 3 M aqueous NaCl (50 mL) and was extracted with Et2O (2 × 30 mL). The aqueous layer was cooled in ice, acidified with 2 N HCl to pH = 2, and extracted with EtOAc (5 × 40 mL). Combined EtOAc layers were washed with saturated NaCl (60 mL). Solvent removal in vacuo furnished 1.49 g of 8 (97%) as a yellow oil: [α] +25.3° (c 0.88). 1H NMR δ 1.73 (s, 3 H), 3.22 (d, 1 H, J = 12.0), 3.41 (s, 3 H), 3.59–3.61 (m, 2 H), 3.72–3.74 (m, 2 H), 3.83 (d, 1 H, J = 11.6), 3.88 (t, 2 H, J = 4.8), 4.19 (t, 2 H, J = 4.4), 6.84 (d, 1 H, J = 2.4), 7.94 (d, 1 H, J = 2.4). 13C NMR δ 24.62, 39.13, 58.98, 67.99, 69.28, 70.63, 71.81, 82.83, 107.67, 126.98, 131.64, 158.15, 158.44, 174.59, 175.94. HRMS m/z calcd for C15H21N2O6S, 357.1115 (M + H); found, 357.1125. Anal. (C15H20N2O6S) C, H, N.
(S)-4,5-Dihydro-2-[3-hydroxy-4-(3,6-dioxaheptyloxy)-2-pyridinyl]-4-methyl-4-thiazolecarboxylic Acid (10)
Compound 14 (0.78 g, 4.58 mmol), pH 6 phosphate buffer (30 mL), and NaHCO3 (0.44 g, 5.23 mmol) were successively added to a solution of 24 (0.78 g, 3.27 mmol) in degassed CH3OH (30 mL). The reaction mixture was heated at 75 °C for 48 h with stirring, cooled and concentrated by rotary evaporation. The residue was dissolved in distilled H2O (25 mL) and the aqueous layer was acidified with cold 2 N HCl to pH <2 followed by extraction with EtOAc (5 × 50 mL). Concentration in vacuo resulted in 1.15 g of 10 (95%) as a light yellow oil: [α] +52.8° (c 0.40). 1H NMR δ 1.73 (s, 3 H), 3.24 (d, 1 H, J = 11.6), 3.39 (s, 3 H), 3.56–3.58 (m, 2 H), 3.73–3.75 (m, 2 H), 3.85 (d, 1 H, J = 11.6), 3.94 (t, 2 H, J = 4.8), 4.27 (t, 2 H, J = 4.8), 6.88 (d, 1 H, J = 5.2), 8.08 (d, 1 H, J = 4.8). 13C NMR δ 24.65, 39.52, 59.08, 68.57, 69.31, 70.87, 71.94, 83.67, 109.81, 133.20, 141.56, 147.16, 154.39, 175.11, 176.26. HRMS m/z calcd for C15H21N2O6S, 357.1115 (M + H); found, 357.1115. Anal. (C15H20N2O6S) C, H, N.
3,5-Bis(4-methoxybenzyloxy)pyridine-2-carbonitrile (12)
Sodium hydride (60%, 3.66 g, 91.5 mmol) was added to 4-methoxybenzyl alcohol (11.5 mL, 92.6 mmol) in DMF (89 mL). The reaction mixture was stirred for 50 min and was cooled in an ice water bath, followed by addition of 11 (5.13 g, 36.6 mmol). After stirring at rt for 30 min and heating at 95–100 °C for 18 h, the reaction was quenched at 0 °C with EtOH and was concentrated by rotary evaporation under high vacuum. The residue was treated with H2O (250 mL) and extracted with warm EtOAc (400 mL, 2 × 100 mL). The organic extracts were washed with saturated NaCl (150 mL). Purification by flash column chromatography using 2% acetone/CH2Cl2 gave 10.11 g of 12 (73%) as a white solid, mp 122–122.5 °C: 1H NMR δ 3.82 (s, 3 H), 3.83 (s, 3 H), 5.04 (s, 2 H), 5.11 (s, 2 H), 6.85 (d, 1 H, J = 2.0), 6.92 (dd, 4 H, J = 8.6, 6.6), 7.31 (t, 4 H, J = 8.6), 8.01 (d, 1 H, J = 2.0). 13C NMR δ 55.45, 55.47, 70.98, 71.00, 106.86, 114.41, 115.63, 116.26, 126.83, 126.94, 129.04, 129.56, 131.98, 158.41, 159.10, 160.00, 160.14. HRMS m/z calcd for C22H21N2O4, 377.1496 (M + H); found, 377.1500. Anal. (C22H20N2O4) C, H, N.
3,5-Dihydroxy-2-pyridinecarbonitrile (13)
TFA (477 g) was added over 26 min to 12 (9.63 g, 25.6 mmol) and pentamethylbenzene (38.2 g, 0.258 mol) with ice bath cooling. The reaction mixture was stirred at rt for 22 h, and volatiles were removed by rotary evaporation. The residue was partitioned between cold 2 N NaOH (180 mL) and Et2O (350 mL) and separated. The Et2O layer was back extracted with 0.5 N NaOH (80 mL). The combined aqueous phase was extracted with Et2O (100 mL), cooled in an ice water bath, and combined with cold 2 M HCl (220 mL) and saturated NaCl (100 mL). The aqueous layer was extracted with EtOAc (250 mL, 2 × 120 mL). The latter organic extracts were washed with saturated NaCl (150 mL) and concentrated in vacuo, giving 3.70 g of 13 (quantitative) as a light tan solid: 1H NMR (DMSO-d6) δ 6.80 (d, 1 H, J = 2.4), 7.74 (d, 1 H, J = 2.0), 10.98 (s, 1 H), 11.39 (s, 1 H). 13C NMR (DMSO-d6) δ 108.69, 111.17, 116.74, 132.54, 158.10, 159.21. HRMS m/z calcd for C6H3N2O2, 135.0200 (M - H); found, 135.0196. An analytical sample was recrystallized from aqueous EtOH. At > 300 °C, the sample was dark but not melted. Anal. (C6H4N2O2) C, H, N.
Ethyl (S)-4,5-Dihydro-2-(3,5-dihydroxy-2-pyridinyl)-4-methyl-4-thiazolecarboxylate (15)
A degassed solution of 0.1 M phosphate buffer (pH 6, 310 mL) and CH3OH (300 mL) was added to 13 (4.04 g, 29.7 mmol) and 14 (6.95 g, 40.5 mmol). The pH of the reaction solution was adjusted to 6.0 with NaHCO3 (4.92 g, 58.6 mmol). The reaction mixture was heated at 73–76 °C for 45 h with stirring, cooled to 0 °C, and reduced in volume by rotary evaporation. The residue was acidified to pH ~ 1 with cold 2 N HCl (61 mL) followed by extraction with EtOAc (300 mL, 2 × 100 mL). The organic layer was washed with saturated NaCl (100 mL), concentrated in vacuo and dried with toluene, resulting in 6.30 g of 6. Iodoethane (3.0 mL, 37.5 mmol) and DIEA (6.5 mL, 37.3 mmol) were successively added to 6 in DMF (130 mL), and the solution was stirred at rt for 47 h. After solvent removal under high vacuum, the residue was treated with 12:5 0.5 M HCl/saturated NaCl (170 mL) followed by extraction with EtOAc (150 mL, 4 × 70 mL). The EtOAc layers were washed with 100 mL portions of 1% NaHSO3 and saturated NaCl, and the solvent was evaporated. Purification by column chromatography using (5% acetone/CH2Cl2) gave 5.88 g of 15 (70%) as a pale yellow solid, mp 85–87.5 °C: [α] +35.6° (c 0.74). 1H NMR δ 1.32 (t, 3 H, J = 7.2), 1.69 (s, 3 H), 3.20 (d, 1 H, J = 11.7), 3.79 (d, 1 H, J = 11.7), 4.27 (q, 2 H, J = 7.2), 6.77 (d, 1 H, J = 2.4), 7.82 (d, 1 H, J = 2.3). 13C NMR δ 14.23, 24.78, 39.60, 62.33, 83.67, 110.11, 127.71, 130.80, 156.17, 157.79, 173.21, 174.02. HRMS m/z calcd for C12H15N2O4S, 283.0747 (M + H), 305.0567 (M + Na); found, 283.0751, 305.0573. Anal. (C12H14N2O4S) C, H, N.
Ethyl (S)-4,5-Dihydro-2-[3-hydroxy-5-(3,6-dioxaheptyloxy)-2-pyridinyl]-4-methyl-4-thiazolecarboxylate (17)
Flame activated K2CO3 (0.72 g, 5.21 mmol) was added to a mixture of 16 (0.96 g, 3.2 mmol) and 15 (0.90 g, 3.19 mmol) in dry acetone (25 mL). The reaction mixture was heated at reflux for 24 h. After cooling to rt the solvent was removed by rotary evaporation. The residue was treated with 1:9 0.2 N HCl/saturated NaCl (50 mL) and was extracted with EtOAc (4 × 30 mL). The organic extracts were washed with saturated NaCl (50 mL) and solvent was removed in vacuo. Column chromatography using 1:2:7 CH3OH/hexane/CH2Cl2 furnished 0.80 g of 17 (65%) as a viscous oil: [α] +30.9° (c 1.12). 1H NMR δ 1.30 (t, 3 H, J = 7.0), 1.67 (s, 3 H), 3.19 (d, 1 H, J = 11.3), 3.40 (s, 3 H), 3.56–3.62 (m, 2 H), 3.70–3.75 (m, 2 H), 3.80 (d, 1 H, J = 11.7), 3.86–3.93 (m, 2 H), 4.19 (t, 2 H, J = 4.7), 4.25 (q, 2 H, J = 7.0), 6.80 (d, 1 H, J = 2.3), 7.95 (d, 1 H, J = 2.3), 12.37 (s, 1 H). 13C NMR δ 14.23, 24.77, 39.45, 59.25, 62.04, 68.11, 69.46, 70.99, 72.01, 83.84, 107.63, 127.72, 131.49, 157.39, 158.22, 172.87, 173.96. HRMS m/z calcd for C17H25N2O6S, 385.1428 (M + H), 407.1247 (M + Na); found, 385.1432, 407.1266. Anal. (C17H24N2O6S) C, H, N.
2-Methyl-3-(benzyloxy)-4-(3,6-dioxaheptyloxy)pyridine (19)
Flame activated K2CO3 (27.6 g, 0.20 mol) and 16 (27.4 g, 0.10 mol) were added to 18 (21.5 g, 0.10 mol) in dry CH3CN (500 mL). The reaction mixture was heated at reflux for 24 h. After cooling to rt, the solvent was evaporated by rotary evaporation. The residue was treated with 1.7 M aqueous NaCl (200 mL) and was extracted with CH2Cl2 (4 × 150 mL). The organic extracts were washed with saturated NaCl (300 mL). After solvent was removed in vacuo, column chromatography using 4:4:2 EtOAc/petroleum ether/acetone furnished 21.5 g of 19 (68%) as a colorless viscous oil: 1H NMR δ 2.42 (s, 3 H), 3.34 (s, 3 H), 3.51–3.53 (m, 2 H), 3.69–3.71 (m, 2 H), 3.91 (t, 2 H, J = 4.8), 4.24 (t, 2 H, J = 4.4), 5.02 (s, 2 H), 6.72 (d, 1 H, J = 5.6), 7.31–7.40 (m, 3 H), 7.44–7.49 (m, 2 H), 8.12 (d, 1 H, J = 5.6). 13C NMR δ 19.34, 59.16, 67.86, 69.45, 70.92, 71.99, 74.57, 106.68, 128.21, 128.45, 128.49, 137.53, 142.32, 145.41, 153.40, 157.64. HRMS m/z calcd for C18H24NO4, 318.1700 (M + H); found, 318.1714. Anal. (C18H23NO4 • 0.2 H2O) C, H, N.
4-(3,6-Dioxaheptyloxy)-3-(benzyloxy)-2-pyridinemethanol (20)
An ice cooled solution of m-CPBA (3.67 g, 36.0 mmol) in CH2Cl2 (75 mL) was added slowly to 19 (10.4 g, 32.8 mmol) in CH2Cl2 (50 mL) over 15 min at 0 °C. The reaction mixture was warmed to rt, stirred for 6 h, and diluted with CH2Cl2 (150 mL). The reaction mixture was washed with 5% Na2CO3 (3 × 100 mL) and saturated NaCl (100 mL) and was concentrated under reduced pressure to give a colorless oil. Acetic anhydride (80 mL, 0.85 mol) was added, and the reaction mixture was heated at 130 °C for 2 h. The solvent was removed under reduced pressure and the residue was dissolved in H2O (100 mL). The pH of the aqueous solution was adjusted to 8 with 2 N NaOH, and the aqueous solution was extracted with CH2Cl2 (3 × 100 mL). The organic fractions were combined and washed with saturated NaCl (100 mL), and concentrated in vacuo. The residue was dissolved in CH3OH, treated with decolorizing charcoal, filtered and concentrated to yield a brown oil, which was dissolved in EtOH (40 mL). Aqueous 1 M NaOH (80 mL) was added and the reaction mixture was refluxed for 4 h and cooled. Extraction with CH2Cl2 (4 × 100 mL), washing with saturated NaCl (100 mL), concentration under reduced pressure, and column chromatography using 10% CH3OH/CHCl3 provided 9.52 g (87%) of 20 as a light brown oil: 1H NMR δ 3.34 (s, 3 H), 3.52−3.54 (m, 2 H), 3.69−3.71 (m, 2 H), 3.92 (t, 2 H, J = 5.2), 4.28 (t, 2 H, J = 4.4), 4.65 (s, 2 H), 5.09 (s, 2 H), 6.83 (d, 1 H, J=5.2) 7.32–7.39 (m, 3 H), 7.40–7.44 (m, 2 H), 8.19 (d, 1 H, J = 5.6). 13C NMR δ 59.20, 60.23, 68.13, 69.44, 70.96, 72.04, 74.79, 107.83, 128.49, 128.54, 128.63, 137.15, 140.53, 144.66, 152.98, 157.52. HRMS m/z calcd for C18H24NO5, 334.1649 (M + H), 356.1468 (M + Na); found, 334.1648, 356.1455. Anal. (C18H23NO5) C, H, N.
4-(3,6-Dioxaheptyloxy)-3-(benzyloxy)pyridine-2-carboxaldehyde (21)
Triethylamine (70 mL, 0.29 mol) followed by DMSO (70 mL) was added to 20 (16.5 g, 49.0 mmol) in CHCl3 (100 mL). Sulfur trioxide-pyridine complex (35 g, 0.22 mol) was slowly added over 35 min to the reaction mixture with ice bath cooling. After warming to rt, the reaction mixture was stirred overnight and was diluted with CHCl3 (200 mL). The organic phase was washed with H2O (3 × 200 mL) and saturated NaCl (100 mL). After solvent was removed in vacuo, column chromatography using 5:5:1 EtOAc/CHCl3/CH3OH furnished 13.61 g of 21 (83%) as a viscous colorless oil: 1H NMR δ 3.34 (s, 3 H), 3.52–3.54 (m, 2 H), 3.70–3.72 (m, 2 H), 3.95 (t, 2 H, J = 4.4), 4.30 (t, 2 H, J = 4.4), 5.24 (s, 2 H), 7.02 (d, 1 H, J = 5.2), 7.32–7.39 (m, 3 H), 7.41–7.46 (m, 2 H), 8.39 (d, 1 H, J = 5.6), 10.25 (s, 1 H). 13C NMR δ 59.17, 68.54, 69.23, 70.94, 71.97, 76.24, 111.69, 128.68, 128.75, 128.87, 136.16, 145.87, 146.90, 148.14, 159.49, 189.87. HRMS m/z calcd for C18H21NNaO5, 354.1312 (M + Na); found, 354.1326. Anal. (C18H21NO5) C, H, N.
4-(3,6-Dioxaheptyloxy)-3-(benzyloxy)pyridine-2-carboxaldehyde Oxime (22)
Hydroxylamine hydrochloride (4.2 g, 60.0 mmol) and NaOAc (5.2 g, 60.0 mmol) were added to a solution of 21 (13.5 g, 40.7 mmol) in CH3OH (50 mL), and the reaction mixture was heated at reflux for 2 h. The reaction mixture was concentrated by rotary evaporation, and the residue was treated with saturated NaCl (100 mL) and 0.1 M aqueous citric acid (100 mL) and then was extracted with EtOAc (2 × 100 mL). The organic layers were washed with H2O (100 mL) and saturated NaCl (100 mL). Solvent was removed in vacuo, providing 12.7 g (90%) of 22 as a pale solid, mp 72–73 °C: 1H NMR δ 3.34 (s, 3 H), 3.46–3.51 (m, 2 H), 3.64–3.71 (m, 2 H), 3.92 (t, 2 H, J = 4.4), 4.27 (t, 2 H, J = 4.4), 5.10 (s, 2 H), 6.85 (d, 1 H, J = 5.6), 7.29–7.46 (m, 5 H), 8.28 (d, 1 H, J = 5.2), 8.46 (s, 1 H). 13C NMR δ 59.14, 68.12, 69.28, 70.86, 71.94, 75.67, 108.62, 128.42, 128.55, 128.59, 136.70, 143.43, 144.81, 145.23, 146.71, 158.66. HRMS m/z calcd for C18H21N2O5, 345.1450 (M - H); found, 345.1426. Anal. (C18H22NO5) C, H, N.
4-(3,6-Dioxaheptyloxy)-3-(benzyloxy)pyridine-2-carbonitrile (23)
Compound 22 (12.15 g, 36.71 mmol) was dissolved in Ac2O (40 mL) and heated at reflux for 8 h under a Drierite tube. The reaction mixture was concentrated by rotary evaporation and was dissolved in 8% aqueous NaHCO3 (100 mL) and extracted with CHCl3 (100 mL, 2 × 50 mL). Combined organic fractions were washed with 4% NaHCO3 (50 mL) and saturated NaCl (100 mL) followed by solvent removal in vacuo. Purification by flash chromatography eluting with 10% CH3OH/CH2Cl2 gave 11.31 g (94%) of 23 as a pale solid, mp 34–35 °C: 1H NMR δ 3.34 (s, 3 H), 3.53–3.55 (m, 2 H), 3.70–3.72 (m, 2 H), 3.93 (t, 2 H, J = 4.8), 4.27 (t, 2 H, J = 4.4), 5.31 (s, 2 H), 6.98 (d, 1 H, J = 5.6), 7.31–7.38 (m, 3 H), 7.49–7.52 (m, 2 H), 8.21 (d, 1 H, J = 5.2). 13C NMR δ 59.14, 68.59, 69.09, 70.92, 71.94, 75.84, 111.27, 115.43, 128.60, 128.66, 128.73, 128.84, 135.78, 147.21, 148.28, 158.42. HRMS m/z calcd for C18H20N2NaO4, 351.1315 (M + Na); found, 351.1325. Anal. (C18H20N2O4) C, H, N.
4-(3,6-Dioxaheptyloxy)-3-hydroxy-2-pyridinecarbonitrile (24)
Palladium on carbon (10%, 0.065 g) was added to a solution of 23 (1.30 g, 3.95 mmol) in CH3OH (15 mL), and the mixture was stirred under H2 at 1 atm for 2 h. The reaction mixture was filtered through Celite, and the solids were washed with CH3OH (3 × 5 mL). The filtrate was concentrated under reduced pressure, and the residue was subjected to column chromatography eluting with 10% CH3OH/EtOAc, furnishing 0.80 g (85%) of 24 as a colorless oil: 1H NMR δ 3.42 (s, 3 H), 3.61–3.63 (m, 2 H), 3.75–3.77 (m, 2 H), 3.92 (t, 2 H, J = 4.8), 4.24 (t, 2 H, J = 4.4), 6.91 (d, 1 H, J = 4.8), 8.10 (d, 1 H, J = 5.6). 13C NMR δ 58.92, 68.66, 69.05, 70.52, 71.78, 110.68, 115.28, 120.29, 143.49, 148.84, 153.72. HRMS m/z calcd for C11H14N2NaO4, 261.0846 (M + Na); found, 261.0849. Anal. (C11H14N2O4) C, H, N.
Job’s Plots for 6, 8 and 10
The stoichiometries of the ligand-Fe(III) complexes of 6, 8 and 10 were determined spectrophotometrically using Job’s plots. Solutions were monitored at the visible λmax of the Fe(III) complexes (498 nm for 6, 484 nm for 8, and 485 nm for 10). A 100 mM Tris HCl buffer was used to maintain the pH at 7.4. Solutions containing different ligand/Fe(III) ratios were prepared by mixing appropriate volumes of 1.0 mM ligand solution and 1.0 mM Fe(III)-nitriloacetate (NTA) in Tris-HCl buffer. The 1.0 mM Fe(III)-NTA solution was prepared immediately prior to use by dilution of a 41.6 mM Fe(III)-NTA stock solution with the Tris HCl buffer, whereas the ligand’s stock solution was prepared by dissolving the ligand as its monosodium salt in Tris HCl buffer at pH 7.4. The Fe(III)-NTA stock solution was prepared by mixing equal volumes of 90 mM of FeCl3 and 180 mM trisodium NTA. The iron content of the Fe(III)-NTA solution was verified by AAS.
Biological Methods
All animal experimental treatment protocols were reviewed and approved by the University of Florida’s Institutional Animal Care and Use Committee.
Cannulation of Bile Duct in Non-iron-overloaded Rats
The cannulation has been described previously.37,48 Bile samples were collected from male Sprague-Dawley rats (400–450 g) at 3 h intervals for up to 48 h. The urine sample(s) was taken at 24 h intervals. Sample collection and handling are as previously described.37,48
Iron Loading of C. apella Monkeys
The monkeys were iron overloaded with intravenous iron dextran as specified in earlier publications48,71 to provide about 500 mg of iron per kg of body weight; the serum transferrin iron saturation rose to between 70 and 80%. At least 20 half-lives, 60 days,72 elapsed before any of the animals were used in experiments evaluating iron-chelating agents.
Primate Fecal and Urine Samples
Fecal and urine samples were collected at 24 h intervals and processed as described previously.37,48,73 Briefly, the collections began 4 days prior to the administration of the test drug and continued for an additional 5 days after the drug was given. Iron concentrations were determined by flame absorption spectroscopy as presented in other publications.48,74
Drug Preparation and Administration: Iron Clearance
In the iron clearing experiments, the rats were given 6, 8 and 10 po at a dose of 300 µmol/kg. The primates were given 6, 8 and 10 po at a dose of 75 µmol/kg; ligand 10 was also given sc at a dose of 75 µmol/kg. The drugs were administered to the rats and primates as their monosodium salts (prepared by the addition of 1 equiv of NaOH to a suspension of the free acid in distilled water). Drug preparation for the rodent urinary Kim-1 excretion studies involving 1, 6, 8, and 10 are described below.
Calculation of Iron Chelator Efficiency
In the text below, the term “iron-clearing efficiency” (ICE) is used as a measure of the amount of iron excretion induced by a chelator. The ICE, expressed as a percent, is calculated as (ligand-induced iron excretion/theoretical iron excretion) × 100. To illustrate, the theoretical iron excretion after administration of one millimole of DFO, a hexadentate chelator that forms a 1:1 complex with Fe(III), is one milli-g-atom of iron. Two millimoles of desferrithiocin (DFT, 1, Table 1), a tridentate iron chelator that forms a 2:1 complex with Fe(III), are required for the theoretical excretion of one milli-g-atom of iron. The theoretical iron outputs of the chelators were generated on the basis of a 2:1 ligand:iron complex. The efficiencies in the rats and monkeys were calculated as set forth elsewhere.38,73 Data are presented as the mean ± the standard error of the mean; p-values were generated via a one-tailed Student’s t-test in which the inequality of variances was assumed; and a p-value of < 0.05 was considered significant.
Drug Preparation and Administration: Rodent Toxicity/ Urinary Kim-1 Excretion Studies
The impact of ligands 1, 6, 8 and 10 on urinary Kim-1 excretion were evaluated in rodents. The chelators were administered to the rats po as their monosodium salts, prepared as described above, twice daily at a dose of 237 µmol/kg/dose (474 µmol/kg/d) for up to 7 d. The studies were performed on rats with normal iron stores. The rats were fasted overnight and were given the first dose of the chelator first thing in the morning. The rats were fed ~3 h post-drug and had access to food for ~5 h before being fasted overnight.
Collection of Urine for Kim-1 Studies
The rats were housed in individual metabolic cages. Urine samples were collected from the metabolic cages at 24 h intervals. A baseline (day 0) urine sample was collected and assessed for its Kim-1 content; each animal served as its own control. The urine was collected chilled as previously described.55
Performance of Urinary Kim-1 Studies
The chilled urine was collected, vortexed, and warmed to room temperature; any sediment in the samples was allowed to settle. Kim-1 content was assessed using a Rat Kim-1 Rapid Test Kit according to the manufacturer’s instructions. The result was read using a ReaScan Test Reader. The quantity of Kim-1 excreted in the urine per day was calculated by multiplying the concentration of Kim-1 (ng/ml urine) × 24-h urine volume, divided by the weight of the animal. The result is expressed as urinary Kim-1 (ng/kg/24 h). Data are presented as the mean ± the standard error of the mean; p-values were generated via a one-tailed Student’s t-test in which the inequality of variances was assumed; and a p-value of < 0.05 was considered significant.
Supplementary Material
Acknowledgments
The project described was supported by grant number R37DK049108 from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Diseases or the National Institutes of Health. We thank Elizabeth M. Nelson and Katie Ratliff-Thompson for their technical assistance, and Miranda E. Coger for her editorial and organizational support. We acknowledge the spectroscopy services in the Chemistry Department, University of Florida, for the mass spectrometry analyses.
Footnotes
Supporting Information Available. Elemental analytical data for synthesized compounds are shown.
Abbreviations: DFO, desferrioxamine B mesylate; DFT, desferrithiocin (S)-4,5-dihydro…; ICE, iron clearing efficiency; DADFT, desazadesferrithiocin (S)-…; SAR, structure-activity relationship; BUN, Blood Urea Nitrogen; SCr, serum creatinine; PR, performance ratio; Kim-1, kidney injury molecule-1; MIC, maximum iron clearance; b.i.d., twice daily
References
- 1.Raymond KN, Carrano CJ. Coordination Chemistry and Microbial Iron Transport. Acc. Chem. Res. 1979;12:183–190. doi: 10.1021/acs.accounts.5b00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byers BR, Arceneaux JE. Microbial Iron Transport: Iron Acquisition by Pathogenic Microorganisms. Met. Ions Biol. Syst. 1998;35:37–66. [PubMed] [Google Scholar]
- 3.Bergeron RJ. Iron: A Controlling Micronutrient in Proliferative Processes. Trends Biochem. Sci. 1986;11:133–136. [Google Scholar]
- 4.Theil EC, Huynh BH. Ferritin Mineralization: Ferroxidation and Beyond. J. Inorg. Biochem. 1997;67:30. [Google Scholar]
- 5.Ponka P, Beaumont C, Richardson DR. Function and Regulation of Transferrin and Ferritin. Semin. Hematol. 1998;35:35–54. [PubMed] [Google Scholar]
- 6.Brittenham GM. Disorders of Iron Metabolism: Iron Deficiency and Overload. In: Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohen HJ, et al., editors. Hematology: Basic Principles and Practice. 3rd ed. New York: Churchill Livingstone; 2000. pp. 397–428. [Google Scholar]
- 7.Olivieri NF, Brittenham GM. Iron-Chelating Therapy and the Treatment of Thalassemia. Blood. 1997;89:739–761. [PubMed] [Google Scholar]
- 8.Vichinsky EP. Current Issues with Blood Transfusions in Sickle Cell Disease. Semin. Hematol. 2001;38:14–22. doi: 10.1016/s0037-1963(01)90056-3. [DOI] [PubMed] [Google Scholar]
- 9.Kersten MJ, Lange R, Smeets ME, Vreugdenhil G, Roozendaal KJ, Lameijer W, Goudsmit R. Long-Term Treatment of Transfusional Iron Overload with the Oral Iron Chelator Deferiprone (L1): A Dutch Multicenter Trial. Ann. Hematol. 1996;73:247–252. doi: 10.1007/s002770050236. [DOI] [PubMed] [Google Scholar]
- 10.Conrad ME, Umbreit JN, Moore EG. Iron Absorption and Transport. Am. J. Med. Sci. 1999;318:213–229. doi: 10.1097/00000441-199910000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Lieu PT, Heiskala M, Peterson PA, Yang Y. The Roles of Iron in Health and Disease. Mol. Aspects Med. 2001;22:1–87. doi: 10.1016/s0098-2997(00)00006-6. [DOI] [PubMed] [Google Scholar]
- 12.Graf E, Mahoney JR, Bryant RG, Eaton JW. Iron-Catalyzed Hydroxyl Radical Formation. Stringent Requirement for Free Iron Coordination Site. J. Biol. Chem. 1984;259:3620–3624. [PubMed] [Google Scholar]
- 13.Halliwell B. Iron, Oxidative Damage, and Chelating Agents. In: Bergeron RJ, Brittenham GM, editors. The Development of Iron Chelators for Clinical Use. Boca Raton, FL: CRC; 1994. pp. 33–56. [Google Scholar]
- 14.Koppenol W. Kinetics and Mechanism of the Fenton Reaction: Implications for Iron Toxicity. In: Badman DG, Bergeron RJ, Brittenham GM, editors. Iron Chelators: New Development Strategies. Ponte Vedra Beach, FL: Saratoga; 2000. pp. 3–10. [Google Scholar]
- 15.Hazen SL, d’Avignon A, Anderson MM, Hsu FF, Heáinecke JW. Human Neutrophils Employ the Myeloperoxidase-Hydrogen Peroxide- Chloride System to Oxidize α-Amino Acids to a Family of Reactive Aldehydes. Mechanistic Studies Identifying Labile Intermediates along the Reaction Pathway. J. Biol. Chem. 1998;273:4997–5005. doi: 10.1074/jbc.273.9.4997. [DOI] [PubMed] [Google Scholar]
- 16.Millán M, Sobrino T, Arenillas JF, Rodriguez-Yáñez M, García M, Nombela F, Castellanos M, de la Ossa NP, Cuadras P, Serena J, Castillo J, Dávalos A. Biological Signatures of Brain Damage Associated with High Serum Ferritin Levels in Patients with Acute Ischemic Stroke and Thrombolytic Treatment. Dis. Markers. 2008;25:181–188. doi: 10.1155/2008/380356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zecca L, Casella L, Albertini A, Bellei C, Zucca FA, Engelen M, Zadlo A, Szewczyk G, Zareba M, Sarna T. Neuromelanin Can Protect Against Iron-Mediated Oxidative Damage in System Modeling Iron Overload of Brain Aging and Parkinson’s Disease. J. Neurochem. 2008;106:1866–1875. doi: 10.1111/j.1471-4159.2008.05541.x. [DOI] [PubMed] [Google Scholar]
- 18.Pietrangelo A. Iron Chelation Beyond Tranfusion Iron Overload. Am. J. Hematol. 2007;82:1142–1146. doi: 10.1002/ajh.21101. [DOI] [PubMed] [Google Scholar]
- 19.Dunaief JL. Iron Induced Oxidative Damage as a Potential Factor in Age-Related Macular Degeneration: The Cogan Lecture. Invest. Ophthalmol. Vis. Sci. 2006;47:4660–4664. doi: 10.1167/iovs.06-0568. [DOI] [PubMed] [Google Scholar]
- 20.Hua Y, Nakamura T, Keep RF, Wu J, Schallert T, Hoff JT, Xi G. Long-Term Effects of Experimental Intracerebral Hemorrhage: The Role of Iron. J. Neurosurg. 2006;104:305–312. doi: 10.3171/jns.2006.104.2.305. [DOI] [PubMed] [Google Scholar]
- 21.Pippard MJ. Iron Overload and Iron Chelation Therapy in Thalassaemia and Sickle Cell Haemoglobinopathies. Acta. Haematol. 1987;78:206–211. doi: 10.1159/000205876. [DOI] [PubMed] [Google Scholar]
- 22.Olivieri NF. Progression of Iron Overload in Sickle Cell Disease. Semin. Hematol. 2001;38:57–62. doi: 10.1016/s0037-1963(01)90060-5. [DOI] [PubMed] [Google Scholar]
- 23.Malcovati L. Impact of Transfusion Dependency and Secondary Iron Overload on the Survival of Patients with Myelodysplastic Syndromes. Leukemia Res. 2007;31:S2–S6. doi: 10.1016/S0145-2126(07)70459-9. [DOI] [PubMed] [Google Scholar]
- 24.Kalinowski DS, Richardson DR. The Evolution of Iron Chelators for the Treatment of Iron Overload Disease and Cancer. Pharm. Rev. 2005;57:547–583. doi: 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]
- 25.Brittenham GM. Iron-Chelating Therapy for Transfusional Iron Overload. New Engl. J. Med. 2011;364:146–156. doi: 10.1056/NEJMct1004810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Desferal. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2008. http://www.pharma.us.novartis.com/product/pi/pdf/desferal.pdf. [Google Scholar]
- 27.Hoffbrand AV, Al-Refaie F, Davis B, Siritanakatkul N, Jackson BFA, Cochrane J, Prescott E, Wonke B. Long-Term Trial of Deferiprone in 51 Transfusion-Dependent Iron Overloaded Patients. Blood. 1998;91:295–300. [PubMed] [Google Scholar]
- 28.Olivieri NF. Long-Term Therapy with Deferiprone. Acta Haematol. 1996;95:37–48. doi: 10.1159/000203854. [DOI] [PubMed] [Google Scholar]
- 29.Olivieri NF, Brittenham GM, McLaren CE, Templeton DM, Cameron RG, McClelland RA, Burt AD, Fleming KA. Long-Term Safety and Effectiveness of Iron-Chelation Therapy with Deferiprone from Thalassemia Major. N. Engl. J. Med. 1998;339:417–423. doi: 10.1056/NEJM199808133390701. [DOI] [PubMed] [Google Scholar]
- 30.Richardson DR. The Controversial Role of Deferiprone in the Treatment of Thalassemia. J. Lab. Clin. Med. 2001;137:324–329. doi: 10.1067/mlc.2001.114105. [DOI] [PubMed] [Google Scholar]
- 31.Nisbet-Brown E, Olivieri NF, Giardini PJ, Grady RW, Neufeld EJ, Séchaud R, Krebs-Brown AJ, Anderson JR, Alberti D, Sizer KC, Nathan DG. Effectiveness and Safety of ICL670 in Iron-Loaded Patients with Thalassemia: A Randomised, Double-Blind, Placebo-Controlled, Dose-Escalation Trial. Lancet. 2003;361:1597–1602. doi: 10.1016/S0140-6736(03)13309-0. [DOI] [PubMed] [Google Scholar]
- 32.Galanello R, Piga A, Alberti D, Rouan M-C, Bigler H, Séchaud R. Safety, Tolerability, and Pharmacokinetics of ICL670, a New Orally Active Iron-Chelating Agent in Patients with Transfusion-Dependent Iron Overload Due to β-Thalassemia. J. Clin. Pharmacol. 2003;43:565–572. [PubMed] [Google Scholar]
- 33.Cappellini MD. Iron-Chelating Therapy with the New Oral Agent ICL670 (Exjade) Best Pract. Res. Clin. Haematol. 2005;18:289–298. doi: 10.1016/j.beha.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Exjade Prescribing Information. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2012. Mar, http://www.pharma.us.novartis.com/product/pi/pdf/exjade.pdf. [Google Scholar]
- 35.Pippard MJ. Desferrioxamine-Induced Iron Excretion in Humans. Bailliere’s Clin. Haematol. 1989;2:323–343. doi: 10.1016/s0950-3536(89)80020-4. [DOI] [PubMed] [Google Scholar]
- 36.Giardina PJ, Grady RW. Chelation Therapy in β-Thalassemia: An Optimistic Update. Semin. Hematol. 2001;38:360–366. doi: 10.1016/s0037-1963(01)90030-7. [DOI] [PubMed] [Google Scholar]
- 37.Bergeron RJ, Streiff RR, Creary EA, Daniels RD, Jr, King W, Luchetta G, Wiegand J, Moerker T, Peter HH. A Comparative Study of the Iron-Clearing Properties of Desferrithiocin Analogues with Desferrioxamine B in a Cebus Monkey Model. Blood. 1993;81:2166–2173. [PubMed] [Google Scholar]
- 38.Bergeron RJ, Wiegand J, McManis JS, McCosar BH, Weimar WR, Brittenham GM, Smith RE. Effects of C-4 Stereochemistry and C-4′ Hydroxylation on the Iron Clearing Efficiency and Toxicity of Desferrithiocin Analogues. J. Med. Chem. 1999;42:2432–2440. doi: 10.1021/jm990058s. [DOI] [PubMed] [Google Scholar]
- 39.Bergeron RJ, Wiegand J, McManis JS, Bussenius J, Smith RE, Weimar WR. Methoxylation of Desazadesferrithiocin Analogues: Enhanced Iron Clearing Efficiency. J. Med. Chem. 2003;46:1470–1477. doi: 10.1021/jm020412d. [DOI] [PubMed] [Google Scholar]
- 40.Bergeron RJ, Wiegand J, Weimar WR, Vinson JRT, Bussenius J, Yao GW, McManis JS. Desazadesmethyldesferrithiocin Analogues as Orally Effective Iron Chelators. J. Med. Chem. 1999;42:95–108. doi: 10.1021/jm980340j. [DOI] [PubMed] [Google Scholar]
- 41.Galanello R, Forni G, Jones A, Kelly A, Willemsen A, He X, Johnston A, Fuller D, Donovan J, Piga A. A Dose Escalation Study of the Pharmacokinetics, Safety, and Efficacy of Deferitrin, an Oral Iron Chelator in Beta Thalassaemia Patients. ASH Annu. Meet. Abstr. 2007;110:2669. [Google Scholar]
- 42.Bergeron RJ, Wiegand J, McManis JS, Vinson JRT, Yao H, Bharti N, Rocca JR. (S)-4,5-Dihydro-2-(2-hydroxy-4-hydroxyphenyl)-4-methyl-4-thiazolecarboxylic Acid Polyethers: A Solution to Nephrotoxicity. J. Med. Chem. 2006;49:2772–2783. doi: 10.1021/jm0508944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bergeron RJ, Wiegand J, Bharti N, Singh S, Rocca JR. Impact of 3,6,9-Trioxadecyloxy Group on Desazadesferrithiocin Analogue Iron Chelators and Organ Distribution. J. Med. Chem. 2007;50:3302–3313. doi: 10.1021/jm070214s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naegeli H-U, Zähner H. Metabolites of Microorganisms. Part 193. Ferrithiocin. Helv. Chim. Acta. 1980;63:1400–1406. [Google Scholar]
- 45.Hahn FE, McMurry TJ, Hugi A, Raymond KN. Coordination Chemistry of Microbial Iron Transport. 42. Structural and Spectroscopic Characterization of Diastereomeric CRIII) and Co(III) Complexes of Desferriferrithiocin. J. Am. Chem. Soc. 1990;112:1854–1860. [Google Scholar]
- 46.Anderegg G, Räber M. Metal Complex Formation of a New Siderophore Desferrithiocin and of Three Related Ligands. J. Chem. Soc., Chem. Commun. 1990:1194–1196. [Google Scholar]
- 47.Bergeron RJ, Wiegand J, Dionis JB, Egli-Karmakka M, Frei J, Huxley-Tencer A, Peter HH. Evaluation of Desferrithiocin and Its Synthetic Analogues as Orally Effective Iron Chelators. J. Med. Chem. 1991;34:2072–2078. doi: 10.1021/jm00111a023. [DOI] [PubMed] [Google Scholar]
- 48.Bergeron RJ, Streiff RR, Wiegand J, Vinson JRT, Luchetta G, Evans KM, Peter H, Jenny H-B. A Comparative Evaluation of Iron Clearance Models. Ann. N.Y. Acad. Sci. 1990;612:378–393. doi: 10.1111/j.1749-6632.1990.tb24325.x. [DOI] [PubMed] [Google Scholar]
- 49.Wolfe LC, Nicolosi RJ, Renaud MM, Finger J, Hegsted M, Peter H, Nathan DG. A Non-Human Primate Model for the Study of Oral Iron Chelators. Br. J. Haematol. 1989;72:456–461. doi: 10.1111/j.1365-2141.1989.tb07732.x. [DOI] [PubMed] [Google Scholar]
- 50.Sangster J. Octanol-Water Partition Coefficients: Fundamentals and Physical Chemistry. Vol. 2. West Sussex, England: John Wiley and Sons; 1997. [Google Scholar]
- 51.Bergeron RJ, McManis JS, Weimar WR, Wiegand J, Eiler-McManis E. Iron Chelators and Therapeutic Uses. In: Abraham DA, editor. Burger's Medicinal Chemistry. 6th. New York: Wiley; 2003. pp. 479–561. [Google Scholar]
- 52.Bergeron RJ, Wiegand J, McManis JS, Bharti N, Singh S. Desferrithiocin Analogues and Nephrotoxicity. J. Med. Chem. 2008;51:5993–6004. doi: 10.1021/jm8003398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bergeron RJ, Wiegand J, McManis JS, Bharti N, Singh S. Design, Synthesis, and Testing of Non-Nephrotoxic Desazadesferrithiocin Polyether Analogues. J. Med. Chem. 2008;51:3913–3923. doi: 10.1021/jm800154m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bergeron RJ, Bharti N, Wiegand J, McManis JS, Singh S, Abboud KA. The Impact of Polyether Chain Length on the Iron Clearing Efficiency and Physiochemical Properties of Desferrithiocin Analogues. J. Med. Chem. 2010;53:2843–2853. doi: 10.1021/jm9018146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bergeron RJ, Wiegand J, Bharti N, McManis JS, Singh S. Desferrithiocin Analogue Iron Chelators: Iron Clearing Efficiency, Tissue Distribution, and Renal Toxicity. Biometals. 2011;24:239–258. doi: 10.1007/s10534-010-9389-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Féau C, Klein E, Kerth P, Lebeau L. Preparation and Optical Properties of Novel 3-Alkoxycarbonyl Aza- and Diazacoumarins. Synth. Commun. 2010;40:3033–3045. [Google Scholar]
- 57.Ornelas MA, Gonzalez J, Sach NW, Richardson PF, Bunker KD, Linton A, Kephart SE, Pairish M, Guo C. An Efficient Synthesis of Highly Functionalized Chiral Lactams. Tetrahedron Lett. 2011;52:4760–4763. [Google Scholar]
- 58.Bergeron RJ, Bharti N, Singh S, McManis JS, Wiegand J, Green LG. Vibriobactin Antibodies: A Vaccine Strategy. J. Med. Chem. 2009;52:3801–3813. doi: 10.1021/jm900119q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marriott JH, Moreno-Barber AM, Hardcastle IR, Rowlands MG, Grimshaw RM, Neidle S, Jarman M. Synthesis of the Farnesyl Ether 2,3,5-Trifluoro-6-hydroxy-4-[(E,E)-3,7,11-trimethyldodeca-2,6,10-trien-1-yloxy]nitrobenzene, and Related Compounds Containing a Substituted Hydroxytrifluorophenyl Residue: Novel Inhibitors of Protein Farnesyltransferase, Geranylgeranyltransferase I and Squalene Synthase. J. Chem. Soc., Perkin Trans. 2000;1:4265–4278. [Google Scholar]
- 60.Piyamongkol S, Liu ZD, Hider RC. Novel Synthetic Approach to 2-(1′-Hydroxyalkyl)- and 2-Amido-3-Hydroxypyridin-4-ones. Tetrahedron. 2001;57:3479–3486. [Google Scholar]
- 61.Li M-J, Kwok W-M, Lam WH, Tao C-H, Yam VW-W, Phillips DL. Synthesis of Coumarin-Appended Pyridyl Tricarbonylrhenium (I) 2,2′-Bipyridyl Complexes with Oligoether Spacer and Their Fluorescence Resonance Energy Transfer Studies. Organometallics. 2009;28:1620–1630. [Google Scholar]
- 62.Bergeron RJ, Wiegand J, Weimar WR, McManis JS, Smith RE, Abboud KA. Iron Chelation Promoted by Desazadesferrithiocin Analogs: An Enantioselective Barrier. Chirality. 2003;15:593–599. doi: 10.1002/chir.10248. [DOI] [PubMed] [Google Scholar]
- 63.Hoffmann D, Fuchs TC, Henzler T, Matheis KA, Herget T, Dekant W, Hewitt P, Mally A. Evaluation of a Urinary Kidney Biomarker Panel in Rat Models of Acute and Subchronic Nephrotoxicity. Toxicology. 2010;277:49–58. doi: 10.1016/j.tox.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 64.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney Injury Molecule-1 (KIM-1): A Novel Biomarker for Human Renal Proximal Tubule Injury. Kidney Int. 2002;62:237–244. doi: 10.1046/j.1523-1755.2002.00433.x. [DOI] [PubMed] [Google Scholar]
- 65.Bonventre JV. Kidney Injury Molecule-1 (KIM-1): A Urinary Biomarker and Much More. Nephro. Dial. Transplant. 2009;24:3265–3268. doi: 10.1093/ndt/gfp010. [DOI] [PubMed] [Google Scholar]
- 66.Zhou Y, Vaidya VS, Brown RP, Zhang J, Rosenzweig BA, Thompson KL, Miller TJ, Bonventre JV, Goering PL. Comparison of Kidney Injury Molecule-1 and other Nephrotoxicity Biomarkers in Urine and Kidney Following Acute Exposure to Gentamicin, Mercury, and Chromium. Toxicol. Sci. 2008;101:159–170. doi: 10.1093/toxsci/kfm260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vaidya VS, Ramirez V, Ichimura T, Bobadilla NA, Bonventre JV. Urinary Kidney Injury Molecule-1: A Sensitive Quantitative Biomarker for Early Detection of Kidney Tubular Injury. Am. J. Physiol. Renal Physiol. 2006;290:F517–F529. doi: 10.1152/ajprenal.00291.2005. [DOI] [PubMed] [Google Scholar]
- 68.Bailly V, Zhang Z, Meier W, Cate R, Sanicola M, Bonventre JV. Shedding of Kidney Injury Molecule-1, A Putative Adhesion Protein Involved in Renal Regeneration. J. Biol. Chem. 2002;277:39739–39748. doi: 10.1074/jbc.M200562200. [DOI] [PubMed] [Google Scholar]
- 69.Vaidya VS, Ford GM, Waikar SS, Wang Y, Clement MB, Ramirez V, Glaab WE, Troth SP, Sistare FD, Prozialeck WC, Edwards JR, Bobadilla NA, Mefferd SC, Bonventre JV. A Rapid Urine Test for Early Detection of Kidney Injury. Kidney Int. 2009;76:108–114. doi: 10.1038/ki.2009.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Antech Diagnostics. [accessed March 2012]; http://www.antechdiagnostics.com/ [Google Scholar]
- 71.Bergeron RJ, Streiff RR, Wiegand J, Luchetta G, Creary EA, Peter HH. A Comparison of the Iron-Clearing Properties of 1,2-Dimethyl-3-Hydroxypyrid-4-one, 1,2-Diethyl-3-Hydroxypyrid-4-one, and Deferoxamine. Blood. 1992;79:1882–1890. [PubMed] [Google Scholar]
- 72.Wood JK, Milner PF, Pathak UN. The Metabolism of Iron-Dextran Given As a Total-Dose Infusion to Iron Deficient Jamaican Subjects. Br. J. Hamaetol. 1968;14:119–129. doi: 10.1111/j.1365-2141.1968.tb01481.x. [DOI] [PubMed] [Google Scholar]
- 73.Bergeron RJ, Wiegand J, Brittenham GM. HBED: A Potential Alternative to Deferoxamine for Iron-Chelating Therapy. Blood. 1998;91:1446–1452. [PubMed] [Google Scholar]
- 74.Bergeron RJ, Wiegand J, Wollenweber M, McManis JS, Algee SE, Ratliff-Thompson K. Synthesis and Biological Evaluation of Naphthyldesferrithiocin Iron Chelators. J. Med. Chem. 1996;39:1575–1581. doi: 10.1021/jm9508752. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.